
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled radical polymerization and in-depth mass-spectrometric characterization of poly(ionic liquid)s and their photopatterning on surfaces†
Jan
Steinkoenig
ab,
Fabian R.
Bloesser
ab,
Birgit
Huber
c,
Alexander
Welle
ab,
Vanessa
Trouillet
d,
Steffen M.
Weidner
e,
Leonie
Barner
c,
Peter W.
Roesky
f,
Jiayin
Yuan
*g,
Anja S.
Goldmann
*ab and
Christopher
Barner-Kowollik
*abc
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany
bInstitut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: christopher.barner-kowollik@kit.edu; anja.goldmann@kit.edu
cSoft Matter Synthesis Laboratory, Institut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dInstitute for Applied Materials (IAM-ESS) and Karlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
eBAM-Federal Institute for Materials Research and Testing, Richard-Willstätter-Straße 11, 12489 Berlin, Germany
fInstitut für Anorganische Chemie, Karlsruhe Institute of Technology (KIT), 76128, Karlsruhe, Germany
gMax-Planck-Institute of Colloids and Interfaces, Research Campus Golm, 14424 Potsdam, Germany. E-mail: Jiayin.Yuan@mpikg.mpg.de
First published on 26th October 2015
Abstract
The preparation and characterization of poly(ionic liquid)s (PILs) bearing a polystyrene backbone via reversible addition fragmentation chain transfer (RAFT) polymerization and their photolithographic patterning on silicon wafers is reported. The controlled radical polymerization of the styrenic ionic liquid (IL) monomers ([BVBIM]X, X = Cl− or Tf2N−) by RAFT polymerization is investigated in detail. We provide a general synthetic tool to access this class of PILs with controlled molecular weight and relatively narrow molecular weight distribution (2000 g mol−1 ≤ Mn ≤ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 with dispersities between 1.4 and 1.3 for p([BVBIM]Cl); 2100 g mol−1 ≤ MP ≤ 14000 g mol−1 for p([BVBIM]Tf2N)). More importantly, we provide an in-depth characterization of the PILs and demonstrate a detailed mass spectrometric analysis via matrix-assisted laser desorption ionization (MALDI) as well as – for the first time for PILs – electrospray ionization mass spectrometry (ESI-MS). Importantly, p([BVBIM]Cl) and p([DMVBIM]Tf2N) were photochemically patterned on silicon wafers. Therefore, a RAFT agent carrying a photoactive group based on ortho-quinodimethane chemistry – more precisely photoenol chemistry – was photochemically linked for subsequent controlled radical polymerization of [BVBIM]Cl and [DMVBIM]Tf2N. The successful spatially-resolved photografting is evidenced by surface-sensitive characterization methods such as X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (ToF-SIMS). The presented method allows for the functionalization of diverse surfaces with poly(ionic liquid)s.
000 g mol−1 with dispersities between 1.4 and 1.3 for p([BVBIM]Cl); 2100 g mol−1 ≤ MP ≤ 14000 g mol−1 for p([BVBIM]Tf2N)). More importantly, we provide an in-depth characterization of the PILs and demonstrate a detailed mass spectrometric analysis via matrix-assisted laser desorption ionization (MALDI) as well as – for the first time for PILs – electrospray ionization mass spectrometry (ESI-MS). Importantly, p([BVBIM]Cl) and p([DMVBIM]Tf2N) were photochemically patterned on silicon wafers. Therefore, a RAFT agent carrying a photoactive group based on ortho-quinodimethane chemistry – more precisely photoenol chemistry – was photochemically linked for subsequent controlled radical polymerization of [BVBIM]Cl and [DMVBIM]Tf2N. The successful spatially-resolved photografting is evidenced by surface-sensitive characterization methods such as X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (ToF-SIMS). The presented method allows for the functionalization of diverse surfaces with poly(ionic liquid)s.
Introduction
Ionic liquids (ILs) are organic salts with a melting point typically below 100 °C. They have found significant applications in various fields ranging from chemistry to physics and materials science.1 Recently, the polymerization of ILs, which forms a new class of ionic polymers, so called poly(ionic liquid)s (PILs), has emerged as a multifunctional platform that combines the advantages of ILs with the general property profile of polymers.2–6 PILs are not only a natural extension of properties and function of ILs in materials science, but significantly advance the research scope of ionic polymers. For example, in spite of a high ion-pair packing density, the favorable solubility in a broad range of organic solvents and the achievable low glass transition temperature of structurally well-defined PILs offer new processing possibilities to fabricate ionic polymers for numerous functional materials.Currently, the chemical structure of polymerizable ILs is dominated by the imidazolium type, which can be further classified into three major subgroups, i.e. the vinylimidazolium, (meth)acrylate, and styrenic ones. In the past several years, a wide range of polymerization techniques have been exploited to synthesize imidazolium-type PILs with defined structure characteristics. Among them, reversible deactivation radical polymerization techniques, such as the atom transfer radical polymerization (ATRP), and reversible addition–fragmentation chain-transfer (RAFT) polymerization are frequently used.7–12 These radical polymerization techniques allow for superior control over the polymer chain characteristics and the endgroup functionalization.
PILs bearing styrenic type IL monomer units have a rigid polystyrene backbone that can provide mechanical stability to lock the highly ionic polymer matrix. These PILs are promising candidates as support for IL networks. For example, covalent porous IL networks are often generated from styrenic type IL monomers.13 Additionally, PIL membrane materials for gas separation or sorption take advantage of the rigid polystyrene backbone to enhance the shape durability of membranes and the gas flux rate.14 Preparation of such PILs from styrenic IL monomers is a synthetic task that usually involves free radical polymerization, which is easy to implement and important for large scale production. Yet, to understand the intrinsic structure–property-function relationship for further optimization of their materials performance, PILs with defined chain length and narrow molecular weight distribution are absolutely necessary. In literature, ATRP of a styrenic IL monomer has been reported by the groups of Matyjaszewski and Gin.8,15 Both groups demonstrated that via ATRP, the PIL chain characteristics are tuneable and their block copolymers are accessible. An inherent problem in the polymerization of IL monomers by ATRP is, however, the inevitable ion exchange process between the required metal catalyst, copper halide and the IL monomer. Further, Cu ions may form complex anions with certain IL anions,16 which makes it even more challenging to be removed from the polymer product. Therefore, in spite of excellent control over the polymerization process via ATRP, the field is motivated to search for alternative polymerization techniques to overcome these challenges. One solution is to apply RAFT polymerization that generally involves no metal salt. A study by Tenhu and coworkers17 reported briefly that it is possible to polymerize a specific styrenic type chiral IL monomer, yet the RAFT polymerization of more representative and commonly used styrenic IL monomers, such as 1-butyl-3-(p-vinylbenzyl)-imidazolium-based IL monomers was not presented. Recently, Detrembleur and coworkers18 demonstrated the preparation of vinylimidazolium-based block PILs featuring two different counter ions via RAFT polymerization.
The precise characterization of PIL chain features, in general, is very challenging. Due to the strong interactions with the column materials, size exclusion chromatography results in broadened traces, or in a complete absorption on the column surface. Different teams pioneered measuring PILs with different counter ions by SEC.19,20 However, to date, only little efforts towards the detailed mass spectrometric analysis of these materials have successfully been carried out.8,9,21 Motivated by the current status, we initiated the project on RAFT polymerization of frequently used styrenic IL monomers and provide a general synthetic protocol to access these PILs with controlled molecular weight, narrow molecular weight distribution, defined chain endgroup and no contamination from metal ion species. More precisely, we investigate the controlled radical polymerization of 1-butyl-3-(p-vinylbenzyl)-1H-imidazolium chloride ([BVBIM]Cl, (1)) and 1-butyl-3-(p-vinylbenzyl)-1H-imidazol-3-ium bis(trifluoromethanesulfonyl)imide ([BVBIM]Tf2N, (2)) (Scheme 1) via RAFT polymerization and provide a comprehensive characterization of the polymers by various (mass spectrometric) analysis methods such as SEC, MALDI-ToF MS as well as ESI-MS. To the best of our knowledge, the mass spectrometric characterization via ESI-MS is shown for the first time in-depth.
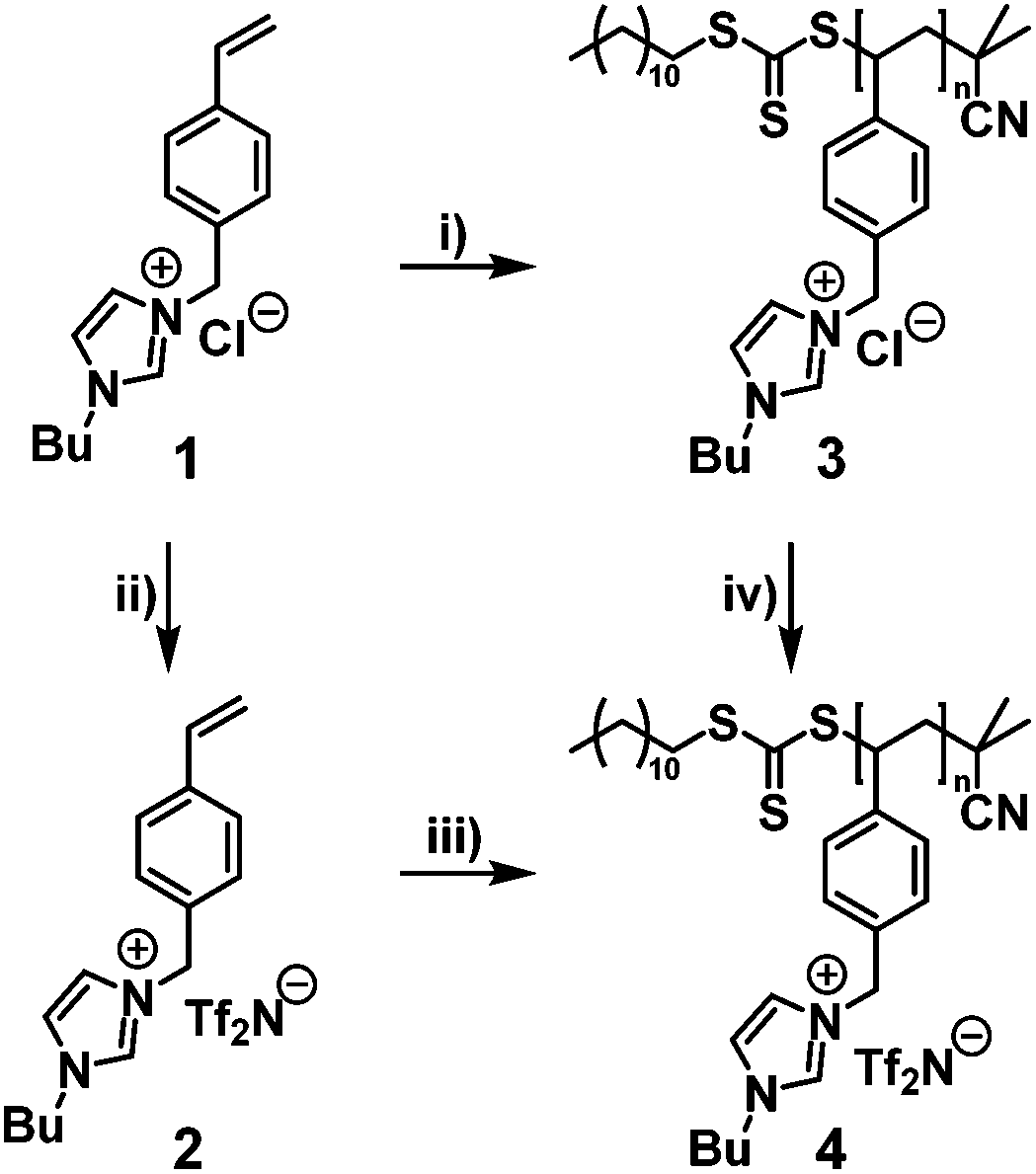 | ||
| Scheme 1 Synthetic approach for the preparation of p([BVBIM]Cl) (3) and p([BVBIM]Tf2N) (4) from the monomers [BVBIM]Cl (1) and [BVBIM]Tf2N (2), respectively. Molar ratio [M]:[CTA]:[I] for (i) 200:1:0.2 and for (iii) 200:1:0.2 (i: RAFT polymerization; ii: Tf2N−vs. Cl− anion exchange; iii: RAFT polymerization; iv: anion exchange). | ||
In addition, surface design is a rapidly growing field in contemporary chemistry. PIL-decorated surfaces can serve as imaging, wetting/dewetting, and anti-biofouling interfaces.8,22,23 Initiated by the pioneering work of Ye et al. on grafted PILs via ROMP on TiO2 substrates and the positive anti-fouling studies,23 Matyjazewski and coworkers thermally attached well-characterized, ATRP-prepared PILs on Si substrates that potentially could show anti-fouling characteristics.8 Furthermore, Drockenmueller and coworkers patterned azide-functionalized copolymerized PILs via UV irradiation and subsequent formation of highly reactive nitrenes on a silicon substrate.24 Herein, we present a polymerization study of p([BVBIM]Cl) and p([BVBIM]Tf2N) in order to investigate the mass spectrometric behavior of PILs in solution. Furthermore, we study the characterization of photochemically patterned p([BVBIM]Cl) and p[1,2-dimethyl-3-(p-vinylbenzyl)-1H-imidazol-3-ium bis(trifluoro methanesulfonyl)imide] (p([DMVIM]Tf2N)) via a surface sensitive mass spectrometric analytical method, i.e. time-of-flight secondary ion mass spectrometry (ToF-SIMS) as well as X-ray photoelectron spectroscopy (XPS).
Results and discussion
Among other versatile features of the RAFT process,25 the high tolerance towards functional monomers e.g. acrylic acids or vinyl esters, is an important characteristic. It is also expected to be a suitable candidate for the controlled radical polymerization of monomeric ILs and thus applied in the presented work. Scheme 1 summarizes the synthetic strategy for the preparation of the polymers p([BVBIM]Cl) (3) and p([BVBIM]Tf2N) (4). It should be noted that the synthesis and properties of 1-alkyl-3-(p-vinylbenzyl)-imidazolium based PILs was widely investigated,8,15,26 yet the RAFT process of the corresponding monomers remains a challenge.The polymerization of these ILs can either be achieved by the direct polymerization of [BVBIM]Tf2N (2) or by the polymerization of [BVBIM]Cl (1) followed by a counter ion exchange with Tf2N−. Both monomers [BVBIM]Cl (1) and [BVBIM]Tf2N (2) were synthesized by a quaternization reaction between 4-vinylbenzyl chloride and n-butylimidazole with a slightly modified synthesis protocol given in the literature.27 A counter ion exchange with Tf2N− resulted in 2. The polymerization of [BVBIM]Cl (1) was performed in DMSO as solvent of choice ensuring a good dissolution of both the IL monomer (1) and 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT) as chain transfer agent (CTA). A molar ratio of the reagents [M]:[CTA]:[I] = 200:1:0.2 (Initiator: 1,1′-azobis(cyclohexanecarbonitrile)) was used. Interestingly, the RAFT polymerization of [BVBIM]Cl (1) is highly affected by the choice of the solvent. For instance, protic solvents, such as ethanol, methanol and water, led to a less controlled polymerization (refer to Table S1 in the ESI†). The polymerization of [BVBIM]Tf2N (2) was performed in butyronitrile as solvent due to its favorable solubility in non-polar/aprotic solvents. In general, the common controlled polymerization techniques, i.e. ATRP, RAFT and NMP, are complex and challenging for IL monomers such as 1 and 2 because of their ionic character.20 The RAFT polymerization of [BVBIM]Cl (1) was carried out employing CPDT as chain transfer agent in DMSO at 85 °C and VAZO-88 (kd = 2.01 × 105 s−1)28 as radical starter. The molecular weight evolution of the SEC traces in Fig. 1(A) verifies the living character. Both the 1H NMR (Fig. S11†) as well as the SEC results are in good agreement with the theoretically obtained molecular weights (Fig. 1(B): solid line). P([BVBIM]Cl) (3) is a charged polymer and thus interacts strongly with the column material and its polymer chains via hydrogen bonding of the chloride counter ion. As a consequence, halide-containing PILs prepared via RAFT polymerization often display a rather broad dispersity in a H2O–SEC system.9
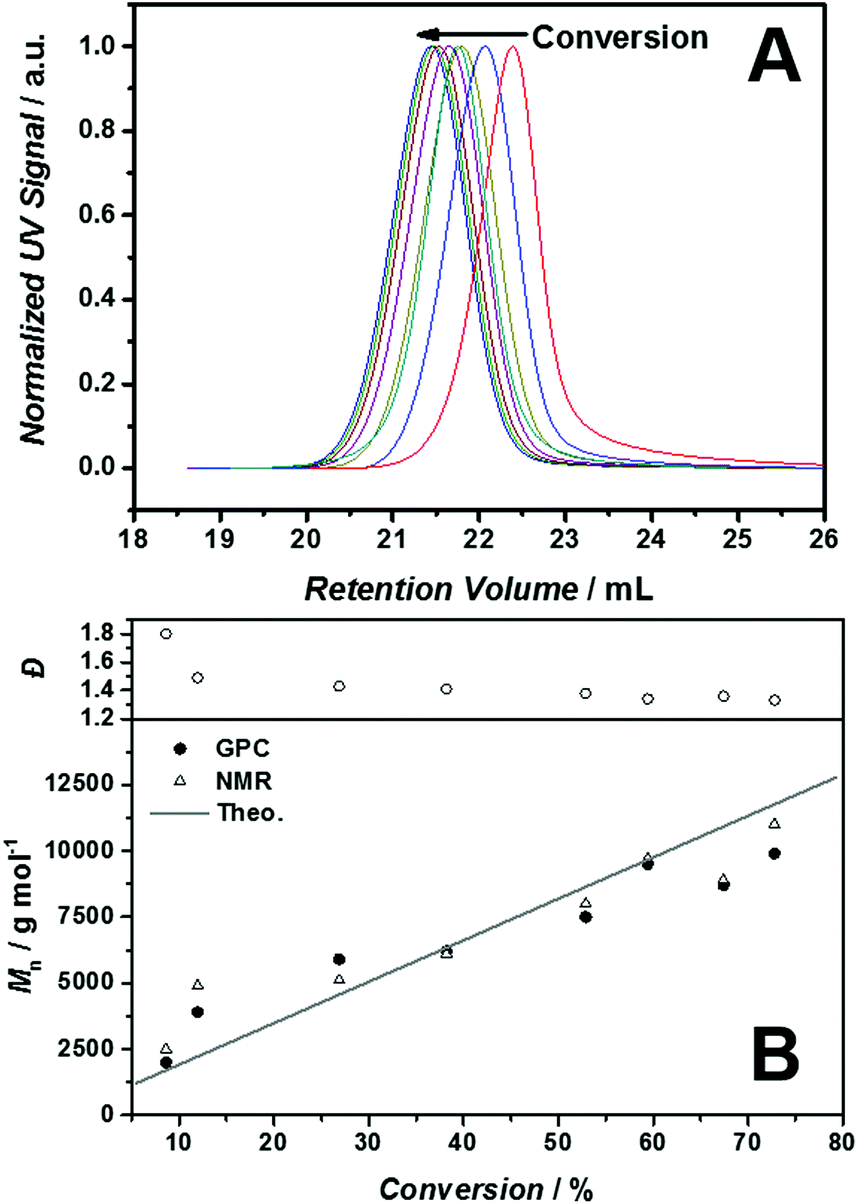 | ||
| Fig. 1 (A) SEC traces of p([BVBIM]Cl) (3) via the RAFT mediated polymerization of [BVBIM]Cl (1) at 85 °C in DMSO. (B) Plot of molecular weight (Mn) and dispersity (Đ) vs. monomer conversion. Molar ratio of monomer:CTA:initiator 200:1:0.2 (CTA = 2-cyano-2-propyl dodecyl trithiocarbonate; initiator = 1,1′-azobis(cyclohexanecarbonitrile)). H2O–SEC system conditions: water/0.3 M formic acid/0.5 g L−1 NaCl, 30 °C, flow rate 1.0 mL min−1. | ||
Thus, the optimum system for the SEC analysis has to be identified to suppress the intermolecular polymer and column interactions. In order to investigate the most appropriate eluent, the SEC measurements were carried out by variation of the polymer concentrations (in mg mL−1) as well as sodium chloride salt concentrations (refer to Fig. S1†). The H2O–SEC system with a water/0.3 M formic acid/0.5 g L−1 NaCl mixture as eluent was identified as ideal system for the polymeric system (3). The system was calibrated against poly(4-vinylpyridine), which forms a polycation under SEC conditions and thus allows for a more accurate estimation of the molecular weight of our PILs. Furthermore, we have demonstrated that the dispersity decreases after successful salt metathesis of the corresponding p([BVBIM]Cl) (3) (Mn = 8900 g mol−1; Đ = 1.55; DPn = 37) against LiTf2N resulting in p([BVBIM]Tf2N) (4) with a dispersity of 1.2 (SEC analysis with an eluent containing 10 mM LiTF2N and 10 mM n-butylimidazole). The calibration against polystyrene was in good agreement to the results obtained with the H2O–SEC system (refer to Fig. S1†).
