
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, regioselective aerobic Pd(II)-catalyzed C–H bond alkenylation and the photophysical properties of pyrenylphenylpyrazoles†
Rafał
Flamholc
a,
Janusz
Zakrzewski
*a,
Anna
Makal
b,
Arnaud
Brosseau
c and
Rémi
Métivier
c
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland. E-mail: janzak@uni.lodz.pl
bUniversity of Warsaw, Biological and Chemical Research Center, Żwirki i Wigury 101, 02-089 Warszawa, Poland
cPPSM, ENS Cachan, CNRS, UniverSud, 61 av President Wilson, 94230 Cachan, France
First published on 21st March 2016
Abstract
This paper reports the synthesis, regioselective aerobic Pd(II)-catalyzed C–H bond alkenylation and the photophysical properties of pyrenylphenylpyrazoles.
Introduction
Pyrene and its derivatives are important and thoroughly investigated organic fluorophores that have found numerous applications in molecular electronics, photovoltaic cells and as fluorescent probes and sensors.1–4 Heterocyclic compounds possessing pyrenyl substituents have also attracted considerable interest due to their unusual luminescence and chemical properties. In the literature there are a number of reports on pyrenylpyrazoles which have displayed interesting metal-coordinating and sensing properties.5–9Taking into account the considerable interest in the chemistry of pyrazoles,10–15 including their fluorescence properties,16–20 we thought it would be of interest to synthesize novel pyrenylpyrazole fluorophores, especially those bearing a phenyl substituent at N1 that could enable easy chemical modification via directed C–H bond activation.21–26 Such compounds are expected to be formed in the reaction of the pyrenyl ynone, 1-(pyren-1-yl)prop-2-yn-1-one (1) with phenylhydrazine.15 In this communication we disclose the synthesis of 1-phenyl-3-(pyren-1-yl)-1H-pyrazole and 1-phenyl-5-(pyren-1-yl)-1H-pyrazole, Pd(II)-catalyzed alkenylation of the former compounds and the photophysical properties of the synthesized compounds.
Results and discussion
Syntheses
The reaction of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 with phenylhydrazine was conducted in methanol containing HCl at room temperature for 5 h. It afforded a mixture of regioisomeric products, 1-phenyl-3-(pyren-1-yl)-1H-pyrazole 2a and 1-phenyl-5-(pyren-1-yl)-1H-pyrazole 2b (Scheme 1), which were easily separated by column chromatography. The isolated yields of 2a and 2b were 58% and 27%, respectively.
27 with phenylhydrazine was conducted in methanol containing HCl at room temperature for 5 h. It afforded a mixture of regioisomeric products, 1-phenyl-3-(pyren-1-yl)-1H-pyrazole 2a and 1-phenyl-5-(pyren-1-yl)-1H-pyrazole 2b (Scheme 1), which were easily separated by column chromatography. The isolated yields of 2a and 2b were 58% and 27%, respectively.
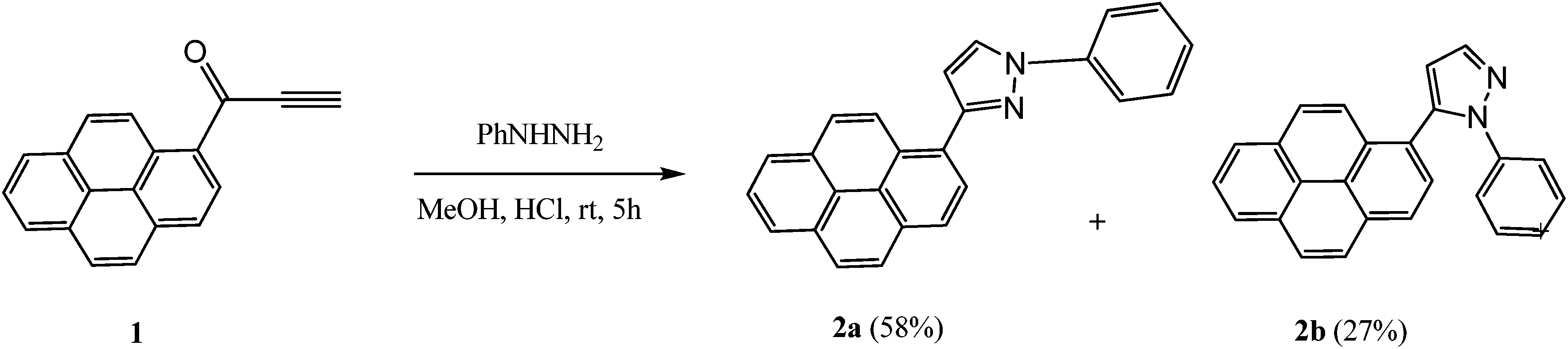 | ||
| Scheme 1 Synthesis of pyrenylphenyl pyrazoles 2a and b. | ||
Compounds 2a and 2b were characterized by 1H and 13C NMR spectroscopy and elemental analyses. Their structures were unambiguously established by single-crystal X-ray diffraction (vide infra).
The presence of the 1-phenylpyrazole moiety in 2a–b offers the possibility of direct functionalization of the phenyl ring via transition metal-catalyzed directed C–H bond activation.21–26 For example, it is known that 1-phenylpyrazole undergoes ruthenium(II)-catalyzed alkenylation,28 arylation29 and rhodium(III)-catalyzed coupling with internal alkynes24 on the phenyl ring.
We performed alkenylation of 2a with n-butyl acrylate under conditions we had described earlier for alkenylation of ferrocene and pyrene.30–32 The reaction was conducted in boiling acetic acid under oxygen (1 atm) in the presence of a catalytic amount (5 mol%) of Pd(OAc)2 and 4,5-diazafluoren-9-one (DAF, 10 mol%) (Scheme 2).
 | ||
| Scheme 2 Aerobic Pd(II)-catalyzed pyrazole-directed alkenylation of 2a. | ||
The reaction afforded selectively, after 6 h of heating, the product of ortho-alkenylation of the phenyl ring 3 in an acceptable isolated yield (42%). The structure of 3 was confirmed by spectroscopic and analytical data. In particular, its 1H NMR spectrum contained four one-proton signals (2 doublets and 2 triplets) in the 7.4–7.85 ppm region, which is a characteristic pattern of a 1,2-disubstituted benzene. The signals of the pyrene moiety (8.0–8.9 ppm) correspond to 9 protons and the splitting pattern is closely similar to that observed for 2a. Finally, the olefinic protons give rise to two doublets (7.95 and 6.48 ppm) with J = 16 Hz confirming a trans configuration.
To the best of our knowledge, the reported reaction constitutes the first example of the alkenylation of a compound bearing the 1-phenylpyrazole moiety under Fujiwara–Moritani conditions (i.e. Pd(II) catalyst and oxidant for the recovery of Pd(II) reduced to Pd(0) in the catalytic cycle). It is worth noting that our system is environmentally benign (acetic acid is one of the solvents that is recommended by the Solvent Selection Guide33) and inexpensive dioxygen is the sole oxidant (aerobic reaction). Furthermore, in our earlier work on alkenylation of pyrene using the same catalytic system32 we found that this reaction also works when oxygen is replaced by air, but the yields of the products were lower.
We also attempted to perform the alkenylation of 2b, but we found that under the aforementioned conditions no reaction took place. In our opinion, this lack of reactivity of 2b may be explained by larger distortions from the planarity of the phenylpyrazole moiety in this compound in comparison with 2a. The generally accepted mechanism of directed C–H activation involves the formation of a cyclometalated intermediate i.e. a compound bearing the Pd atom bridging nitrogen and an ortho-carbon. We believe that the formation of such an intermediate from 2b would be less favorable than from 2a due the larger twist of the phenyl ring from the pyrazole plane in the former compounds (according to X-ray data 46° as compared with 27° found in 2a).
Photophysical properties of 2a–b and 3
The electronic absorption spectra of 2a–b and 3 in CHCl3 (10−5 M, Fig. 1a and Table 1) display features typical for a monomeric pyrene chromophore. They reveal only a weak solvent effect (see ESI†) suggesting a small transition moment. The absorption spectra of 2a and 3 are very similar. The absorption spectrum of 2b shows a more pronounced vibrational structure than 2a and 3, which may suggest a more rigid structure of the former compound in solution. A comparison of the λmax values of 2a and 3 (352 nm and 353 nm, respectively) shows that the introduced electron-poor alkenyl group practically does not influence the excitation energy of the fluorophore. | ||
| Fig. 1 Synthesis of pyrenylphenylpyrazoles 2a–b. | ||
| Compound | Absorption | Emission (λexcit = 345 nm) | |
|---|---|---|---|
| λ max/nm (εmax/M−1 cm−1) | λ max/nm | Φ Ar (air)a | |
| a Determined using quinine bisulfate in 1 M sulfuric acid as a standard. | |||
| 2a | 283 (50500), 352 (45570) |
392, 415, 438 (sh) | 0.69 (0.62) |
| 2b | 270 (31750), 281 (44680), 333 (28480), 346 (39150) |
386, 399 | 0.28.(0.21) |
| 3 | 273 (64650), 282 (74630), 353 (49640) |
395, 414, 445 (sh), 495 | 0.42 (0.31) |
Compounds 2a–b and 3 emit fluorescence in solution and in the solid state. Fig. 1b shows their emission spectra recorded in deaerated, diluted (10−6 M) chloroform solutions. The spectroscopic data are gathered in Table 1.
The more or less resolved vibrational structure of the absorption and emission bands of 2a–b may be due to various reasons such as different conformational rigidity, different deviations from planarity, different solvatation (e.g. formation of hydrogen bonds to pyrazole nitrogens). These bands may be attributed to a local excitation (LE) of the pyrenyl fluorophore. In contrast, 3 displays dual fluorescence. Its emission spectrum in CHCl3 displays a structured band that is similar to that of 2a and a broad, featureless band with a maximum at 495 nm. Dual fluorescence is a phenomenon that is encountered for some donor–acceptor π-systems, and is usually interpreted as a result of emission from an LE state and an excited state with intramolecular charge transfer (ICT).34 It was observed for some biphenyl pyrene derivatives bearing electron withdrawing ester groups.35 ICT emission was also observed for some pyrene-heterocycle systems and their metal complexes.36–39 This means that the introduction of the lateral acrylic ester chain endowed 3 with an emissive low-lying ICT excited state. As expected, the relative contributions of the LE and ICT bands strongly depend on the polarity of the solvent (Fig. 2).
 | ||
| Fig. 2 Emission spectra of 3 in various solvents (c = 10−6 M, λexcit = 345 nm). | ||
In a nonpolar solvent, hexane, only the LE band is observed with maxima at 395 and 405 nm. The emission quantum yield in this solvent reaches 0.59. In more polar solvents (chloroform and dichloromethane) the intensity of the LE band decreases and the ICT band appears. A careful analysis of the ICT band observed in chloroform and dichloromethane reveals the presence of two contributions, located at ∼500 and 550 nm, respectively. They can be tentatively assigned to two distinct conformations of the ICT excited state of 3. Finally, in highly polar solvents (methanol, acetonitrile) fluorescence is weak and the ICT band becomes very broad and hardly detectable. Since the largest difference is observed between hexane and chloroform we also measured the fluorescence of 3 in mixed hexane–chloroform solvents. The spectra are shown in Fig. 3.
 | ||
| Fig. 3 Fluorescence spectra of 3 in hexane–chloroform mixtures. c = 10−6 M, λexcit = 345 nm. | ||
It can be observed that addition of up to 30% of chloroform to hexane brings about sharp changes in the emission spectra of 3. The intensity of the LE band drops down and the broad ICT band appears. However, the intensity of the ICT band decreases and the two relative contributions of the ICT bands evolve at higher chloroform contents which may be due to the decrease of the fluorescence quantum yield (0.59 in hexane and 0.42 in chloroform) and a change of the relative proportion of the different conformations of the ICT state.
We also performed a time-resolved study of fluorescence of the synthesized compounds in CHCl3 solution. The fluorescence decay curves are shown in Fig. 4.
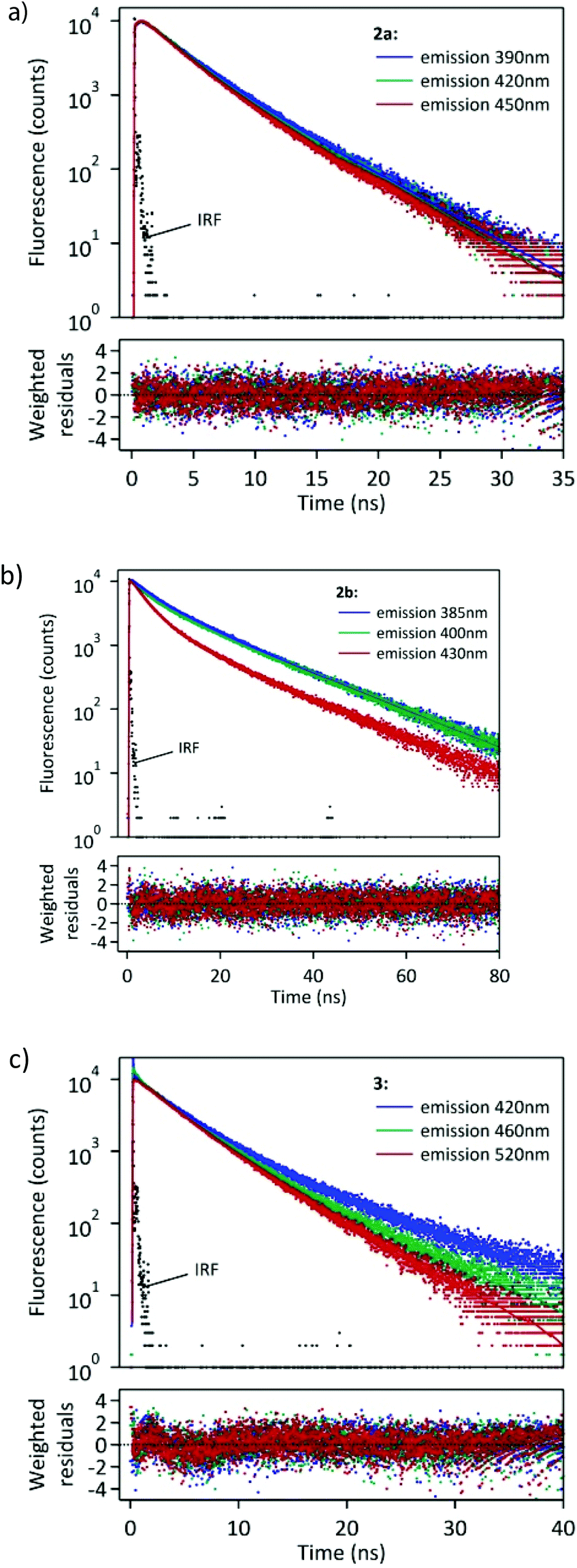 | ||
| Fig. 4 Fluorescence decay curves of 2a–b and 3 in CHCl3 solution monitored at three different wavelengths. λexcit = 360 nm. | ||
The fitting of these curves (Table 2) revealed multiexponential decays, which is not surprising when taking into account the various possible conformations of the excited 2a–b and 3.
| λ em (nm) | τ 1 (ns) | τ 2 (ns) | τ 3 (ns) | A 1 (f1) | A 2 (f2) | A 3 (f3) | χ 2 R | |
|---|---|---|---|---|---|---|---|---|
| a A fourth time-constant τ4 ∼ 0.008 ns with a fraction of intensity lower than 0.01 was necessary to obtain satisfactory fitting curves, probably due to residual scattering signal; therefore, we have neglected this very small contribution here. | ||||||||
| 2a | 390 | 0.39 | 2.68 | 4.68 | −0.27 (−0.02) | 0.76 (0.47) | 0.51 (0.56) | 1.12 |
| 420 | 0.39 | 2.68 | 4.68 | −0.30 (−0.03) | 0.86 (0.55) | 0.43 (0.48) | 1.04 | |
| 450 | 0.39 | 2.68 | 4.68 | −0.28 (−0.03) | 0.94 (0.63) | 0.34 (0.40) | 1.09 | |
| 2b | 385 | 2.86 | 6.56 | 14.78 | 0.17 (0.05) | 0.39 (0.27) | 0.44 (0.68) | 1.05 |
| 400 | 2.86 | 6.56 | 14.78 | 0.31 (0.10) | 0.24 (0.17) | 0.45 (0.73) | 1.07 | |
| 430 | 2.86 | 6.56 | 14.78 | 0.60 (0.30) | 0.24 (0.28) | 0.16 (0.42) | 1.05 | |
| 3 | 420 | 0.32a | 3.75 | 8.38 | 0.26 (0.03) | 0.62 (0.69) | 0.12 (0.28) | 1.14 |
| 460 | 0.32a | 3.75 | 8.38 | 0.38 (0.05) | 0.57 (0.81) | 0.05 (0.14) | 1.13 | |
| 520 | 0.32a | 3.75 | 8.38 | 0.12 (0.01) | 0.85 (0.92) | 0.03 (0.07) | 1.17 | |
Interestingly, compound 2a showed a fast rise (τ ∼ 0.39 ns) of fluorescence following the laser pulse, which was observed at 390, 420 and 450 nm, thus suggesting that the emissive state is formed by a fast relaxation of the initial Franck–Condon state. In our opinion, the rise time of 2a is probably due to some geometry reorganisation of the molecule in the excited state (such as planarisation of some conjugated substituent in the excited state).
At a longer time period two emissive components are clearly distinguishable with lifetimes of 2.68 and 4.68 ns. Two main decay times, 6.56 and 14.78 ns, were observed for 2b at 385 nm. However, at longer emission wavelengths (400 and 430 nm) a third component at 2.86 ns became important (f1 = 0.10–0.30).
Fitting of the fluorescence decay of 3 revealed three decay components. The two main ones (f2 + f3 > 0.95) have been determined to be τ2 = 3.75 and τ3 = 8.38 ns. The contribution of the faster component τ2 increased along with the increase in the monitoring wavelength and reached f2 = 0.92 at 520 nm. Therefore, this component can be identified as corresponding to the ICT excited state. On the other hand, the slower component had the largest contribution in emission observed at 420 nm, thus corresponding to emission originating mainly from the LE state. The above data suggest the formation of both LE and ICT excited states of 3 during the laser pulse. The lifetime of the LE state is longer than that of the ICT state.
The data presented here show that dual fluorescence of 3 is sensitive to the polarity of the environment surrounding the fluorophore and, therefore, that this compound can be used as a fluorescent micropolarity probe, especially in weakly polar media.
The solid-state emission spectra of 2a, 2b and 3 are shown in Fig. 5. The emission maxima are at 503, 484 and 513 nm, and the emission quantum yields are respectively 0.05, 0.05 and 0.07.
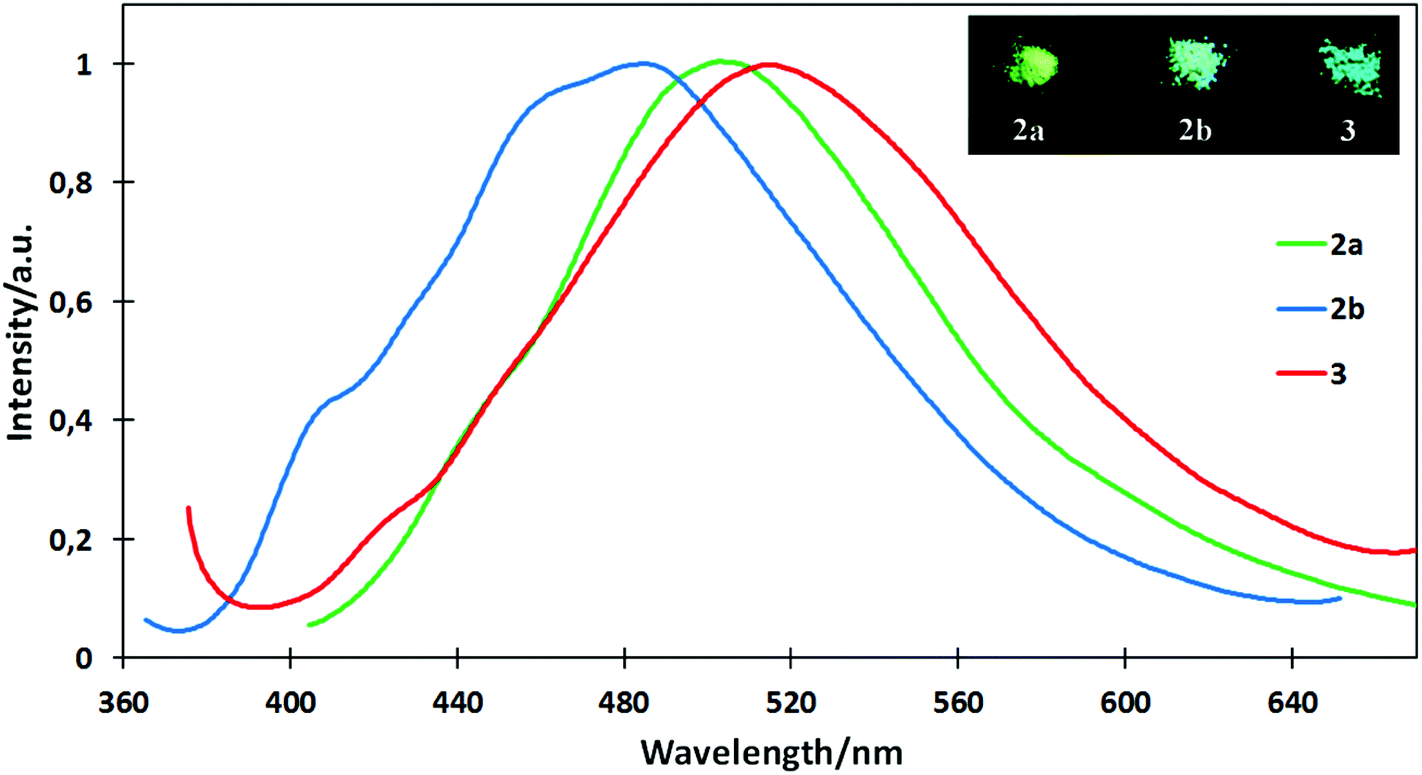 | ||
| Fig. 5 Normalized fluorescence spectra of 2a, 2b and 3 in the solid state. λexcit = 380 nm (2a); 370 nm (2b); 375 nm (3). | ||
The spectra are closely similar to the maxima in the region of the ICT transition of 3 in solution. The presence of the unsaturated substituent in 3 has only a slight influence on the emission maximum. The vibrational structure that is present in the spectrum of 2b suggests emission originating from the monomers, whereas the broad structureless bands observed for 2a and 3 may be assigned to solid-state aggregates or excimers. This hypothesis was confirmed by X-ray data, which reveal different crystal packings in 2a and 2b (unfortunately all of our attempts to obtain crystals of 3 that would be suitable for the X-ray diffraction study failed).
X-ray diffraction study of 2a and 2b
Compounds 2a and 2b crystallize from dichloromethane–hexane in the centrosymmetric P2(1)/c space group in the monoclinic system, unattended by solvent molecules. Their molecular structures are shown in Fig. 6. In terms of molecular geometry, both compounds show great similarity. The only significant difference in the bond lengths is visible in the C17–C18 bond in the pyrazole moiety, which in the case of 2a is longer by ∼0.05 Å. The relative orientations of the pyrene moiety, the pyrazole ring and the phenyl ring are, however, quite different. In particular, the twist of the phenyl ring with respect to the pyrazole moiety is much more pronounced in the case of 2b (as stated above). The values of the important dihedral angles are reported in Table 3. In the case of 2b, a short C1–C20 contact (3.101(2) Å) between the anchor atoms of the pyrene moiety and the phenyl ring is present. This is in fact the minimal possible distance for these two atoms among all of the possible conformations of 2b. The C–C bonds between the pyrene and pyrazole fragments and the C–N bond between the pyrazole and the phenyl group are essentially single, when judged by their lengths, thus suggesting no strong conjugation between these fragments. The pyrene moiety is essentially flat in the case of 2a, while in the case of 2b it is slightly bent.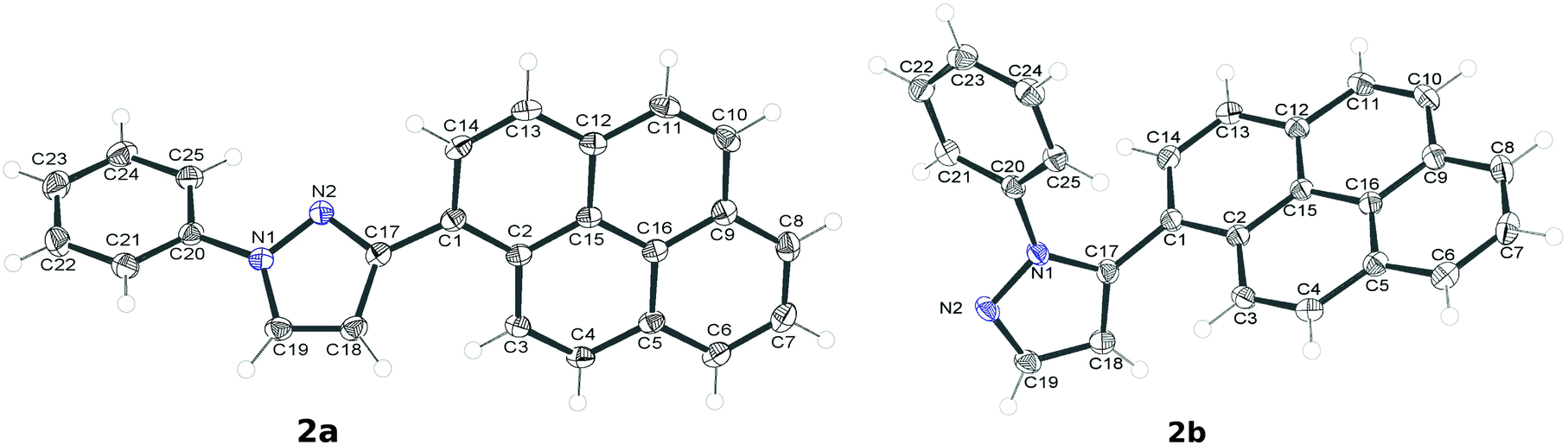 | ||
| Fig. 6 Molecular structures (ORTEP's, 50% probability) of 2a and 2b. | ||
| 2a | 2b | |
|---|---|---|
| Bond lengths [Å] | ||
| C1–C17 | 1.479(2) | 1.480(2) |
| C17–C18 | 1.418(2) | 1.375(2) |
| C18–C19 | 1.367(2) | 1.397(2) |
| C19–N1 | 1.358(2) | 1.328(2) |
| N1–N2 | 1.362(2) | 1.367(1) |
| N2–C17 | 1.341(2) | 1.362(1) |
| N1–C20 | 1.421(2) | 1.427(1) |
| Dihedral angles [°] | ||
| C2–C1–C17–C18 | 49.2(2) | −67.5(2) |
| N2–N1–C20–C21 | −154.3(1) | −46.5(1) |
However, the compounds differ significantly in terms of the intermolecular interactions. In the case of 2a the molecules display close and extensive π⋯π stacking interactions between the pyrenyl moieties related by crystallographic inversion (Fig. 7a). In addition, the phenyl rings of the molecules related by the other crystallographic inversion are also showing π⋯ interactions (there is a short contact of 3.394(4) Å). On the other hand, N2 is effectively shielded from any C–H⋯N interactions. The dimers of 2a interact in the crystal lattice mainly by C–H⋯π contacts; in particular, the phenyl moieties are oriented in such a way as to form short C–H⋯π interactions with the pyrenyl moieties.
 | ||
| Fig. 7 The most important intermolecular interactions in the crystal lattices of 2a and 2b; (a) 2a: π⋯π interactions of the pyrenyl moieties: (b) 2b: network of the short C–H⋯N contacts; atoms directly involved in these interactions are represented as ellipsoids, the remaining atoms as wireframe. | ||
There are no π⋯π interactions in the crystal lattice of 2b. On the other hand, N2 is involved in the C–H⋯N interaction with the aromatic C–H group from the pyrenyl group in an adjacent molecule (the H⋯N distance is relatively short, 2.659(6) Å). A network of such short contacts stretches along the crystallographic [001] direction (Fig. 7b). The only other types of interactions present in the case of 2b are numerous C–H⋯π interactions of the pyrenyl moieties with one another and with the phenyl moieties.
Conclusions
We have synthesized new fluorophores having a 1-phenylpyrazole moiety attached to the pyrenyl group and we demonstrated feasibility of regioselective C–H functionalization (alkenylation) of the phenyl ring in these compounds. The introduction of the electron-withdrawing acrylic substituent endowed the (phenylpyrazolyl)pyrene fluorophore with polarity-sensitive dual fluorescence, tentatively explained as originating from LE and ICT excited states. Therefore, it appears that directed C–H functionalization offers an easy route for tuning the emissive properties of this fluorophore.Experimental
Solvents were purified before use by reported methods. All reagents were purchased from Sigma-Aldrich and used without further purification. Column chromatography was carried out on silica gel 60 (0.040–0.063 mm, 230–400 mesh, Fluka). 1H and 13C NMR spectra were recorded at room temperature (291 K) in CDCl3 on a Bruker ARX 600 MHz (600 MHz for 1H and 151 MHz for 13C). The chemical shifts are expressed in ppm. IR spectra were run on a FT-IR Nexus spectrometer in KBr pellets. Elemental analyses were performed at Laboratory of Microanalysis at The Centre of Molecular and Macromolecular Studies in Łódź, Poland.Synthesis of 2a and 2b
A suspension of 1 (254 mg, 1 mmol) and phenylhydrazine hydrochloride (288 mg, 2 mmol) in methanol (95 ml) containing conc. aq. HCl (5.5 ml) was stirred for 5 h at room temperature. The mixture was poured into water and extracted several times with dichloromethane. The extracts were washed with aqueous NaHCO3, water, dried over Na2SO4 and evaporated to dryness. Compounds 2a and 2b were separated by column chromatography (eluent:hexane–dichloromethane 1:1).
2a. Yield 58%. Bright orange solid. Mp 169–170 °C. 1H NMR: δ 8.97 (d, J = 9.3 Hz, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.24 (d, J = 8.0 Hz, 1H), 8.20 (d, J = 7.7 Hz, 2H), 8.14 (d, J = 9.1 Hz, 1H), 8.13 (d, J = 2.5 Hz, 1H), 8.10 (s, 2H), 8.02 (t, J = 7.5 Hz, 1H), 7.89 (d, J = 8.0 Hz, 2H), 7.52 (t, J = 8.1 Hz, 2H), 7.34 (t, J = 7.2 Hz, 1H), 6.94 (d, J = 2.4 Hz, 1H). 13C NMR: δ 153.38, 140.34, 131.90, 131.51, 131.24, 131.07, 129.50, 128.83, 128.48, 127.87, 127.63, 127.45, 127.40, 126.44, 125.96, 125.67, 125.24, 125.17, 124.95, 124.91, 124.80, 119.14, 109.29. IR (cm−1): 3136, 3120, 3043, 2920, 2850, 2182, 1598, 1505, 1389. Elemental analysis: found: C, 87.01; H, 4.51; N, 8.47%. Molecular formula C25H16N2 requires C, 87.18; H, 4.68; N, 8.13%.
2b. Yield 27%. Bright yellow solid. Mp 167–168 °C. 1H NMR: δ 8.24 (d, J = 7.5 Hz, 1H), 8.20 (d, J = 7.5 Hz, 1H), 8.14 (d, J = 9.1 Hz, 1H), 8.13 (d, J = 2.2 Hz, 1H), 8.11 (s, 1H), 8.07 (d, J = 9.0 Hz, 1H), 8.06 (d, J = 1.6 Hz, 1H), 8.04 (t, J = 3.4 Hz, 1H), 7.98 (d, J = 1.7 Hz, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.29–7.27 (m, 2H), 7.14–7.10 (m, 3H), 6.76 (d, J = 1.9 Hz, 1H). 13C NMR: δ 141.37, 140.31, 140.12, 131.58, 131.30, 130.86, 129.79, 128.73, 128.32, 127.28, 126.90, 126.28, 125.76, 125.64, 125.50, 124.82, 124.59, 124.56, 124.47, 124.13, 124.32, 123.73, 110.58. IR (cm−1): 3133, 3042, 2953, 2922, 2852, 2161, 1595, 1496, 1384. Elemental analysis: found: C, 86.92; H, 4.96; N, 7.83%. Molecular formula C25H16N2 requires C, 87.18; H, 4.68; N, 8.13%.
Alkenylation of 2a with n-butyl acrylate
A suspension of 2a (344 mg, 1 mmol), n-butyl acrylate (2 mmol), 4,5-diazafluoren-9-one (18.2 mg, 10% mol) and Pd(OAc)2 (11.2 mg, 5% mol) in acetic acid (5 ml) was refluxed for 6 h under 1 atm of oxygen (balloon). The reaction mixture was poured into water and extracted several times with dichloromethane. The extracts were washed with aqueous NaHCO3, water, dried over Na2SO4 and evaporated to dryness. Compound 3 was isolated by column chromatography (eluent:dichloromethane–hexane 2:3). Yield 32%. Yellow solid. Mp 91.5–92.5 °C. 1H NMR: δ 8.94 (d, J = 9.4 Hz, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.19 (t, J = 6.2 Hz, 2H), 8.13 (d, J = 9.4 Hz, 1H), 8.10 (s. 2H), 8.02 (t, J = 7.8 Hz, 1H), 7.95 (d, J = 16.0 Hz, 1H), 7.86 (d, J = 2.2 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.55 (t, J = 7.15 Hz, 1H), 7.47 (t, J = 7.3 Hz, 1H), 6.98 (d, J = 2.3 Hz, 1H), 6.48 (d, J = 16.0 Hz, 1H), 4.21 (t, J = 6.6 Hz, 2H), 1.66–1.61 (m, 2H), 1.39–1.33 (m, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): 166.63, 153.70, 140.42, 139.98, 132.16, 131.46, 131.22, 131.05, 130.64, 130.08, 128.83, 128.41, 128.24, 127.95, 127.87, 127.63, 127.54, 127.44, 126.19, 125.93, 125.63, 125.18, 125.14, 124.95, 124.88, 124.78, 121.05, 109.01, 64.63, 30.68, 19.11, 13.66. IR (cm−1): 3437, 3107, 3043, 2951, 2929, 2874, 1707, 1638. Elemental analysis: found: C, 81.39; H, 5.57; N, 5.62%. Molecular formula C32H26N2O2 requires C, 81.68; H, 5.57; N, 5.95%.
X-ray diffraction study
The data for crystals of 2a and 2b were collected on an Agilent Supernova 4 circle diffractometer system equipped with a copper and molybdenum microsource and an Atlas CCD detector. The data were collected using molybdenum radiation for 2a and copper radiation for 2b with CrysAlis171 software and integrated with the CrysAlisPRO software. Data were corrected for absorption effects using the multi-scan method (SCALE3 ABSPACK).
The structures were solved by direct methods using SXELXS and refined by a full-matrix least squares procedure with SHELXL within OLEX2 graphical interface. Figures were produced with Ortep3v2 and Mercury_3.3 software.
CCDC 1054146 (2a) and 1054147 (2b) contain the supplementary crystallographic data for the structures.
For further details of the X-ray diffraction study, see ESI.†
Acknowledgements
Financial support from the National Science Centre Poland (NCN, Grant Harmonia UMO-2012/04/M/ST5/00712) is gratefully acknowledged.References
- T. M. Figueira-Duarte and K. Muellen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- G. Bains, A. B. Patel and V. Narayanaswami, Molecules, 2011, 16, 7909–7935 CrossRef CAS PubMed.
- C. X. Yao, H. B. Kraatz and R. P. Steer, Photochem. Photobiol. Sci., 2005, 4, 191–199 CAS.
- M. Ottonelli, M. Piccardo, D. Duce, S. Thea and G. Dellepiane, J. Phys. Chem. A, 2012, 116, 611–630 CrossRef CAS PubMed.
- A. Jouaiti, M. W. Hosseini and N. Kyritsakas, Eur. J. Inorg. Chem., 2003, 2003, 57–61 CrossRef.
- A. Ciupa, M. F. Mahon, P. A. De Bank and L. Caggiano, Org. Biomol. Chem., 2012, 10, 8753–8757 CAS.
- Z. Jin, J. Wu, C. Wang, G. Dai, S. Liu, J. Lu and H. Jiang, Spectrochim. Acta, Part A, 2014, 117, 527–534 CrossRef CAS PubMed.
- T. Ren, J. Wang, G. Li and Y. Li, J. Fluoresc., 2014, 1–9 Search PubMed.
- F. Reviriego, P. Navarro, E. García-España, M. T. Albelda, J. C. Frías, A. Domènech, M. J. R. Yunta, R. Costa and E. Ortí, Org. Lett., 2008, 10, 5099–5102 CrossRef CAS PubMed.
- S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed.
- A. Schmidt and A. Dreger, Curr. Org. Chem., 2011, 15, 1423–1463 CrossRef CAS.
- A. Chauhan, P. K. Sharma and N. Kaushik, Int. J. ChemTech Res., 2011, 3, 11–17 Search PubMed.
- A. P. Sadimenko, in Advances in Heterocyclic Chemistry, 2001, pp. 157–240 Search PubMed.
- K. Makino, H. S. