
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA well-defined block copolymer synthesis via living cationic polymerization and nitroxide-mediated polymerization using carboxylic acid-based alkoxyamines as a dual initiator†
Dao
Le
,
Trang N. T.
Phan
*,
Laurent
Autissier
,
Laurence
Charles
and
Didier
Gigmes
Aix-Marseille Université, CNRS, Institut de Chimie Radicalaire UMR 7273, 13397, Marseille, France. E-mail: trang.phan@univ-amu.fr
First published on 25th January 2016
Abstract
A series of carboxylic acid-based alkoxyamines associated with SnBr4, a Lewis acid, have been used as protonic acids of a binary initiating system to control the cationic polymerization of vinyl ether. The living character of the homopolymerization of isobutyl vinyl ether was investigated under various conditions (solvent, amount of catalyst, initiator structure etc.). Among the different studied dual initiators, TEMPO based-alkoxyamine showed high efficiency to lead the polymerization of isobutyl vinyl ether (IBVE) in a controlled manner. The prepared polymers exhibited high TEMPO functionality (>0.85) and low dispersity (1.2) which is essential for the synthesis of well-defined block copolymers thereafter. TEMPO-functionalized PIBVE was used as a macro-initiator controller in the nitroxide-mediated polymerization (NMP) of styrene at 130 °C leading successfully to well-defined block copolymers of poly(isobutyl vinyl ether)-b-polystyrene (PIBVE-b-PS) with a narrow dispersity (1.2).
Introduction
Due to their applications in a wide range of domains such as surface modifiers, compatibilizers, coating materials, antistatic agents, adhesives, drug delivery and information storage systems, block copolymers have always been attracting researchers’ interest.1,2 Usually, block copolymers are prepared by the coupling reaction of homopolymers, sequential addition of monomers in a living polymerization system, chain-end transformation approach or using a dual initiator.2–4 Among them, dual initiator systems show many advantages compared to other approaches because they provide the opportunity to combine mechanistically incompatible monomer units into the same macromolecule.4 Basically, a dual initiator is a component, which contains two different initiation functions, which are selective for a different polymerization mechanism. The use of such an initiator requires however that each initiating group remains stable in both types of polymerization conditions.5Following the discovery of a hydrogen iodide/iodine initiating system by Higashimura and Sawamoto,6 a variety of binary initiating systems consisting of protonic acid/Lewis acid were developed to polymerize vinyl ether monomers in a living/controlled manner.7–13 Hence, various functional block copolymers composed of vinyl ether monomer units were synthesized acting as polymer surfactants,14,15 antibacterial agents,16–21 glycopolymer stimuli-responsive micelles and gels,16,18–21 thermoplastic elastomers,22,23 or optical plastics.24 By combination those binary initiating systems with living/controlled radical polymerization techniques such as reversible addition–fragmentation transfer polymerization (RAFT) or atom transfer radical polymerization (ATRP), block-, statistical- and graft copolymers of vinyl ether monomers with radically polymerizable monomers have been subsequently achieved using the chain-end chemical transformation strategy or the use of a dual initiator.25–38 Sugihara et al.31,32 used mono- and dicarboxylic RAFT agents for the synthesis of AB and ABA block copolymers by the transformation of living cationic polymerization (LCP) into RAFT polymerization. In such a process, the LCP was initiated from the proton generated by a carboxylic RAFT agent to form a poly(vinyl ether)-bearing RAFT group as a counteranion. The latter was then used in the RAFT process for the synthesis of the second block. On the other hand, Kamigaito et al.34,35,38 demonstrated that the thioester bond of a RAFT agent can be activated by Lewis or Brønsted acid to mediate the LCP of a vinyl ether monomer. The ability of generating cationic and radical intermediates of the RAFT agent allows the synthesis of diblock- or multiblock copolymers of vinyl ether and meth(acrylate) or vinyl acetate monomers by subsequent or simultaneous LCP and RAFT polymerization. Du Prez et al.36 reported the synthesis of a dual initiator containing Br and acetal end groups that can be used both sides to prepare AB and ABC block copolymers by ATRP and LCP. As far as we are aware, the combination of protonic acid/Lewis acids/additive-base initiation system and nitroxide-mediated polymerization (NMP) using a multi-functional initiator for the synthesis of block copolymers has not been reported yet. NMP is historically the first and perhaps the easiest controlled/living radical polymerization method to apply. It allows the polymerization, in a controlled manner, a large range of monomers both in homogeneous and heterogeneous media and the synthesis of block copolymers and complex architectures.39
The desire to learn more about the feasibility of the NMP process in combination with a cationic polymerization system, we studied, in this work, the synthesis of poly(vinyl ether)-based block copolymers with a radically polymerizable monomer having a well-defined structure and controlled molecular weight. The synthesis was achieved by LCP and subsequent NMP using carboxylic-based alkoxyamines as a dual initiator, as shown in Scheme 1. It is well-known that due to its relatively low acidity, the carboxylic acid can be part of initiators40,41 or chain transfer agents42 without affecting the CRP process. This functionality could be useful for the post chemical modification of the produced polymers. Moreover, carboxylic acids are one of the most versatile protonic acids that have been employed as the initiating systems for vinyl ether LCP.9,10,13,37,43 Besides the acidity of carboxylic and Lewis acids, the steric crowding of the carboxylate counteranion is also an important factor to control the cationic polymerization. It has been reported that it influences the dynamic equilibrium between active and dormant species, hence, the polymerization behavior. For instance, Hashimoto et al.11–13 studied the steric effects of the counteranion on the cationic polymerization of isobutyl vinyl ether using a series of carboxylic acids (RCOOH) with R = CH3CH2, (CH3)2CH, (CH3)3C, C6H5CH2, etc. They found that all studied carboxylic acids gave living polymers but the steric hindrance of carboxylate counteranions significantly affected the polymerization rate and broadened the molecular weight distributions of the produced polymers. In order to obtain well-defined block copolymers, in this work, we have also studied the influence of the initiator structure on the living/controlled characters of LCP. To get a better insight on how the structure of a bifunctional initiator could affect the living/controlled characters of cationic polymerization, a series of carboxylic acid-based alkoxyamines were prepared and used as initiators of LCP and NMP (Scheme 1). Among them, the alkoxyamine BlocBuilder40 (A1) which enables to control the polymerization of a wide range of monomers such as styrene, acrylate and acrylamide by NMP.39,44,45 However, considering the steric crowding of the carboxylic group of A1, the use of the latter as the initiator in cationic polymerization of vinyl ether could significantly influence the polymerization in terms of reaction kinetics and broadening of the polymer molecular weight distributions. Moreover, the presence of a diethyl phosphate group in A1 may interact as a Lewis base with Lewis acid, thereby decreasing the living/controlled characters.10 Another series of initiators consisting of TEMPO-based alkoxyamines have been also considered. Typically, the alkoxyamines A2 and A3 bearing a benzoic acid derivative and a methyl-propanoic acid respectively have been prepared and investigated.
