
Pentablock star shaped polymers in less than 90 minutes via aqueous SET-LRP†
R.
Aksakal
a,
M.
Resmini
*b and
C. R.
Becer
*a
aSchool of Biological and Chemical Sciences, Queen Mary University of London, London E1 4NS, UK. E-mail: r.becer@qmul.ac.uk
bSchool of Engineering and Materials Science, Queen Mary University of London, London E1 4NS, UK. E-mail: m.resmini@qmul.ac.uk
First published on 19th October 2015
Abstract
The synthesis of multi-block star-shaped copolymers via SET-LRP in aqueous solution has been reported for the first time. This aqueous polymerization technique allows rapid and direct access to acrylamide based star-shaped polymers. It is possible to synthesize an A-B-A-B-C penta-block copolymer in <90 minutes in total. To achieve this, we have investigated a water-soluble 3-arm initiator based on a glycerol structure for the first time. Using N-isopropylacrylamide 3-arm star-shaped polymers were prepared with DP = 60 to 240 with full conversions in <30 minutes with polydispersities <1.11. The scope of the reaction was demonstrated by synthesizing diblock copolymers using a combination of NIPAM, DMA and HEAm in different ratios. In addition a sequence controlled 3-arm pentablock copolymer has been obtained with excellent control over molecular weight distribution (PDI < 1.14) as evidenced by GPC, 1H NMR, and MALDI-TOF MS.
Synthesis of structurally complex precision polymers has been an essential requirement in order to mimic successfully biomacromolecules and biological systems.1,2 More recently the main focus has moved to the regulation of the building block sequence and folding in linear or branched polymers, to obtain control over their secondary structure, all of which allow researchers to design and tailor unique features to enable biologically inspired functionalities as in DNA, RNA, peptides or proteins.3,4 Star shaped polymers exhibit distinct physical and biological properties that can be used in a broad range of applications including drug delivery,5,6 lectin recognition,7,8 treatment of cancer,9 as well as photonics.10
Star-shaped polymers can be prepared by arm-first, coupling-onto and core-first approaches. In the arm-first approach, linear polymer chains are synthesized before crosslinking with a difunctional monomer. However, this technique allows the formation of multi-arm star-shaped polymers with large number of arms (>100) and the conversion to core cross-linked stars (CCS) is often incomplete, which results in a broad distribution of number of arms per molecule.11 Additional purification steps are usually required to separate unreacted chains from the desired end product.12 Similarly, the coupling-onto approach also requires pre-synthesis of arms and then conjugation to a multifunctional core via efficient coupling reactions (e.g. “click” reaction).13,14 Although it is possible to obtain stars with pre-defined number of arms, this approach requires complete modification of the active chain end groups prior to conjugation to the core. On the other hand, in the core-first approach a core molecule is functionalized into a multifunctional initiator, where the arms are grown directly from. This approach is not only highly efficient, reaching nearly always quantitative monomer conversion, but also the number of arms is predefined, which allows control over the repeating units per arm and total size. In addition, core-first stars retaining high chain-end fidelity can be further used to polymerize other monomers to obtain star block copolymers, therefore introducing more complexity to the polymer.
Although all three methodologies are well established, numerous studies were carried out to optimize the synthesis of star-shaped polymers using controlled radical polymerization techniques. Due to the highly reactive nature of radicals, the main challenge has been to avoid undesired side reactions such as star–star coupling or termination of growing arms.15,16 The key approach has been to reduce the number of active radicals present in the solution at any time to suppress the termination reactions. However, this resulted in extended reaction times and/or highly diluted reactions solutions. Yet the problem to use highly polar organic solvents, such as DMSO, together with multistep purifications in order to obtain multi block star copolymers, remains unsolved.
Current methods published in the literature mainly consist of either homo-/diblock copolymers with short reaction times or multi-block star copolymers with reaction times up to days, even with blocks consisting of as few as 5 repeating units (DPn).17,18 For this reason there has been a drive towards developing more efficient protocols with fewer steps to obtain high DPn multi-block star shaped copolymers.
In recent years, aqueous Cu(0)-mediated single electron transfer living radical polymerization (SET-LRP) has gained great popularity, as nearly 100% chain end fidelity is retained at full conversion.19 Utilizing water as the reaction solvent has a great advantage not only because of water being an environmentally friendly and cheap solvent, but also to provide fast polymerization kinetics for acrylamides.20 Therefore, aqueous SET-LRP has been investigated to overcome the above mentioned drawbacks associated with the core first approach and even take this approach to the next level by preparing sequence controlled polymers. We report here our synthesis of a hydrophilic star-core with 3 symmetrical initiating sites and utilized it to obtain well-defined core-first multi-block stars (Scheme 1) in less than 90 minutes with well defined monomer sequences.
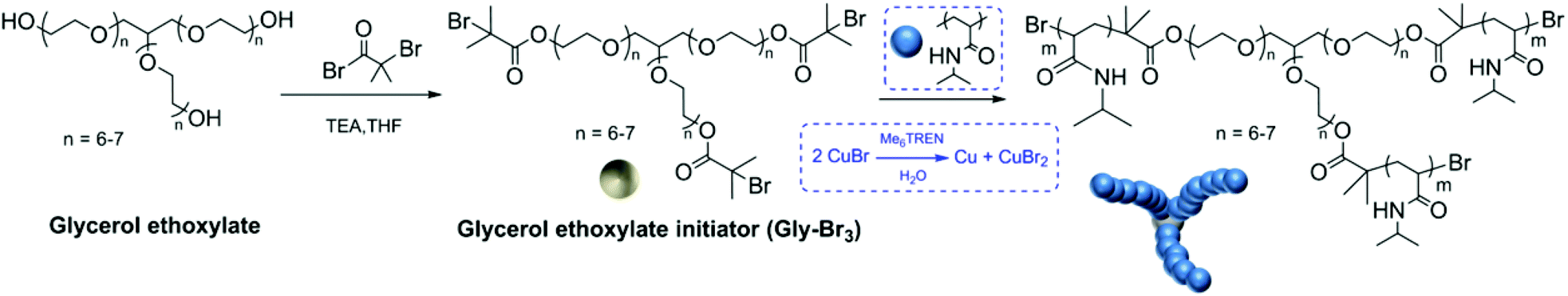 | ||
| Scheme 1 Schematic representation of the 3-arm initiator synthesis (Gly-Br3) and the polymerisation of NIPAM using CuBr. | ||
Recently, SET-LRP in acetone has been performed by Zhu et al. to synthesize pH-responsive A2B2 and fluorescent A3B type mikto-arm stars using a combination of SET-LRP and RAFT.21 Similarly, the combination of SET-LRP and NMP to polymerize star shaped acrylates was reported by Save et al.22 Moreover, Whittaker et al. reported the synthesis of low molecular weight 4-arm poly(methyl acrylate) and also demonstrated high chain-end fidelities, however with broad PDi, which was attributed to star–star coupling, yet the degree of coupling was not fully investigated. The technique was optimized later, when a 5-arm glucose core was used to polymerize different acrylates, with each individual block polymerising for 24 h.17,23 Haddleton et al. reported successful synthesis of 8-arm acrylate stars with high molecular weight and narrow molecular weight distribution.24 Furthermore, they observed phase separation of the polymer from the reaction media, which was deemed to be beneficial in reducing star–star coupling in certain cases.
