
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monomolecular pyrenol-derivatives as multi-emissive probes for orthogonal reactivities†‡
Björn
Finkler
a,
Iris
Riemann
b,
Michael
Vester
a,
Andreas
Grüter
a,
Frank
Stracke
b and
Gregor
Jung
*a
aBiophysical Chemistry, Saarland University, Campus B2 2, 66123 Saarbrücken, Germany. E-mail: g.jung@mx.uni-saarland.de; Fax: +49 (0)681 302 64846; Tel: +49 (0)681 302 64848
bFraunhofer-IBMT, Ensheimer Straße 48, 66386 St. Ingbert, Germany
First published on 28th October 2016
Abstract
Photoacids on the basis of pyrenol have been extensively studied in the past 60 years. As their photophysical properties strongly depend on the substituents at the aromatic scaffold, we introduced two reactive moieties with different electronic coefficients thus creating multi-wavelength fluorescent probes. One probe is capable of monitoring two orthogonal transformations by four fluorescence colors, distinguishable even by the naked human eye. Another derivative can act as a three-color sensor for a wide range of different pH values. Both the presented compounds allow for mimicking of fundamental and advanced two-input logic operations due to the multi-wavelength emission. Furthermore, these compounds can process information in a logically reversible way (Feynman gate).
Introduction
Information processing with optical probes enables rapid and sensitive detection of different analytes in biochemical and supramolecular systems.1–3 Fluorogenic substrates, which represent a logic YES-gate, are widely used for the detection and activity determination of enzymes due to the high sensitivity of fluorescence based methods.1,4–9 Yet, measurements with on–off probes impose complications in quantification,10,11 which are largely overcome by the use of dual-emissive probes, i.e. a YES and a NOT gate in a single molecule.2,3,11–16 Enzymes with their fundamental importance for the function of biological systems are one of the key objectives of these probes. Since multianalyte systems are promising tools for “smart” medical diagnostics,17–20 probes for multiple enzymes are of particular interest. So far, only a few single probes for two enzymes have been reported.17,21–23 Most of these examples are fluorogenic since they emit only fluorescence if both enzymes are present and can monitor only a combined activity of both enzymes. When only one or the other enzyme is present, these systems provide no information. A separate single enzyme probe would be necessary to address this scenario. One notable exception that enables resolution of individual enzyme activity with a cleavable FRET-pair is presented by Li et al.21 While the fluorogenic probes can be seen as AND gates, this latter example “has to be regarded as an OR logic gate”,22 since a clear assignment of the corresponding metabolization states to the emission spectra is demanding for this probe. Here we present the ratiometric, monomolecular dual enzyme probe 1. The probe utilizes the spectral differences of a photoacid and the corresponding base as well as the sensitivity of the pyrene based photoacids to modifications which alter the electron-withdrawing strength of the aromatic substituents. Our system exhibits four distinct metabolization states denoted by four easily distinguishable fluorescence colors. Since phosphatase activity is an important marker for several human diseases13,24,25 and carboxyl esterase plays a key role in detoxification processes and drug targeting,12,16,26 these enzymes were chosen as a target. Furthermore, a multi-wavelength sensor for a wide range of different pH values is introduced which is also based on the same system as probe 1. Also, both derivatives can mimic Boolean operations like AND or INHIB27–29 and even more advanced logic operations.Results and discussion
Synthesis
Scheme 1 illustrates the synthetic pathway yielding compounds 1–4. In the first step 1-pyrenol was selectively mono-brominated at the distal ring. The mixture of both regioisomers 5 was subsequently transformed into the corresponding disulfonamides 6. After the reaction with benzyl bromide, the two isomers could be separated by preparative column chromatography. For further synthesis, solely derivative 7 was used, the substitution pattern of which was confirmed by X-ray crystallographic analysis (see ESI S17–S20‡). Subsequently, the bromine atom was transformed in a palladium-catalyzed carbonylation into the carboxylic ethyl ester 8. The hydroxyl group was then deprotected via palladium-catalyzed transfer hydrogenation yielding 2. The dual enzyme sensor 1 was synthesized by the reaction of 2 with phosphoroxy chloride and subsequent hydrolysis of the resulting dichloride in a water/acetone mixture in an overall yield of 10% over six steps. Additionally, the expected products of an enzymatic hydrolysis (3 and 4) were synthesized for comparison. The multi-wavelength pH-sensor compound 4 was obtained upon alkaline hydrolysis of 2 with potassium tert-butoxide in the presence of water according to the procedure of Gassman et al.30 and compound 1 was hydrolyzed in a weakly alkaline aqueous solution to yield 3. | ||
| Scheme 1 Synthesis of 1–4. | ||
Since the hydroxyl group and the opposite position of the aromatic system are the substitution sites inducing spectral changes to distinctly different extents, derivatives for other analytes can be created by variation of two of the six reaction steps (Scheme 1). Furthermore, compounds 1–4 exhibit high photostability (see ESI S38‡), long fluorescence lifetimes and quantum yields of at least 85% (Tables 1 and 3) which qualifies them as powerful tools for ultrasensitive fluorescence spectroscopy.
Dual enzyme substrate
Pyrenol and especially its sulfonated derivatives are classified as photoacids.31–33 If these molecules are electronically excited, the observed fluorescence mainly results from the conjugated base formed by excited-state proton transfer (ESPT) and is bathochromically shifted compared to the conjugated acid for electronic reasons. The corresponding base can also be excited directly at pH values above the ground state pKa. Introduction of electron-withdrawing groups into the aromatic system is a common way to tune the photophysical properties of photoacids.34–43 Hence, we introduced a cleavable carboxylic ester into the pyrene scaffold (1 and 2). The carboxylic ethyl ester (R-CO2Et) exhibits a Hammett σp-value, a convenient measure of electron-withdrawing properties, of 0.45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 whereas carboxylate (R-CO2−) at a physiological pH is less electron-withdrawing (σp = 0.00).44 Thus, fluorescence emission of the hydrolysis products (3 and 4) is blue shifted compared to 1 and 2, respectively (Fig. 1). A substrate for phosphatases is obtained by conversion of the hydroxyl-group to a phosphate monoester (1 and 3). At physiological pH, hydrolysis of this group results in a strongly red shifted emission since the emission arises from the corresponding base of compound 2 or 4, respectively (Fig. 1 and Table 1). Remarkably, the effects of phosphorylation and esterification are roughly additive on the wavelength scale.
44 whereas carboxylate (R-CO2−) at a physiological pH is less electron-withdrawing (σp = 0.00).44 Thus, fluorescence emission of the hydrolysis products (3 and 4) is blue shifted compared to 1 and 2, respectively (Fig. 1). A substrate for phosphatases is obtained by conversion of the hydroxyl-group to a phosphate monoester (1 and 3). At physiological pH, hydrolysis of this group results in a strongly red shifted emission since the emission arises from the corresponding base of compound 2 or 4, respectively (Fig. 1 and Table 1). Remarkably, the effects of phosphorylation and esterification are roughly additive on the wavelength scale.
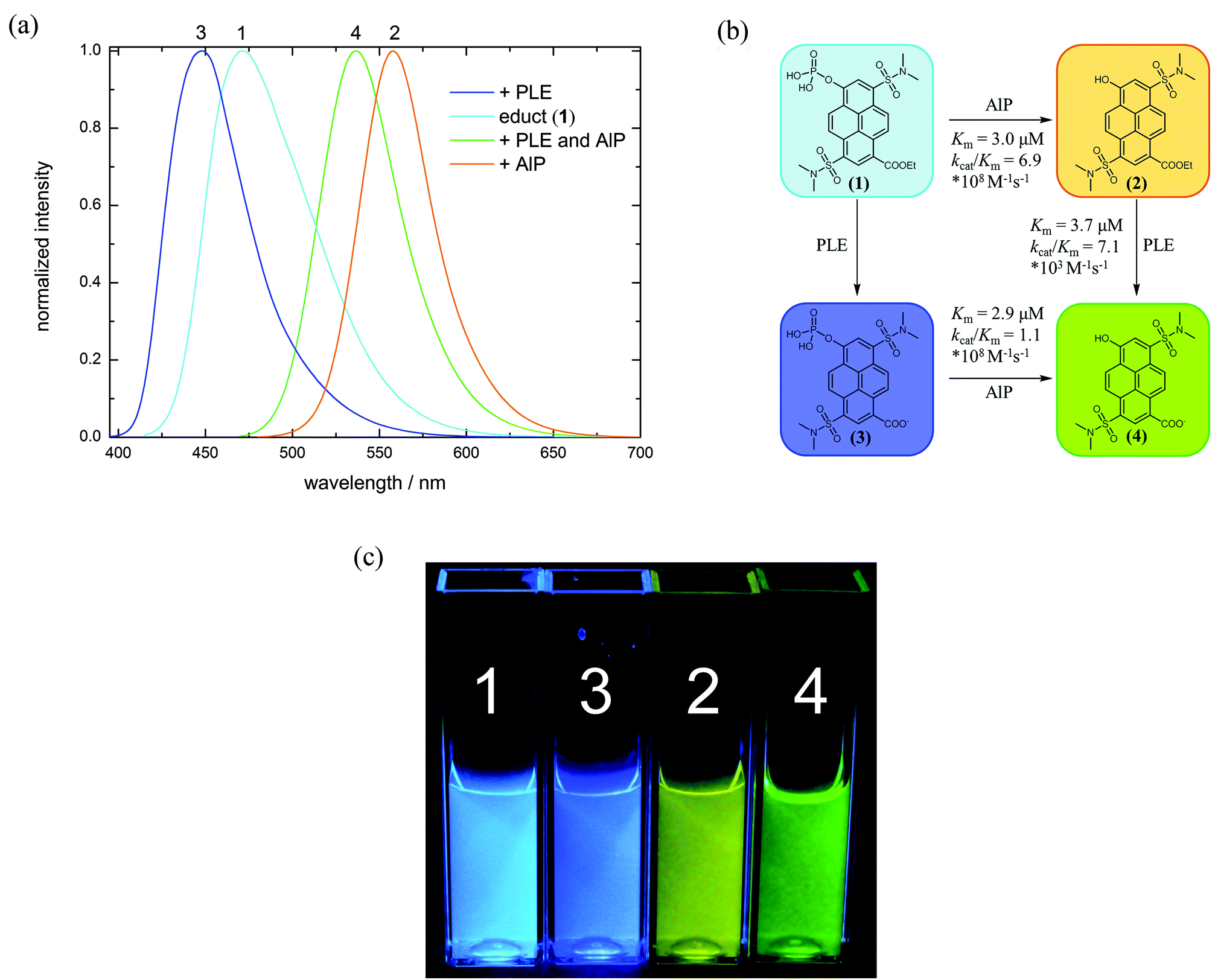 | ||
| Fig. 1 (a) Normalized fluorescence emission of 1 (10−5 M, pH = 8) recorded 3 hours after incubation (enzymes in nanomolar concentration). (b) Schematic overview of spectral and molecular changes upon enzymatic conversion and the corresponding Km and kcat/Km values. (c) Different metabolic states of 1, visualized by backlight fluorescence excitation (λex = 365 nm). | ||
Enzymatic hydrolysis of 1 by porcine liver esterase (PLE) shifts the emission from λem = 472 nm to λem = 448 nm. Upon dephosphorylation by alkaline phosphatase (AlP), fluorescence is shifted from λem = 472 to λem = 558 nm. When both enzymes are present, their combination induces an emission maximum at λem = 536 nm. Precise comparison of the spectroscopic data with the reference compounds 2–4 confirms that indeed these products are formed by the intended enzymatic reactions. Kinetic parameters of the enzyme reactions were determined via fluorescence spectroscopy using a Lineweaver–Burk plot (see ESI S39–S44‡). As reference substrates, commercially available 4-methylumbelliferyl phosphate (4-MUP)45 and butyrate (4-MUBu),46 respectively, were used. The determined Km values for the enzymatic hydrolysis of 4-MUP by AlP (Table 2) under the used assay conditions are in a similar range as those determined by Fernley et al.45
| Compound | 1 | 3 | 4-MUP | 1 | 3 | 2 | 4-MUBu |
|---|---|---|---|---|---|---|---|
| Enzyme | Alkaline phosphatase | Acid phosphatase | Porcine liver esterase | ||||
| K m [μM] | 3.0 | 2.9 | 2.2 | 40.8 | 48.6 | 3.7 | 16.9 |
| k cat/Km [M−1 s−1] | 6.9 × 108 | 1.1 × 108 | 1.8 × 107 | 6.0 × 102 | 3.9 × 104 | 7.1 × 103 | 5.7 × 103 |
The determined Km values (Table 2) characterize 1 (Km = 3.0 × 10−6 M) and 3 (Km = 2.9 × 10−6 M) as good substrates for alkaline phosphatase comparable to the fluorogenic reference compound 4-MUP under the same assay conditions (Km = 2.2 × 10−6 M). The enzymatic efficiency of these probes, i.e. kcat/Km are also in a similar range as that found for 4-MUP (Table 2). Furthermore, the phosphate hydrolysis of 1 and 3 can also be achieved by acid phosphatase (AP). Km values are generally about ten times higher compared to enzymatic hydrolysis with alkaline phosphatase, and kcat/Km is reduced by several orders of magnitude (Table 2).