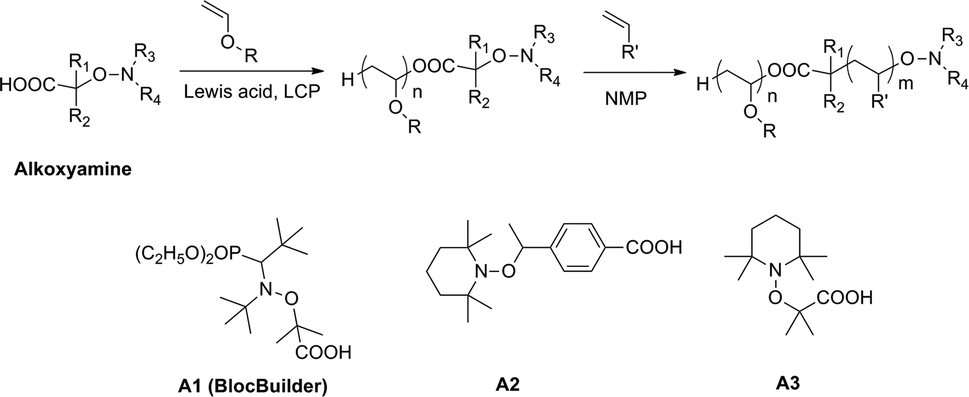 | ||
| Scheme 1 Combination of living cationic polymerization and nitroxide-mediated polymerization using carboxylic-based alkoxyamine and structure of alkoxyamines used in this study. | ||
Experimental section
Materials
Isobutyl vinyl ether (IBVE) (Sigma-Aldrich; 99%) was washed with water and distilled over calcium hydride and stored under nitrogen at −20 °C. Anhydrous toluene (>99.8%), anhydrous dioxane (>99.8%), anhydrous tetrahydrofuran (THF, >99.8%), SnBr4 (99%), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO, 98%), 2-bromo-2-methylpropionic acid (98%), 4-(1-bromoethyl)benzoic acid (97%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%) and other compounds were purchased from Sigma-Aldrich and used without further purification. BlocBuilder® (>99%), an alkoxyamine based on the nitroxide SG1 (N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)]nitroxide) was kindly provided by Arkema (France). All solvents and other reagents were purchased from commercial sources and used as received. Salts (lithium chloride, sodium chloride, ammonium acetate) used as cationizing agents in ESI-MS experiments were purchased from Sigma-Aldrich.Characterization techniques
1H and 31P NMR spectra in CDCl3 were recorded on a Bruker Advance 400 spectrometer. Chemical shifts are given in ppm relative to tetramethylsilane in the case of 1H NMR spectra.Polymer molecular weights and dispersities were determined by size exclusion chromatography (SEC). The used system was an EcoSEC (Tosoh, Japan) equipped with a PL Resipore Precolumn (4.6 × 50 mm, Agilent) and two linear M columns (4.6 × 250 mm, Agilent) with a gel particle diameter of 3 μm. These columns were thermo-stated at 40 °C. Detection was made by using an UV/visible detector operated at λ = 254 nm and a dual flow differential refractive index detector, both from Tosoh, and a viscometer ETA2010 from PSS. Measurements were performed in THF at a flow rate of 0.3 mL min−1. Calibration was based on polystyrene standards from Polymer Laboratories (ranging from 370 g mol−1 to 371![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1).
100 g mol−1).
High-resolution MS mass spectrometry (HR-MS) and MS/MS experiments were performed using a QStar Elite mass spectrometer (AB SCIEX, Concord, ON, Canada) equipped with an electrospray ionization source operated in the positive mode. The capillary voltage was set at +5500 V and the cone voltage at +75 V. In this hybrid instrument, ions were measured using an orthogonal acceleration time-of-flight (oa-TOF) mass analyzer. A quadrupole was used for selection of precursor ions to be further subjected to collision-induced dissociation (CID) in MS/MS experiments. In the MS mode, accurate mass measurements were performed using reference ions from a poly(ethylene glycol) internal standard. The precursor ion was used as the reference for accurate measurements of product ions in the MS/MS mode. In this instrument, air was used as the nebulizing gas (10 psi) while nitrogen was used as the curtain gas (20 psi) as well as the collision gas. Instrument control, data acquisition and data processing of all experiments were achieved using Analyst software (QS 2.0) provided by AB Sciex. Sample solutions were prepared in methanol supplemented with LiCl (1.0 mM), NaCl (1.0 mM) or ammonium acetate (3.0 mM) and were introduced in the ionization source using a syringe pump at a 5 μL min−1 flow rate.
Alkoxyamine synthesis
TEMPO (1 g, 6.4 mmol), 2-bromo-2-methylpropionic acid or 4-(1-bromoethyl)benzoic acid (1.2 eq.), Cu (1.2 eq.), CuBr (1.2 eq.), THF (30 mL) and a stir bar were added into a two-neck round flask. The flask was sealed with a rubber septum and purged with argon for 20 minutes. Degassed PMDETA (2.4 eq.) was added into the glass flask using dry medical syringes. The resulting solution was subsequently stirred at room temperature for 24 h. The reaction mixture was diluted with dichloromethane (DCM) (100 mL) and then washed with 3 × 100 mL of an aqueous disodium ethylenediamine–tetraacetate solution to remove the catalyst. The organic layer was dried over MgSO4, filtered, and evaporated. The alkoxyamines were then purified by chromatography on a silica gel column using a mixture of DCM/ethyl acetate (2/1 v/v) as the eluent.4-(1-((2,2,6,6-Tetramethylpiperidin-1-yl)oxy)ethyl)benzoic acid, A2
White powder, yield: 40%.1H NMR (400 MHz, CDCl3): δ (ppm) = 8.00 (d, J = 8.3 Hz, 2H, ArH), 7.35 (d, J = 8.2 Hz, 2H, ArH), 4.79 (d, J = 6.7 Hz, 1H, CH3CH), 1.42 (d, J = 6.7 Hz, 3H, CH3CH), 1.31–0.57 (m, 18H, CH3–C, CH2–CH2–CH2), (Fig. S1†).
13C NMR (CDCl3): δ (ppm) = 171.46, 152.12, 130.18, 127.71, 126.59, 83.00, 59.78, 40.32, 34.18, 23.59, 20.33, 17.17, (Fig. S2†).
ESI-HRMS. Calc. for [C18H27NO3 + H]+: m/z 306.2064, found: m/z 306.2068.
2-Methyl-2-((2,2,6,6-tetramethylpiperidin-1-yl)oxy)propanoic acid, A3
White powder, yield: 35%.1H NMR (400 MHz, CDCl3): δ (ppm) = 1.71 (s, 6H, CH2), 1.64 (s, 6H, CH3), 1.33 (s, 6H, CH3), 1.27 (s, 6H, CH3), (Fig. S3†).
13C NMR (CDCl3): δ (ppm) = 176.92, 82.34, 62.95, 39.69, 31.17, 28.37, 20.73, 16.22, (Fig. S4†).
ESI-HRMS. Calc. for [C13H25NO3 + H]+: m/z 244.1907, found: m/z 244.1907.
Cationic polymerization procedure
All cationic polymerizations were carried out under an argon atmosphere at 20 °C in a dry round bottom flask. The alkoxyamine was first added into the flask and dried under vacuum (10−7 bar) for 60 minutes at room temperature. Solvent, IBVE and dioxane were then added into the flask using dry syringes under an argon atmosphere. The solution was cooled to 20 °C and the polymerization was initiated by addition of 1 M SnBr4 solution under an argon atmosphere. After the desired time, aliquots were removed and rapidly cooled off below −40 °C. The reaction was quenched with cooled methanol containing a small amount of aqueous ammonia solution (0.1%). Monomer conversion was determined by 1H NMR directly from aliquot solutions. For SEC analysis, polymers were purified by dilution of aliquots with DCM, washed successively with 0.1 M HCl solution, 0.1 M NaOH solution and water. The organic layer was dried over MgSO4, filtered, evaporated and polymer was dried under vacuum.Nitroxide-mediated polymerization procedure
Typically, styrene and PIBVE-based macroalkoxyamine were dissolved in ethylbenzene (60% v/v). The mixture was then loaded in a three-neck flask equipped with a reflux condenser and a magnetic stir bar, purged with argon for 20 min at room temperature to remove oxygen. Polymerization was performed at 130 °C under an argon atmosphere. After desired times, aliquots were removed and rapidly quenched by freezing the reaction mixture in a liquid nitrogen bath. Portions of these aliquots were directly analyzed by 1H NMR for determination of monomer conversion or dried over vacuum for SEC analysis.Results and discussion
Synthesis of carboxylic acid-based alkoxyamines
Alkoxyamines were mainly prepared through two strategies, via nucleophilic substitution or via coupling reaction between an alkyl bromide and a nitroxide radical according to the Atom Transfer Radical Addition (ATRA) process.46,47 Herein, carboxylic acid-based alkoxyamines A2 and A3 have been prepared via coupling reaction (Scheme 2). Halogen transfer between Cu(I) complexes and organic halides generates the alkyl radicals which are rapidly trapped by TEMPO radicals to afford the corresponding alkoxyamines. The chemical structure of the alkoxyamines was confirmed by 1H, 13C NMR and HR-MS (ESI S1–4†).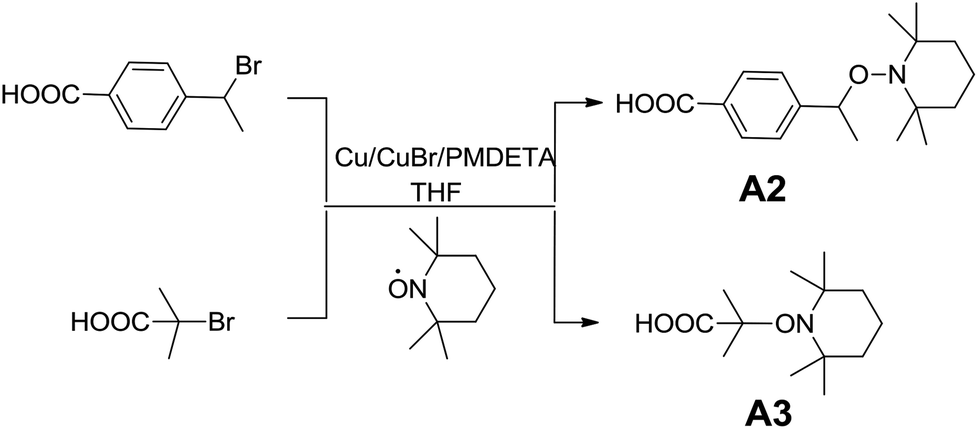 | ||
| Scheme 2 Synthesis of carboxylic acid-based alkoxyamines by ATRA. | ||
Effect of alkoxyamine structure on the cationic polymerization
Previous studies in the literature showed a high efficiency of the carboxylic acid/SnBr4/additive-based initiation system toward the cationic polymerization of vinyl ether monomers.11,32,48 Herein, we used similar conditions to assess the cationic polymerization of IBVE using carboxylic acid-based akoxyamines as the initiator. All polymerizations of IBVE were carried out at 20 °C in the presence of dioxane, a weak base and SnBr4, a Lewis acid. The roles of dioxane are now well known to stabilize the growing carbocations, to adjust the acidity of (free) Lewis acids and to form monomeric Lewis acids.49–51 Consequently, the addition of weak bases allows to get a better polymerization control. Table 1 summarizes the results for various polymerizations carried out under different conditions. In the case of A1, a first test was performed with 0.1 equivalent of Lewis acid with respect to the amount of carboxylic acid-based alkoxyamine; however, no polymerization occurred (entry 1, Table 1). A higher amount of Lewis acid (0.3 eq.) was then used for the polymerization, but it also failed to produce a polymer (entry 2, Table 1). Doubling the amount of SnBr4, i.e. 0.6 eq., allowed the polymerization to occur with a relatively acceptable dispersity (1.46) (entry 3, Table 1). However, after quenching the polymerization with methanol at low temperature (<−40 °C), the dormant polymer with terminated-nitroxide was not obtained. Instead, all polymer chains were terminated by a methoxy group. In similar experiments, polymerization of IBVE using RCOOH with R = CH3, CH3CH2, (CH3)2CH, C6H5CH2 or (C6H5)2CH led to the formation of living polymers with low dispersities (1.1–1.2) and high ester-terminated functionality in the presence of only 0.1 eq. of SnBr4 as Lewis acid.11–13 Such behaviour of A1 is probably related to the presence of diethyl phosphate groups on the nitroxide moiety which enhances the interactions with Lewis acid10 decreasing the catalytic efficiency of the carboxylate counter-anions of the propagating species. To evidence these interactions, 0.6 eq. of SnBr4 was added to the solution of A1. Fig. 1B shows the 31P NMR spectrum of this mixture, in which we observed several peaks shifted towards low frequency including two intense broad peaks at 25 and 22 ppm and three sharp small peaks at 16, 15.5 and 9 ppm respectively. The presence of these peaks is probably the result of the interactions between nitroxide moieties of A1 and SnBr4 and different decomposition reactions caused by SnBr4. Subsequent addition of methanol to the solution of A1 and SnBr4 to neutralize SnBr4 did not allow initial peaks of A1 to be obtained (Fig. 1C). This indicates that the electrophilic interactions were irreversible and/or that the alkoxyamine was decomposed in the presence of Lewis acid.52 Thus, the use of A1/SnBr4 as the initiator allowed performing the cationic polymerization of IBVE, but the produced polymer chains did not contain any nitroxyl end-group, as a consequence of low stability of A1 in the Lewis acid solution.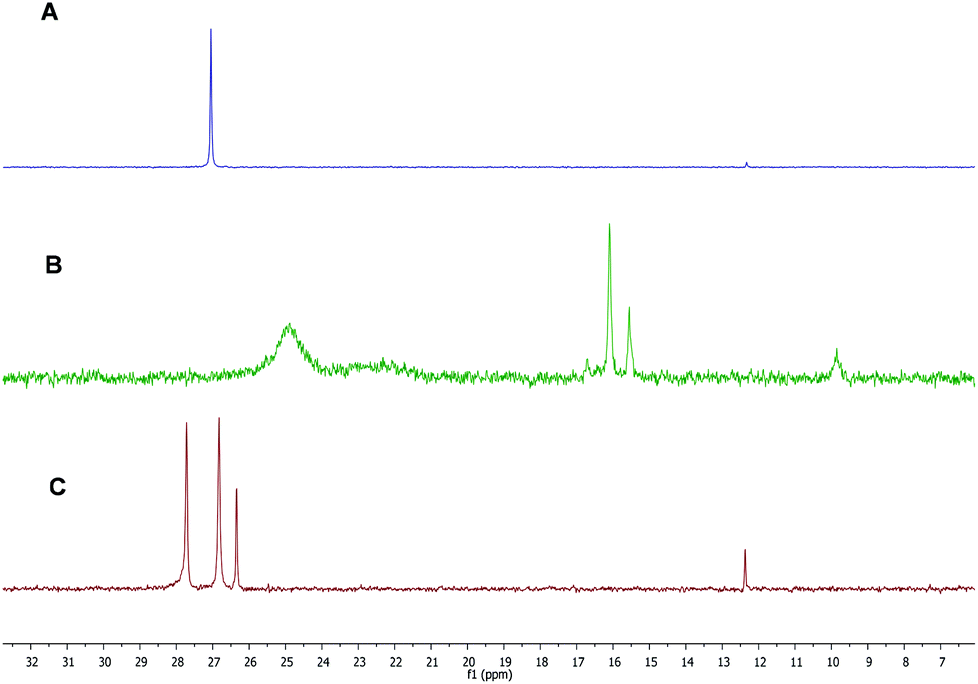 | ||