In a more recent study, Qiao et al. demonstrated the synthesis of core crosslinked star polymers in a one-pot two-step reaction, where MA was polymerized and crosslinked in a second step with ethylene glycol diacrylate in DMSO.25 In a later work, they have shown the synthesis of stimuli-responsive heteroarm star polymers by the arm-first approach, where a poly(ethylene glycol) methyl ether (PEG) macro initiator was used to polymerize N-isopropyl acrylamide (NIPAM) and 2-hydroxyethyl acrylate (HEA).12
In order to investigate the ideal reaction conditions, SET-LRP of NIPAM was initiated using a water-soluble 3-arm initiator. The amount of CuBr/Me6TREN amount was optimized. (ESI,†Table 2) The reactions were carried out either at 25 °C or at 0 °C as the temperature has a critical effect on disproportionation of CuBr.26 Glycerol ethoxylate was functionalized into a 3-arm ATRP initiator (Gly-Br3) and employed as the water-soluble core for the first time. Initially, homopolymerization of NIPAM was carried out using the ratio of [NIPAM]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Gly-Br3]:[CuBr]:[Me6TREN] = 60:1:0.8:0.4, which are established for high DPn polymerizations using monofunctional initiators.19 Typically, the required amount of CuBr was weighed out into a Schlenk tube fitted with a magnetic stirrer bar, which was subsequently sealed and deoxygenated with argon. Then, a degassed solution of Me6TREN in H2O was carefully transferred via degassed syringe to the Schlenk tube. The resulting mixture was allowed to fully disproportionate under vigorous stirring. Meanwhile, a mixture of monomer and Gly-Br3 was degassed and carefully transferred to the Schlenk tube to initiate the polymerization. The low conversions obtained were attributed to insufficient amount of in situ generated Cu(0), thus the CuBr concentration was increased and the Me6TREN concentration adjusted accordingly. We have found the ratio of [I]:[CuBr]:[Me6TREN] = 1:1.8:1.2 to be the most suitable for the SET-LRP of NIPAM due to very low polydispersity and theoretical number average molecular weights (Mn,theo) being close to Mn,GPC. Although no difference was observed when carrying out the reactions at 25 °C or at 0 °C using this ratio, all further reactions were carried out at 0 °C to avoid possible termination events by the hydrolysis of the terminal bromine during further chain extensions. (ESI,†Table 2) To demonstrate the applicability of these ratios to a range of repeating units we have targeted stars with relatively high molecular weights (Table 1, P1–P7, DPn = 60–240). Even at DPn = 240, quantitative conversion was reached in less than 30 minutes.
:[Gly-Br3]:[CuBr]:[Me6TREN] = 60:1:0.8:0.4, which are established for high DPn polymerizations using monofunctional initiators.19 Typically, the required amount of CuBr was weighed out into a Schlenk tube fitted with a magnetic stirrer bar, which was subsequently sealed and deoxygenated with argon. Then, a degassed solution of Me6TREN in H2O was carefully transferred via degassed syringe to the Schlenk tube. The resulting mixture was allowed to fully disproportionate under vigorous stirring. Meanwhile, a mixture of monomer and Gly-Br3 was degassed and carefully transferred to the Schlenk tube to initiate the polymerization. The low conversions obtained were attributed to insufficient amount of in situ generated Cu(0), thus the CuBr concentration was increased and the Me6TREN concentration adjusted accordingly. We have found the ratio of [I]:[CuBr]:[Me6TREN] = 1:1.8:1.2 to be the most suitable for the SET-LRP of NIPAM due to very low polydispersity and theoretical number average molecular weights (Mn,theo) being close to Mn,GPC. Although no difference was observed when carrying out the reactions at 25 °C or at 0 °C using this ratio, all further reactions were carried out at 0 °C to avoid possible termination events by the hydrolysis of the terminal bromine during further chain extensions. (ESI,†Table 2) To demonstrate the applicability of these ratios to a range of repeating units we have targeted stars with relatively high molecular weights (Table 1, P1–P7, DPn = 60–240). Even at DPn = 240, quantitative conversion was reached in less than 30 minutes.
