
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic and reactive primary amine-stabilised nanoparticles via RAFT aqueous dispersion polymerisation†
M.
Williams
,
N. J. W.
Penfold
and
S. P.
Armes
*
Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
First published on 2nd November 2015
Abstract
The synthesis of primary amine-functionalised diblock copolymer nanoparticles via polymerisation-induced self-assembly (PISA) using a RAFT aqueous dispersion polymerisation formulation is reported. The primary amine steric stabiliser is a macromolecular chain transfer agent (macro-CTA) based on 2-aminoethyl methacrylate AMA, which can be readily polymerised in its hydrochloride salt form with good control (Mw/Mn < 1.30) using RAFT aqueous solution polymerisation. Subsequent chain extension of this macro-CTA with 2-hydroxypropyl methacrylate (HPMA) leads to the formation of relatively monodisperse spherical nanoparticles (68 to 288 nm) at pH 6. However, worms or vesicles could not be obtained, because strong lateral repulsion between the highly cationic PAMA stabiliser chains impedes the formation of these higher order copolymer morphologies. Deprotonation of the primary amine stabiliser chains at or above pH 9 results in flocculation of these spherical nanoparticles as the PAMA block becomes uncharged. Diblock copolymer spheres, worms or vesicles can be synthesised that remain stable at pH 9 by supplementing the PAMA macro-CTA with a poly(glycerol monomethacrylate) (PGMA) macro-CTA, since this non-ionic block confers effective steric stabilisation in alkaline media. A series of diblock copolymer nanoparticles with the general formula ([1 − n]PGMAx + nPAMAy)–PHPMAz can be synthesised by optimising: (i) the mean degree of polymerisation (DP, or x) of the PGMA block, (ii) the PHPMA core-forming DP (or z); (iii) the mol fraction (n) of the PAMA stabiliser; and (iv) the copolymer concentration. These spheres, worms and vesicles are both cationic at low pH and colloidally stable at high pH. Furthermore, deprotonation of the protonated primary amine groups on the PAMA stabiliser chains at high pH renders these particles susceptible to epoxy-amine conjugation. This is demonstrated by the reaction between the primary amine groups on (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 diblock copolymer spheres, and epoxide-functionalised diblock copolymer nanoparticles in aqueous solution at pH 8.
Introduction
Primary amine-based polymers are of great interest as the result of their use in a wide range of applications such as drug delivery,1,2 biomedical applications,3,4 polypeptide synthesis5–7 and biomineralisation.8,9 Two important characteristics are their cationic character and their reactivity. For example, the cationic nature of primary amines has been utilised to facilitate their occlusion into calcium carbonate,10 as well providing organic templates for silica deposition.11–13 Furthermore, post-polymerisation reactions such as the ring-opening of cyclic esters14 and Michael addition15 can be exploited to derivatise primary amine polymers, as well as allowing conjugation to other molecules such as proteins and polypeptides.16Despite considerable interest, the synthesis of well-defined primary amine methacrylate-based polymers with precise molecular weights and low polydispersities has proven to be a difficult synthetic challenge. This is because 2-aminoethyl methacrylate (AMA) rapidly undergoes molecular rearrangement to form 2-hydroxyethyl methacrylamide in its deprotonated form (i.e. at high pH).17 In principle, this problem can be overcome by protecting the monomer prior to its polymerisation; however, this approach requires additional protection/deprotection steps and hence is both time-consuming and atom-inefficient.18
The development of reversible-addition fragmentation chain transfer polymerisation (RAFT)19–22 has enabled the controlled synthesis of a wide range of water-soluble polymers. Furthermore, polymers with neutral,23–26 cationic,27–29 anionic,30–32 and zwitterionic33–36 character can be synthesised directly in aqueous solution. Despite this, there are very few reports of the RAFT polymerisation of 2-aminoethyl methacrylate (AMA). He et al. demonstrated that AMA can be polymerised in its hydrochloride salt form using RAFT solution polymerisation in DMSO.37 Moreover, it was also shown that linear PAMA homopolymers and AB diblock copolymers (where A = AMA and B = 2-(diisopropylamino)ethyl methacrylate, or DPA) could be prepared with reasonably narrow polydispersities (Mw/Mn = 1.2–1.3). More recently, Li et al. showed that PAMA homopolymers could be prepared in acidic aqueous solution using dioxane as a co-solvent.38 Performing reactions at low pH eliminated monomer degradation problems17 and allowed the synthesis of polymers with low polydispersities (Mw/Mn = 1.10). Alidedeoglu et al. also reported the synthesis of well-defined PAMA homopolymers by aqueous RAFT, with double-hydrophilic AB diblock copolymers being subsequently obtained via chain extension of PAMA with N-(2-hydroxypropyl)methacrylamide.39 Recent advances in polymerisation-induced self-assembly (PISA) via RAFT aqueous dispersion polymerisation has enabled diblock copolymer nanoparticles to be prepared directly in aqueous solution at high solids, with various copolymer morphologies such as spheres, worms, vesicles and framboidal vesicles.40–44 In a typical aqueous PISA formulation, the water-soluble steric stabiliser block is prepared first, with subsequent growth of the water-insoluble block driving in situ phase separation.45 For a given degree of polymerisation (DP) of the stabiliser block, the final copolymer morphology is mainly dictated by the DP of the core-forming block and the overall copolymer concentration.46
Very recently, Bauri et al. reported a range of particle morphologies using a primary amine-based macro-CTA based on a poly(Boc-L-alanine methacryloyloxyethyl ester) via RAFT-mediated PISA conducted in methanol.47 In principle, the preparation of a range of primary amine-functionalised diblock copolymer nanoparticles should also be feasible via PISA using a RAFT aqueous dispersion polymerisation formulation. However, Semsarilar and co-workers reported that use of polyelectrolytic macro-CTAs only led to spherical morphologies, even in the presence of added salt. This is believed to be the result of strong lateral electrostatic repulsive forces between the charged stabiliser chains, which impedes the formation of higher order morphologies such as worms and vesicles during PISA.48–51 In principle, this problem can be overcome by reducing the charge density within the coronal stabiliser layer by either statistically copolymerising the desired charged monomer with a non-ionic comonomer or by using a binary mixture of polyelectrolytic and non-ionic macro-CTAs. In practice, the latter approach allows convenient fine-tuning of the non-ionic/ionic stabiliser molar ratio and has proved to be particularly useful in allowing access to the full range of copolymer morphologies.50,51
Herein the synthesis of primary amine-functionalised diblock copolymer nanoparticles via RAFT aqueous dispersion polymerisation is reported. More specifically, a PAMA macro-CTA is chain-extended with 2-hydroxypropyl methacrylate (HPMA) to form a hydrophobic core-forming PHPMA block. Higher order morphologies such as worms and vesicles were obtained in the absence of salt by using a binary mixture of macro-CTAs, with a non-ionic poly(glycerol monomethacrylate) (PGMA) macro-CTA being utilised to dilute the coronal charge density arising from the cationic PAMA macro-CTA (see Fig. 1). Furthermore, the pH-dependent colloidal stability and reactivity of the resulting new diblock copolymer nanoparticles is investigated.