| Fig. 1 31P NMR in CDCl3 for (A) A1, (B) A1 + 0.6 eq. of SnBr4 and (C) A1 + 0.6 eq. of SnBr4 and quenching by MeOH. | ||
| Entry | Alkoxyamine | Solvent | SnBr4/A ratio | Time (h) | Conversion (%) | M n, calcdb (g mol−1) |
M
n,NMRc (g mol−1) |
M n,SEC (g mol−1) | Dispersity (Đ) | F (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Cationic polymerization conditions: [IBVE]0 = 1.5 M, [A]0 = 0.015M, [dioxane]0 = 0.14 M in toluene at 20 °C. b M n, calcd = Makoxyamine + MIBVE × conversion × ([IBVE]0/[A]0). c Number-average molecular weight determined by 1H NMR. d Calculated based on the peak intensity ratio of the methine proton on the hemiacetal ester to that of the methine proton on the hemiacetal ester and the methine proton produced by methanol termination. | ||||||||||
| 1 | A1 | Toluene | 0.1 | 20 | — | 10384 |
— | — | — | — |
| 2 | 0.3 | 4 | — | 10384 |
— | — | — | — | ||
| 3 | 0.6 | 20 | ∼100 | 10384 |
— | 13600 |
1.46 | 0 | ||
| 4 | A2 | Toluene | 0.1 | — | — | 10306 |
— | — | — | — |
| 5 | THF | 0.1 | 24 | ∼100 | 10306 |
— | 6090 | 1.95 | ∼40 | |
| 6 | A3 | Toluene | 0.1 | 0.75 | 19 | 2144 | 1844 | 3200 | 1.58 | ∼85 |
| 7 | 0.1 | 5 | 31 | 3344 | 3000 | 5100 | 1.46 | ∼82 | ||
| 8 | 0.1 | 24 | ∼100 | 10244 |
9544 | 10500 |
1.30 | ∼80 | ||
The high functionality of final polymers with a nitroxide end-group is an important factor that allows performing the next NMP step efficiently for the synthesis of block copolymers. To investigate the high end-group functionality of polymers, the cationic polymerization has been examined using TEMPO-based alkoxyamines as initiators. First, A2 with an aromatic substituent of a carboxylic group has been employed for the LCP of IBVE. In toluene, the polymerization could not be performed because of the precipitation of the initiator after addition of Lewis acid (entry 4, Table 1). Then a more polar solvent, THF, was used. The precipitation was no longer observed, but the polymerization did not occur under the truly living/controlled conditions as indicated by the broad dispersity (1.9) and low chain-end functionality (40%, entry 5, Table 1). The non-living/controlled polymerization of a protonic acid/Lewis acid system in a polar solvent7 as well as benzoic acid/zinc chloride initiating systems9 was also reported. The reason could be that the propagation rate was much faster than the initiation, which led to a low initiator efficiency.7 We next polymerized IBVE with alkoxyamine A3 with dimethyl substituents of carboxylic acid under the same condition as above, i.e. in toluene, 0.1 eq. of SnBr4 (entries 6–8 Table 1). The polymerization was well-controlled producing polymers with a relatively low dispersity (1.30, entry 8, Table 1) without precipitation in the early period. Furthermore, the nitroxide end-chain functionality of the final polymer was high (∼0.80). Fig. 2 shows the proton NMR of the final polymer with two kinds of terminated polymer structures, acetal and ester.
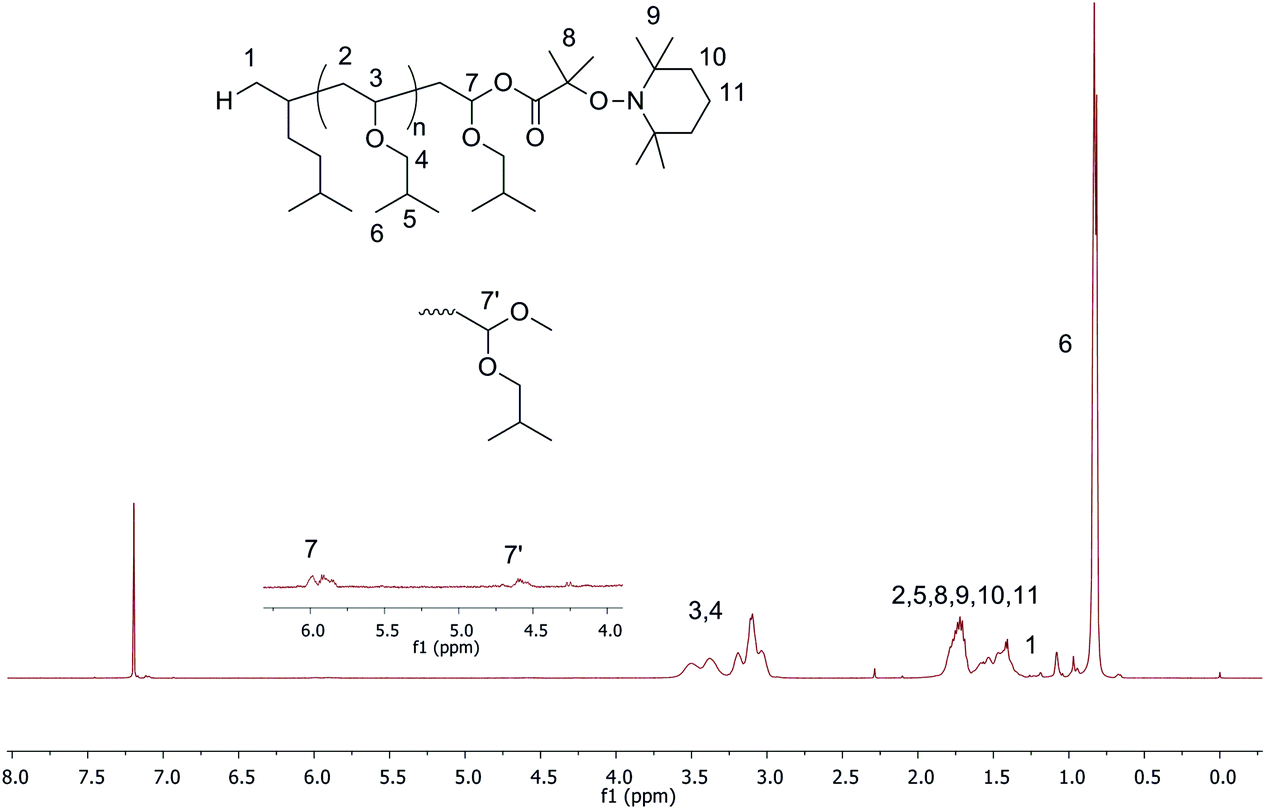 | ||
| Fig. 2 1H NMR in CDCl3 of PIBVE synthesized using A3 as the initiator. The inset shows the estimated chemical structure of the PIBVE-macroalkoxyamine with full peak assignments. | ||
To get further evidence of nitroxide chain end functionality, a PIBVE-TEMPO with a low molar mass (entry 6, Table 1) was analyzed by ESI-MS. ESI is known as a soft ionization technique allowing mass analysis of intact polymer ions with no or little fragmentation. The ESI mass spectrum shown in Fig. 3 was obtained after dilution of the polymer sample in a methanolic solution of LiCl. Experiments conducted with alternative salts in the polymer solution (data not shown) confirmed that all species observed in Fig. 3 were lithiated molecules, all in the +1 charge state. The major distribution of singly-charged molecules (annotated with white stars) spaced by 100.1 m/z, that is, the mass of the IBVE monomeric unit, was assigned to the targeted species with n ranging from 3 to 17, as supported by accurate mass measurements (inset of Fig. 3). This assignment was further validated by MS/MS experiments, where dissociation reactions observed upon collisional activation of these lithiated oligomers provided unambiguous evidence of the ω end-group structure. Indeed, as exemplified with the case of the m/z 650.6 ion in Fig. S5,† the main dissociation reaction consisted of the release of TEMPO as a radical (156 Da), followed by the elimination of the HOOC–(CH3)2C˙ radical (87 Da). The Mn and Mw values of the obtained product was calculated from data of the ESI mass spectrum, using the peak intensity as Ni in the equations Mn = (∑NiMi)/(∑Ni) and Mw = (∑NiMi2)/(∑NiMi), where Ni is the number of chains with mass Mi. The so-obtained Mn and Mw values were 781 g mol−1 and 852 g mol−1, respectively, far below the data obtained by NMR and SEC but consistent with low mass-to-charge ions dominating the mass spectrum, as usually observed in ESI-MS.
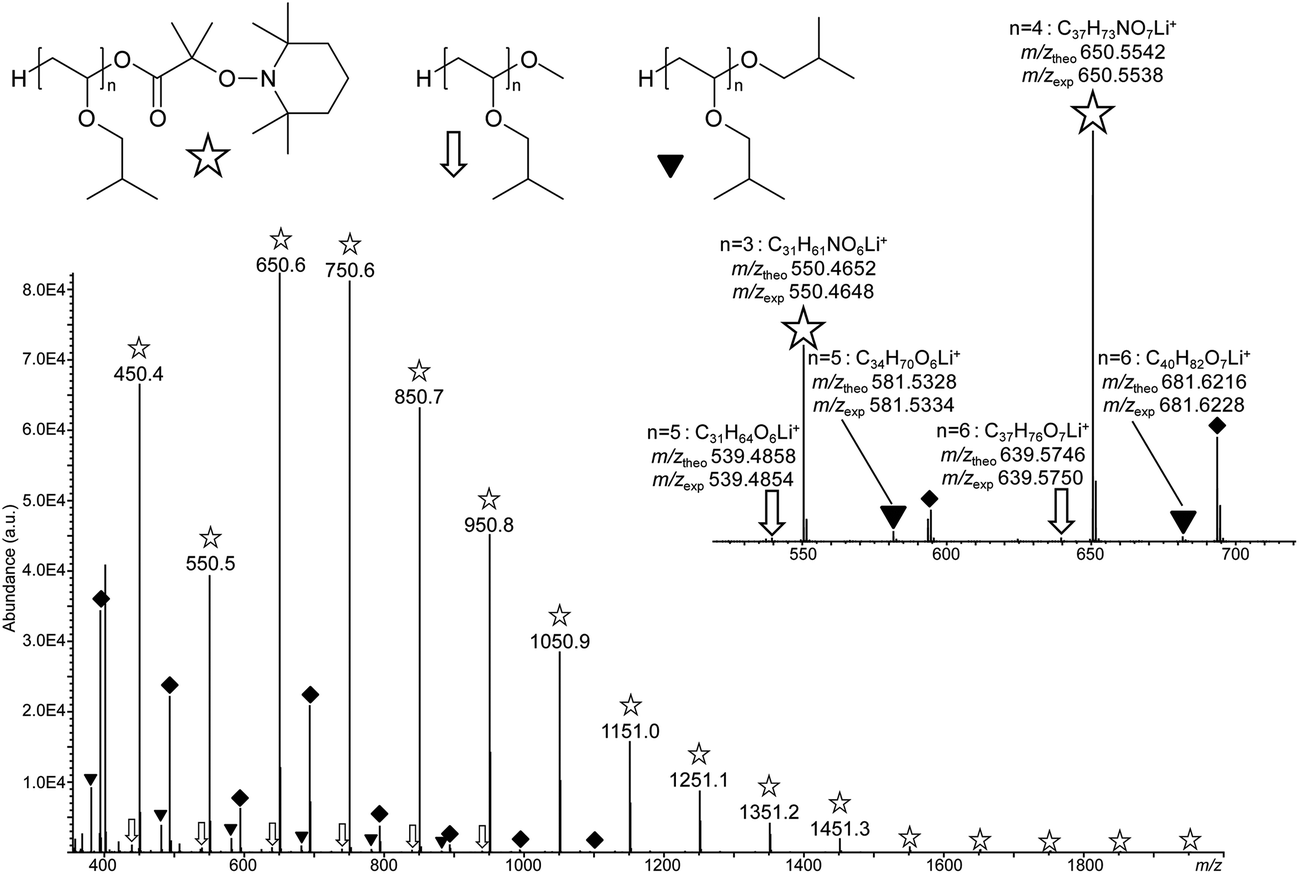 | ||
| Fig. 3 ESI-MS spectrum of PIBVE-macroalkoxyamine (Table 1, entry 6), showing a major distribution of the targeted oligomers (designated with a white star), together with two minor polymer species, respectively annotated with a white arrow and a black triangle, all detected as lithiated molecules. The peak series annotated with a black diamond corresponds to non-covalent complexes formed between the PIBVE-macroalkoxyamine and residual A3. Inset: details of the mass spectrum in the m/z 520–620 range, with elemental composition of the ions as determined from their accurately measured m/z values (m/zexp) and structure proposed for the three observed PIBVE polymers. | ||
Three other minor PIBVE series were also observed in Fig. 3. Signals designated by black diamonds were found to be non-covalent complexes generated in the ESI source between the abundant PIBVE-macroalkoxyamine and either A3 or the 1-mer of the main polymer, as revealed by MS/MS data (Fig. S6†). As a result, this peak series did not correspond to a new PIBVE polymer. In contrast, ions designated with white arrows were found to be lithium adducts of PIBVE polymeric impurities holding a methoxy ω end-group. This assignment was supported by accurate mass data with errors below 2 ppm (inset of Fig. 3), as well as by the loss of a methanol molecule as a diagnostic MS/MS reaction experienced by precursor ions containing a methoxy moiety (Fig. S7†). The presence of this impurity in the PIBVE-TEMPO sample could be explained by the use of methanol as a quenching agent at the end of the polymerization. Finally, a structure was also proposed for the second minor polymeric by-products (annotated with black triangles in Fig. 3). In this PIBVE polymer, the ω end-group would have the same structure as the pendant moiety (–OiBu) of the monomeric unit, as suggested by accurate mass measurements (inset of Fig. 3) and MS/MS data (Fig. S8†). This structure was generated by quenching with isobutanol and has already been evidenced by other authors.53,54 Obviously, the presence of isobutanol was not expected in the initial polymerization solution; its formation was most probably generated via the decomposition of an unstable hemiacetal (IVBE–H2O) formed by addition of a double bond of IBVE with H2O (Fig. S9†).53–56
In order to get more information about the control character of this polymerization, a kinetic study of IBVE cationic polymerization using A3 as the initiator was performed with a molar ratio of [IBVE]/[A3] = 245/1. During the polymerization, the ln([M]o/[M]t) versus time plot (Fig. 4A) clearly showed linearity with monomer conversion indicating a constant concentration of propagating species. Fig. 4B shows the evolution of Mn,SEC and dispersity during the polymerization. Molecular weight, Mn,SEC, determined by SEC in THF with an RI detector, increased linearly with conversion. The Mn,SEC values are somewhat slightly higher than those predicted by the theory. It is noted that the dispersities of polymers were broad in early stage of the polymerization (1.93), and then became much narrower at high conversion (∼1.2). The SEC chromatograms of polymers are shown in the ESI (Fig. S10†). All these results showed that the alkoxyamine A3/SnBr4 initiating system led to the living/controlled cationic polymerization of vinyl ether with high functionality of the nitroxide end-group of polymer, to be further employed as a macro-alkoxyamine for NMP of styrene.