| Entry | DPn | M n,theo [g mol−1] |
M
n,GPCa [g mol−1] |
PDia | ρ [%] |
T
cpc [°C] |
|---|---|---|---|---|---|---|
| a DMF eluent, PMMA standards. b Conversion (ρ) measured by 1H NMR. c Cloud point calculated from the 50% transmittance point in the heating cycle using 1 mg mL−1 sample concentration. | ||||||
| P1 | 60 | 8200 | 9000 | 1.11 | 100 | 49 |
| P2 | 90 | 11600 |
11700 |
1.09 | 100 | 46 |
| P3 | 120 | 15000 |
14400 |
1.08 | 100 | 43 |
| P4 | 150 | 18400 |
16600 |
1.07 | 100 | 40 |
| P5 | 180 | 21800 |
19900 |
1.11 | 100 | 40 |
| P6 | 210 | 25200 |
21400 |
1.09 | 100 | 39 |
| P7 | 240 | 28600 |
25100 |
1.10 | 100 | 39 |
Contrary to what has been reported in the literature, we were able to maintain good control over the molecular weight distribution (Đ < 1.11) without the need of DPn dependent adjustment of the ratios (Fig. 1).27 In addition, all isolated polymers were assessed via GPC and the differences beween measured and theoretical molar mass were becoming more pronounced. This is most probably due to the difference in the linear hydrodynamic volume increase with higher molar mass between used PMMA linear calibration standards and PNIPAM.
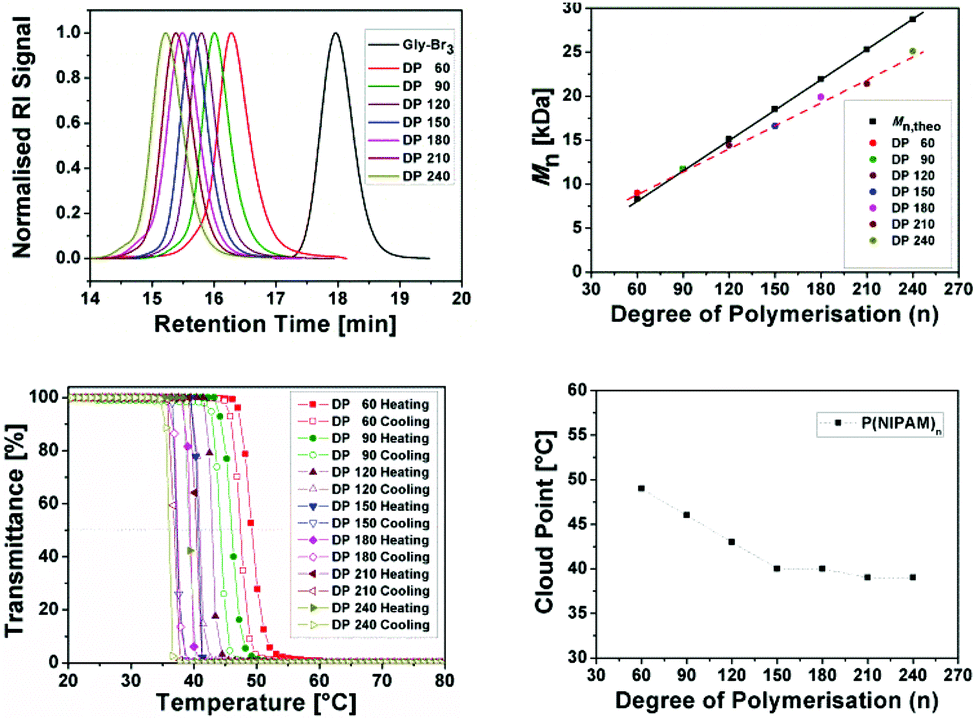 | ||
| Fig. 1 General overview of the obtained results for the homopolymers of NIPAM (P1–P7). GPC traces of 3-arm star shaped poly(NIPAM) with various DP (top left), comparison of Mn,theo and Mn,GPC (top right), turbidity curves of aqueous solutions of P1–P7 in water (c = 1 mg mL−1) (bottom left) and dependence of the cloud point temperature (Tcp) of P1–P7 on the degree of polymerization (bottom right). | ||
Furthermore, we have measured the cloud points (Tcp) of 3-arm star-shaped PNIPAM with different DPnvia UV/VIS spectroscopy. No hysteresis was observed for P1–P7 and all polymer samples redissolved fully upon cooling. The cloud points were found to be in the range of 39 to 49 °C, decreasing with increasing chain lengths. The LCST of PNIPAM homopolymer is usually reported to be around 32 °C and the difference of 8–10 °C could be due to the effect of hydrophilic core, which has around 20 ethyleneglycol units.28,29 For higher molecular weight stars (DPn ≥ 150), all cloud points observed were at elevated human body temperatures (e.g. fever temperature 38–42 °C). It appears that the effect of the hydrophilic core is less significant in cloud point depression for high DPn stars compared to smaller stars. This being another advantage of using a hydrophilic core, these PNIPAM polymers can also be used as an amphiphilic star polymer with a hydrophilic core.