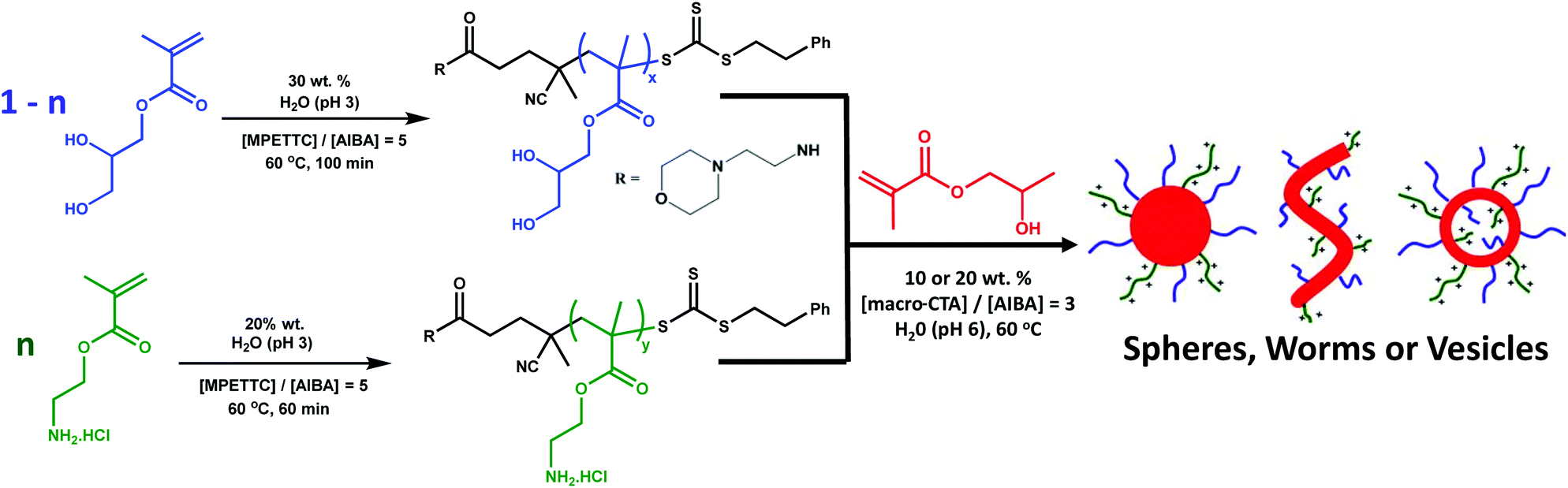 | ||
| Fig. 1 Synthesis of diblock copolymer particles with the general formula ([1 − n]PGMAx + nPAMAy)–PHPMAz by RAFT aqueous dispersion polymerisation using a binary mixed macro-CTA approach. Optimisation of parameters n, x, y, and z result in the formation of spheres, worms or vesicles that are cationic at low pH, and both colloidally stable and reactive at high pH. | ||
Experimental
Materials
Glycerol monomethacrylate (GMA; 99%) was donated by GEO Specialty Chemicals (Hythe, UK) and was used without further purification. A water-soluble trithiocarbonate-based RAFT agent known as MPETTC was synthesised as previously reported.52 2-Aminoethyl methacrylate hydrochloride (AMA; 90%) was purchased from Sigma-Aldrich (UK) and purified by washing extensively with THF. 2-Hydroxypropyl methacrylate (HPMA; 97%), glycidyl methacrylate (GlyMA; 97%), azobis(2-methylpropionamidine) dihydrochloride (AIBA; 97%), methanol, dichloromethane and D2O were all purchased from Sigma-Aldrich (UK) and were used as received. THF and DMF were purchased from Fisher Scientific (Loughborough, UK). Deuterated methanol (CD3OD) was purchased from Goss Scientific Instruments Ltd (Cheshire, UK). Deionised water was used for all experiments.1H NMR spectroscopy
All NMR spectra were recorded using a 400 MHz Bruker Avance-400 spectrometer (64 scans averaged per spectrum).DMF gel permeation chromatography (GPC)
Molecular weights and polydispersities of PGMA macro-CTAs were determined using a DMF GPC set-up operating at 60 °C and comprising two Polymer Laboratories PL gel 5 μm Mixed-C columns connected in series to a Varian 390-LC multi-detector suite (only the refractive index detector was utilised in this study) and a Varian 290-LC pump injection module. The GPC eluent was HPLC-grade DMF containing 10 mM LiBr at a flow rate of 1.0 mL min−1. DMSO was used as a flow rate marker. Calibration was conducted using a series of ten near-monodisperse poly(methyl methacrylate) standards (Mn = 625 to 618![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). Chromatograms were analysed using Varian Cirrus GPC software (version 3.3).
000 g mol−1). Chromatograms were analysed using Varian Cirrus GPC software (version 3.3).
Aqueous gel permeation chromatography (GPC)
Aqueous gel permeation chromatography (GPC) was used to characterise the PAMA macro-CTA. The GPC protocol involved using an Agilent 1260 Infinity series degasser and pump, two 8 μm Agilent PL Aquagel-OH 30 columns and both a UV and RI detector. The eluent was an acidic aqueous buffer (pH 3.3) comprising 0.50 M acetic acid, 0.30 M NaH2PO4 and acidified with HCl at a flow rate of 1.0 ml min−1. Calibration was achieved using a series of near-monodisperse poly(ethylene oxide) standards ranging from 1080 to 905000 g mol−1.
Dynamic light scattering (DLS)
The intensity-average hydrodynamic diameter was determined for selected diblock copolymer particles by DLS using a Malvern Zetasizer NanoZS instrument. Aqueous solutions of 0.01 w/v % copolymer dispersions were analysed using disposable plastic cuvettes, and the results were averaged over three consecutive runs. Deionised water was used to dilute all dispersions, which were ultra-filtered through a 0.20 μm membrane to remove dust prior to analysis.Aqueous electrophoresis
Zeta potentials were recorded in the presence of 1 mM KCl for diblock copolymer particles using a Malvern Zetasizer Nano ZS instrument. The solution pH was adjusted using either dilute NaOH or HCl as required.Transmission electron microscopy (TEM)
Copper/palladium TEM grids (Agar Scientific, UK) were surface-coated to yield a thin film of amorphous carbon. The grids were then plasma glow-discharged for 30 seconds to create a hydrophilic surface. A small volume (10 μL) of a dilute aqueous copolymer solution was placed on the freshly prepared grids for 20 seconds and then blotted with filter paper to remove excess solution. To stain the aggregates, a 0.75% w/v uranyl formate solution (10 μL) was placed on the sample-loaded grid for 15 seconds and then carefully blotted to remove excess stain. The grids were then dried using a vacuum hose. Imaging was performed using a FEI Tecnai Spirit microscope fitted with a Gatan 1KMS600CW CCD camera operating at 80 KV.Synthesis of a poly(2-aminoethyl methacrylate hydrochloride) (PAMA) macro-CTA
The synthesis of a PAMA macro-CTA was conducted as follows. A water-soluble morpholine-based RAFT agent (MPETTC) was obtained by amidation of 4-cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanylpentanoic acid using 4-aminoethylmorpholine, as previously reported.52 MPETTC (0.60 mmol, 0.265 g, AIBA initiator (0.12 mmol, 0.033 g, CTA/AIBA molar ratio = 5.0) and AMA monomer (0.07 mol, 12.0 g) were weighed into a 100 mL round-bottom flask. Deionised water (49.2 mL, affording a 20% w/w AMA solution) was added and the resulting solution was adjusted to pH 3 using 0.1 M HCl before being purged with nitrogen for 30 min. The sealed flask was immersed into an oil bath set at 60 °C for 60 min and the resulting RAFT polymerisation was subsequently quenched by immersion in liquid nitrogen (final AMA conversion = 85% as judged by 1H NMR). The crude PAMA was purified by exhaustive dialysis (SpectraPor membrane; molecular weight cut-off = 3.5 kDa) against an aqueous acidic solution (pH 3), followed by freeze-drying overnight. 1H NMR analysis indicated a mean degree of polymerisation of 96 for this PAMA macro-CTA. Its Mn and Mw/Mn were 27700 g mol−1 and 1.30 respectively, as judged by aqueous GPC.
Self-blocking chain extension experiments with PAMA96
A typical protocol for the synthesis of PAMA96–PAMA300 at 10% w/w solids is given below: PAMA96 macro-CTA (0.075 g, 0.004 mmol), AIBA (0.23 mg, 0.0008 mmol, CTA/AIBA molar ratio = 5.0) and AMA monomer (0.207 g, 1.2 mmol; target DP = 300) were weighed into a 25 mL round-bottomed flask. Deionised water (2.6 mL, producing a 10% w/w aqueous solution) was then added and the resulting solution was adjusted to pH 3 using 0.1 M HCl before being degassed for 30 min prior to immersion in an oil bath set at 60 °C. The reaction solution was stirred for 5 h to ensure complete AMA monomer conversion (as judged by 1H NMR) and the RAFT polymerisation was quenched by exposure to air. The Mn and Mw/Mn values for the resulting chain-extended PAMA were 68100 g mol−1 and 1.21 respectively, as judged by aqueous GPC.
Synthesis of a poly(glycerol monomethacrylate) (PGMA) macro-CTA
The synthesis of a PGMA101 macro-CTA was conducted as follows. MPETTC (0.70 mmol, 0.293 g, AIBA initiator (0.14 mmol, 0.036 g, CTA/AIBA molar ratio = 5.0) and GMA monomer (0.09 mol, 15.0 g) were weighed into a 100 mL round-bottom flask. Deionised water (35.8 mL) was added to afford a 30% w/w GMA solution and the resulting solution was adjusted to pH 3 using 0.1 M HCl before being purged with nitrogen for 30 min. The sealed flask was immersed into an oil bath set at 60 °C for 90 min and the RAFT polymerisation was subsequently quenched by immersion in liquid nitrogen (90% GMA conversion as judged by 1H NMR). The crude PGMA was freeze-dried from water overnight, before being redissolved in methanol (100 mL) followed by precipitation into a ten-fold excess of dichloromethane. This precipitation process was repeated prior to dissolution into deionised water, freeze-drying overnight afforded the purified PGMA macro-CTA. 1H NMR analysis indicated a mean degree of polymerisation of 101, while DMF GPC studies indicated an Mn of 36200 g mol−1 and an Mw/Mn of 1.13. Essentially the same protocol was used to prepare a PGMA macro-CTA with a mean degree of polymerisation of 60 (Mn = 17900 g mol−1 and Mw/Mn = 1.16).