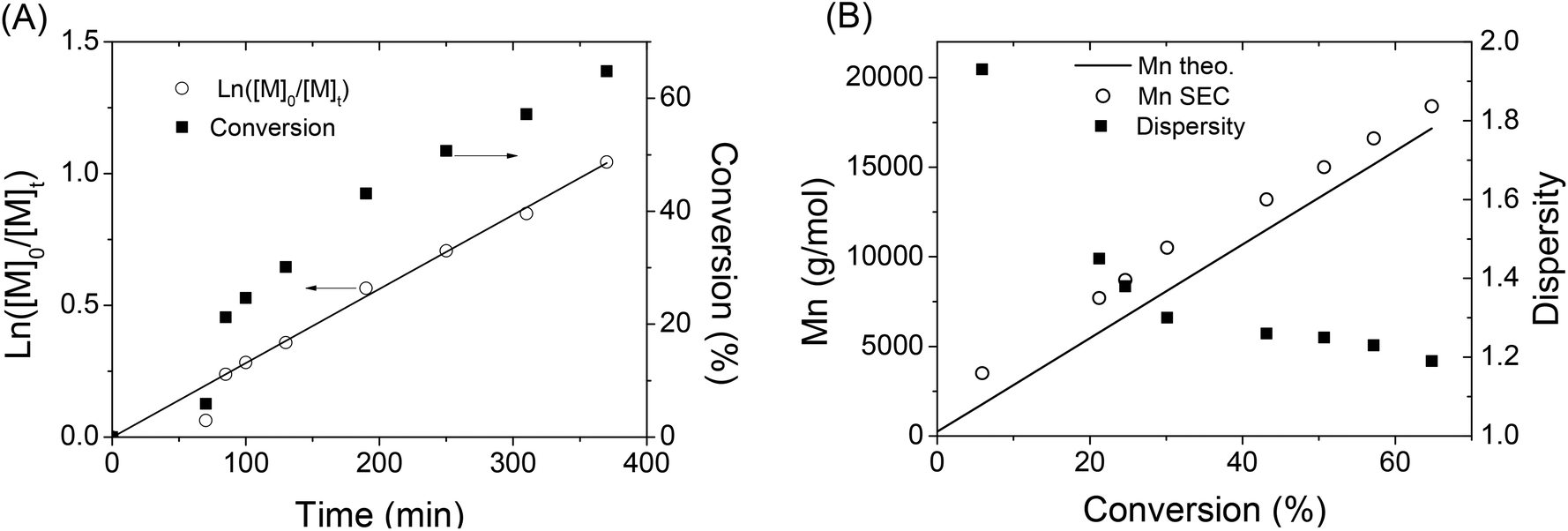 | ||
| Fig. 4 (A) First-order kinetic plot and (B) evolution of molecular weight and dispersity of polymer with conversion during the CLP of IBVE using alkoxyamine A3 as the dual initiator. | ||
Synthesis of block copolymer using PIBVE-TEMPO as macro-alkoxyamine
Once the cationic polymerization was optimized, a TEMPO-functionalized PIBVE (entry 7, Table 1) was used for the NMP of styrene. In order to check the livingness of the NMP, a kinetic study of the polymerization of styrene was performed in ethylbenzene at 130 °C with a molar ratio of [St]/[PIBVE-TEMPO] = 576/1. The pseudo-first-order kinetic plot remained mostly linear indicating an approximately constant radical concentration over the polymerization of St mediated by macro-alkoxyamine (Fig. 5A). For short reaction time (<8 h) or low monomer conversion (<50%), we observed a linearity of molar mass with monomer conversion (Fig. 5B) and these values were very close to the calculated ones. Furthermore, the dispersity decreased with conversion to reach low values (1.20 at 41% conversion). However, there is small tailing in the lower molar mass region in SEC chromatograms (Fig. 6, chromatogram 2), which was due to the low-end functionality of PIBVE produced via living cationic polymerization. Indeed, TEMPO-end functionality of the PIBVE used in this polymerization was about 82% (Table 1, entry 7). For longer polymerization time (20 h) or higher monomer conversion (70%), the small tailing in the region of low molar mass previously observed became larger (Fig. 6, chromatogram 3). This shoulder is superimposed to the chromatogram of PIBVE-macroalkoxyamine (Fig. 6, chromatogram 1) and thus assigned to PIBVE species. The increase of PIBVE homopolymer in the polymerization mixture for a long polymeriszation time at high temperature can be explained by the cleavage of hemiacetal ester group that links PIBVE and PS blocks. The ester bond in the hemiacetal ester group is considered as weak bond and several authors have studied its thermal stability.57–59 They found the dependence of the dissociation temperature of the ester bond with the electron-donating ability of the ester substituent, in the other words, the relative stability of the resulting cation after decomposition.57,58 Endo et al. showed that the thermal stability of bulky polymer attached with three different types of alkyl hemiacetal ester side groups varied as 1° > 2° > 3° with expected order of dissociation temperature of 191, 166 and 140 °C respectively.58 Therefore, the increasing presence of PIBVE homopolymer during the polymerization process at 130 °C was justified. The labile character of this hemiacetal ester group could be advantageously used in stimuli-responsive systems. Aoshima et al. demonstrated a complete and rapid degradation of hemiacetal group under acidic conditions.21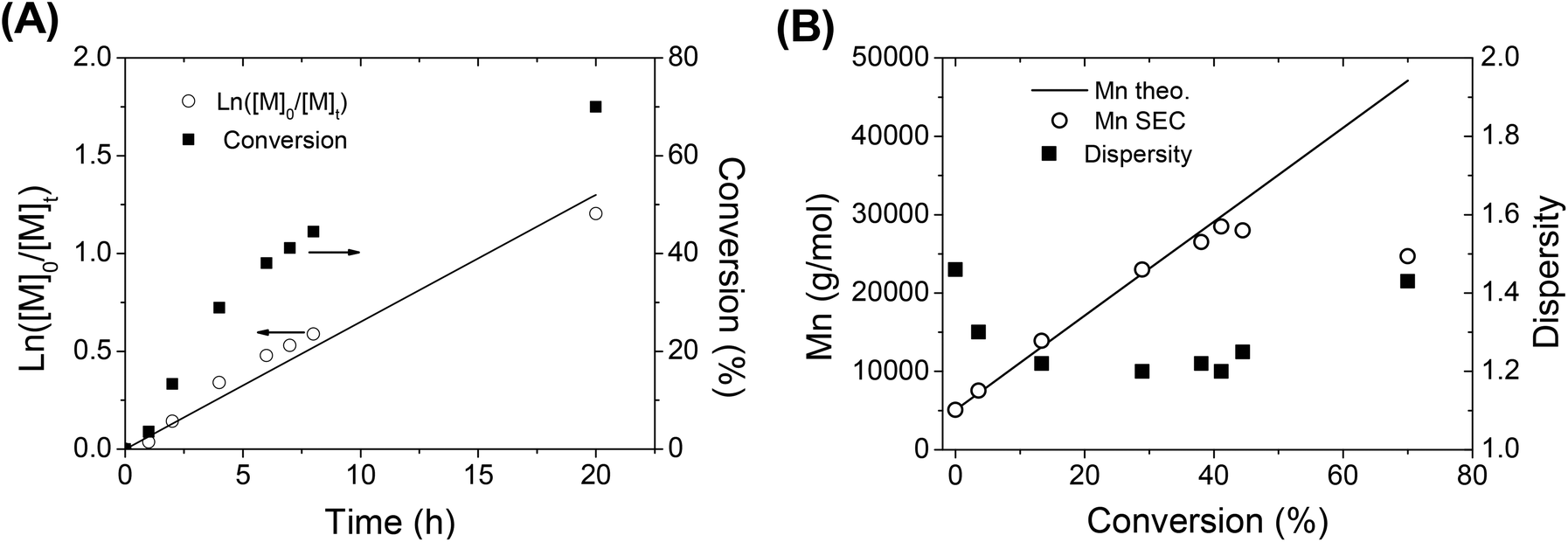 | ||
| Fig. 5 (A) First-order kinetics plot and (B) evolution of molecular weight and dispersity of PIBVE-b-PS copolymer with conversion during the NMP of styrene using PIBVE-macroalkoxyamine as the initiator. | ||
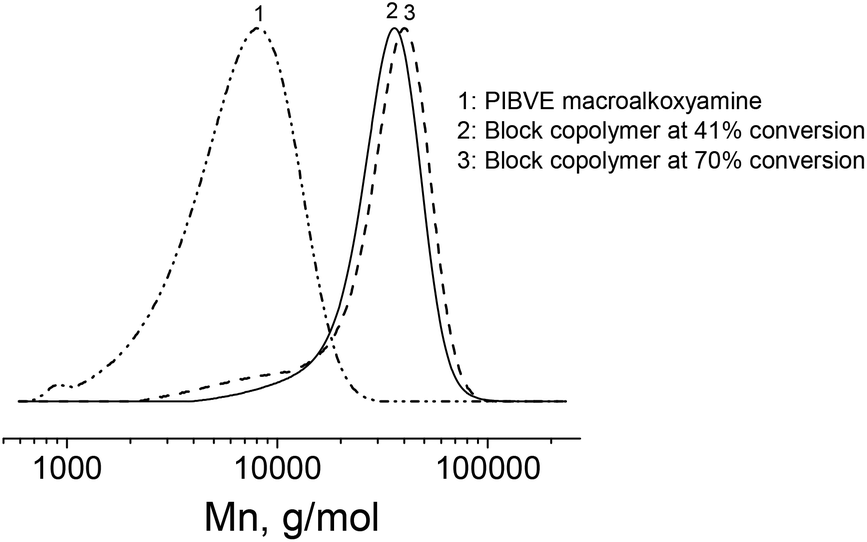 | ||
| Fig. 6 SEC chromatograms of (1) PIBVE macroalkoxyamine, PS-b-PIBVE copolymers at (2) 41% conversion and (3) 70% conversion of St. | ||
As a typical example, the structure of the purified block copolymer at 13.3% conversion was determined by 1H NMR spectroscopy as shown in Fig. 7. The copolymer was first purified by precipitation in cool ethanol to eliminate all PIBVE homopolymers. The composition of block copolymer determined by NMR, i.e. St/IBVE = 78/27.5, was in good agreement with the monomer feed ratio of St to PIBVE at this conversion, which was calculated by comparing the characteristic peak intensities of the methylene and methine protons of PIBVE and of aromatic proton of PS. This result reinforces that almost PIBVE macroalkoxyamine mediated the polymerization of St to obtain block copolymer.
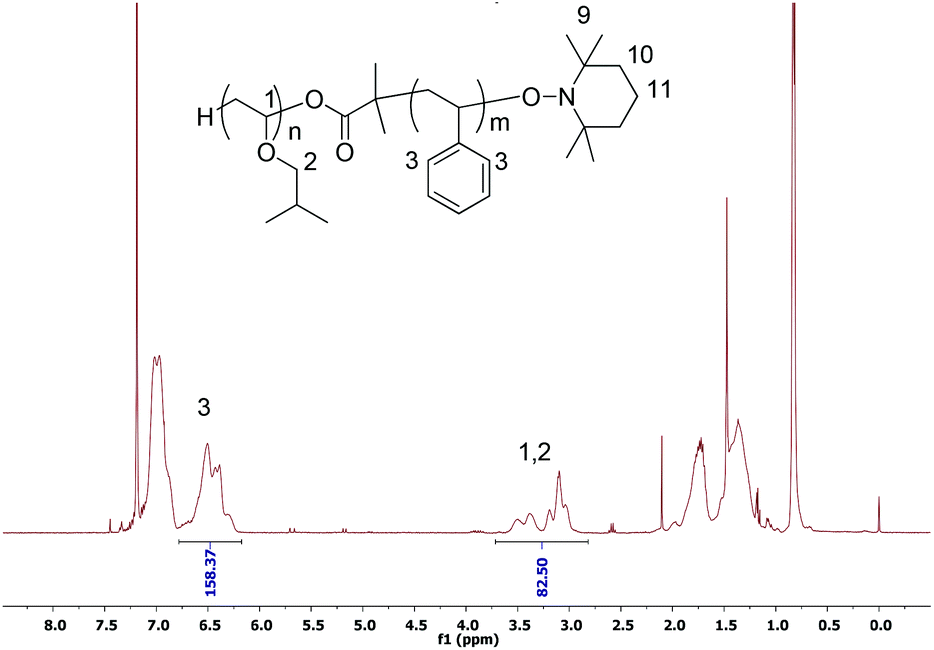 | ||
| Fig. 7 1H NMR spectrum in CDCl3 of purified PIBVE-b-PS. | ||
Conclusion
Several carboxylic acid-functionalized alkoxyamines were successfully synthesized by atom transfer radical addition reaction and used as a dual initiator for the preparation of block copolymers composed of PIBVE and PS via the combination of living cationic polymerization and nitroxide-mediated polymerization routes. The results showed that the structure of the dual initiator has a strong influence on the living/controlled characters of LCP. We demonstrated that the key to succeed cationic polymerization was the use of a suitable alkoxyamine, which has high solubility in an apolar solvent, and the latter should not cause interactions between a nitroxide moiety and Lewis acid. After the cationic polymerization of IBVE, the polymerization was quenched and the alkoxyamine group as a counteranion was concurrently recovered with high yield. The chain extension of the TEMPO-terminated PIBVE by NMP of styrene led to a well-defined diblock copolymer. Although both IBVE and styrene can be polymerized by using LCP techniques as well as the preparation of their block copolymers by sequential monomer addition have been reported,60 we are confident that the use of a carboxylic-based alkoxyamine as a dual initiator can be applied in the synthesis of block copolymers with a well-defined structure, which cannot be polymerized by using the LCP method such as styrene derivatives (sodium 4-vinylbenzenesulfonate, 4-nitrostyrene or 4-aminostyrene, etc.).Acknowledgements
This work was financially supported by Aix-Marseille University and CNRS.References
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898 CrossRef CAS PubMed.