To investigate the efficiency of this approach for the preparation of multi-block copolymers, the first block PNIPAM60 was chain extended with twice as much acrylamide, equivalent to DP 120. (Table S5, see ESI†) Full conversion was reached within less than 30 minutes with excellent control for PNIPAM60-PNIPAM120 (Fig. 2, P8, Mn,GPC = 24200 g mol−1, PDI = 1.11). As predicted, no unwanted termination reactions were observed on the low or high molecular weight region of the GPC spectrum, indicating very high retention of the active end groups and no occurrence of star–star coupling. To assess the limits of applicability of this approach, two more chain extensions were carried out with aliquots of N,N-dimethyl acrylamide (PDMA120) (Fig. 2, P9, Mn,GPC = 24900 g mol−1, PDI = 1.14) and N-hydroxyethyl acrylamide (HEAm120), where in both cases full conversion was reached within 30 minutes with excellent control over the diblock star formation. (Fig. 2, P10, Mn,GPC = 23700 g mol−1, PDI = 1.14).
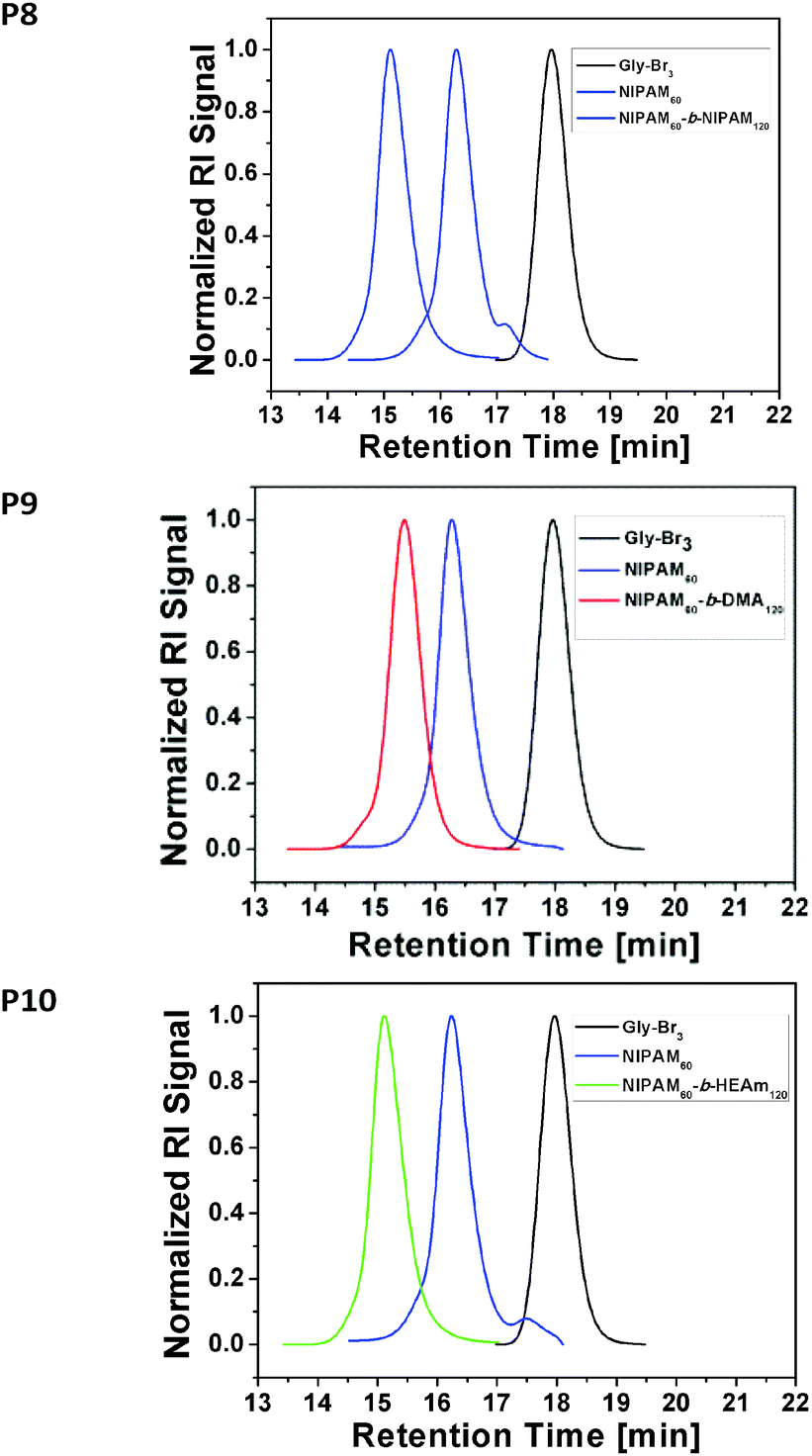 | ||
| Fig. 2 GPC traces of the 3-arm star initiator, NIPAM block and respective HEAm or DMA blocks (P8–P10) with full conversion according to 1H NMR (Fig. S3–S5). | ||
Based on the encouraging results obtained for diblock copolymerizations, we have performed multi-block copolymerizations in order to investigate the limits of this system. For this purpose, aliquots of NIPAM, DMA and HEAm were injected alternatingly and a pentablock 3-arm star-shaped polymer was obtained. Initially, at high monomer concentrations the rate of propagation was high enough that the substitution of the chain end was found to be negligible.
After reaching complete monomer conversion, prolonged reaction times may cause the loss of bromine end groups due to possible side reactions, such as hydrolysis or coupling with the amine-based ligand.30 In order to retain high chain end fidelity, conversion of each chain extension was monitored closely, to chain extend with the next block before monomer concentration reached zero, ideally at ρ ≥ 95% (ESI, Table S4†). This procedure was carried out for each block until the desired pentablock core-first star polymer (P11.5, Mn,GPC = 29700 g mol−1, PDI = 1.14) was obtained via iterative chain extension (Fig. 3).
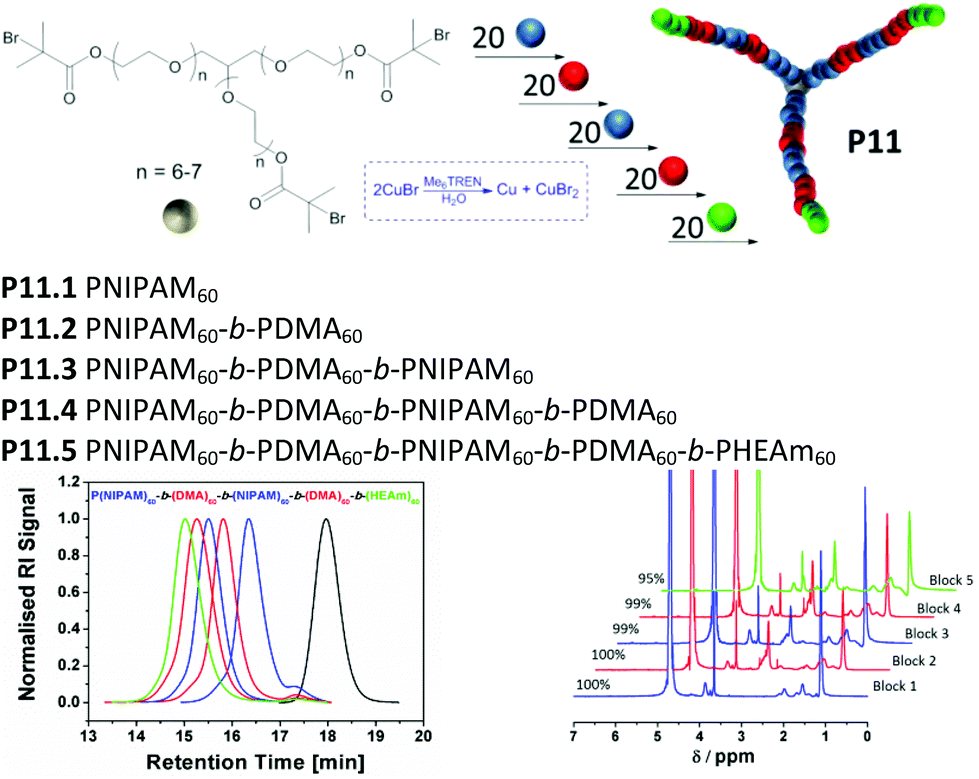 | ||
| Fig. 3 Representative scheme for the obtained pentablock star copolymer P11 using [M]:[I]:[CuBr]:[Me6TREN] = 60:1:1.8:1.2 at 0 °C (top), obtained molecular weight distributions of P11 blocks (middle) and 1H NMR spectra displaying full conversion for each block (bottom). | ||
The multi-block copolymerization was started by using 3-arm water soluble glycerol initiator (Table 2, P0). As the first block 20 repeating units of NIPAM per arm has been polymerized in 9 minutes (P11.1). Following to the first block, 20 repeating units of DMA per arm has been reacted in only 5 minutes due to the higher propagation rate constant of DMA in comparison to NIPAM.31 (P11.2). These two steps were repeated to get PNIPAM60-b-PDMA60-b-PNIPAM60-b-PDMA60 tetra-block copolymer (P11.4). Finally, the fifth block has been polymerized using 20 repeating units of PHEAm per arm (P11.5). As the total polymer chain length increased, the reaction time required for the next block increased as well. This can be due the decrease in initiator concentration or increase in CuBr2 concentration as a result of termination reactions.32
| Entry |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n,theo [g mol−1]
n,theo [g mol−1] |
n,SECa [g mol−1] |
PDia | ρ [%] | Timec [min] |
|---|---|---|---|---|---|
| a DMF eluent, PMMA standards. b Conversion (ρ) measured by 1H-NMR spectroscopy. c Cumulative times stated in parenthesis. | |||||
| P0 | 1450 | 1900 | 1.04 | — | — |
| P11.1 | 8200 | 8100 | 1.14 | 100 | 9 (9) |
| P11.2 | 14100 |
14400 |
1.10 | 100 | 5 (14) |
| P11.3 | 20900 |
19200 |
1.10 | 99 | 13 (27) |
| P11.4 | 28600 |
24000 |
1.14 | 99 | 12 (39) |
| P11.5 | 35500 |
29700 |
1.14 | 95 | 45 (84) |
Despite this, every block has reached to full conversion already in less than 15 minutes, except for the fifth block reached to 95% conversion in 45 minutes according to 1H NMR measurement. No significant side reaction or unreacted initiator was observed according to GPC spectra.
In conclusion, an aqueous polymerization technique has been investigated and by employing optimized reaction conditions, thermoresponsive core-first 3-arm star shaped polymers (P1–P7) and sequence controlled multi-block core-first 3-arm star shaped polymers based on acrylamides (P8–P11) utilizing a hydrophilic initiator have been reported for the first time. Core-first star shaped polymers can be synthesized via iterative chain extension up to a pentablock star, within a very short time period. Furthermore by systematically probing the optimum conditions for the polymerization, time consuming and costly purification steps in between block copolymerizations can be avoided. Kinetic investigations have shown to be useful to obtain high chain end fidelity to retain and give excellent control over the molecular weight distribution. Importantly, the reactions were carried out in water; a “green”, safe and cheaper alternative to organic solvents. This method can be easily up-scaled and be potentially utilized as a drug-delivery vehicle in biomedical applications.
Acknowledgements
The authors are grateful to the QMUL Materials Research Institute for the financial support.Notes and references
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143 CrossRef CAS PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189 CrossRef CAS PubMed.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341 CrossRef CAS PubMed.
- H. Colquhoun and J.-F. Lutz, Nat. Chem., 2014, 6, 455 CrossRef CAS PubMed.
- S. Pearson, H. Lu and M. H. Stenzel, Macromolecules, 2015, 48, 1065 CrossRef CAS.
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavía-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. P. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056 CrossRef CAS PubMed.
- Y. Chen, M. S. Lord, A. Piloni and M. H. Stenzel, Macromolecules, 2015, 48, 346 CrossRef CAS.
- Q. Zhang, L. Su, J. Collins, G. Chen, R. Wallis, D. A. Mitchell, D. M. Haddleton and C. R. Becer, J. Am. Chem. Soc., 2014, 136, 4325 CrossRef CAS PubMed.
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141 CrossRef CAS PubMed.
- W. Shi, A. L. Hamilton, K. T. Delaney, G. H. Fredrickson, E. J. Kramer, C. Ntaras, A. Avgeropoulos and N. A. Lynd, J. Am. Chem. Soc., 2015, 137, 6160 CrossRef CAS PubMed.
- H. Gao, S. Ohno and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 15111 CrossRef CAS PubMed.
- T. G. McKenzie, E. H. H. Wong, Q. Fu, S. J. Lam, D. E. Dunstan and G. G. Qiao, Macromolecules, 2014, 47, 7869 CrossRef CAS.
- Q. Zhang, G.-Z. Li, C. R. Becer and D. M. Haddleton, Chem. Commun., 2012, 48, 8063 RSC.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176 RSC.
- S. Harrisson and J. Nicolas, ACS Macro Lett., 2014, 3, 643 CrossRef CAS.
- S. Sinnwell, M. Lammens, M. H. Stenzel, F. E. Du Prez and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2207 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128 CrossRef CAS PubMed.
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. R. Whittaker, Macromolecules, 2011, 44, 8028 CrossRef CAS.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355 CrossRef CAS PubMed.
- Q. Zhang, P. Wilson, A. Anastasaki, R. McHale and D. M. Haddleton, ACS Macro Lett., 2014, 3, 491 CrossRef CAS.
- W. Zhang, W. Zhang, Z. Zhang, Z. Cheng, Y. Tu, Y. Qiu and X. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4268 CrossRef CAS.
- S. Paillet, A. Roncin, G. Clisson, G. Pembouong, L. Billon, C. Derail and M. Save, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2967 CrossRef CAS.
- C. Boyer, A. Derveaux, P. B. Zetterlund and M. R. Whittaker, Polym. Chem., 2012, 3, 117 RSC.
- C. Waldron, A. Anastasaki, R. McHale, P. Wilson, Z. Li, T. Smith and D. M. Haddleton, Polym. Chem., 2014, 5, 892 RSC.
- E. H. H. Wong, A. Blencowe and G. G. Qiao, Polym. Chem., 2013, 4, 4562 RSC.
- S. R. Samanta, V. Nikolaou, S. Keller, M. J. Monteiro, D. A. Wilson, D. M. Haddleton and V. Percec, Polym. Chem., 2015, 6, 2084 RSC.
- A. Anastasaki, A. J. Haddleton, Q. Zhang, A. Simula, M. Droesbeke, P. Wilson and D. M. Haddleton, Macromol. Rapid Commun., 2014, 35, 965 CrossRef CAS PubMed.
- C. Weber, T. Neuwirth, K. Kempe, B. Ozkahraman, E. Tamahkar, H. Mert, C. R. Becer and U. S. Schubert, Macromolecules, 2012, 45, 20 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138 CrossRef CAS.
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672 RSC.
- N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1752 CrossRef CAS.
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Polymerization kinetics, chemical analysis. See DOI: 10.1039/c5py01623a |
| This journal is © The Royal Society of Chemistry 2016 |