Synthesis of PAMA–PHPMA diblock copolymer nanoparticles via RAFT aqueous dispersion polymerisation of HPMA
The synthesis of PAMA96–PHPMA500 at 10% w/w solids was conducted as follows: PAMA96 macro-CTA (0.10 g, 0.006 mmol), AIBA (0.57 mg, 0.002 mmol, CTA/AIBA molar ratio = 3.0) and HPMA monomer (0.43 g, 3.1 mmol; target DP = 500) were weighed into a 25 mL round-bottomed flask. Deionised water (4.9 mL) was added to produce a 10% w/w aqueous solution and the resulting solution was adjusted to pH 6 using 0.1 M NaOH before being degassed for 30 min prior to immersion in an oil bath set at 60 °C. The reaction solution was stirred for 5 h to ensure complete HPMA monomer conversion (>99% conversion as judged by 1H NMR) and then quenched by exposure to air.Synthesis of PGMA–PHPMA diblock copolymer nanoparticles via RAFT aqueous dispersion polymerisation of HPMA
The synthesis of PGMA101–PHPMA1000 diblock copolymer nanoparticles at 10% w/w solids was conducted as follows. PGMA96 macro-CTA (0.10 g, 0.006 mmol), AIBA (0.57 mg, 0.002 mmol, CTA/AIBA molar ratio = 3.0) and HPMA monomer (0.87 g, 6.1 mmol; target DP = 1000) were weighed into a 25 mL round-bottomed flask. Deionised water (8.7 mL) was added to produce a 10% w/w aqueous solution and the resulting solution was adjusted to pH 6 using 0.1 M NaOH before being degassed for 30 min prior to immersion in an oil bath set at 60 °C. The reaction solution was stirred for 5 h to ensure complete HPMA monomer conversion (>99% conversion as judged by 1H NMR) and quenched by exposure to air.Synthesis of ([1−n]PGMAx + nPAMAy)–PHPMAz diblock copolymer nanoparticles by RAFT aqueous dispersion polymerisation of HPMA using a binary mixture of macro-CTAs
The synthesis of (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 is representative and was conducted at 10% w/w solids as follows. PGMA101 macro-CTA (0.10 g, 6.0 μmol), PAMA96 macro-CTA (0.245 g, 0.0015 mmol), AIBA (0.68 mg, 0.0025 mmol, CTA/AIBA molar ratio = 3.0) and HPMA monomer (1.08 g, 7.5 mmol; target DP = 1000) were weighed into a 25 mL round-bottomed flask. Deionised water (10.8 mL) was then added to produce a 10% w/w aqueous solution, which was adjusted to pH 6 using 0.1 M NaOH before being degassed for 30 min prior to immersion in an oil bath set at 60 °C. The reaction solution was stirred for 5 h to ensure complete HPMA monomer conversion (>99% conversion as judged by 1H NMR) and quenched by exposure to air.Synthesis of epoxy-functionalised PGMA–PGlyMA diblock copolymer nanoparticles by RAFT aqueous emulsion polymerisation
The synthesis of PGMA60–PGlyMA100 diblock copolymer nanoparticles at 10% w/w solids was achieved via RAFT aqueous emulsion polymerisation of glycidyl methacrylate (GlyMA). Briefly, PGMA60 macro-CTA (0.47 g, 0.047 mmol), AIBA (2.54 mg, 0.009 mmol, CTA/AIBA molar ratio = 5.0) and GlyMA monomer (0.66 g, 4.7 mmol; target DP = 100) were weighed into a 25 mL round-bottomed flask. Deionised water (10.2 mL) was then added to produce a 10% w/w aqueous solution, which was degassed for 30 min prior to immersion in an oil bath set at 60 °C. This reaction solution was stirred for 4 h to ensure complete HPMA monomer conversion (>99% conversion as judged by 1H NMR) and then quenched by exposure to air.Epoxy-amine reaction between (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 spheres and PGMA60–PGlyMA100 spheres
A 10 mL aqueous dispersion containing 1.0% w/w (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 and 1.4% w/w PGMA60–PGlyMA100 diblock copolymer nanoparticles was prepared. The solution pH was adjusted to pH 8 using 0.1 M NaOH, followed by heating at 60 °C for 16 h. As a control experiment, an identical solution was prepared, adjusted to pH 3 using 0.1 M HCl and heated at 60 °C for 16 h.Results and discussion
The aim of this work was to develop a robust synthetic route to primary amine-functionalised diblock copolymer nanoparticles via polymerisation-induced self-assembly (PISA) using a RAFT aqueous dispersion polymerisation formulation. In order to prepare such cationic nanoparticles, a PAMA macro-CTA was prepared at 20% w/w solids by RAFT aqueous solution polymerisation at pH 3. To produce a water-soluble RAFT agent that dissolved at low pH, 4-cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanylpentanoic acid (PETTC) was modified as previously reported52 to yield a morpholine-functionalised RAFT agent known as MPETTC. 2,2′-Azobis(2-methylpropionamidine) dihydrochloride (AIBA) was used as initiator at 60 °C and an MPETTC/AIBA molar ratio of 5.0 was employed. Fig. 2A shows both the conversion and semi-logarithmic plot of monomer concentration vs. time, as determined by 1H NMR spectroscopy. Inspecting the conversion vs. time data (red triangles), the polymerisation reaches 75% conversion after 1 h and is almost complete within 2 h. The semi-logarithmic plot of monomer concentration vs. time (blue circles) is reasonably linear, which suggests that the RAFT polymerisation is well-controlled. For such PISA syntheses via RAFT aqueous dispersion polymerisation, a large batch of purified macro-CTA is initially prepared to enable a series of chain extension experiments targeting a wide range of core-forming block DPs. Typically, the initial RAFT solution polymerisation is terminated at intermediate conversion to minimise loss of RAFT end-group functionality, which can otherwise result in a reduction in living character and molecular weight control. Using the kinetic data shown in Fig. 2A, a 40 gram scale synthesis of PAMA macro-CTA was conducted under the same conditions and terminated after 60 min. A mean degree of polymerisation (DP) of 96 was determined by 1H NMR, while aqueous GPC studies (vs. PEO calibration standards) indicated an Mw/Mn of 1.30 (see Fig. 2B). Following the synthesis of this primary amine macro-CTA, chain-extension with 2-hydroxypropyl methacrylate (HPMA) could be achieved via RAFT aqueous dispersion polymerisation at pH 6 and 60 °C, using AIBA initiator and a CTA/initiator molar ratio of 3.0. Unfortunately, the living character and blocking efficiency of the PAMA macro-CTA could not be assessed by GPC as there is no appropriate eluent that dissolves both the hydrophilic PAMA and hydrophobic PHPMA blocks. Therefore a self-blocking experiment with AMA monomer was conducted in order to assess the blocking efficiency and living character of the PAMA96 macro-CTA, as described by Semsarilar and co-workers for a different cationic macro-CTA.50Fig. 2B shows the GPC chromatograms obtained for the PAMA96 macro-CTA before and after chain extension targeting a further 300 units of AMA. Clearly, reasonably good living character is maintained as the Mw/Mn value of the final copolymer remains below 1.30. Furthermore, the blocking efficiency is reasonably high, since the GPC chromatogram indicates little or no presence of the original PAMA96 macro-CTA. Given these data and bearing in mind various literature reports,43,46,50,50 it is reasonable to assume that chain extension of the PAMA96 macro-CTA with HPMA under RAFT aqueous dispersion polymerisation conditions should be similarly well controlled and efficient. Fig. 3 shows the resulting hydrodynamic particle diameters determined by DLS and the corresponding transmission electron microscopy (TEM) images obtained for a series of PAMA96–PHPMAz diblock copolymer nanoparticles synthesised at 10% w/w solids (where z = 100 to 1000). The hydrodynamic DLS diameter increases monotonically as higher z values are targeted, while the DLS polydispersity is gradually reduced, see Fig. 3A and S1.†Fig. 3B shows the corresponding TEM images of these diblock copolymer particles, which confirms this particle size trend. Furthermore, TEM confirms that only spherical nanoparticles are formed in all cases. Semsarilar and co-workers reported similar observations when using polyelectrolytes as the sole stabiliser block for the PISA synthesis of diblock copolymer nanoparticles. This kinetically-trapped morphology was attributed to the strong electrostatic repulsion between the charged stabiliser chains.50,51 PAMA has a pKa of approximately 7.6 in dilute aqueous solution.17 Given that these PAMA-based diblock copolymers are synthesised at pH 6, the primary amine groups on the stabiliser chains have a high degree of protonation and are therefore strongly cationic, which explains why only spherical morphologies are produced in the present study. This high cationic charge density is confirmed in Fig. 4, which shows the pH-dependence of the hydrodynamic diameter and zeta potential for PAMA96–PHPMA1000 diblock copolymer nanoparticles. At pH 3, these nanoparticles have a zeta potential of +45 mV because of the protonated nature of the PAMA stabiliser chains. As the pH is raised towards the pKa of the PAMA, gradual deprotonation occurs such that the zeta potential is reduced to +30 mV at pH 7.5. An isoelectric point is observed at pH 9, and weakly anionic particles are obtained at higher pH. Fig. 4 also shows the effect that this variation in electrophoretic behaviour has on the particle size, as determined by DLS (blue triangles). At pH 3, the mean particle diameter is around 250 nm. As the pH approaches the approximate pKa of the stabiliser chains, there is a slight reduction in size (to 201 nm diameter at pH 7.5). This indicates gradual contraction of the stabiliser chains as they gradually become less protonated. At around the isoelectric point, flocculation occurs as the PAMA stabiliser chains become neutral and steric stabilisation between neighbouring particles is no longer effective. Previous reports have demonstrated that a binary mixture of a polyelectrolytic and a non-ionic macro-CTA allows fine-tuning of the coronal charge density.50,51 | ||
| Fig. 2 (A) Conversion vs. time data derived from 1H NMR spectroscopy studies of a RAFT solution polymerisation of 2-aminoethyl methacrylate hydrochloride (AMA) at 60 °C in water using MPETTC CTA and AIBA initiator at 20% w/w solids with a CTA/initiator molar ratio = 5. (B) Aqueous gel permeation chromatograms (pH 3.3 vs. PEO standards) obtained for a PAMA96 macro-CTA, and a subsequent PAMA300 homopolymer formed by chain extension with further AMA monomer. | ||
 | ||
| Fig. 3 (A) Hydrodynamic diameter and polydispersity vs. PHPMA core DP, as measured by DLS, and (B) TEM images of PAMA96–PHPMAz (z = 100–1000) diblock copolymer particles synthesised at 10% w/w solids by RAFT aqueous dispersion polymerisation of HPMA. | ||
 | ||
| Fig. 4 DLS intensity-average diameter and zeta potential vs. pH curves for PAMA96–PHPMA1000 diblock copolymer spheres in the presence of 1 mM KCl background salt. The isoelectric point observed at pH 9 coincides with particle flocculation. | ||
The convenient synthesis of both cationic and anionic diblock copolymer nanoparticles can be achieved via this convenient approach, with entropic mixing being confirmed by mobility data.50,51 Moreover, introducing a non-ionic stabiliser into the corona should provide the particles with additional steric stabilisation when the deprotonated PAMA chains collapse, thus preventing flocculation at around the isoelectric point and hence producing colloidally stable particles over the entire pH range. Furthermore, using a binary mixture of macro-CTAs under appropriate conditions can result in the formation of so-called ‘higher order’ copolymer morphologies such as worms and vesicles. To investigate this approach, a non-ionic poly(glycerol monomethacrylate) (PGMA) macro- CTA was synthesised at 30% w/w solids, at pH 3 using MPETTC and AIBA using a CTA/initiator molar ratio of 5.0. Again, a kinetic study was performed to assess the living character of the reaction. A linear semi-logarithmic plot of monomer concentration vs. time and a monotonic evolution of molecular weight with conversion indicated good living character (see Fig. S2†). Subsequently, PGMA macro-CTAs with mean DPs of 60 and 101 were prepared with final Mw/Mn values of 1.16 and 1.13 respectively, as determined by DMF GPC analysis (see Fig. S3†).
Initially, diblock copolymer nanoparticles of the general formula ([1 − n]PGMA101 + nPAMA96)–PHPMA1000 were prepared at 10% w/w solids by RAFT aqueous dispersion polymerisation (see Fig. 1). Utilising a relatively long non-ionic stabiliser should confer effective steric stabilisation at high pH. The mean DP of the PHPMA core-forming block was fixed to enable direct comparisons to be made between nanoparticles with differing coronal stabiliser compositions. First, PGMA101–PHPMA1000 (n = 0) diblock copolymer nanoparticles were prepared as a reference material. Fig. 5 shows zeta potential vs. pH curves and the corresponding TEM images obtained for a series of diblock copolymer nanoparticles formed using a binary mixture of macro-CTAs. PGMA101–PHPMA1000 particles are weakly cationic (+12 mV) at pH 3 as a result of protonation of the morpholine-based RAFT end-groups. At pH 6 and above, approximately neutral nanoparticles are obtained that remain stable over the entire pH range, showing no signs of flocculation (see Fig. S4†). TEM studies indicate a spherical morphology, and DLS studies indicate a mean particle diameter of 96 nm (see Fig. S5†). Indeed, the TEM images shown in Fig. 5 confirm that a solely spherical morphology is obtained for all with the general formula ([1 − n]PGMA101 + nPAMA96)–PHPMA1000 diblock copolymer nanoparticles prepared at 10% w/w solids. Purely non-ionic stabilised particles (i.e. n = 0) have a mean droplet diameter of 97 nm while wholly cationic stabilised particles (i.e. n = 1) have a mean droplet diameter of 280 nm. Clearly, there is a linear increase in diameter over this particle size range as the proportion of PAMA stabiliser is increased (see Fig. S5†). Moreover, there is a concomitant increase in cationic character for the resulting diblock copolymer nanoparticles between pH 3 and pH 8, see Fig. 5. However, the isoelectric point of these particles is not affected: all of the diblock copolymer nanoparticles become neutral and weakly anionic above pH 9. Hence controlling the proportion of the PAMA stabiliser chains within the diblock copolymer nanoparticles enables fine-tuning of the cationic character at low pH. Crucially, up to 20 mol% (i.e. n = 0.20) PAMA stabiliser can be incorporated into the nanoparticles before flocculation is observed. Weak aggregation occurs at 30 mol% and flocculation comparable to PAMA96–PHPMA1000 particles can be detected at 40 mol% PAMA (see Fig. S4†). Thus spherical diblock copolymer nanoparticles prepared using a binary mixture of macro-CTAs with the general formula (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 are not only cationic at low pH (+32 mV at pH 3), but also stable to flocculation at high pH where the PAMA chains become deprotonated and collapsed. Following the successful formation of spherical diblock copolymer nanoparticles that remain stable at high pH, binary mixtures of macro-CTAs were further examined in order to target worm and vesicle morphologies. In the context of wholly non-ionic PISA formulations, Blanazs et al. showed that two important parameters are the copolymer concentration46 and the DP of the stabiliser macro-CTA.43 In order to target higher order copolymer morphologies, both higher copolymer concentrations and a lower PGMA DP are required. Drawing on published phase diagrams,43 the effect of addition of the PAMA96 stabiliser on the copolymer morphology was studied by preparing two series of ([1 − n]PGMA60 + nPAMA96)–PHPMA200 and ([1 − n]PGMA60 + nPAMA96)–PHPMA500 nanoparticles at 20% w/w solids. Fig. 6A shows the TEM images obtained for the first series when n is increased from 0 to 0.20. When no PAMA stabiliser is present (i.e. n = 0), the target PGMA60–PHPMA200 composition produced a pure worm morphology, as expected. Incorporation of 5 mol% (i.e. n = 0.05) PAMA96 stabiliser also produced worms, but 10 mol% PAMA96 gave a worm plus sphere mixed phase, while only spheres are produced at either 15 or 20 mol% PAMA96. This gradual change in copolymer morphology is also observed for the ([1 − n]PGMA60 + nPAMA96)–PHPMA500 diblock copolymer nanoparticles, see Fig. 6B. A pure vesicle phase is obtained when using either zero or 5 mol% PAMA stabiliser, but incorporating 10 mol% PAMA96 stabiliser results in a vesicle plus sphere mixed phase, while only spheres are produced in the presence of 20 mol% PAMA96. This loss of higher order morphologies is a result of progressively stronger lateral interactions between the cationic PAMA chains within the coronal layer, which leads to a higher stabiliser volume fraction and hence a lower packing parameter, P.53,54 A higher proportion of PAMA96 stabiliser conferred greater cationic character on the spherical diblock copolymer nanoparticles at pH 3–8, with an isoelectric point being identified at pH 9 (see Fig. 5). Similar electrophoretic behavior was observed for worms and vesicles prepared using 5 mol% PAMA96 (see Fig. S6 and S7†). Up to 20 mol% PAMA96 stabiliser can be incorporated into ([1 − n]PGMA60 + nPAMA96)–PHPMAz diblock copolymer nanoparticles without flocculation being observed. Thus diblock copolymer nanoparticles with spherical, worm-like or vesicular morphologies can be synthesised using the general formula ([1 − n]PGMAx + nPAMAy)–PHPMAz that are not only cationic at low pH, but also stable to flocculation at high pH (when the deprotonated PAMA chains undergo collapse).
 | ||
| Fig. 5 Zeta potential vs. pH curves and TEM images obtained for diblock copolymer particles synthesised at 10% w/w solids with the formula ([1 − n]PGMA101 + nPAMA96)–PHPMA1000. The scale bar shown in the first TEM image applies to all three TEM images. | ||
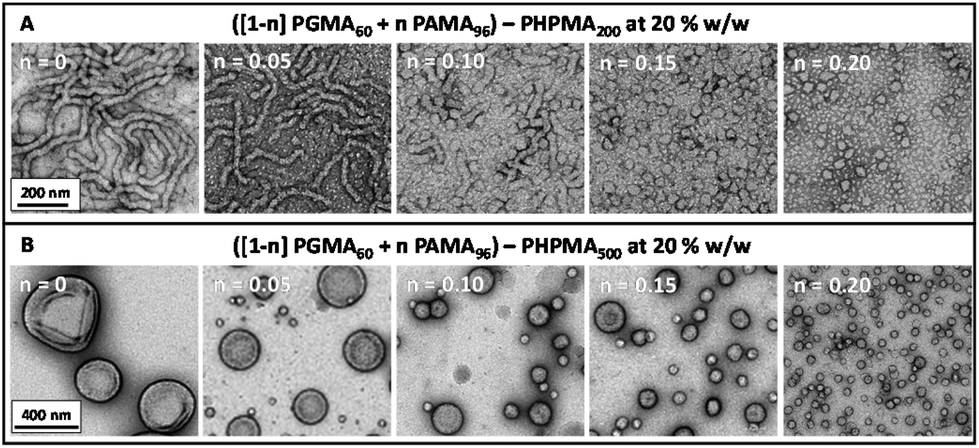 | ||
| Fig. 6 (A) TEM images obtained for diblock copolymer nano-objects synthesised at 20% w/w solids with the formula ([1 − n]PGMA60 + nPAMA96)–PHPMA200. Increasing the mol fraction of PAMA96 results in a change in copolymer morphology from pure worms (n = 0–0.05) to a worm plus sphere mixed phase (n = 0.10) to pure spheres (n = 0.15–0.20). (B) TEM images obtained for diblock copolymer nanoparticles synthesised at 20% w/w solids with the formula ([1 − n]PGMA60 + nPAMA96)–PHPMA500. Increasing the mol fraction of PAMA96 results in a change in copolymer morphology from pure vesicles (n = 0–0.05) to a vesicles plus spheres mixed phase (n = 0.10–0.15) to pure spheres (n = 0.20). | ||
In principle, the primary amine groups on the collapsed PAMA96 chains located at the nanoparticle surface should become deprotonated and hence reactive above pH 8.15 To test this hypothesis, epoxy-functional spherical nanoparticles were prepared via RAFT aqueous emulsion polymerisation of glycidyl methacrylate (GlyMA) at 10% w/w solids using a protocol similar to that recently reported by Cunningham et al.55 More specifically, a PGMA60 macro-CTA was utilised to prepare PGMA60–PGlyMA100 nanoparticles with a mean DLS diameter of 33 nm. A 1.4% w/w aqueous dispersion of these nanoparticles was added to a 1.0% w/w aqueous dispersion of (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 spherical diblock copolymer nanoparticles with a DLS diameter of 87 nm and the resulting mixture was heated at 60 °C overnight. It is well known that epoxides readily react with free amines in aqueous solution.56 Thus, if the solution pH is adjusted to pH 8, the deprotonated primary amine group on the (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 nanoparticles can react with the epoxy-functional PGMA60–PGlyMA100 nanoparticles. Conversely, no epoxy-amine conjugation is expected at pH 3, since protonated amines are unreactive under these conditions. Fig. 7A shows the mean DLS diameters for the two types of nanoparticles determined at pH 3 prior to mixing, and also for the binary particle mixtures following attempted reaction at pH 3 and pH 8. Prior to mixing, the (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 and PGMA60–PGlyMA100 nanoparticles each exhibit narrow particle size distributions. After mixing at pH 3, a hydrodynamic diameter of 75 nm is observed, which lies between the two initial particle diameters. This is because DLS is a relatively low resolution technique that is unable to resolve the two non-interacting populations and hence reports a weighted average in size. Following reaction at pH 8, DLS studies (determined at pH 3 to maximise repulsion between any unreacted particles) indicate a unimodal particle size distribution with a mean diameter of 97 nm. This increase in particle size suggests that the expected epoxy-amine reaction between the two types of particles has occurred, leading to the formation of composite particles. Given that the smaller nanoparticles are in excess, a core–shell type morphology might be anticipated, with the epoxy-functional nanoparticles forming the shell. To investigate this further, Fig. 7B shows the mobilities (determined at pH 3 by aqueous electrophoresis) obtained for the two types of nanoparticles prior to mixing, and also for the nanoparticle mixtures following reaction at pH 3 and pH 8. Prior to mixing, the (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 and PGMA60–PGlyMA100 nanoparticles exhibit unimodal mobilities of +2.2 μm and +1.5 μm cm V−1 s−1, respectively. Following attempted reaction at pH 3, a bimodal mobility distribution is observed, with the two peaks corresponding to the individual mobilities of the original nanoparticles. This suggests that no reaction has occurred under these conditions, as expected. In contrast, a single unimodal mobility of +1.4 μm cm V−1 s−1 is observed after reaction at pH 8. This is comparable to that of the PGMA60–PGlyMA100 nanoparticles alone, suggesting that these small epoxy-functional spheres now coat the surface of the larger primary amine-functional spheres. TEM images of the nanoparticle dispersions were recorded after drying at either pH 3 or pH 8. Following attempted reaction at pH 3, Fig. 7C clearly shows two distinct populations of essentially non-interacting nanoparticles, indicating that no epoxy-amine reaction has occurred. Fig. 7D shows the TEM image obtained after reacting at pH 8: the smaller PGMA60–PGlyMA100 spheres are clearly adsorbed at the surface of the larger (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 spheres. In summary, the primary amine-functionalised nanoparticles are cationic and unreactive at pH 3, whereas deprotonation at pH 8 leads to neutral nanoparticles that can be readily reacted with epoxy-functionalised nanoparticles.
 | ||
| Fig. 7 (A) Intensity-average particle size distributions and (B) electrophoretic mobilities obtained for PGMA60–PGlyMA100 (red trace) and (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 (blue trace) diblock copolymer spheres both separately and after mixing at pH 3 (black trace) and pH 8 (green trace). The corresponding TEM images obtained at (C) pH 3 and (D) pH 8 are also shown. The epoxy-amine reaction was conducted at 60 °C for 16 h at pH 8. | ||
Conclusions
The PISA synthesis of primary amine-functionalised diblock copolymer nanoparticles is reported. A poly(2-aminoethyl methacrylate) (PAMA) macro-CTA (mean DP = 96; Mw/Mn = 1.30) was chain-extended with 2-hydroxypropyl methacrylate (HPMA) at pH 6 using a RAFT aqueous dispersion polymerisation formulation. Systematic variation of the degree of polymerisation (DP = 100–1000) of the core-forming PHPMA block at 10% w/w solids leads to the formation of cationic spherical nanoparticles, with mean particle diameters ranging from 68 to 288 nm. At or above pH 9, deprotonation of the PAMA stabiliser chains occurs, which results in flocculation. This problem can be overcome by utilising a binary mixture of a cationic PAMA macro-CTA and a non-ionic PGMA macro-CTA, since the latter component confers steric stabilisation in alkaline media. Systematic variation of the molar ratio of these two macro-CTAs enables the cationic character of the resulting spherical nanoparticles to be fine-tuned. Up to 20 mol% PAMA stabiliser can be incorporated into the diblock copolymer nano-objects before flocculation is observed at high pH. Furthermore, synthesis of diblock copolymer nanoparticles with the general formula ([1 − n]PGMA60 + nPAMA96)–PHPMAz using this binary mixture of macro-CTAs enabled the reproducible formation of worms and vesicles at 20% w/w solids that are both cationic at low pH and colloidally stable at high pH. Increasing the PAMA96 mol fraction in these diblock copolymer nanoparticles results in greater cationic character but also favors the spherical morphology; incorporation of just 5 mol% (n = 0.05) PAMA stabiliser is sufficient to prevent access to the worm and vesicle morphologies. The reactivity of (0.8PGMA101 + 0.2PAMA96)–PHPMA1000 particles is demonstrated by the addition of model epoxy-functional nanoparticles prepared by RAFT aqueous emulsion polymerisation. No inter-particle reaction proceeds at pH 3 since the primary amine groups remain protonated under these conditions, but epoxy-amine reaction occurs on deprotonation of the PAMA stabiliser chains at pH 8, as evidenced by DLS, aqueous electrophoresis and TEM studies.Acknowledgements
EPSRC is thanked for a Platform grant (EP/J007846/1) which supported MW. SPA acknowledges a five-year ERC Advanced Investigator grant (PISA 320372). The three reviewers are thanked for their helpful comments, which have undoubtedly improved this manuscript.Notes and references
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- J. F. KukowskaLatallo, A. U. Bielinska, J. Johnson, R. Spindler, D. A. Tomalia and J. R. Baker, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4897–4902 CrossRef CAS.
- T. Basinska and S. Slomkowski, Colloid Polym. Sci., 1995, 273, 431–438 CAS.
- L. Nabzar, D. Duracher, A. Elaissari, G. Chauveteau and C. Pichot, Langmuir, 1998, 14, 5062–5069 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS PubMed.
- M. S. Wong, J. N. Cha, K. S. Choi, T. J. Deming and G. D. Stucky, Nano Lett., 2002, 2, 583–587 CrossRef CAS.
- E. P. Holowka, D. J. Pochan and T. J. Deming, J. Am. Chem. Soc., 2005, 127, 12423–12428 CrossRef CAS PubMed.
- R. H. Jin and J. J. Yuan, Chem. Commun., 2005, 1399–1401 RSC.
- J. J. Yuan and R. H. Jin, Adv. Mater., 2005, 17, 885–888 CrossRef CAS.
- A. S. Schenk, B. Cantaert, Y.-Y. Kim, Y. Li, E. S. Read, M. Semsarilar, S. P. Armes and F. C. Meldrum, Chem. Mater., 2014, 26, 2703–2711 CrossRef CAS.
- J. Cornelissen, E. F. Connor, H. C. Kim, V. Y. Lee, T. Magibitang, P. M. Rice, W. Volksen, L. K. Sundberg and R. D. Miller, Chem. Commun., 2003, 1010–1011 RSC.
- J.-J. Yuan, P.-X. Zhu, N. Fukazawa and R.-H. Jin, Adv. Funct. Mater., 2006, 16, 2205–2212 CrossRef CAS.
- J. J. Yuan and R. H. Jin, Langmuir, 2005, 21, 3136–3145 CrossRef CAS PubMed.
- R. Narain and S. P. Armes, Biomacromolecules, 2003, 4, 1746–1758 CrossRef CAS PubMed.
- E. S. Read, K. L. Thompson and S. P. Armes, Polym. Chem., 2010, 1, 221–230 RSC.
- W. N. E. van Dijk-Wolthuis, P. van de Wetering, W. L. J. Hinrichs, L. J. F. Hofmeyer, R. M. J. Liskamp, D. J. A. Crommelin and W. E. Hennink, Bioconjugate Chem., 1999, 10, 687–692 CrossRef CAS PubMed.
- K. L. Thompson, E. S. Read and S. P. Armes, Polym. Degrad. Stab., 2008, 93, 1460–1466 CrossRef CAS.
- M. H. Dufresne and J. C. Leroux, Pharm. Res., 2004, 21, 160–169 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- D. B. Thomas, B. S. Sumerlin, A. B. Lowe and C. L. McCormick, Macromolecules, 2003, 36, 1436–1439 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724–1730 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- A. Favier, M. T. Charreyre, P. Chaumont and C. Pichot, Macromolecules, 2002, 35, 8271–8280 CrossRef.
- Y. A. Vasilieva, D. B. Thomas, C. W. Scales and C. L. McCormick, Macromolecules, 2004, 37, 2728–2737 CrossRef CAS.
- A. W. York, C. W. Scales, F. Huang and C. L. McCormick, Biomacromolecules, 2007, 8, 2337–2341 CrossRef CAS PubMed.
- A. W. York, Y. Zhang, A. C. Holley, Y. Guo, F. Huang and C. L. McCormick, Biomacromolecules, 2009, 10, 936–943 CrossRef CAS PubMed.
- B. S. Sumerlin, M. S. Donovan, Y. Mitsukami, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 6561–6564 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, D. B. Thomas and C. L. McCormick, Macromolecules, 2003, 36, 5982–5987 CrossRef CAS.
- S. Yusa, Y. Shimada, Y. Mitsukami, T. Yamamoto and Y. Morishima, Macromolecules, 2003, 36, 4208–4215 CrossRef CAS.
- M. S. Donovan, T. A. Sanford, A. B. Lowe, B. S. Sumerlin, Y. Mitsukami and C. L. McCormick, Macromolecules, 2002, 35, 4570–4572 CrossRef CAS.
- M. S. Donovan, B. S. Sumerlin, A. B. Lowe and C. L. McCormick, Macromolecules, 2002, 35, 8663–8666 CrossRef CAS.
- M. S. Donovan, A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Macromolecules, 2002, 35, 4123–4132 CrossRef CAS.
- K. E. B. Doncom, A. Pitto-Barry, H. Willcock, A. Lu, B. E. McKenzie, N. Kirby and R. K. O'Reilly, Soft Matter, 2015, 11, 3666–3676 RSC.
- L. He, E. S. Read, S. P. Armes and D. J. Adams, Macromolecules, 2007, 40, 4429–4438 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2009, 42, 939–945 CrossRef CAS.
- A. H. Alidedeoglu, A. W. York, C. L. McCormick and S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5405–5415 CrossRef CAS.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS PubMed.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jerome, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- K. Bauri, A. Narayanan, U. Haidar and P. De, Polym. Chem., 2015, 6, 6152–6162 RSC.
- S. Boisse, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- S. Boisse, J. Rieger, G. Pembouong, P. Beaunier and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3346–3354 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- N. J. W. Penfold, J. R. Lovett, N. J. Warren, J. Smets and S. P. Armes, Polym. Chem., 2015 10.1039/C5PY01510C.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- G. H. Posner and D. Z. Rogers, J. Am. Chem. Soc., 1977, 99, 8208–8214 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Dynamic light scattering data for a series of PAMA96–PHPMAz diblock copolymer particles, conversion vs. time curve for a PGMA macro-CTA synthesis, evolution of the number-average molecular weight with conversion of a PGMA macro-CTA, DMF GPC curves for PGMA macro-CTAs, hydrodynamic diameter vs. pH curves for a series of diblock copolymer particles with the general formula ([1 − n]PGMA101 + nPAMA96)–PHPMA1000, and zeta potential vs. pH curves for diblock copolymer particles with the formula ([1 − n]PGMA60 + nPAMA96)–PHPMA200 and ([1 − n]PGMA60 + nPAMA96)–PHPMA500. See DOI: 10.1039/c5py01577d |
| This journal is © The Royal Society of Chemistry 2016 |