- N. Hadjichristidis, S. Pispas and G. Floudas, Block copolymers: synthetic strategies, physical properties, and applications, John Wiley & Sons, 2003 Search PubMed.
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133 CrossRef CAS.
- K. V. Bernaerts and F. Du Prez, Prog. Polym. Sci., 2006, 31, 671 CrossRef CAS.
- C. J. Hawker, J. L. Hedrick, E. E. Malmström, M. Trollsås, D. Mecerreyes, G. Moineau, P. Dubois and R. Jérôme, Macromolecules, 1998, 31, 213 CrossRef CAS.
- T. Higashimura, M. Miyamoto and M. Sawamoto, Macromolecules, 1985, 18, 611 CrossRef CAS.
- H. Bouchekif, A. Sulhami, R. Alghamdi, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2015, 6, 1236 RSC.
- M. Sawamoto, Prog. Polym. Sci., 1991, 16, 111 CrossRef CAS.
- M. Kamigaito, K. Yamaoka, M. Sawamoto and T. Higashimura, Macromolecules, 1992, 25, 6400 CrossRef CAS.
- M. Kamigaito, M. Sawamoto and T. Higashimura, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2987 CrossRef CAS.
- T. Hashimoto, T. Iwata, N. Uchiyama and T. Kodaira, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2923 CrossRef CAS.
- T. Hashimoto, T. Iwata, A. Minami and T. Kodaira, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 3173 CrossRef CAS.
- T. Hashimoto, T. Takahashi and T. Kodaira, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 781 CrossRef CAS.
- M. Minoda, M. Sawamoto and T. Higashimura, Macromolecules, 1987, 20, 2045 CrossRef CAS.
- S. Zhou, Y. Oda, A. Shimojima, T. Okubo, S. Aoshima and A. Sugawara-Narutaki, Polym. J., 2015, 47, 128 CrossRef CAS.
- S. Sugihara, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2601 CrossRef CAS.
- Y. Oda, S. Kanaoka, T. Sato, S. Aoshima and K. Kuroda, Biomacromolecules, 2011, 12, 3581 CrossRef CAS PubMed.
- S. Sugihara, K. Hashimoto, S. Okabe, M. Shibayama, S. Kanaoka and S. Aoshima, Macromolecules, 2004, 37, 336 CrossRef CAS.
- S. Sugihara, S. Kanaoka and S. Aoshima, Macromolecules, 2005, 38, 1919 CrossRef CAS.
- S. Sugihara, S. Ito, S. Irie and I. Ikeda, Macromolecules, 2010, 43, 1753 CrossRef CAS.
- S. Aoshima, Y. Oda, S. Matsumoto, Y. Shinke, A. Kanazawa and S. Kanaoka, ACS Macro Lett., 2013, 3, 80 CrossRef.
- J. Pretula, K. Kaluzynski, B. Wisniewski, R. Szymanski, T. Loontjens and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 830 CrossRef CAS.
- T. Hashimoto, T. Imaeda, S. Irie, M. Urushisaki and T. Sakaguchi, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1114 CrossRef CAS.
- T. Namikoshi, T. Hashimoto, Y. Makino, T. Imaeda, M. Urushisaki and T. Sakaguchi, Polym. Bull., 2014, 71, 1389 CrossRef CAS.
- E. Mishima, T. Yamada, H. Watanabe and S. Yamago, Chem. – Asian J., 2011, 6, 445 CrossRef CAS PubMed.
- E. Mishima and S. Yamago, Macromol. Rapid Commun., 2011, 32, 893 CrossRef CAS PubMed.
- A. H. Ma'Radzi, S. Sugihara, S. Miura, N. Konegawa and Y. Maeda, Polymer, 2014, 55, 1920 CrossRef.
- K. Yamada, M. Miyazaki, K. Ohno, T. Fukuda and M. Minoda, Macromolecules, 1999, 32, 290 CrossRef CAS.
- O. Nuyken, H. Kröner and S. Aechtner, Makromol. Chem., Rapid Commun., 1988, 9, 671 CrossRef CAS.
- J. Lu, H. Liang, A. Li and Q. Cheng, Eur. Polym. J., 2004, 40, 397 CrossRef CAS.
- S. Sugihara, K. Yamashita, K. Matsuzuka, I. Ikeda and Y. Maeda, Macromolecules, 2011, 45, 794 CrossRef.
- S. Sugihara, K. Iwata, S. Miura, A. H. Ma'Radzi and Y. Maeda, Polymer, 2013, 54, 1043 CrossRef CAS.
- S. Sugihara, N. Konegawa and Y. Maeda, Macromolecules, 2015, 48, 5120 CrossRef CAS.
- H. Aoshima, M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2014, 53, 10830 CrossRef.
- M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2015, 54, 1924 CrossRef CAS PubMed.
- K. V. Bernaerts and F. E. Du Prez, Polymer, 2005, 46, 8469 CrossRef CAS.
- M. Ouchi, A. Konishi, M. Takenaka and M. Sawamoto, Polym. Chem., 2012, 3, 2193 RSC.
- S. Kumagai, K. Nagai, K. Satoh and M. Kamigaito, Macromolecules, 2010, 43, 7523 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63 CrossRef CAS.
- J.-L. Couturier, O. Guerret, D. Bertin, D. Gigmes, S. Marque, P. Tordo, F. Chauvin and P. E. Dufils, WO2004/014926, 2004 Search PubMed.
- X. Zhang and K. Matyjaszewski, Macromolecules, 1999, 32, 7349 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754 CrossRef CAS.
- Y. Kishimoto, S. Aoshima and T. Higashimura, Macromolecules, 1989, 22, 3877 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485 CrossRef CAS.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34 CrossRef CAS.
- K. Matyjaszewski, Macromol. Symp., 1996, 111, 47 CrossRef CAS.
- K. Matyjaszewski, B. E. Woodworth, X. Zhang, S. Gaynor and Z. Metzner, Macromolecules, 1998, 31, 5955 CrossRef CAS.
- H. Ito and D. C. Miller, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1468 CrossRef CAS.
- A. Kanazawa, Y. Hirabaru, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5795 CrossRef CAS.
- S. Aoshima, T. Yoshida, A. Kanazawa and S. Kanaoka, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1801 CrossRef CAS.
- S. Aoshima and T. Higashimura, Macromolecules, 1989, 22, 1009 CrossRef CAS.
- N. Chagneux, T. Trimaille, M. Rollet, E. Beaudoin, P. Gerard, D. Bertin and D. Gigmes, Macromolecules, 2009, 42, 9435 CrossRef CAS.
- H. Katayama, M. Kamigaito and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1249 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3702 CrossRef CAS.
- Q. Huang, Y. Wu and J. Dan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 546 CrossRef CAS.
- A. V. Radchenko, S. V. Kostjuk and F. Ganachaud, Polym. Chem., 2013, 4, 1883 RSC.
- H. Otsuka and T. Endo, Macromolecules, 1999, 32, 9059 CrossRef CAS.
- B. Barkakaty, K. Matsumoto and T. Endo, Macromolecules, 2009, 42, 9481 CrossRef CAS.
- M. Shirai, K. Mitsukura and H. Okamura, Chem. Mater., 2008, 20, 1971 CrossRef CAS.
- T. Ohmura, M. Sawamoto and T. Higashimura, Macromolecules, 1994, 27, 3714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The 1H, and 13C spectra of A2 and A3 alkoxyamines, SEC chromatograms and MS spectra of PIBVE-macroalkoxyamine. See DOI: 10.1039/c5py01934f |
| This journal is © The Royal Society of Chemistry 2016 |