The polymerization of the Tf2N− containing monomeric IL 2 can be achieved in a similar synthetic approach. CPDT was used as chain transfer agent in butyronitrile at 85 °C and 1,1′-azobiscyclohexane-carbonitrile as initiator.
Fig. 2(A) shows the molecular weight evolution for the RAFT polymerization of [BVBIM]Tf2N (4) (10 mmol LiTf2N, 10 mmol n-butylimidazole, 35 °C, flow rate 1.0 mL min−1). The controlled molecular weight evolution is depicted in Fig. 2(B). The determination of molecular weights of the polymers (Mn < 10000 g mol−1) is hampered by the presence of residual monomer. Precipitation in methanol/water was not successful, also dialysis in water (3 d at ambient temperature as well as for 14 d at elevated temperatures of close to 40 °C) did not result in pure PILs. The dialysis in THF or acetone according to a literature method8 may work for high-molecular weight PILs, however failed for our low-molecular weight polymers (due to diffusion out of the membrane). The dialysis in DMSO was successful for certain polymer samples, however, we were not able to implement the DMSO purification as a standard dialysis for Tf2N− containing PILs. Thus, the polymer signals and the monomer signal overlap in the SEC trace. The overlap is most likely due to the SEC column system in our laboratory that differs slightly from the SEC column system employed by Matyjaszewski and coworkers.8,20
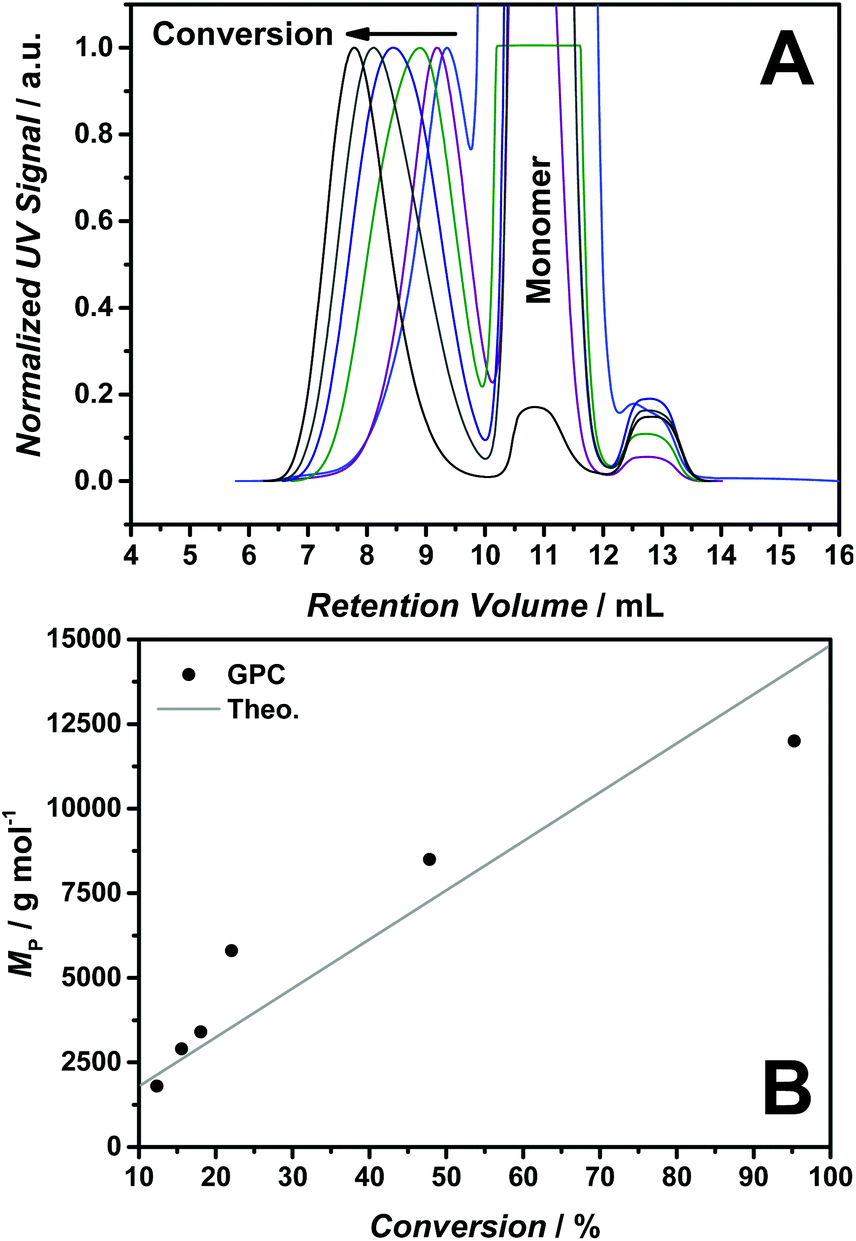 | ||
| Fig. 2 (A) Molecular weight evolution of the RAFT polymerization of [BVBIM]Tf2N (2) at 85 °C in butyronitrile. (B) Molecular weight evolution (MP) with conversion for the RAFT mediated polymerization of 2. Molar ratio [M]:[CTA]:[I] = 200:1:0.2, 85 °C, butyronitrile (CTA: 2-cyano-2-propyl dodecyl trithiocarbonate; I = 1,1′-azobiscyclohexane carbonitrile). THF–SEC system conditions: 10 mmol LiTf2N, 10 mmol n-butylimidazole, 35 °C, flow rate 1.0 mL min−1. | ||
Thus, the peak of the molecular weight (MP) was correlated with the conversion indicating a linear trend. However, the dispersity (Đ) (due to the overlap of the polymer signal and the monomer signal) was not determined and also the Mn determination via NMR spectroscopy was hampered by the presence of the residual monomer.
Table 1 provides an overview of the molecular weights calculated according to the monomer conversion, as well as the molecular weights obtained by NMR spectroscopy and SEC for 3 and 4. Salt exchange against bis(trifluoromethanesulfonyl)imide (Tf2N−) converts the water-soluble polymers into highly hydrophobic ones that are soluble in solvents such as THF, DMAc and acetone.29 Matyjaszewski and co-workers have shown that such PILs can easily be characterized by SEC with THF as eluent containing a small concentration of the appropriate counter ion.20
:[CTA]:[I] = 200:1:0.2, DMSO. Conditions for 4: 85 °C, CPDT, VAZO-88, [M]:[CTA]:[I] = 200:1:0.2, butyronitrile
| Sample | Time [h] | M n theo. [g·mol−1] | M n NMR [g·mol−1] |
M
n
SEC* [g·mol−1] |
Đ SEC | Conv.NMR [%] |
|---|---|---|---|---|---|---|
| *For samples 3: MnSEC [g mol−1]; for samples 4: MpSEC [g mol−1].a H2O–SEC, eluent: water/0.3 M formic acid/0.5 g L−1 NaCl; temperature: 30 °C; flow rate: 1.0 mL min−1.b THF–SEC, eluent: THF/10 mM LiTf2N/10 mM n-butylimidazole; temperature: 35 °C; flow rate: 1.0 mL min−1. | ||||||
| 3-1 | 1 | 1700 | 2500 | 2000a | 1.80a | 9 |
| 3-2 | 2 | 2300 | 4900 | 3900a | 1.49a | 12 |
| 3-3 | 3 | 4500 | 5100 | 5900a | 1.43a | 27 |
| 3-4 | 4 | 6400 | 6100 | 6200a | 1.41a | 38 |
| 3-5 | 5 | 8600 | 8000 | 7500a | 1.38a | 53 |
| 3-6 | 6 | 9600 | 8900 | 8700a | 1.36a | 59 |
| 4-1 | 3 | 2100 | n.d. | 1800b | n.d. | 12 |
| 4-2 | 4 | 2600 | n.d. | 2900b | n.d. | 16 |
| 4-3 | 6 | 3000 | n.d. | 3400b | n.d. | 18 |
| 4-4 | 7 | 3500 | n.d. | 5800b | n.d. | 22 |
| 4-5 | 8 | 7300 | n.d. | 8500b | n.d. | 48 |
| 4-6 | 24 | 14000 |
n.d. | 12000b |
n.d. | 95 |
In contrast to SEC, mass spectrometry is often employed in order to characterize the polymer chain structure. Nevertheless, mass spectrometric analysis of PILs is a considerable challenge because of their ionic character.4 In particular when endgroup analysis is required, the determination of the exact mass is a crucial step for a detailed characterization of the synthesized materials, i.e. for subsequent post-modification reactions.30 Electrospray ionization mass spectrometry (ESI-MS)31 and matrix assisted laser desorption ionization (MALDI)32 are the methods of choice for an in-depth characterization of the molecular structure of 3 and 4. Tenhu and coworkers17 used MALDI-ToF MS characterization to identify the molecular weight as well as the dispersity of their prepared PILs.
Although we could not reproduce the MALDI-ToF MS measurements for our systems with the conditions given (refer to the ESI Fig. S3†), we obtained a mass spectrum of polymer 3-1 (Mn = 2000 g mol−1) by employing the non-acidic matrix material trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malonitrile (DCTB) (Fig. 4) and methanol as solvent. In contrast to ESI-MS, MALDI-ToF MS has the advantage to form mainly single charged species. The formation of single charged species is crucial for the detection of polymer (3) as during the ionization process different side reactions can occur (Fig. 3).
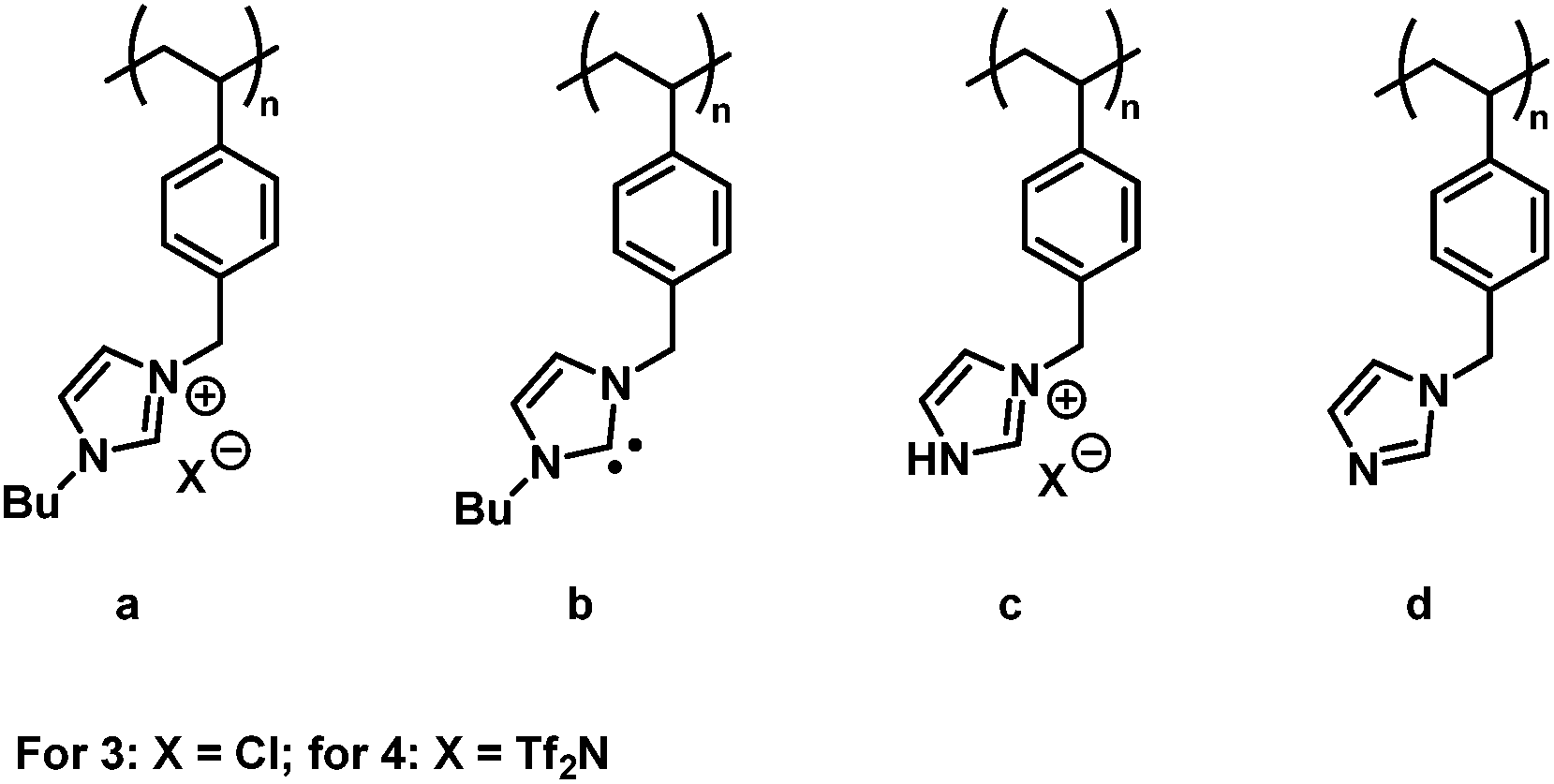 | ||
| Fig. 3 Representation of possible different side chain transformations occurring during the ESI or MALDI ionization process for 3: (3a) the side chain still represents the IL repeating unit; (3b) hydrogen chloride is released forming a N-heterocyclic carbene (NHC); (3c) degradation of the n-butyl side chain; and (3d) degradation of the n-butyl side chain by release of hydrogen chloride forming an imidazole. | ||
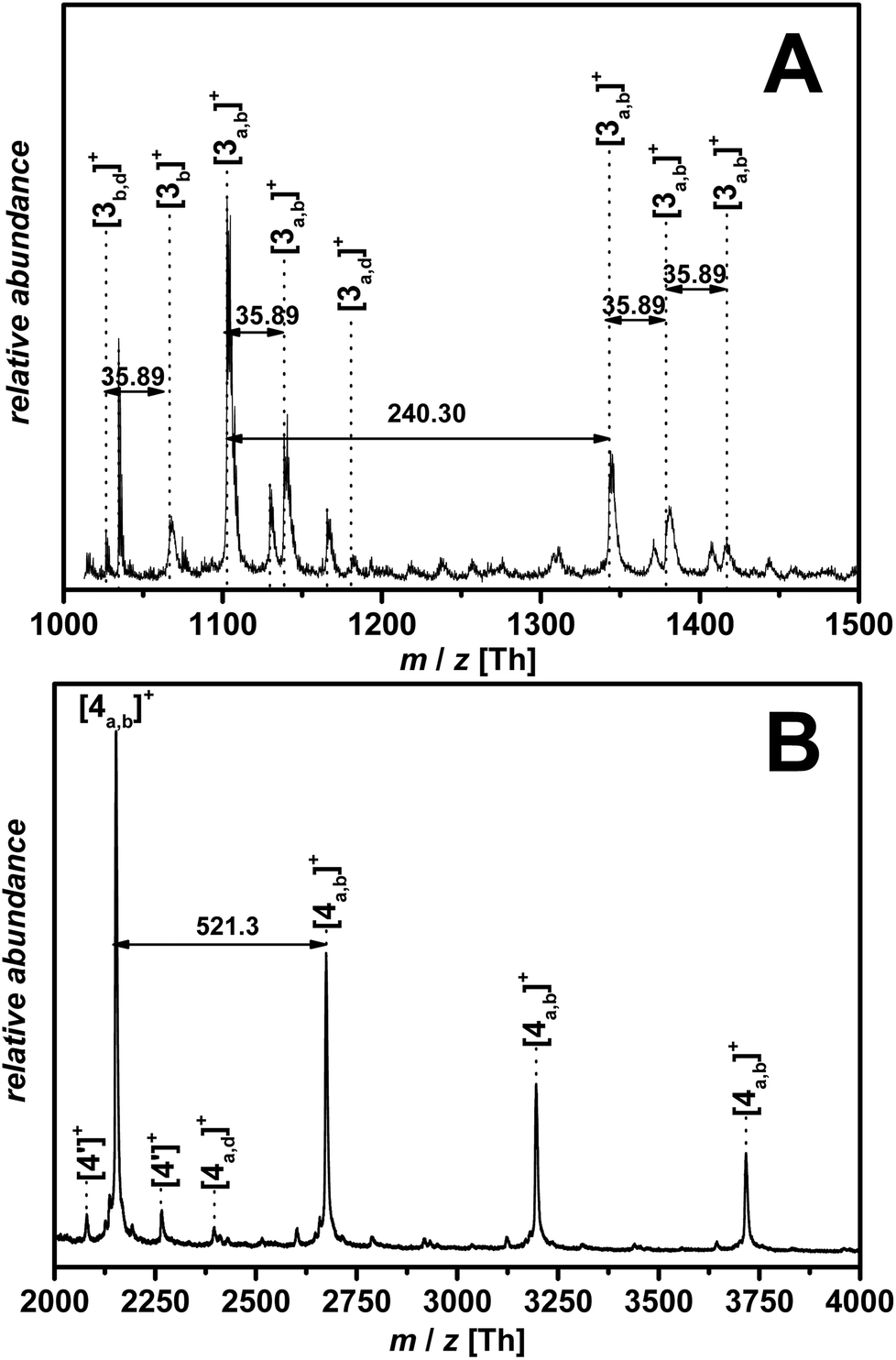 | ||
| Fig. 4 Zoom of the MALDI-ToF spectrum of (A) p([BVBIM]Cl) (3-1, Table 1) (Mw = 1900 g mol−1) and (B) p([BVBIM]Tf2N) (4) in the representative area of the mass spectrum with the assigned peaks employing DCTB as matrix material, respectively. Please refer to Tables S2 and S3 in the ESI† for the list of assignments. | ||
The high affinity of chloride acting as a H-bond acceptor is responsible for the unexpected ionization process.33 According to the mass spectrometric data, the chloride counter ion is prone to be separated from the polymer chain, resulting in a N-heterocyclic carbene as illustrated in (3b) with m/z = 240.2 Th.34 Furthermore, in accordance with the work by Kroon et al.,35 ILs bearing chloride as counter ion release alkyl chloride at elevated temperature (approx. 250 °C in the case of [BMIM]Cl). Thus, 3c and 3d were detected as minor products during mass spectrometric analysis. The signals of the highest intensity (for single charged species) can be assigned to a mixture of species a and b (refer to Fig. 3). The detection of the polymer is attributed – in most of the cases – to the protonation of only one protonated carbene species b in the polymer chain while the other repeating units stay in the carbene form. In some cases, however, the ionization of only one repeating unit of the neutral polymer chain is induced by the sodium ion (refer to the structures in Table S4 and S5†). In contrast to ESI-MS analysis, the molecular weight distribution obtained by MALDI-ToF MS analysis is limited by the dispersity of the polymer (Đ < 1.2).36 Thus, only low molecular weight PIL polymers of rather narrow dispersities are characterized with MALDI-ToF MS. Interestingly, the linear mode of MALDI-ToF MS revealed a better resolution compared to the reflectron mode. This observation may be due to the broad dispersity of the sample whereas the reflectron mode normally enhances the resolution in the case of a narrowly distributed polymer.
Fig. 4(A) and 6(A) illustrate the MALDI-ToF as well as the ESI-MS spectrum obtained for p([BVBIM]Cl) (3). Due to the different side products formed during the measurements, the mass spectrometric analysis of PILs bearing chloride counter ions is highly restricted to the chain length. Thus, only very low molecular weights (approx. Mw = 2000 Da) were observed in spectra with appropriately separated signals.
Due to the preference to form single charged species during MALDI-ToF MS analysis, the separation of hydrogen chloride from the polymer chain can be found in a MALDI-ToF spectrum (m/z = 35.8 Th). Furthermore, also the endgroup of the polymer (initiator of RAFT agent) or the degradation of the trithiocarbonate to the corresponding thiol can be found in the ESI-MS spectrum (marked with ′ for a thiol and * for the initiator as endgroup, Fig. 6(A)). In order to identify the different endgroup combinations of a RAFT polymerization mixture, it is necessary to analyze the polymers with MALDI-ToF MS. The value of the repeating unit (m/z = 240.3 Th) between two [3a,b]+ peaks is in good agreement with the theoretical value (m/z(theo.) = 240.2 Th). As already discussed, the chloride degrades the alkyl chain ([3b,d]+ and [3a,d]+), however, these signals are of minor intensity (Fig. 4(A)).
Also, a RAFT endgroup conversion was studied dispersing the polymer in THF for three days. After three days, THF was evaporated, the polymer dissolved in methanol and mixed with the matrix in a ratio of 10:1 (matrix: sample). Clearly, THF reacted with the RAFT moiety38 as indicated by the MALDI-ToF MS measurements with the identified peaks [3 OH] and [3 SO2] (refer to the ESI Fig. S4† and Fig. 5).
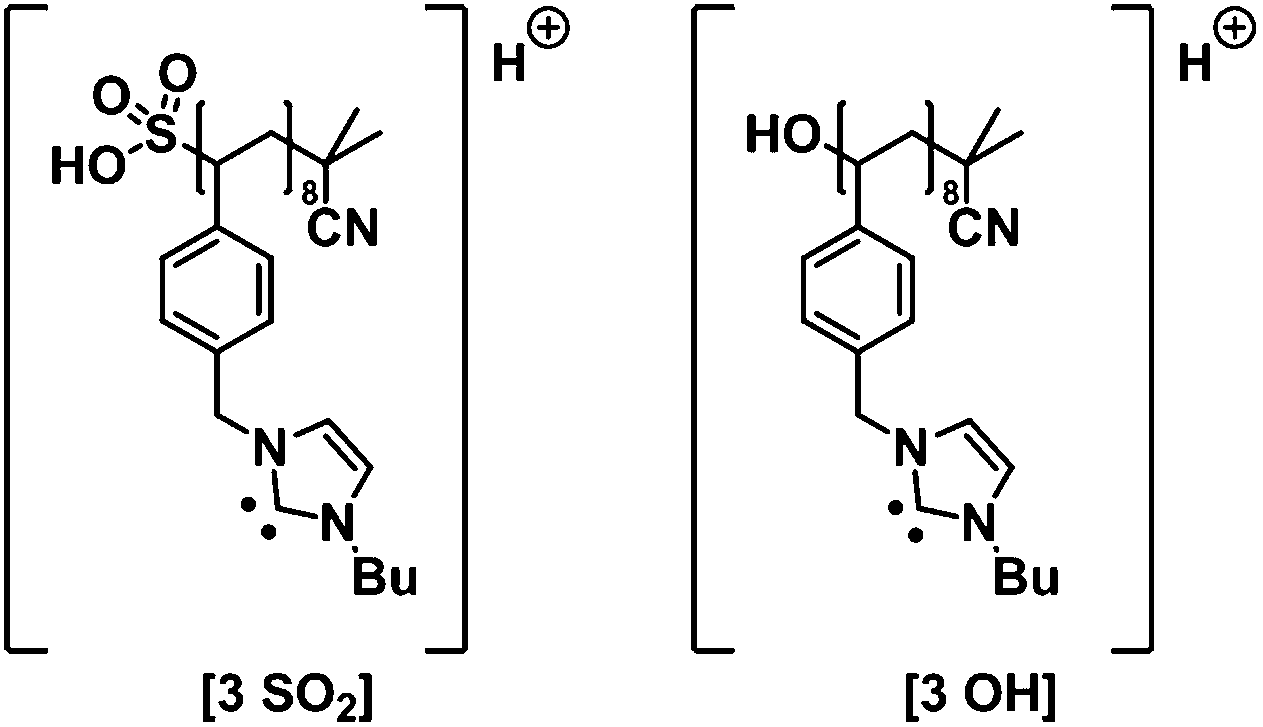 | ||
| Fig. 5 Chemical structures assigned in the MALDI spectrum of p([BVBIM]Cl) (3) (Mw = 1900 g mol−1). PIL structure with a sulfonic acid endgroup and a hydroxyl endgroup.37 The m/z difference of both peaks originates from the loss of SO2. | ||
Most evident are two adjacent peaks with a m/z difference of 64 Th. According to Benicewicz and coworkers,37 trithiocarbonates are prone to be oxidized to the corresponding sulfonic acid in THF after storage for 48 h, and to release gaseous SO2 (64 Th) to form the hydroxyl endgroup (as matrix the team employed dithranol doped with 0.01 M AgTFA). As observed for methanol as solvent, gaseous HCl is released during the ionization process, resulting in an uncharged polymer.
Fig. 4(B) and 6(B) illustrate the MALDI-ToF as well as the ESI-MS spectrum obtained for p([BVBIM]Tf2N) 4. In contrast to the counter ion Cl− of (3) that shows a high H-bond acceptor affinity, the weakly coordinating counter ion Tf2N− of 4 does not interact with the protons of the imidazolium ring. The repeating unit between two [4]+ peaks (m/z = 521.1 Th) agrees well with the theoretical value (m/z(theo.) = 521.1 Th).
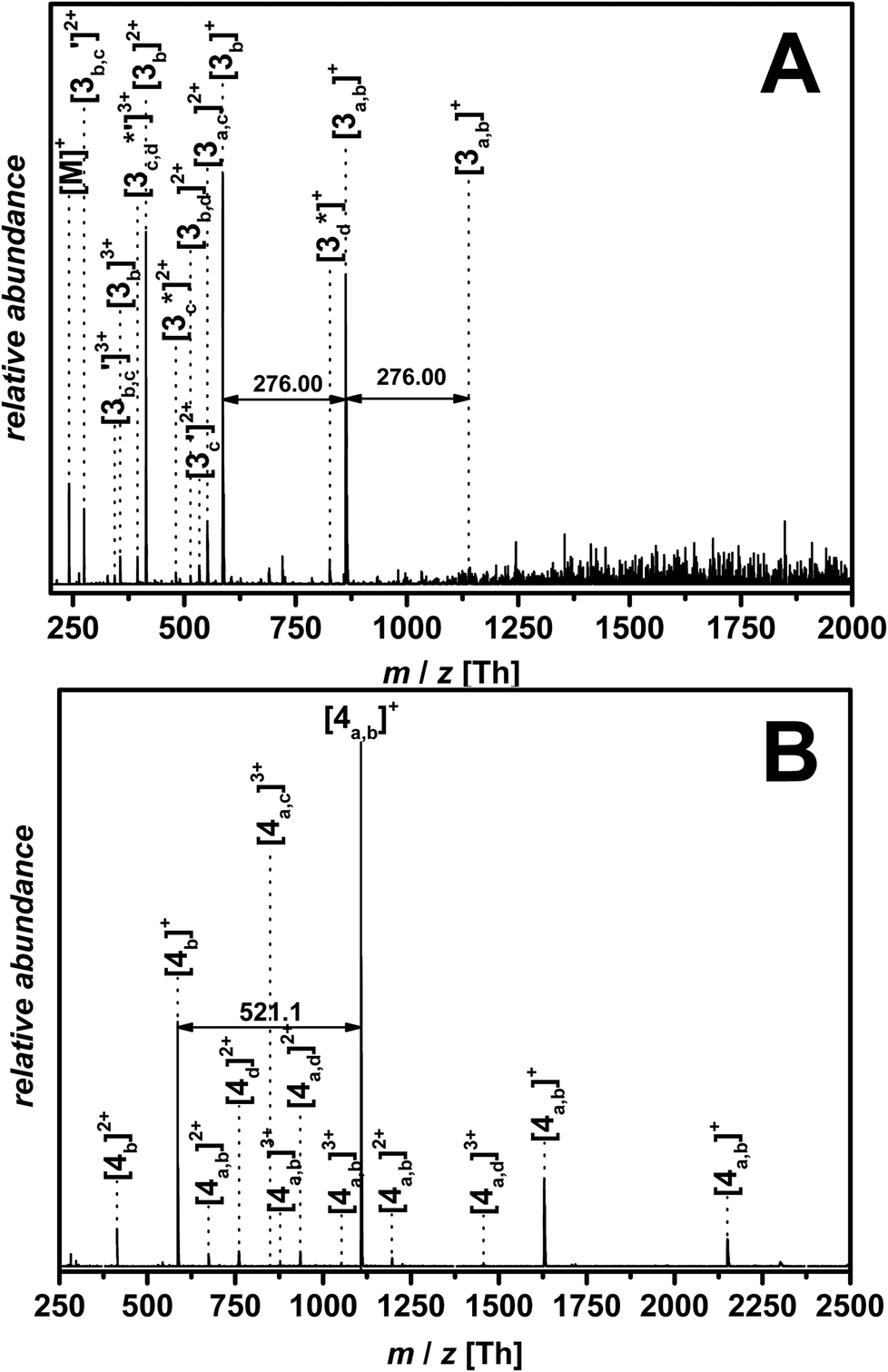 | ||
| Fig. 6 Zoom of ESI-MS spectra of (A) p([BVBIM]Cl) (3) with a molecular weight of Mw = 1900 g mol−1 measured in THF/MeOH (3:2, v/v) and (B) p([BVBIM]Tf2N) (4) measured in a mixture of acetonitrile/water (1:1, v/v). Please refer to the ESI† for a list of the assignments (Tables S4 and S5†). | ||
As observed in the MALDI-ToF measurements as well, the quality of the ESI-MS measurement correlates with the molecular weight of the polymer. The repeating unit of p([BVBIM]Cl) (3) is present in four species (please refer to Fig. 3). The permutation of these processes together with the increasing chain length, multiple charges, and whether a proton or a sodium ion ionizes the polymer, results in high amounts of detectable ions. However, the degradation of the alkyl side chain (Fig. 3c and d) is observed with a limited probability. Thus, it is not surprising that – especially for high-molecular weight polymers – the spectrum is complex. The assignment of the dominant peaks is summarized in Table S4 in the ESI.†
Furthermore, the Coulomb interactions of Tf2N− appear to be stronger compared to Cl− and thus ESI-MS measurements lead to clear spectra (Fig. 6(B)). The signal difference of m/z = 521.1 Th is assigned to the molecular weight of the monomer unit (m/z(theo.) = 521.1 Th). The thiocarbonyl thio endgroup of 4 is clearly identified (see Table S5†). The ESI-MS analysis of the PIL bearing Tf2N− as counter ion is easier to evaluate since there are far less side events affecting the polymer chain during the ionization process compared to p([BVBIM]Cl) (3) polymers.
To demonstrate the versatility of the employed approach, [BVBIM]Cl (1) and another monomer, i.e. 1,2-dimethyl-3-(p-vinylbenzyl)-1H-imidazol-3-ium bis(trifluoromethane-sulfonyl)imide ([DMVBIM]Tf2N (5), were applied for spatially resolved light-induced surface modification. Fig. 7 details the grafting-from approach of the RAFT polymerization of 1 and 5. The photoenol RAFT agent 7′ was photopatterned via photoenol chemistry onto maleimide-functionalized silicon wafers 6. Subsequently, 1 and 5 were polymerized on the surfaces 7 by a grafting-from approach leading to highly functionalized patterned structures (8a, 8b).
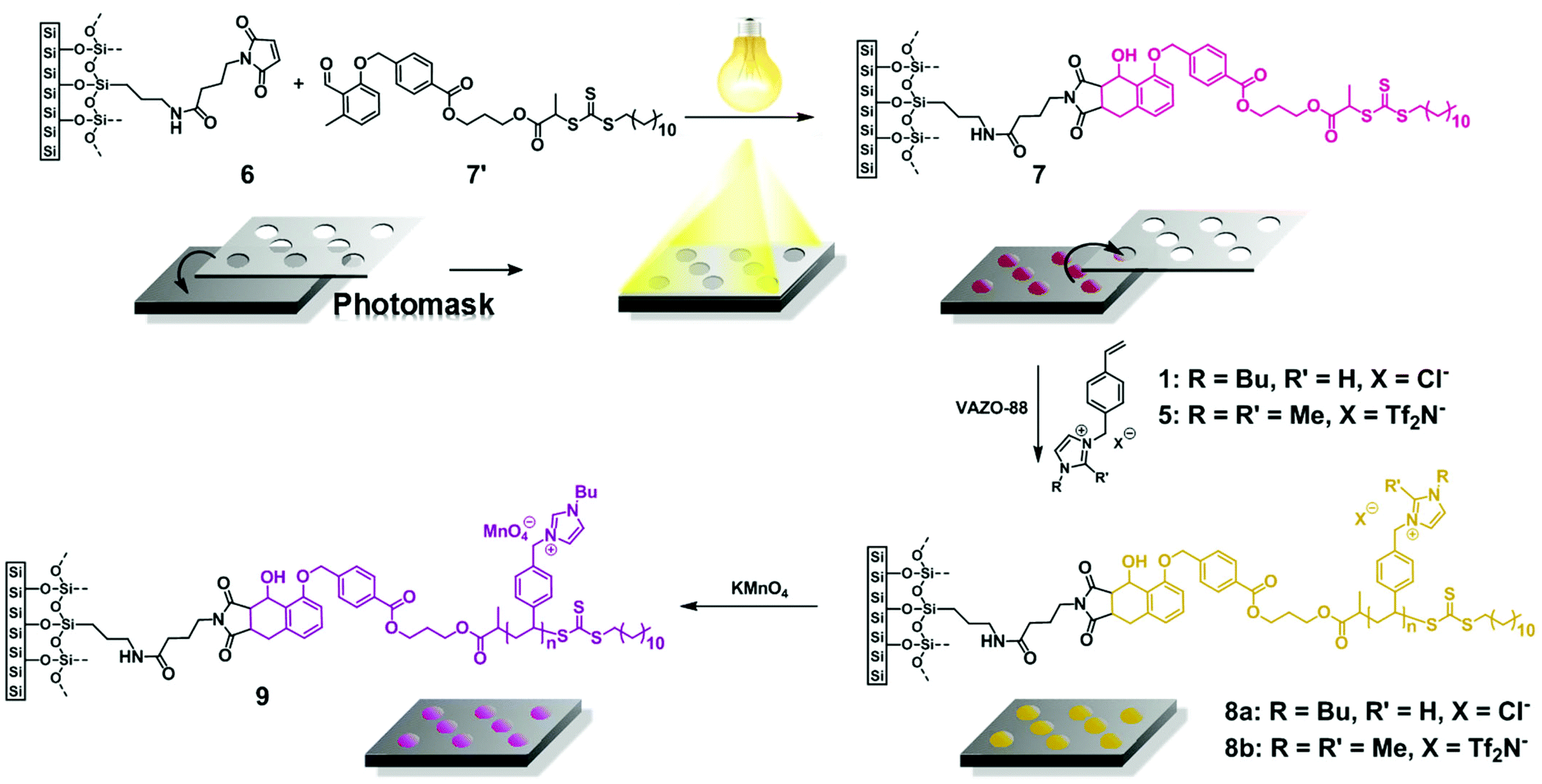 | ||
| Fig. 7 Schematic illustration of the surface functionalization for the spatially resolved surface functionalization of silicon wafers with (1) and (5). (3-Aminopropyl)triethoxysilane (APTES)-coated silicon wafers were coated with 4-maleimidobutyroyl chloride to achieve maleimide-functionalized surfaces (6). After irradiation, the surfaces covered with a photomask with an ARIMED B6 lamp (λmax = 320 nm) in a solution of the photoenol functional CTA to result in (7) followed by RAFT polymerization of (1) from the surface. Subsequent salt metathesis of (8a) with KMnO4 resulted in a highly violet colored meander pattern of p([BVBIM]MnO4) (9). | ||
First, APTES-coated surfaces,39 thoroughly cleaned by rinsing them with toluene, acetone, methanol and Milli Q water, were placed in a solution of freshly prepared 4-maleimidobutyroyl chloride in dichloromethane to obtain maleimide-coated surface 6 acting as dienophile in the subsequent photoenol reaction with the CTA (7′) in acetonitrile (due to its low absorption in the UV/Vis area) for 2.5 hours.
Therefore, a photomask is placed on the maleimide-coated surfaces 6, and irradiated with an ARIMED B6 lamp (λmax = 320 nm) for spatially resolved photopatterning of the samples with a RAFT agent for subsequent RAFT polymerization of 1 and 5. The surface was rinsed thoroughly with THF, acetone, methanol and Milli Q water, dried under a flow of nitrogen, and analyzed via XPS and ToF-SIMS, respectively.
The polymerization of [BVBIM]Cl (1) was subsequently carried out in a DMSO/DMF mixture (1:1, v/v) with a monomer concentration of approx. 0.6 mol L−1. After addition of 2-(dodecylthiocarbonothioyl-thio)propionic acid (DoPAT) as sacrificial RAFT agent and VAZO-88 as radical starter, the mixture was degassed via three consecutive freeze–pump–thaw cycles. As demonstrated by Zamfir et al.,40 the grafting-from approach is best carried out in a 1:1 ratio of CTA:initiator. The high initiator concentration used is necessitated by the hopping migration mechanism explaining the steady termination of adjacent radicals on the surface. The polymerization was allowed to proceed for 7 h (50% conversion), ensuring the formation of high-molecular weight polymer on the surface. The residual monomer as well as potential polymer attached via physisorption was removed by thorough rinsing of the functionalized surface 8a with Milli Q water, methanol, acetone, and Milli Q water followed by immersing the surface in methanol for 12 h. Subsequently, the surface was ultrasonically treated in a solution containing 0.5 g L−1 ammonium chloride and rinsed thoroughly with acetone.
Since p([DMVBIM]Tf2N (55% conversion after 7 h) is highly hydrophobic, the residual monomer as well as potential polymer attached via physisorption was removed by thorough rinsing of the functionalized surface 8b with acetone, dichloromethane, methanol and Milli Q water followed by treating the surface in a solution containing 1.0 g L−1 LiTf2N. Residual salt was removed via ultrasonification in acetone.
Fig. 8 depicts the corresponding XP spectra. The C 1s spectrum shows the (C–C, C–H) component at 285.0 eV, (C–O, C–N) at 286.4 eV and (COO, NCO) at 288.5 eV, which are present in the maleimide (surface 6) and the photoenol (surface 7).41 The photo-ligation of 7 was evidenced by the sulfur signal at 163.6 eV indicating the presence of the trithiocarbonate group (refer to Fig. S13 in the ESI†).42
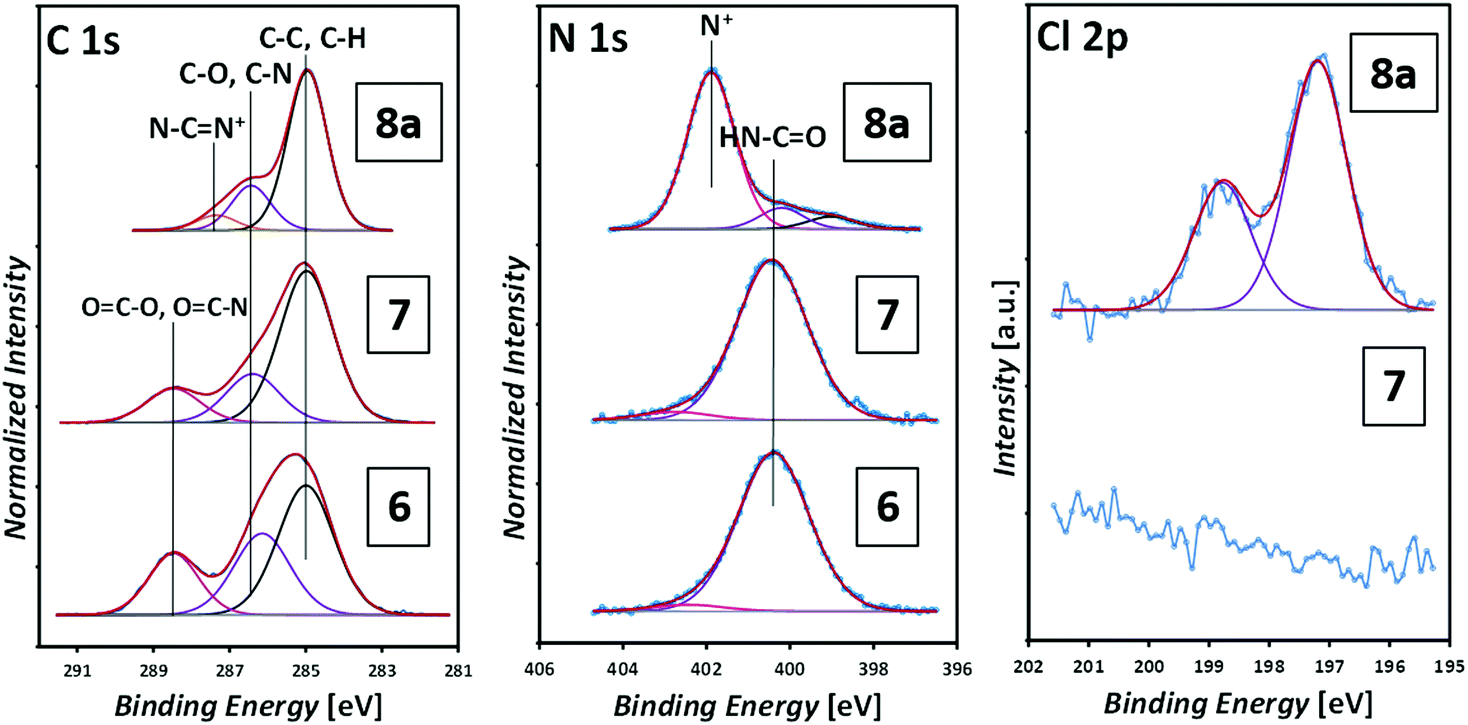 | ||
| Fig. 8 XP spectra of the maleimide functionalization (6), photoenol functionalization (7) and the grafting-from polymerization (8a) with [BVBIM]Cl (1). | ||
After the surface-initiated polymerization to obtain 8a, the formation of the signal at 401.8 eV, which is characteristic for the imidazole group (N+), is detected in the N 1s spectrum. The (protonated) amine signal at 401.8 eV in the XPS increases from 0.3 at% (surface 7) obviously stemming from a contamination (e.g. unreacted APTES) to 8.9 at% (surface 8a), which can be assigned to the formation of the imidazole group. Most characteristically, the presence of a signal originating from Cl 2p indicates the counter ion, which is absent on the APTES/maleimide-coated surface 6 as well as on the RAFT agent-functionalized surface 7. ToF-SIMS images of the C12H25S−, S−, C16H21N2+ (surface 8a) and C14H17N2+ (surface 8b) fragments are shown in Fig. 9. ToF-SIMS images clearly confirm the covalent attachment of 7′ precisely following the dotted shape of the photomask. As expected, in the areas covered by the photomask, no photoenol reaction proceeded and thus no characteristic signals stemming from 7 were observed. After successful grafting-from of [BVBIM]Cl (1), ToF-SIMS measurements in the positive mode confirm the presence of the repeating unit (C16H21N2+; m/z = 241.20 Th) on the surface 8a. Furthermore, successful grafting of [DMVBIM]Tf2N (5) was also confirmed by its characteristic repeating unit (C14H17N2+; m/z = 213.13 Th) on the surface 8a. Since this surface was prepared as proof-of-principle for the ToF-SIMS measurement, no XPS measurements were carried out on surface 8b. In addition, in order to confirm the presence of the ionic nature of the PIL attached to the surface, the substrate was immersed in KMnO4 (refer to Fig. 7). After dipping the substrate into a 50 mM KMnO4 solution for only two to three seconds, the surface was thoroughly rinsed with methanol and Milli Q water. Clearly, the meander structure – assigned to the successful photopatterning – became visible immediately. The stability of PILs with a redox-active counter ion was shown by Mecerreyes and coworkers.43
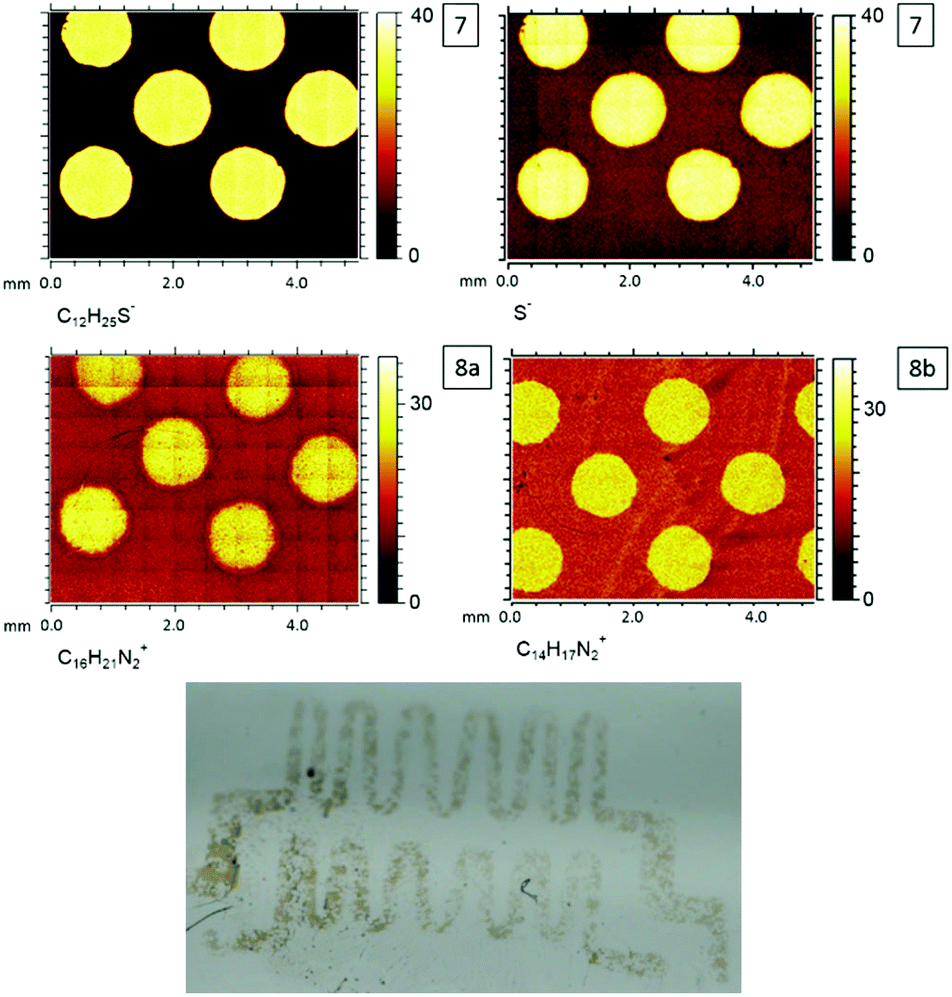 | ||
| Fig. 9 ToF-SIMS images of photoenol functionalization (7), grafting-from polymerization (8a) with [BVBIM]Cl (1), grafting-from polymerization (8b) with [DMVBIM]Tf2N (5) and detail of a photograph of the violet colored meander pattern of p([BVBIM]MnO4) (9) resulting from the salt metathesis of 8a with KMnO4. Please refer to Fig. S16† for the complete photograph. | ||
Thus, it is believed that the ion exchange of Cl− against MnO4− proceeded on the surface 9.44 It has to be noted that the brownish color of the pattern can be assigned to MnO2 that was formed due to the rapid degradation of MnO4− (Fig. 9). The meander structure was used for this experiment to show the versatility of the presented photochemically induced approach.
Experimental
Synthesis of 1-butyl-3-(4′-vinylbenzyl)-1H-imidazol-3-ium chloride ([BVBIM]Cl, 1)
The IL was prepared by a slightly modified procedure by Bara et al.27 9.00 g n-butylimidazole (72.5 mmol, 1.00 eq.) was dissolved in 100 mL chloroform and cooled to 0 °C. Subsequently, 15.4 mL vinylbenzyl chloride (16.6 g, 108.7 mmol, 1.50 eq.) was added. The mixture was stirred at 50 °C for 24 h. The mixture was extracted with water (3 × 25 mL), evaporated at reduced pressure to get rid of the residue organic solvents, and freeze–dried. 17.5 g of product 1 (63.1 mmol, 87%) was received as a colorless, highly viscous liquid.
1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.72 (s, 1 H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N), 7.91 (d, 3J = 13.8 Hz, 2 H,
CH–N), 7.91 (d, 3J = 13.8 Hz, 2 H, 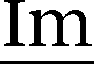 –H), 7.50 (d, 3J = 8.2 Hz, 2 H,
–H), 7.50 (d, 3J = 8.2 Hz, 2 H,  –H), 7.46 (d, 3J = 8.2 Hz, 2 H,
–H), 7.46 (d, 3J = 8.2 Hz, 2 H,  –H), 6.72 (dd, 3J(Z) = 11.0 Hz, 3J(E) = 17.7 Hz, 1 H, H2CCH), 5.85 (d, 3J(E) = 17.7 Hz, 1 H, HC(H)CH), 5.49 (s, 2 H,
–H), 6.72 (dd, 3J(Z) = 11.0 Hz, 3J(E) = 17.7 Hz, 1 H, H2CCH), 5.85 (d, 3J(E) = 17.7 Hz, 1 H, HC(H)CH), 5.49 (s, 2 H, 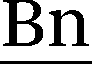 –H), 5.27 (d, 3J(Z) = 11.0 Hz, 1 H, HC(H)CH), 4.20 (t, 3J = 7.2 Hz, 2 H, N–CH2–CH2), 1.92–1.60 (m, 2 H, CH2–CH2–CH2), 1.43–1.02 (m, 2 H, CH2–CH2–CH3), 0.87 (t, 3J(Z) = 7.5 Hz, 3 H, CH2–CH3) ppm.
–H), 5.27 (d, 3J(Z) = 11.0 Hz, 1 H, HC(H)CH), 4.20 (t, 3J = 7.2 Hz, 2 H, N–CH2–CH2), 1.92–1.60 (m, 2 H, CH2–CH2–CH2), 1.43–1.02 (m, 2 H, CH2–CH2–CH3), 0.87 (t, 3J(Z) = 7.5 Hz, 3 H, CH2–CH3) ppm.
13C-NMR (100 MHz, DMSO-d6, 298 K): δ = 137.56 (C), 136.17 (C), 135.93 (CH), 134.30 (CH), 128.67 (2 CH), 126.67 (2 CH), 122.79 (CH), 122.54 (CH), 115.29 CH2), 51.65 (CH2), 48.67 (CH2), 31.24 (CH2), 18.79 (CH2), 13.26 (CH3) ppm.
Analytical data is in accordance to the results obtained by He et al.20
1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.29 (s, 1 H, NCH–N), 7.77 (s, 2 H,  –H), 7.52(d, 3J = 8.1 Hz, 2 H,
–H), 7.52(d, 3J = 8.1 Hz, 2 H,  –H), 7.43 (d, 3J = 8.2 Hz, 2 H,
–H), 7.43 (d, 3J = 8.2 Hz, 2 H,  –H), 6.74 (dd, 3J(Z) = 11.0 Hz, 3J(E) = 17.7 Hz, 1 H, H2CCH), 5.86 (d, 3J(E) = 17.7 Hz, 1 H, HC(H)CH), 5.42 (s, 2 H,
–H), 6.74 (dd, 3J(Z) = 11.0 Hz, 3J(E) = 17.7 Hz, 1 H, H2CCH), 5.86 (d, 3J(E) = 17.7 Hz, 1 H, HC(H)CH), 5.42 (s, 2 H,  –H), 5.29 (d, 3J(Z) = 11.0 Hz, 1 H, HC(H)CH), 4.19 (t, 3J = 7.2 Hz, 2 H, N–CH2–CH2), 1.79 (quin, 3J = 7.5 Hz, 2 H, CH2–CH2–CH2), 1.28 (m, 2 H, CH2–CH2–CH3), 0.90 (t, 3J = 7.5 Hz, 3 H, CH2–CH3) ppm.
–H), 5.29 (d, 3J(Z) = 11.0 Hz, 1 H, HC(H)CH), 4.19 (t, 3J = 7.2 Hz, 2 H, N–CH2–CH2), 1.79 (quin, 3J = 7.5 Hz, 2 H, CH2–CH2–CH2), 1.28 (m, 2 H, CH2–CH2–CH3), 0.90 (t, 3J = 7.5 Hz, 3 H, CH2–CH3) ppm.
13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 139.32 (C), 135.90 (C), 135.83 (CH), 131.52 (CH), 129.36 (2 CH), 127.49 (2 CH), 122.30 (CH), 122.08 (CH), 119.95 (q, 1J = 318.0 Hz, CF3), 115.88 (CH2), 53.59 (CH2), 50.31 (CH2), 32.07 (CH2), 19.49 (CH2), 13.39 (CH3) ppm.
Analytical data is in accordance to the results obtained by He et al.20
1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.67 (d, 3J = 20.2 Hz, 2 H, Im–H), 7.51 (d, 3J = 8.2 Hz, 2 H, Ph–H), 7.32 (d, 3J = 8.2 Hz, 2 H, Ph–H), 6.74 (dd, 3J(Z) = 11.0 Hz, 3J(E) = 17.7 Hz, 1 H, H2CCH), 5.86 (d, 3J(E) = 17.7 Hz, 1 H, HC(H)CH), 5.41 (s, 2 H, Bn–H), 5.29 (d, 3J(Z) = 11.0 Hz, 1 H, HC(H)CH), 3.77 (s, 3 H, N–CH3), 2.60 (s, 3 H, N–C(CH3)–N) ppm.
13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 144.66 (C), 137.42 (C), 135.94 (C), 134.04 (CH), 128.12 (2 CH), 124.36 (2 CH), 122.69 (CH), 121.24 (CH), 119.57 (q, 1J = 321.8 Hz, CF3), 115.07 (CH2), 50.46 (CH2), 34.78 (CH3), 9.40 (CH3) ppm.
ESI-MS for C14H17N2+: m/z = 213.14.
:H2O2, 4:1 v/v) for 3 h. Subsequently, the Si wafers were rinsed thoroughly with Milli Q water and dried under a stream of nitrogen. In a further step, the activated surfaces were placed in a solution of 5.0 mL APTES, 70.0 mL toluene and 0.1 vol% triethylamine and allowed to react overnight at 80 °C. The substrates were rinsed with toluene, acetone and methanol, before being dried under a stream of nitrogen.
Conclusions
In summary, we provide a useful and general synthetic tool to access imidazolium poly(ionic liquids) with styrenic IL monomer units i.e. p([BVBIM]Cl) and p([BVBIM]Tf2N) via the RAFT polymerization technique. The mass spectrometric in-depth characterization of poly(ionic liquid)s is described giving a detailed account of the molecular structure of these polyelectrolytes. In addition, we present a light-induced photoenol driven avenue to provide spatially resolved functional interfaces. The results of our contribution will facilitate access to the discussed class of poly(ionic liquid)s to further understand their molecular weight dependent physical properties and functions. The surface patterning method can be applied to develop task-specific interfaces.Acknowledgements
C. B.-K. acknowledges continued funding from the Karlsruhe Institute of Technology (KIT), the Helmholtz association via the BioInterfaces and the Science and Technology of Nanosystems programs, and the German Research Council (DFG). J. Y. thanks the Max Planck Society for the financial support. Dr Maria Schneider, Katrin Kockler and Andrea Hufendiek (all KIT) are thanked for fruitful discussions regarding SEC measurements.Notes and references
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- M. D. Green and T. E. Long, Polym. Rev., 2009, 49, 291–314 CrossRef CAS.
- O. Green, S. Grubjesic, S. Lee and M. A. Firestone, Polym. Rev., 2009, 49, 339–360 CrossRef CAS.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- Y. Gu and T. P. Lodge, Macromolecules, 2011, 44, 1732–1736 CrossRef CAS.
- H. He, S. Averick, E. Roth, D. Luebke, H. Nulwala and K. Matyjaszewski, Polymer, 2014, 55, 3330–3338 CrossRef CAS.
- H. Mori, M. Yahagi and T. Endo, Macromolecules, 2009, 42, 8082–8092 CrossRef CAS.
- J. Texter, V. A. Vasantha, R. Crombez, R. Maniglia, L. Slater and T. Mourey, Macromol. Rapid Commun., 2012, 33, 69–74 CrossRef CAS PubMed.
- K. Vijayakrishna, S. K. Jewrajka, A. Ruiz, R. Marcilla, J. A. Pomposo, D. Mecerreyes, D. Taton and Y. Gnanou, Macromolecules, 2008, 41, 6299–6308 CrossRef CAS.
- J. Yuan, H. Schlaad, C. Giordano and M. Antonietti, Eur. Polym. J., 2011, 47, 772–781 CrossRef CAS.
- A. Wilke, J. Yuan, M. Antonietti and J. Weber, ACS Macro Lett., 2012, 1, 1028–1031 CrossRef CAS.
- J. E. Bara, C. J. Gabriel, E. S. Hatakeyama, T. K. Carlisle, S. Lessmann, R. D. Noble and D. L. Gin, J. Membrane Sci., 2008, 321, 3–7 CrossRef CAS.
- Z. Shi, B. S. Newell, T. S. Bailey and D. L. Gin, Polymer, 2014, 55, 6664–6671 CrossRef CAS.
- F. Jiang, C. Li, H. Fu, Y. Wang, X. Guo and G. Wu, Sci. China Chem., 2015, 58, 716–721 CrossRef CAS.
- E. Karjalainen, D. F. Izquierdo, V. Marti-Centelles, S. V. Luis, H. Tenhu and E. Garcia-Verdugo, Polym. Chem., 2014, 5, 1437–1446 RSC.
- D. Cordella, A. Kermagoret, A. Debuigne, C. Jérôme, D. Mecerreyes, M. Isik, D. Taton and C. Detrembleur, Macromolecules, 2015, 48, 5230–5243 CrossRef CAS.
- P. Coupillaud, J. Pinaud, N. Guidolin, J. Vignolle, M. Fèvre, E. Veaudecrenne, D. Mecerreyes and D. Taton, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4530–4540 CAS.
- H. He, M. Zhong, B. Adzima, D. Luebke, H. Nulwala and K. Matyjaszewski, J. Am. Chem. Soc., 2013, 135, 4227–4230 CrossRef CAS PubMed.
- D. Cordella, A. Kermagoret, A. Debuigne, R. Riva, I. German, M. Isik, C. Jérôme, D. Mecerreyes, D. Taton and C. Detrembleur, ACS Macro Lett., 2014, 3, 1276–1280 CrossRef CAS.
- D. England, N. Tambe and J. Texter, ACS Macro Lett., 2012, 1, 310–314 CrossRef CAS.
- Q. Ye, T. Gao, F. Wan, B. Yu, X. Pei, F. Zhou and Q. Xue, J. Mater. Chem., 2012, 22, 13123–13131 RSC.
- I. Abdelhedi-Miladi, D. Montarnal, M. M. Obadia, H. Ben Romdhane and E. Drockenmuller, ACS Macro Lett., 2014, 3, 1187–1190 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- Y. Li, G. Li, X. Wang, Z. Zhu, H. Ma, T. Zhang and J. Jin, Chem. Commun., 2012, 48, 8222–8224 RSC.
- J. E. Bara, S. Lessmann, C. J. Gabriel, E. S. Hatakeyama, R. D. Noble and D. L. Gin, Ind. Eng. Chem. Res., 2007, 46, 5397–5404 CrossRef CAS.
- E. H. I. J. Brandrup and E. A. Grulke, Polymer Handbook, John Wiley and Sons, Weinheim, Germany, 1999 Search PubMed.
- R. Marcilla, J. A. Blazquez, R. Fernandez, H. Grande, J. A. Pomposo and D. Mecerreyes, Macromol. Chem. Phys., 2005, 206, 299–304 CrossRef CAS.
- A. S. Goldmann, M. Glassner, A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810–849 CrossRef CAS PubMed.
- J. Fenn, M. Mann, C. Meng, S. Wong and C. Whitehouse, Science, 1989, 246, 64–71 CAS.
- A. Calderaro, M.-C. Arcangeletti, I. Rodighiero, M. Buttrini, C. Gorrini, F. Motta, D. Germini, M.-C. Medici, C. Chezzi and F. De Conto, Sci. Rep., 2014, 4, 6803–6813 CrossRef CAS PubMed.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- J. Pinaud, J. Vignolle, Y. Gnanou and D. Taton, Macromolecules, 2011, 44, 1900–1908 CrossRef CAS.
- M. C. Kroon, W. Buijs, C. J. Peters and G.-J. Witkamp, Thermochim. Acta, 2007, 465, 40–47 CrossRef CAS.
- H. C. M. Byrd and C. N. McEwen, Anal. Chem., 2000, 72, 4568–4576 CrossRef CAS.
- D. V. Zagorevskii, M. J. Nasrullah, V. Raghunadh and B. C. Benicewicz, Rapid Commun. Mass Spectrom., 2006, 20, 178–180 CrossRef CAS PubMed.
- M. Dietrich, M. Glassner, T. Gruendling, C. Schmid, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 634–644 RSC.
- B. Yameen, C. Rodriguez-Emmenegger, C. M. Preuss, O. Pop-Georgievski, E. Verveniotis, V. Trouillet, B. Rezek and C. Barner-Kowollik, Chem. Commun., 2013, 49, 8623–8625 RSC.
- M. Zamfir, C. Rodriguez-Emmenegger, S. Bauer, L. Barner, A. Rosenhahn and C. Barner-Kowollik, J. Mater. Chem. B, 2013, 1, 6027–6034 RSC.
- C. M. Preuss, A. S. Goldmann, V. Trouillet, A. Walther and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 640–644 CrossRef CAS PubMed.
- M. Kaupp, T. Tischer, A. F. Hirschbiel, A. P. Vogt, U. Geckle, V. Trouillet, T. Hofe, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2013, 46, 6858–6872 CrossRef CAS.
- R. Gracia, K. Vijayakrishna and D. Mecerreyes, React. Funct. Polym., 2014, 79, 54–58 CrossRef CAS.
- Y.-J. Lu, W.-L. Wong and C.-F. Chow, Catal. Commun., 2015, 69, 25–28 CrossRef CAS.
- A. Herrmann, G. Mihov, G. W. M. Vandermeulen, H.-A. Klok and K. Müllen, Tetrahedron, 2003, 59, 3925–3935 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization methods, materials and additional characterization data. See DOI: 10.1039/c5py01320h |
| This journal is © The Royal Society of Chemistry 2016 |