K m values for the ester hydrolysis of 2 by porcine liver esterase are lower by the factor 5 whereas kcat/Km is roughly 1.3 times higher compared to the 4-methylumbelliferyl substrate 4-MUBu (Table 2), which suggests that 2 is a somewhat better substrate than the commercial fluorogenic probe. While the hydrolysis of 2 to 4 by PLE is rather fast, hydrolysis of 1 to 3 by PLE is decelerated, thus disabling determination of Km. Hydrolysis rates were similarly slow with carboxylic acid esterase from Paenibacillus barcinonensis, which suggests that 1 is not an ideal substrate for carboxylesterases. According to the widely used empirical model proposed by Jones et al.47,48 only the large hydrophobic pocket provides enough space for binding the aromatic core of 1 and 2. Compared to 2, 1 is not only sterically more bulky, but also twice negatively charged, which likely causes repulsion from the hydrophobic pocket. As a result, substrate 1 might not be optimally coordinated at the active site and hence, the enzymatic hydrolysis may not be very effective. However, kcat/Km of AlP and PLE are ordered in such a way that the conversion of 1 → 2 → 4 can be easily tracked by fluorescence spectroscopy.
Live-cell application
The dual enzyme sensor 1 was applied to L929 cells (Fig. 2; for T84 cells, see ESI S45‡). Excitation of 1 and the corresponding metabolites was performed, as previously, with two-photon excitation (λex = 800 nm).43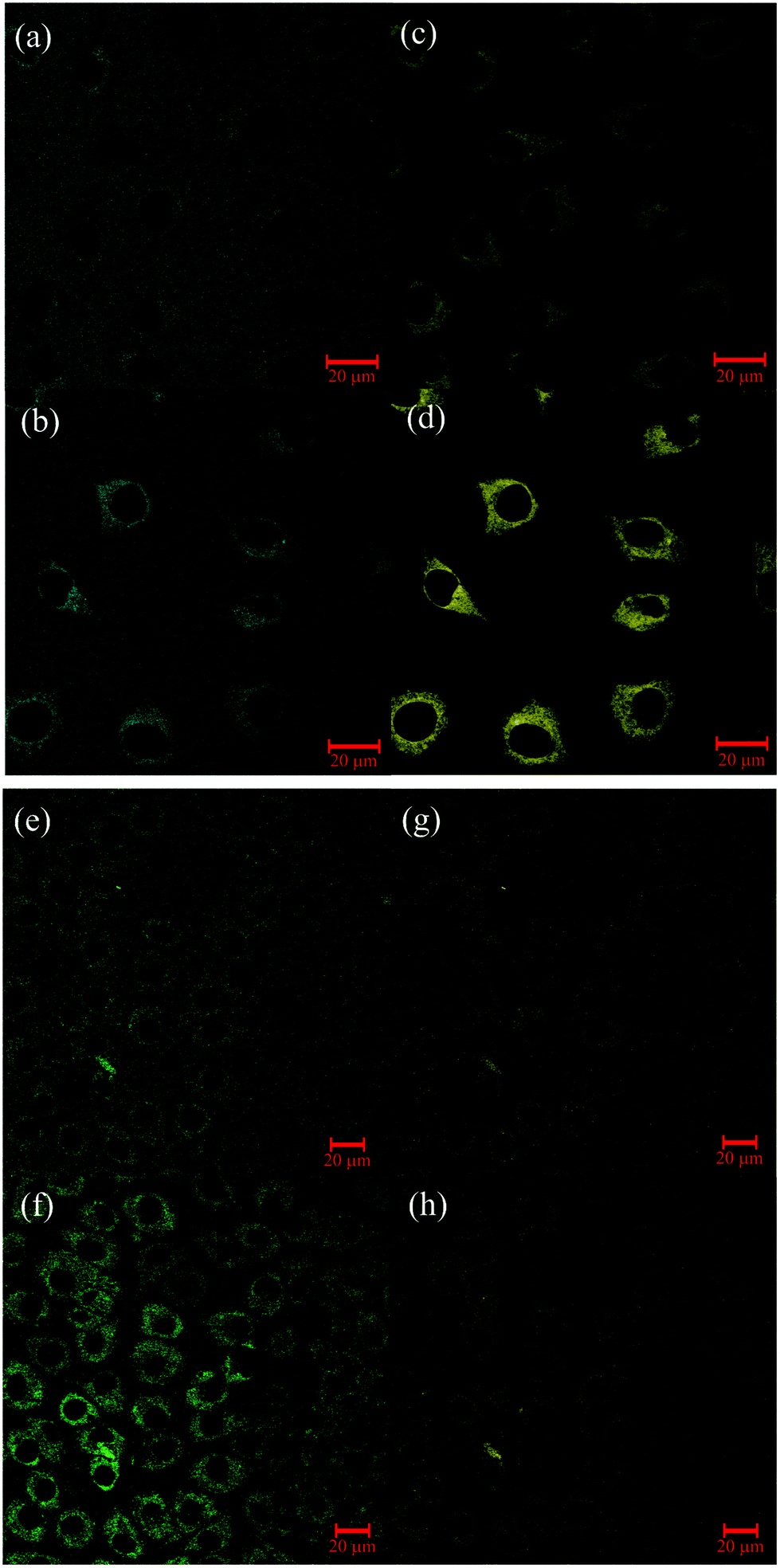 | ||
| Fig. 2 Pseudo-colored fluorescence microscopic images of several cell clusters incubated with 1 and excited at λex = 800 nm: (a–d) L929 cells, 1 h (a, c) and 3 h (b, d) after incubation, blue channel (a, b): λdet = 435–485 nm (detection of compound 1), yellow channel (c, d): λdet = 535–612 nm (detection of compound 2); (e–h) L929 cells, 1 h (e, g) and 3 h (f, h) after incubation, green channel (e, f): λdet = 500–530 nm (detection of compound 4), yellow channel (g, h): λdet = 565–615 nm (detection of compound 2). | ||
As control, cells were incubated with the different possible products of the enzymatic transformation (see ESI S46 and S47‡). Even 24 hours after incubation, no permeation in cells was found for compounds with the free carboxylate group (3 and 4), presumably due to the negative charge at this substituent. Besides the absence of membrane penetration of 4, the corresponding image further shows that no significant autofluorescence was detectable under the used setup conditions. Interestingly, uptake of the negatively charged phosphoric acid monoester 1 into the cell plasma was detectable in each measurement. The transport inside might be facilitated by transporter proteins which are naturally specialized in the exchange of nucleosides49,50 through the cell membranes.
Fluorescence filters in Fig. 2(a, b) and (c, d) were selected to exclusively monitor the phosphatase activity. In the extracellular medium only emission of 1 is detected. After three hours, fluorescence emission of 1 is still detectable in the medium outside the cell, indicating an incomplete uptake and, moreover, the absence of segregated phosphatase. Detection of 1 within the cells suggests that metabolization occurs only after internalization. Emission of the dephosphorylated product 2 measured in the cytosol rises over time and reflects the intracellular phosphatase activity.
After conversion of 1 to 2, the esterase activity was monitored by observation of the hydrolysis of 2 (λdet = 565–615 nm) to 4 (λdet = 500–530 nm). One hour after incubation with 1, fluorescence from 2 is clearly discernible and emission of the hydrolyzed product 4 is only weakly detectable. While emission intensity of 2 was about the same after 3 hours (Fig. 2(g and h)), the fluorescence emission of 4 is much more pronounced (Fig. 2(e and f)). Similar behavior was previously observed in HeLa cells by use of a carboxyl BODIPY derivative.16 Beneficially, kcat/Km of AlP and PLE (Table 2 and Fig. 1(b)) are ordered in such a way that the uptake and the subsequent conversion of 1 → 2 → 4 can be easily monitored by microscopy. It should be mentioned that 3 can only be detected when phosphatase activity is completely lacking. This is observed in selected areas of T84 cells (ESI Fig. S74‡).
The possible monitoring of the four distinct fluorescent products in the cell clusters enables the application of the above described concepts of molecular logic. For example, after incubation with 1, the output of the green channel (λdet = 500–530 nm, Fig. 2(e and f)) in combination with a suitable threshold value and the esterase/phosphatase activity as inputs represents an AND gate within the cell cluster similarly as previously shown.22 In contrast to fluorogenic systems22,51 our multi-wavelength probe additionally enables the read-out of more gates like INHIB (λdet = 565–615 nm, Fig. 2(g and h)) without further effort, thus providing additional information. The use of all four detection wavelengths enables distinction of each metabolization state and allows for sensitive multiparameter sensing. As physicians often evaluate clinical parameters by binary read-out to draw a first diagnosis, the range of different molecular logic gates qualifies our device for multipurpose use in medical diagnostics.52 This is actually possible without further technical tools as color changes are visible even by the naked human eye. In summary these first preliminary tests hint the use of 1 as a valuable tool for live-cell experiments, and other substrates with a broader application range are conceivable on the basis of Scheme 1.
Fluorescent multi-wavelength pH-sensor 4
Compound 4 contains two pH-sensitive groups which affect its spectral properties in a specific way (Fig. 3(b) and 5 and Table 3). The spectral variation is caused by the different emission of the photoacid (ROH) and the corresponding base (RO−) and by the change of the electron-withdrawing properties by protonation/deprotonation of the carboxylic acid group (σp(R-CO2H) = 0.45;44σp(R-CO2−) = 0.0044). In contrast to compound 1, where all the mutual combinations of substituents at the two orthogonal reaction sites are easily discernible (Fig. 1(a and c)), only three states are visible in the emission spectra of 4 (Fig. 3(a)). The assignment of protonation states of 4a–d to the emissive species relies on similar spectroscopic features of compounds 1–3, where the substituents exhibit similar Hammett coefficients. Moreover, our interpretation also takes into account that the ordering of the acidities of the –OH and the –CO2H groups likely is reversed upon excitation due to the pronounced photoacidity of aromatic alcohols and the photobasicity of aromatic carboxylates.53–56 This peculiarity leads to the situation that 4d is the stable monovalent form in the ground state whereas isomeric 4b is only found in the excited state. Förster-calculations to estimate lacking acidity constants e.g. of 4d or 4b are therefore not yet possible.
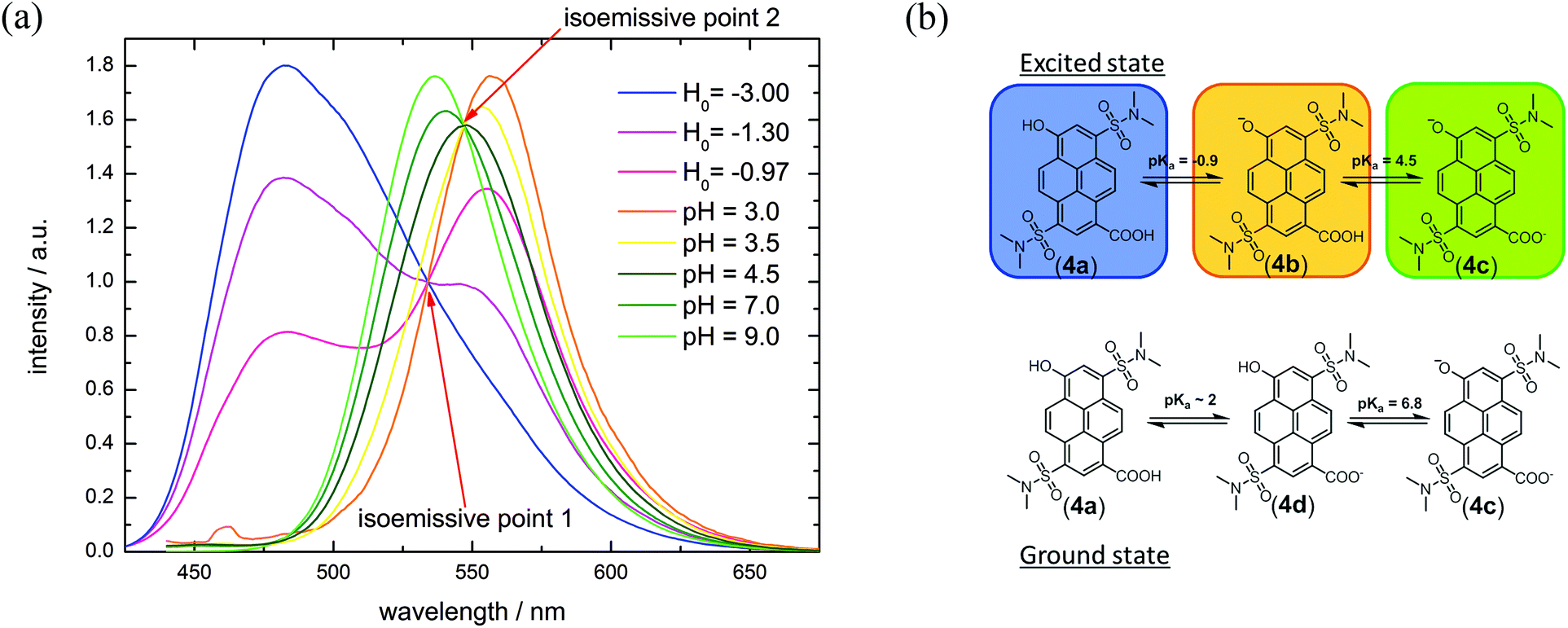 | ||
| Fig. 3 (a) pH-Titration of compound 4 (λex = 395 nm). (b) Different protonation states of 4 in the ground and excited states. The color refers to the spectra in (a). | ||
| 4 | |
|---|---|
| a Determined via absorption titration (for details, see ESI S32–S35). b Determined by fluorescence titration (for details, see ESI S32–S35). | |
| λ max,abs [nm] | 418 (4a); 415 (4d); 463 (4c) |
| λ max,em [nm] | 482 (4a); 557 (4b); 536 (4c) |
| pKaa |
6.8 (ROHCO2−); ∼2.0 (RO−CO2H) |
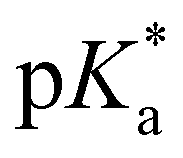
|
4.5 (ROHCO2H); −0.9 (ROHCO2H) |
| Φ | 0.85 (4c) |
| τ fl, RO− [ns] | 5.58 |
At pH ≪ 0, fluorescence emission of compound 4 at λem = 482 nm (Fig. 3(b) and 5) results from the completely protonated excited species (4a). The excited-state acidity behind (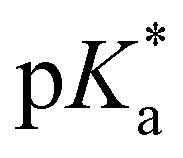 = −0.9) agrees well with the value of 2 (
= −0.9) agrees well with the value of 2 (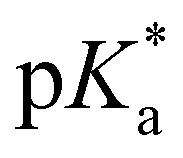 = −0.8; Tables 1 and 3), evaluated by fluorescence titration (ESI S32 and S30‡),57,58 and also lies in the range of that of HPTA (N,N,N′,N′,N′′,N′′-hexamethyl-1,3,6-trisulfonamide) (
= −0.8; Tables 1 and 3), evaluated by fluorescence titration (ESI S32 and S30‡),57,58 and also lies in the range of that of HPTA (N,N,N′,N′,N′′,N′′-hexamethyl-1,3,6-trisulfonamide) (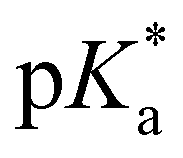 = −0.3).43 Ongoing ESPT at pH values > 0 delivers the red shifted spectrum of the corresponding base (4b, λem = 557 nm). In this pH-range, i.e. 0 < pH < 4, no color change in the emission by conversion of 2 → 4 would be distinguishable because of the identical Hammett coefficients of the –CO2H and –CO2Et substituents. At pH > 5, the fluorescence emission shifts hypsochromically due to deprotonation of the carboxylic acid thus reducing the electron-withdrawing strength (4c, λem = 536 nm). Please note that the exact titration of the latter
= −0.3).43 Ongoing ESPT at pH values > 0 delivers the red shifted spectrum of the corresponding base (4b, λem = 557 nm). In this pH-range, i.e. 0 < pH < 4, no color change in the emission by conversion of 2 → 4 would be distinguishable because of the identical Hammett coefficients of the –CO2H and –CO2Et substituents. At pH > 5, the fluorescence emission shifts hypsochromically due to deprotonation of the carboxylic acid thus reducing the electron-withdrawing strength (4c, λem = 536 nm). Please note that the exact titration of the latter 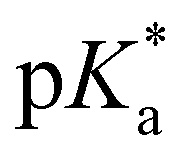 -values using fluorescence intensities is hampered by convolution of the respective molar extinction coefficients with the ground state equilibria. Anyway, the two mentioned underlying
-values using fluorescence intensities is hampered by convolution of the respective molar extinction coefficients with the ground state equilibria. Anyway, the two mentioned underlying 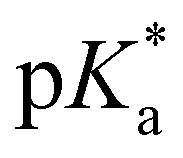 -values are experimentally accessible and are obtained from the trend of the depicted fluorescence titrations (Fig. 3(a); see also ESI Fig. S54, S55, S59 and S60‡), whereas the
-values are experimentally accessible and are obtained from the trend of the depicted fluorescence titrations (Fig. 3(a); see also ESI Fig. S54, S55, S59 and S60‡), whereas the 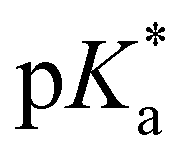 -values of the unstable isomeric form 4d are not yet experimentally obtainable.
-values of the unstable isomeric form 4d are not yet experimentally obtainable.
Conventional absorption titration experiments allow for the determination of the ground state pKa-values (ESI Fig. S57 and S58‡). Above pH 6.8, 4 is primarily present in the double deprotonated form (4c) and the excitation maximum shifts from λex = 415 nm (4d) to λex = 463 nm (4c, Table 3). The fluorescence intensity at λdet = 548 nm therefore drops from pH 6.5 to 11 due to weak excitation with light of the wavelength λex = 395 nm (Fig. 5). A comparison with other pyrenol-derivatives provides insight into the electronic effects and supports our interpretation: structurally and electronically, 4 (as well as 2) is a derivative of the photoacid HPTA.42,43,59–62 This molecule contains a third N,N′-dimethyl sulfonamide group (σp = 0.65) which is more electron-withdrawing than the carboxylate resp. the ester in dye 4d and 2. As the influence of σp is reflected in the ground state acidity, the hydroxyl's pKa of 2 and 4d (pKa = 6.6 and 6.8; Table 3) is about one logarithmic unit higher than that found for HPTA (pKa = 5.6) but almost one unit below that of HPTS (8-hydroxypyrene-1,3,6-trisulfonic acid).43,63 It should be mentioned that the difference of hydroxyl's pKa-values between 2 and 4d is less than expected, since the Hammett coefficients of the substituents (σp(R-CO2Et) = 0.45; σp(R-CO2−) = 0.00) point to higher distinction. Indeed, the slightly lower pKa-values obtained from FCS-experiments provide a larger difference (see ESI S37‡).
The second protonation equilibrium of 4a due to deprotonation of the –CO2H moiety is less clearly defined in the absorption spectra. Only a minor spectral shift between pH 0 and 3 from λabs = 418 to 415 nm (ESI Fig. S56‡) prevents a reliable pKa evaluation with absorption titration and provides only an estimate for the ground state pKa-value of 4a. Further experiments, including less ambiguous NMR-titrations with their demand for larger amounts of 4 and following the model by Ullmann,64 represent current research. In particular, detection of the isomeric form 4b, which appears to be less populated than in other systems,65 and the associated protonation equilibria in the electronic ground state deserve exhaustive investigations. It is, however, yet unclear whether the excited-state acidity constant of 4d can be experimentally found by time-resolved spectroscopy.
As the pKa and 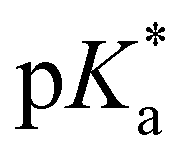 of the hydroxyl and carboxyl groups are significantly different and the prevalent protonation states can be identified via fluorescence and/or absorption spectroscopy, compound 4 can be used as an optical sensor for a wide range of different pH values from highly acidic (pH < −1) to basic conditions (pH > 7). Four different pH-stages can be discriminated by optical spectroscopy due to the different spectral properties of species 4a–d. While the pH values below −1 and between −0.5 and 4 resp. pH 5 and 6 can be identified via fluorescence spectroscopy, the pH-variation from 6 to 7 can be followed by a drop in fluorescence intensity (Fig. 5). This idea can also be transferred to the concept of molecular logic.
of the hydroxyl and carboxyl groups are significantly different and the prevalent protonation states can be identified via fluorescence and/or absorption spectroscopy, compound 4 can be used as an optical sensor for a wide range of different pH values from highly acidic (pH < −1) to basic conditions (pH > 7). Four different pH-stages can be discriminated by optical spectroscopy due to the different spectral properties of species 4a–d. While the pH values below −1 and between −0.5 and 4 resp. pH 5 and 6 can be identified via fluorescence spectroscopy, the pH-variation from 6 to 7 can be followed by a drop in fluorescence intensity (Fig. 5). This idea can also be transferred to the concept of molecular logic.
Molecular logic
Although many examples of molecular logic are already known, we demonstrate the diversity of our systems which can act as different gates or mimic more complex operations. As each metabolization state of 1 is characterized by a distinct fluorescence color and thus several different output channels can be observed, probe 1 in combination with different enzyme inputs represents a versatile tool for molecular logic. This system is able to perform simple Boolean logic operations like AND (O4), NOR (O1) and INHIB (O2, O3) (Fig. 4(a) and Table 4, for details see ESI S50–S58‡).27–29,66–68 Additionally, OR and XOR gates can be constructed by the use of negative logic (see ESI S51 and S57‡).69 The logic gate which is mimicked by the system is determined by selection of a suitable output channel wavelength and threshold value. In any of the mentioned examples, a buffer solution of 1 without any enzyme is defined as the initial state. By combination of several output channels and the two enzymes as input a 2:4 decoder,70–72 1:2 demultiplexer,70,73,74 a half-adder75–79 and -subtractor69,78,79 and a transfer gate70 (see ESI S49, S48, S59, S60 and S56‡) can be implemented. Furthermore, a molecular keypad lock70,80–86 is realized by the use of acid phosphatase, potassium hydroxide and excitation light as input (see ESI S62‡).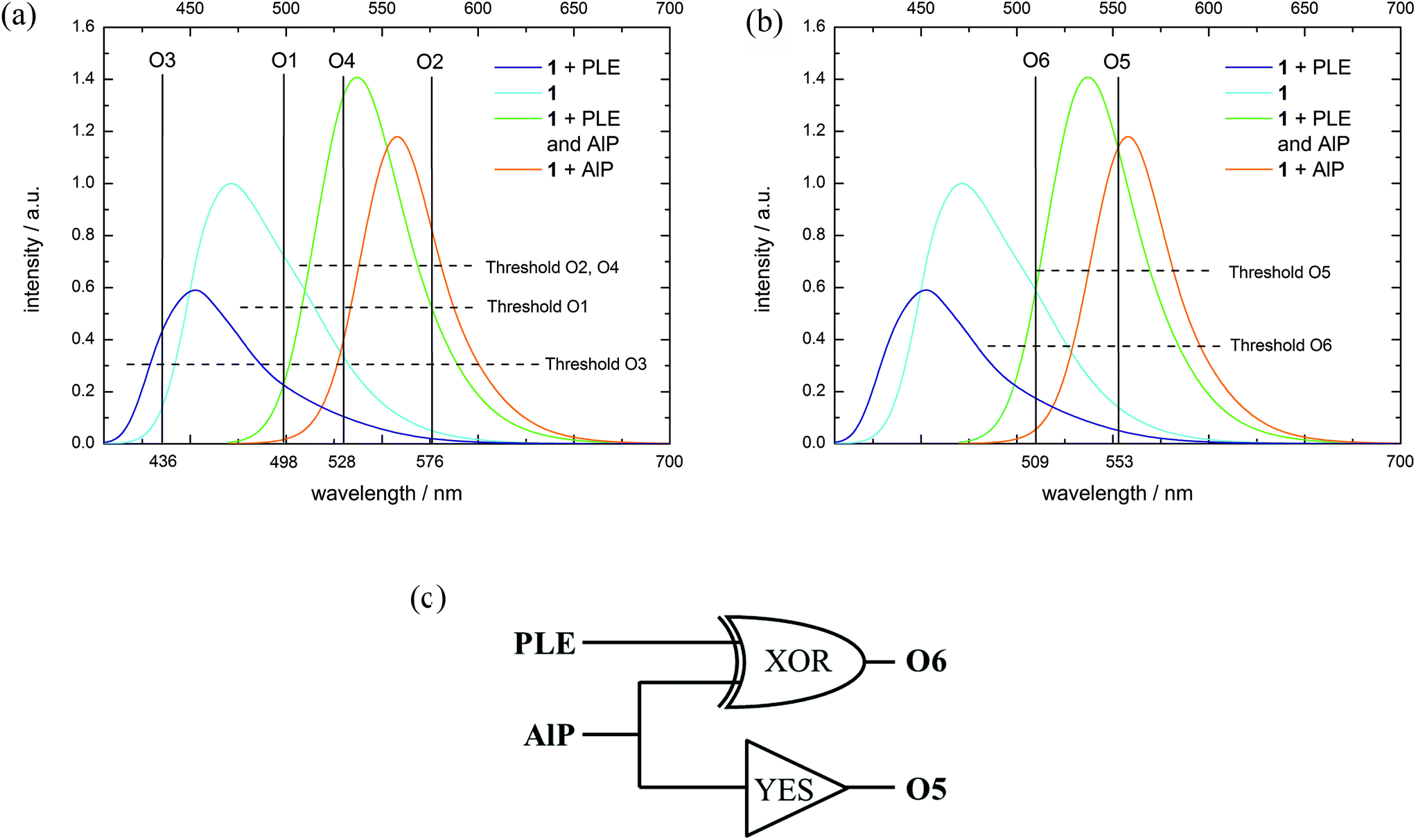 | ||
| Fig. 4 (a) Fluorescence spectra (normalized to substrate fluorescence, λex = 395 + 460 nm (see ESI S48‡), substrate concentration: 10−5 M, Tris-HCl buffer solution at pH = 8) with the corresponding output channels and threshold levels for (a) several logic operations (O1–O4) and (b) Feynman gate (O5, O6). (c) Logic circuit for the Feynman gate. | ||
| In1 (AlP) | In2 (PLE) | O1 (λ = 498 nm) | O3 (λ = 436 nm) | O2 (λ = 576 nm) | O4 (λ = 528 nm) | O5 (λ = 553 nm) | O6 (λ = 509 nm, negative logic) |
|---|---|---|---|---|---|---|---|
| 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 | 1 | 0 | 1 | 1 |
| 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 |
| Gate (isolated) | NOR | INHIB2 | INHIB1 | AND | YES | XOR | |
To exemplify some logic operations, the use of 1 as a 2:4 decoder is explained in more detail. A 2:4 decoder translates binary information from two encoded (In1, In2) inputs to four unique outputs (O1–O4).71 The truth table for this information processor is shown in Table 4. In the initial state, only O1 is on due to the fluorescence emission of 1. When either AlP (In1) or PLE (In2) is added, the substrate is converted into the corresponding product, which means that the substrate (1) fluorescence vanishes (O1 = off) and solely the fluorescence of the formed dye is monitored (O2 = on resp. O4 = on). If both inputs are present, compound 4 is generated and the fluorescence at output channel O4 exceeds the threshold value, while all other output channels are off.
Reversible molecular logic was achieved with a Feynman gate (Fig. 4(b and c) and Table 4, see ESI S61‡). Here, O5 represents the control output for the AlP input (YES-gate) and O6 acts as an XOR gate for AlP and PLE (target output). While the fluorescence emission at O5 only exceeds the corresponding threshold value when AlP is present, the simultaneous absence or presence of both PLE and AlP yields an emission at O6 above the corresponding threshold. As output of O6 is treated as negative logic, the XOR-like behavior is realized. The combination of these both gates to the Feynman gate generates a unique output pattern for each input.
Although the presented system can process information in a logically reversible way, it is not resettable since 1 is irreversibly metabolized by the enzymes. Many resettable systems with a chelating motif use ions as input. However, resetting the system in its initial state usually requires removal of the ions with additional effort.78,87 Alternative resettable systems use photons70,71,81,88 or pH-variation as input.77–79,89 In the latter case, H+ and OH− are used as inputs and the system becomes resettable, since these inputs can annihilate each other.78
In combination with definite amounts of H+ and/or OH− as input and a particular output wavelength, the pH-dependent fluorescence changes of 4 allow for mimicking nearly every simple logic operation (see ESI S63–S71‡). Moreover, the system can be applied as a half-adder, half-subtractor and comparator80,90 (see ESI S72–S74‡). In each of these applications, the system can be reset by addition of acid or base.
In the mentioned operation examples, the inputs are either chemically indistinguishable (the same amounts of H+or OH−) or can fully annihilate each other (the same amounts of H+and OH−). However, a reversible logic operation is achieved by choosing two different concentrations of base as input to create a Feynman gate (Fig. 5 and Table 5). While O I in the negative logic mode represents the YES gate for In I (low amount of OH−), O II mimics an XOR gate for In I and In II (higher amount of OH−). In the initial state at pH < 0, the emission above the threshold value at O I causes an on-response which is inverted by the negative operation to off. The same is true for the addition of In II, which sets the system to pH 6.5. Each other scenario causes an off response which is converted to on. O II only processes an on-signal after In I or In II addition (pH 3.5 or ∼6.5, Fig. 5 and Table 5).
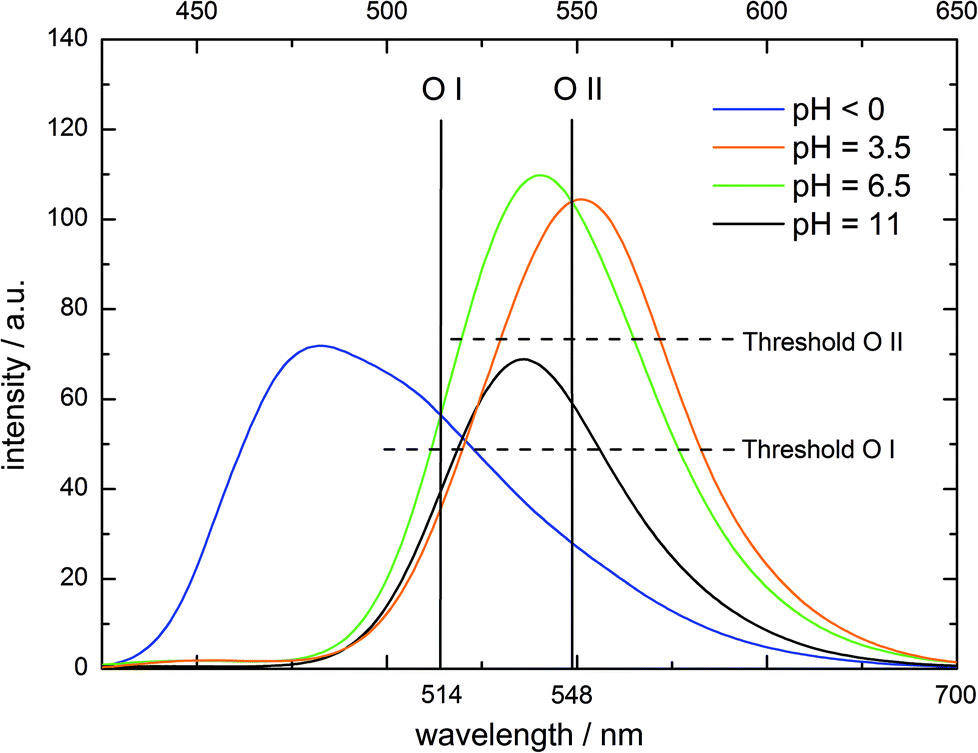 | ||
| Fig. 5 Fluorescence emission of 4 at different pH values (λex = 395 nm), output channels and the corresponding threshold values for molecular logic. | ||
| In I (low amount OH−) | In II (high amount OH−) | O I (λ = 514 nm, neg. logic) | O II (λ = 548 nm) |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 1 | 0 | 1 | 1 |
| 0 | 1 | 0 | 1 |
| 1 | 1 | 1 | 0 |
| Gate (isolated) | YES | XOR | |
Conclusion and outlook
The dual enzyme probe 1 constitutes an efficient tool for monitoring orthogonal enzymatic activities producing four distinct emission colors which are distinguishable even by the naked human eye. It can act as an individual probe for two different enzymes, allowing for their identification on the basis of different fluorescence colors, and can indicate the presence of both enzymes as well. Internal referencing in a ratiometric way is enabled by the fluorescent substrate.Compound 4 can be used as a multi-wavelength sensor for a wide range of different pH values as the protonation states can be distinguished by their spectral properties. Furthermore, the two multi-wavelength ratiometric pyrene derivatives can be used for different applications of molecular logic. Beyond simple Boolean logic, both derivatives 1 and 4 are capable of more complex operations. Moreover, both systems can operate in a logically reversible way thus preventing information loss, and 4 can even be reset. Suchlike operation of 4 as a non-trivial Feynman gate appears promising in view of our own attempts to combine chemistry with quantum optics.91,92 The described synthesis allows for a modification of pyrenol into a multi-wavelength probe for other analytes or macromolecules. Since the color of the fluorescence emission strongly relies on the substitution of the hydroxyl group (position 6) by enabling or disabling deprotonation and to a minor extent on the opposite position (position 1), more derivatives with orthogonal reactivities will be available. In summary, the described pyrenol derivatives present a versatile, flexible and chameleonic platform for the multianalyte probing and molecular logic in life science and chemistry.
Experimental section
UV/Vis and fluorescence spectroscopy
Absorption spectra were recorded with a Jasco Spectrophotometer V-650, and fluorescence emission and excitation spectra were recorded with a Jasco Spectrofluorometer FP-6500. Concentrations of the measured solutions were in the micromolar range if not stated otherwise.Enzyme kinetics
All enzymatic in vitro reactions were examined at 25 °C in aqueous buffer solution (0.1 M Tris-HCl at pH 8.0 for alkaline phosphatase (AlP) and porcine liver esterase (PLE); 0.1 M acetate at pH 5.0 for acid phosphatase (AP)). Esterase from porcine liver (activity = 18 units per mg solid) and acid phosphatase from potato (3.3 units per mg solid) were obtained as lyophilized powder from Sigma-Aldrich. Alkaline phosphatase from bovine intestinal mucosa in buffered aqueous solution (3.0 M NaCl, 0.2 mM MgCl2, 30 mM triethanolamine, pH 7.6) with an activity of 3408 units per mg protein was also obtained from Sigma-Aldrich. Kinetic parameters were determined via fluorescence spectroscopy using a Lineweaver–Burk plot. Substrate concentrations were in the range of 0.1–25 μM and enzyme concentration was in the picomolar range in the case of AlP and in the nanomolar range in that of PLE and AP. For determination of the kinetics, the decrease of the substrate fluorescence was monitored. When evolution of the product fluorescence is recorded, the observed kinetics were found to be nearly the same. Details can be found in the ESI S39–S45.‡Two-photon excitation laser scanning microscopy
Laser scanning microscopy was performed with a confocal laser microscope (LSM510 META, Zeiss; Objective: Plan-Neofluar 40×/1.3, Zeiss). Excitation was performed with a Ti:Sa laser (Chameleon XR, Coherent) operating at λ = 800 nm.Cell cultures
T84 cells were grown in a 1:1 mixture of Ham's F12 medium and Dulbecco's modified Eagle's medium with 2.5 mM 95% L-glutamine and 5% fetal bovine serum before incubation at 37 °C, 5% CO2 for 1–2 days. Before the experiment, the cells were washed with a 0.25% (w/v) trypsin–0.53 mM EDTA solution to remove all traces of serum that contains a trypsin inhibitor. L929 cells were grown in Eagle's Minimum Essential Medium modified to contain Earle's Balanced Salt Solution with non-essential amino acids, 2 mM L-glutamine, 1 mM sodium pyruvate, and 1500 mg L−1 sodium bicarbonate containing horse serum to a final concentration of 10% prior incubation at 37 °C, 5% CO2 for 1–2 days. Before the experiment, the cells were washed with a 0.25% (w/v) trypsin–0.53 mM EDTA solution to remove all traces of serum that contains a trypsin inhibitor. For incubation, compounds 1 and 3 were dissolved in water and dyes 2 and 4 were dissolved in DMSO.
Synthetic procedures: general
All reagents and solvents were obtained from Sigma-Aldrich, Merck or Acros Organics and used without further purification. For the chromatographic purification of compound 4, silica gel was washed prior to use with an 8:2 (v/v) mixture of methylene chloride/methanol and then dried in vacuo. Pyrenol was synthesized according to a modified procedure of Sehgal et al.93
1H- and 13C-NMR spectra were recorded with a Bruker AM 400 spectrometer operating at 400 and 100 MHz, respectively. Alternatively, a Bruker AM 500 spectrometer operating at 500 and 125 MHz, respectively, was used.
HRMS spectra were recorded with a Bruker solariX 7 Tesla instrument equipped with an Infinity Cell.
Data for X-ray structure determination were collected on an X8 ApexII X-ray diffractometer operating with MoKα radiation. Further details of the crystal data and structure refinement are gathered in Tables S1 and S2 in the ESI.‡
:6 (v/v)) and a mixture of 6- and 8-bromopyren-1-ol was obtained as a white solid (142.0 mg, 0.48 mmol, 86%). UV/Vis (DMSO): λmax = 358 nm, (DMSO + NaOH): λmax = 472 nm; fluorescence (DMSO): λmax = 399 nm, (DMSO + NaOH): λmax = 477 nm. 6-Bromopyren-1-ol: 1H-NMR (400 MHz, acetone-d6, 25 °C): δ = 9.68 (1H, s, OH), 8.58 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 8.30 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 8.22–8.14 (2 H, m, Ar–H), 8.05–8.01 (2 H, m, Ar–H), 7.88 (1 H, d, 3J(H,H) = 9.0 Hz, Ar–H), 7.69 ppm (1 H, d, 3J(H,H) = 8.5 Hz, Ar–H). 13C-NMR (100 MHz, acetone-d6, 25 °C): δ = 153.5, 132.6, 131.1, 131.0, 130.3, 128.9, 128.1, 127.2, 126.3, 125.8, 124.8, 124.7, 124.3, 119.5, 118.6, 114.8 ppm. 8-Bromopyren-1-ol: 1H-NMR (400 MHz, acetone-d6, 25 °C): δ = 9.67 (1H, s, OH), 8.48 (1 H, d, 3J(H,H) = 9.3 Hz, Ar–H), 8.22–8.14 (4 H, m, Ar–H), 8.05–8.01 (2 H, m, Ar–H), 7.68 ppm (1 H, d, 3J(H,H) = 8.3 Hz, Ar–H). 13C-NMR (100 MHz, acetone-d6, 25 °C): δ = 153.7, 132.5, 131.1, 131.0, 127.8, 127.1, 126.4, 126.3, 125.6, 125.5, 125.4, 123.2, 122.8, 119.7, 118.9, 114.8 ppm. MS (ESI): m/z calc. for C16H9BrO: 294.975852 [M − H]−, found: 294.97555.
:4 (v/v)) to yield the isomer mixture 6 (96.1 mg, 0.19 mmol, 56%) as a yellow powder. UV/Vis (DMSO + TFA): λmax = 432 nm, (DMSO): λmax = 532 nm; fluorescence (DMSO; DMSO + TFA): λmax = 450 nm, (DMSO): λmax = 532 nm. 3-Bromo-6-hydroxy-N,N,N′,N′-tetramethylpyrene-1,8-disulfonamide: 1H-NMR (400 MHz, acetone-d6, 25 °C): δ = 10.54 (1H, s, OH), 9.36 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 9.23 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 8.86 (1 H, s, Ar–H), 8.79 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 8.50 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 8.38 (1 H, s, Ar–H), 2.90 (6 H, s, 2 CH3), 2.87 ppm (6 H, s, 2 CH3). 13C-NMR (100 MHz, acetone-d6, 25 °C): δ = 153.9, 134.1, 133.8, 133.3, 131.5, 130.3, 129.4, 127.3, 126.8, 125.7, 125.1, 124.4, 122.7, 122.5, 119.5, 116.9, 38.1 (2C), 38.0 (2C) ppm. 3-Bromo-8-hydroxy-N,N,N′,N′-tetramethylpyrene-1,6-disulfonamide: 1H-NMR (400 MHz, acetone-d6, 25 °C): δ = 10.54 (1H, s, OH), 9.27 (1 H, d, 3J(H,H) = 10.0 Hz, Ar–H), 9.08 (1 H, d, 3J(H,H) = 10.0 Hz, Ar–H), 8.86 (1 H, s, Ar–H), 8.85 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 8.61 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 8.38 (1 H, s, Ar–H), 2.91 (6 H, s, 2 CH3), 2.88 ppm (6 H, s, 2 CH3). 13C-NMR (100 MHz, acetone-d6, 25 °C): δ = 153.9, 134.6, 134.3, 133.9, 131.5, 133.4, 131.7, 129.3, 127.5, 127.3, 126.8, 126.5, 123.5, 122.1, 119.5, 116.9, 38.1 (2C), 38.0 (2C) ppm. MS (ESI): m/z calc. for C20H19BrN2O5S2: 508.984052 [M − H]−, found: 508.98384.
:1 (v/v)) was used to purify the crude mixture of the resulting regioisomers and yielded 7 as a yellow powder (116.7 mg, 0.19 mmol, 45%). UV/Vis (DMSO): λmax = 420 nm; fluorescence (DMSO): λmax = 436 nm. 1H-NMR (400 MHz, chloroform-d, 25 °C): δ = 9.26 (1 H, d, 3J(H,H) = 9.8 Hz), 9.27 (1 H, d, 3J(H,H) = 9.19 Hz, Ar–H), 8.93 (1 H, s, Ar–H), 8.82 (1 H, d, 3J(H,H) = 9.8 Hz, Ar–H), 8.49 (1 H, d, 3J(H,H) = 9.8 Hz, Ar–H), 8.32 (1 H, s, Ar–H), 7.5 (5 H, m, Ar–H), 5,58 (2 H, s, CH2), 2.89 (6 H, s, 2 CH3), 2.72 ppm (6 H, s, 2 CH3). 13C-NMR (100 MHz, chloroform-d, 25 °C): δ = 152.8, 135.8, 132.9, 132.8, 131.8, 130.6, 128.9 (2C), 128.8, 128.5, 127.9, 127.5 (3C), 126.2, 125.6, 125.4, 123.5, 123.3, 122.7, 119.8, 112.9, 71.3, 37.6 (2C), 37.5 (2C) ppm. MS (ESI): m/z calc. for C27H25BrN2O5S2: 623.028043 [M + Na]+, found: 623.02891.
:1 (v/v)) in a yield of 80% (48.9 mg, 0.09 mmol). UV/Vis (DMSO): λmax = 420 nm; fluorescence (DMSO): λmax = 462 nm. 1H-NMR (400 MHz, chloroform-d, 25 °C): δ = 9.24 (4 H, m, Ar–H), 8.91 (1 H, 3J(H,H) = 9.6 Hz Ar–H), 8.32 (1 H, s, Ar–H), 7.47 ppm (5 H, m, Ar–H), 5.59 (2 H, s, CH2), 4.61 (2H, q, 3J(H,H) = 7.3 Hz, CH2), 2.90 (6 H, s, 2 CH3), 2.73 (6 H, s, 2 CH3), 1.55 ppm (3H, t, 3J(H,H) = 7.3 Hz, CH3). 13C-NMR (100 MHz, chloroform-d, 25 °C): δ = 166.6, 153.2, 135.7, 135.0, 133.9, 132.5, 131.9, 131.1, 130.5, 129.5, 128.9, 128.5, 128.0, 127.5, 125.8, 125.7, 125.2, 125.0, 124.2, 123.9, 123.2, 122.1, 112.7, 71.3, 62.0, 37.5 (4C), 14.4 ppm. MS (ESI): m/z calc. for C30H30N2O7S2: 617.138661 [M + Na]+, found: 617.13953.
:4 (v/v)) of the raw product yielded 2 as a yellow powder (143.0 mg, 0.30 mmol, 85%). UV/Vis (DMSO + TFA): λmax = 426 nm, (DMSO): λmax = 559 nm, (H2O + HCl): λmax = 419 nm, (H2O): λmax = 494 nm; ε(494) (H2O, RO−) = 25000 L mol−1 cm−1; fluorescence (DMSO + TFA): λmax = 482/576 nm, (DMSO): λmax = 576 nm, (H2O + HCl): λmax = 478 nm, (H2O): λmax = 558 nm. 1H-NMR (500 MHz, acetone-d6, 25 °C): δ = 9.34 (1 H, d, 3J(H,H) = 10.0 Hz, Ar–H), 9.28 (1 H, d, 3J(H,H) = 9.4 Hz, Ar–H), 9.25 (1 H, 3J(H,H) = 10.0 Hz, Ar–H), 9.21 (1 H, s, Ar–H), 8.88 (1 H, d, 3J(H,H) = 9.4 Hz, Ar–H), 8.38 (1 H, s, Ar–H), 4.60 (2H, q, 3J(H,H) = 7.3 Hz, CH2), 2.89 (6 H, s, 2 CH3), 2.88 (6 H, s, 2 CH3), 1.53 ppm (3 H, t, 3J(H,H) = 7.3 Hz, CH3). 13C-NMR (125 MHz, acetone-d6, 25 °C): δ = 167.3, 154.3, 134.7 (2C), 132.2, 131.8, 130.4, 129.2, 127.1, 126.8, 126.0, 125.6, 124.6, 124.0, 122.4, 122.1, 116.8, 62.6, 38.1 (2C), 38.0 (2C), 14.8 ppm. MS (ESI): m/z calc. for C23H24N2O7S2: 503.09467 [M − H]−, found: 503.09282.
:1 (v/v)) to give a yellow powder (12.6 mg, 0.03 mmol, 72%). UV/Vis (DMSO + TFA): λmax = 425 nm, (DMSO): λmax = 424/557 nm, (H2O + HCl): λmax = 415/418 nm, (H2O): λmax = 463 nm; ε(463) (H2O, RO−) = 17000 L mol−1 cm−1; fluorescence (DMSO + TFA): λmax = 476/573 nm, (DMSO): λmax = 573 nm, (H2O + HCl): λmax = 482 nm, (H2O): λmax = 536/557 nm. 1H-NMR (500 MHz, acetone-d6, 25 °C): δ = 9.38 (1 H, d, 3J(H,H) = 10.0 Hz, Ar–H), 9.24 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 9.20 (1 H, 3J(H,H) = 10.0 Hz, Ar–H), 9.16 (1 H, s, Ar–H), 8.79 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 8.34 (1 H, s, Ar–H), 2.88 (6 H, s, 2 CH3), 2.85 ppm (6 H, s, 2 CH3). 13C-NMR (125 MHz, acetone-d6, 25 °C): δ = 179.3, 153.8, 134.1, 133.7, 132.1, 131.7, 130.0, 128.8, 127.9, 127.2, 126.0, 125.6, 125.4, 125.0, 122.4, 122.2, 116.3, 38.1 (2C), 38.0 (C2). MS (ESI): m/z calc. for C21H20N2O7S2: 475.06337 [M − H]−, found: 475.06494.
:1 (v/v)) of the crude product yielded 1 as a yellow powder (37.6 mg, 0.07 mmol, 68%). UV/Vis (H2O): λmax = 410 nm; ε(410) (H2O) = 19300 L mol−1 cm−1; fluorescence (H2O): λmax = 472 nm. 1H-NMR (500 MHz, methanol-d4, 25 °C): δ = 9.35 (1 H, d, 3J(H,H) = 10.0 Hz, Ar–H), 9.30 (1 H, d, 3J(H,H) = 9.6 Hz, Ar–H), 9.24 (1 H, 3J(H,H) = 10.0 Hz, Ar–H), 9.19 (1 H, s, Ar–H), 8.92 (1 H, d, 3J(H,H) = 10.0 Hz, Ar–H), 8.86 (1 H, s, Ar–H), 4.61 (2H, q, 3J(H,H) = 7.0 Hz, CH2), 2.93 (6 H, s, 2 CH3), 2.86 (6 H, s, 2 CH3), 1.55 ppm (3H, t, 3J(H,H) = 7.0 Hz, CH3). 13C-NMR (125 MHz, methanol-d4, 25 °C): δ = 168.0, 150.2, 134.9, 134.7, 133.3, 132.1, 131.1, 129.4, 127.1, 127.0, 126.9, 126.6, 125.8, 125.5, 124.9, 121.3 (C2), 63.3, 38.4 (C2), 38.0 (C2), 14.9 ppm. MS (ESI): m/z calc. for C23H25N2O10PS2: 583.06155 [M − H]−, found: 583.06245.
:3 (v/v)) and yielded 3 as a yellow powder (3.1 mg, 0.006 mmol, 81%). UV/Vis (H2O): λmax = 407 nm; ε(407) (H2O) = 15000 L mol−1 cm−1; fluorescence (H2O): λmax = 448 nm. 1H-NMR (500 MHz, methanol-d4, 25 °C): δ = 9.25 (1 H, d, 3J(H,H) = 9.8 Hz, Ar–H), 9.24 (1 H, d, 3J(H,H) = 9.8 Hz, Ar–H), 8.97 (1 H, 3J(H,H) = 10.0 Hz, Ar–H), 8.89 (1 H, s, Ar–H), 8.87 (1 H, s, Ar–H), 8.86 (1 H, d, 3J(H,H) = 10.0 Hz, Ar–H), 2.90 (6 H, s, 2 CH3), 2.86 ppm (6 H, s, 2 CH3). 13C-NMR (125 MHz, methanol-d4, 25 °C): δ = 175.5, 149.7, 136.9, 133.1, 132.4, 130.9, 130.7, 129.8, 127.6, 127.5, 127.1, 127.0, 126.8, 126.7, 125.4, 125.3, 121.1, 38.4 (C2), 38.1 (C2) ppm. MS (ESI): m/z calc. for C21H21N2O10PS2: 555.03025 [M − H]−, found: 555.03196.
Acknowledgements
Financial support by the German Science Foundation (DFG, JU650/7-1) is gratefully acknowledged. Furthermore we thank Tobias Dier for recording the mass spectra, Volker Huch for X-ray crystallography measurements and Alexander Schiller for the helpful discussion of our manuscript.Notes and references
- A. Baruch, D. A. Jeffery and M. Bogyo, Enzyme activity – it's all about image, Trends Cell Biol., 2004, 14, 29–35 CrossRef CAS PubMed.
- K. Kikuchi, Design, synthesis and biological application of chemical probes for bio-imaging, Chem. Soc. Rev., 2010, 39, 2048–2053 RSC.
- M. H. Lee, J. S. Kim and J. L. Sessler, Small molecule-based ratiometric fluorescence probes for cations, anions, and biomolecules, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- I. Johnson and M. T. Z. Spence, The Molecular Probes Handbook, 2010 Search PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Design strategies for water-soluble small molecular chromogenic and fluorogenic probes, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed.
- D. J. Yee, V. Balsanek, D. R. Bauman, T. M. Penning and D. Sames, Fluorogenic metabolic probes for direct activity readout of redox enzymes: Selective measurement of human AKR1C2 in living cells, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13304–13309 CrossRef CAS PubMed.
- H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, Real-time monitoring of cell apoptosis and drug screening using fluorescent light-up probe with aggregation-induced emission characteristics, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS PubMed.
- M. Kamiya, D. Asanuma, E. Kuranaga, A. Takeishi, M. Sakabe, M. Miura, T. Nagano and Y. Urano, β-galactosidase fluorescence probe with improved cellular accumulation based on a spirocyclized rhodol scaffold, J. Am. Chem. Soc., 2011, 133, 12960–12963 CrossRef CAS PubMed.
- A. P. Demchenko, Practical aspects of wavelength ratiometry in the studies of intermolecular interactions, J. Mol. Struct., 2014, 1077, 51–67 CrossRef CAS.
- A. P. Demchenko, The concept of λ-ratiometry in fluorescence sensing and imaging, J. Fluoresc., 2010, 20, 1099–1128 CrossRef PubMed.
- Z.-M. Liu, L. Feng, G.-B. Ge, X. Lv, J. Hou, Y.-F. Cao, J.-N. Cui and L. Yang, A highly selective ratiometric fluorescent probe for in vitro monitoring and cellular imaging of human carboxylesterase 1, Biosens. Bioelectron., 2014, 57, 30–35 CrossRef CAS PubMed.
- Z. Song, R. T. K. Kwok, E. Zhao, Z. He, Y. Hong, J. W. Y. Lam, B. Liu and B. Z. Tang, A ratiometric fluorescent probe based on ESIPT and AIE processes for alkaline phosphatase activity assay and visualization in living cells, ACS Appl. Mater. Interfaces, 2014, 6, 17245–17254 CAS.
- Y. Kurishita, T. Kohira, A. Ojida and I. Hamachi, Rational design of FRET-based ratiometric chemosensors for in vitro and in cell fluorescence analyses of nucleoside polyphosphates, J. Am. Chem. Soc., 2010, 132, 13290–13299 CrossRef CAS PubMed.
- L. Chen, et al., The first ratiometric fluorescent probes for aminopeptidase N cell imaging, Org. Biomol. Chem., 2013, 11, 378–382 CAS.
- T. Komatsu, Y. Urano, Y. Fujikawa, T. Kobayashi, H. Kojima, T. Terai, K. Hanaoka and T. Nagano, Development of 2,6-carboxy-substituted boron dipyrromethene (BODIPY) as a novel scaffold of ratiometric fluorescent probes for live cell imaging, Chem. Commun., 2009, 7015–7017 RSC.
- S. Debieu and A. Romieu, Dual enzyme-responsive ‘turn-on’ fluorescence sensing systems based on in situ formation of 7-hydroxy-2-iminocoumarin scaffolds, Org. Biomol. Chem., 2015, 13, 10348–10361 CAS.
- J. Halámek, et al., Multiplexing of injury codes for the parallel operation of enzyme logic gates, Analyst, 2010, 135, 2249–2259 RSC.
- L. Halámková, J. Halámek, V. Bocharova, S. Wolf, K. E. Mulier, G. Beilman, J. Wang and E. Katz, Analysis of biomarkers characteristic of porcine liver injury–from biomolecular logic gates to an animal model, Analyst, 2012, 137, 1768–1770 RSC.
- A. Romieu, ‘AND’ luminescent ‘reactive’ molecular logic gates: a gateway to multi-analyte bioimaging and biosensing, Org. Biomol. Chem., 2015, 13, 1294–1306 CAS.
- Y. Li, H. Wang, J. Li, J. Zheng, X. Xu and R. Yang, Simultaneous intracellular β-D-glucosidase and phosphodiesterase I activities measurements based on a triple-signaling fluorescent probe, Anal. Chem., 2011, 83, 1268–1274 CrossRef CAS PubMed.
- M. Prost and J. Hasserodt, ‘Double gating’ - a concept for enzyme-responsive imaging probes aiming at high tissue specificity, Chem. Commun., 2014, 50, 14896–14899 RSC.
- S.-Y. Li, L.-H. Liu, H. Cheng, B. Li, W.-X. Qiu and X.-Z. Zhang, A dual-FRET-based fluorescence probe for the sequential detection of MMP-2 and caspase-3, Chem. Commun., 2015, 51, 14520–14523 RSC.
- T.-I. Kim, H. Kim, Y. Choi and Y. Kim, A fluorescent turn-on probe for the detection of alkaline phosphatase activity in living cells, Chem. Commun., 2011, 47, 9825–9827 RSC.
- J. L. Millán and W. H. Fishman, Biology of human alkaline phosphatases with special reference to cancer, Crit. Rev. Clin. Lab. Sci., 1995, 32, 1–39 CrossRef PubMed.
- Y. Zhang, W. Chen, D. Feng, W. Shi, X. Li and H. Ma, A spectroscopic off-on probe for simple and sensitive detection of carboxylesterase activity and its application to cell imaging, Analyst, 2012, 137, 716–721 RSC.
- J. Andréasson and U. Pischel, Molecules with a sense of logic: a progress report, Chem. Soc. Rev., 2015, 1053–1069 RSC.
- A. P. de Silva, Molecular logic-based computation, RSC Publishing, 2013 Search PubMed.
- K. Szaciłowski, Infochemistry: Information Processing at the Nanoscale, Wiley-VCH, 2012 Search PubMed.
- P. Gassman and W. Schenk, A general procedure for the base-promoted hydrolysis of hindered esters at ambient temperatures, J. Org. Chem., 1977, 42, 918–920 CrossRef CAS.
- T. Förster, Elektrolytische Dissoziation angeregter Moleküle, Z. Elektrochem., 1950, 54, 42–46 Search PubMed.
- E. Pines, The Chemistry of Phenols, John Wiley & Sons Ltd, 2003 Search PubMed.
- N. Agmon, Elementary steps in excited-state proton transfer, J. Phys. Chem. A, 2005, 109, 13–35 CrossRef CAS PubMed.
- L. M. Tolbert and J. E. Haubrich, Enhanced photoacidities of cyanonaphthols, J. Am. Chem. Soc., 1990, 112, 8163–8165 CrossRef CAS.
- L. M. Tolbert and J. E. Haubrich, Photoexcited proton transfer from enhanced photoacids, J. Am. Chem. Soc., 1994, 116, 10593–10600 CrossRef CAS.
- D. Huppert, L. M. Tolbert and S. Linares-Samaniego, Ultrafast excited-state proton transfer from cyano-substituted 2-naphthols, J. Phys. Chem. A, 1997, 101, 4602–4605 CrossRef CAS.
- C. Clower, K. M. Solntsev, J. Kowalik, L. M. Tolbert and D. Huppert, Photochemistry of ‘ super ‘ photoacids. 3. excited-state proton transfer from perfluoroalkylsulfonyl-substituted 2-naphthols, J. Phys. Chem. A, 2002, 106, 3114–3122 CrossRef CAS.
- T.-H. Tran-Thi, C. Prayer, P. Millié, P. Uznanski and J. T. Hynes, Substituent and solvent effects on the nature of the transitions of pyrenol and pyranine. identification of an intermediate in the excited-state proton-transfer reaction, J. Phys. Chem. A, 2002, 106, 2244–2255 CrossRef CAS.
- K. M. Solntsev, E. N. Sullivan, L. M. Tolbert, S. Ashkenazi, P. Leiderman and D. Huppert, Excited-state proton transfer reactions of 10-hydroxycamptothecin, J. Am. Chem. Soc., 2004, 126, 12701–12708 CrossRef CAS PubMed.
- M. Prémont-Schwarz, T. Barak, D. Pines, E. T. J. Nibbering and E. Pines, Ultrafast excited state proton transfer reaction of 1-naphthol-3,6-disulfonate and several 5-substituted 1-naphthol derivatives, J. Phys. Chem. B, 2013, 117, 4593–4594 CrossRef PubMed.
- C. Spies, B. Finkler, N. Acar and G. Jung, Solvatochromism of pyranine-derived photoacids, Phys. Chem. Chem. Phys., 2013, 15, 19893–19905 RSC.
- C. Spies, S. Shomer, B. Finkler, D. Pines, E. Pines, G. Jung and D. Huppert, Solvent dependence of excited-state proton transfer from pyranine-derived photoacids, Phys. Chem. Chem. Phys., 2014, 16, 9104–9114 RSC.
- B. Finkler, et al. Highly photostable ‘super’-photoacids for ultrasensitive fluorescence spectroscopy, Photochem. Photobiol. Sci., 2014, 13, 548–562 CAS.
- C. Hansch, A. Leo and R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- H. N. Fernley and P. G. Walker, Kinetic behaviour of calf-intestinal alkaline phosphatase with 4-methylumbelliferyl phosphate, Biochem. J., 1965, 97, 95–103 CrossRef CAS PubMed.
- S. Lun and W. R. Bishai, Characterization of a novel cell wall-anchored protein with carboxylesterase activity required for virulence in mycobacterium tuberculosis, J. Biol. Chem., 2007, 282, 18348–18356 CrossRef CAS PubMed.
- L. Provencher and J. B. Jones, A concluding specification of the dimensions of the active site model of pig liver esterase, J. Org. Chem., 1994, 59, 2729–2732 CrossRef CAS.
- P. D. De María, C. A. García-Burgos, G. Bargeman and R. W. Van Gemert, Pig liver esterase (PLE) as biocatalyst in organic synthesis: From nature to cloning and to practical applications, Synthesis, 2007, 10, 1439–1452 CrossRef.
- J. L. Ward and C. M. Tse, Nucleoside transport in human colonic epithelial cell lines: evidence for two Na+-independent transport systems in T84 and Caco-2 cells, Biochim. Biophys. Acta, Biomembr., 1999, 1419, 15–22 CrossRef CAS.
- M. Pastor-Anglada, P. Cano-Soldado, M. Molina-Arcas, M. P. Lostao, I. Larráyoz, J. Martínez-Picado and F. J. Casado, Cell entry and export of nucleoside analogues, Virus Res., 2005, 107, 151–164 CrossRef CAS PubMed.
- M. N. Win and C. D. Smolke, Higher-order cellular information processing with synthetic RNA devices, Science, 2008, 322, 456–460 CrossRef CAS PubMed.
- M. Elstner, J. Axthelm and A. Schiller, Sugar-based molecular computing by material implication, Angew. Chem., Int. Ed., 2014, 53, 7339–7343 CrossRef CAS PubMed.
- J. Ditkovich, T. Mukra, D. Pines, D. Huppert and E. Pines, Bifunctional photoacids: Remote protonation affecting chemical reactivity, J. Phys. Chem. B, 2015, 119, 2690–2701 CrossRef CAS PubMed.
- B. Zelent, J. M. Vanderkooi, R. G. Coleman, I. Gryczynski and Z. Gryczynski, Protonation of excited state pyrene-1-carboxylate by phosphate and organic acids in aqueous solution studied by fluorescence spectroscopy, Biophys. J., 2006, 91, 3864–3871 CrossRef CAS PubMed.
- N. V. Nucci, B. Zelent and J. M. Vanderkooi, Pyrene-1-carboxylate in water and glycerol solutions: Origin of the change of pK upon excitation, J. Fluoresc., 2008, 18, 41–49 CrossRef CAS PubMed.
- B. Zelent, J. M. Vanderkooi, N. V. Nucci, I. Gryczynski and Z. Gryczynski, Phosphate assisted proton transfer in water and sugar glasses: A study using fluorescence of pyrene-1-carboxylate and IR spectroscopy, J. Fluoresc., 2009, 19, 21–31 CrossRef CAS PubMed.
- A. Weller, Outer and inner mechanism of reactions of excited molecules, Discuss. Faraday Soc., 1959, 27, 28–33 RSC.
- A. Weller, Fast reactions of excited molecules, Prog. React. Kinet., 1961, 1, 187–214 CAS.
- D. B. Spry and M. D. Fayer, Observation of slow charge redistribution preceding excited-state proton transfer, J. Chem. Phys., 2007, 127, 204501 CrossRef CAS PubMed.
- E. Pines, D. Pines, Y.-Z. Ma and G. R. Fleming, Femtosecond pump–probe measurements of solvation by hydrogen-bonding interactions, ChemPhysChem, 2004, 5, 1315–1327 CrossRef CAS PubMed.
- D. B. Spry and M. D. Fayer, Observation of slow charge redistribution preceding excited-state proton transfer, J. Chem. Phys., 2007, 127, 204501 CrossRef CAS PubMed.
- C. Spies, B. Finkler, N. Acar and G. Jung, Solvatochromism of pyranine-derived photoacids, Phys. Chem. Chem. Phys., 2013, 15, 19893–19905 RSC.
- H. Offenbacher, O. S. Wolfbeis and E. Furlinger, Fluorescence optical sensors for continuous determination of near-neutral pH values, Sens. Actuators, 1986, 9, 73–84 CrossRef CAS.
- G. M. Ullmann, Relations between protonation constants and titration curves in polyprotic acids: A critical view, J. Phys. Chem. B, 2003, 107, 1263–1271 CrossRef CAS.
- R. Bizzarri, et al. Green fluorescent protein ground states: The influence of a second protonation site near the chromophore, Biochemistry, 2007, 46, 5494–5504 CrossRef CAS PubMed.
- A. P. de Silva, N. H. Q. Gunaratne and C. P. McCoy, A molecular photoionic AND gate based on fluorescent signalling, Nature, 1993, 364, 42–44 CrossRef.
- J. Andréasson and U. Pischel, Smart molecules at work-mimicking advanced logic operations, Chem. Soc. Rev., 2010, 39, 174–188 RSC.
- A. P. de Silva and N. D. McClenaghan, Molecular-scale logic gates, Chem. – Eur. J., 2004, 10, 574–586 CrossRef PubMed.
- A. Coskun, E. Deniz and E. U. Akkaya, Effective PET and ICT switching of boradiazaindacene emission: A unimolecular, emission-mode, molecular half-subtractor with reconfigurable logic gates, Org. Lett., 2005, 7, 5187–5189 CrossRef CAS PubMed.
- J. Andréasson, U. Pischel, S. D. Straight, T. a. Moore, A. L. Moore and D. Gust, All-photonic multifunctional molecular logic device, J. Am. Chem. Soc., 2011, 133, 11641–11648 CrossRef PubMed.
- J. Andréasson, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, Molecular all-photonic encoder-decoder, J. Am. Chem. Soc., 2008, 130, 11122–11128 CrossRef PubMed.
- D. Kang, R. J. White, F. Xia, X. Zuo, A. Vallée-Bélisle and K. W. Plaxco, DNA biomolecular-electronic encoder and decoder devices constructed by multiplex biosensors, NPG Asia Mater., 2012, 4, e1 CrossRef.
- R. Orbach, F. Remacle, R. D. Levine and I. Willner, DNAzyme-based 2:1 and 4:1 multiplexers and 1:2 demultiplexer, Chem. Sci., 2014, 5, 1074–1081 RSC.
- L. Zhao, W. Xia and C. Yang, Fluorescent 1:2 demultiplexer and half-subtractor based on the hydrolysis of N-salicylidene-3-aminopyridine, Spectrochim. Acta, Part A, 2014, 117, 397–401 CrossRef CAS PubMed.
- J. Andréasson, G. Kodis, Y. Terazono, P. A. Liddell, S. Bandyopadhyay, R. H. Mitchell, T. A. Moore, A. L. Moore and D. Gust, Molecule-based photonically switched half-adder, J. Am. Chem. Soc., 2004, 126, 15926–15927 CrossRef PubMed.
- F. Remacle, R. Weinkauf and R. D. Levine, Molecule-based photonically switched half and full adder, J. Phys. Chem. A, 2006, 110, 177–184 CrossRef CAS PubMed.
- H. Pei, L. Liang, G. Yao, J. Li, Q. Huang and C. Fan, Reconfigurable three-dimensional DNA nanostructures for the construction of intracellular logic sensors, Angew. Chem., Int. Ed., 2012, 51, 9020–9024 CrossRef CAS PubMed.
- D. Margulies, G. Melman and A. Shanzer, Fluorescein as a model molecular calculator with reset capability, Nat. Mater., 2005, 4, 768–771 CrossRef CAS PubMed.
- D. Margulies, G. Melman and A. Shanzer, A molecular full-adder and full-subtractor, an additional step toward a moleculator, J. Am. Chem. Soc., 2006, 128, 4865–4871 CrossRef CAS PubMed.
- D. Margulies, C. E. Felder, G. Melman and A. Shanzer, A molecular keypad lock: A photochemical device capable of authorizing password entries, J. Am. Chem. Soc., 2007, 129, 347–354 CrossRef CAS PubMed.
- P. Remón, M. Hammarson, S. Li, A. Kahnt, U. Pischel and J. Andréasson, Molecular implementation of sequential and reversible logic through photochromic energy transfer switching, Chem. – Eur. J., 2011, 17, 6492–6500 CrossRef PubMed.
- Q. Li, Y. Yue, Y. Guo and S. Shao, Fluoride anions triggered ‘OFF–ON’ fluorescent sensor for hydrogen sulfate anions based on a BODIPY scaffold that works as a molecular keypad lock, Sens. Actuators, B, 2012, 173, 797–801 CrossRef CAS.
- X.-J. Jiang and D. K. P. Ng, Sequential logic operations with a molecular keypad lock with four inputs and dual fluorescence outputs, Angew. Chem., Int. Ed., 2014, 53, 10481–10484 CrossRef CAS PubMed.
- B. Rout, P. Milko, M. A. Iron, L. Motiei and D. Margulies, Authorizing multiple chemical passwords by a combinatorial molecular keypad lock, J. Am. Chem. Soc., 2013, 135, 15330–15333 CrossRef CAS PubMed.
- G. Strack, M. Ornatska, M. Pita and E. Katz, Biocomputing security system: concatenated enzyme-based logic gates operating as a biomolecular keypad lock, J. Am. Chem. Soc., 2008, 130, 4234–5235 CrossRef CAS PubMed.
- J. Chen, S. Zhou and J. Wen, Concatenated logic circuits based on a three-way DNA junction: A keypad-lock security system with visible readout and an automatic reset function, Angew. Chem., Int. Ed., 2014, 54, 446–450 Search PubMed.
- A. P. de Silva and N. D. McClenaghan, Proof-of-principle of molecular-scale arithmetic, J. Am. Chem. Soc., 2000, 122, 3965–3966 CrossRef CAS.
- P. Remón, M. Bälter, S. Li, J. Andréasson and U. Pischel, An all-photonic molecule-based D flip-flop, J. Am. Chem. Soc., 2011, 133, 20742–20745 CrossRef PubMed.
- Y. Liu, W. Jiang, H. Y. Zhang and C. J. Li, A multifunctional arithmetical processor model integrated inside a single molecule, J. Phys. Chem. B, 2006, 110, 14231–14235 CrossRef CAS PubMed.
- R. Gotor, A. M. Costero, S. Gil, M. Parra, P. Gaviña and K. Rurack, Boolean operations mediated by an ion-pair receptor of a multi-readout molecular logic gate, Chem. Commun., 2013, 49, 11056–11058 RSC.
- M. Vester, T. Staut, J. Enderlein and G. Jung, Photon antibunching in a cyclic chemical reaction scheme, J. Phys. Chem. Lett., 2015, 6, 1149–1154 CrossRef CAS PubMed.
- M. Vester, A. Grueter, B. Finkler, R. Becker and G. Jung, Biexponential photon antibunching: Recombination kinetics within the Forster-cycle in DMSO, Phys. Chem. Chem. Phys., 2016, 18, 10281–10288 RSC.
- R. K. Sehgal and S. Kumar, A simple preparation of 1-hydroxypyrene, Org. Prep. Proced. Int. New J. Org. Synth., 1989, 21, 223–225 CrossRef CAS.
Footnotes |
| † This work is dedicated to Andreas Zumbusch on the occasion of his 50th birthday. |
| ‡ Electronic supplementary information (ESI) available: Further compound characterization, determination of enzyme kinetics, further preliminary live-cell experiments and details for molecular logic. CCDC 1443123. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6pp00290k |
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |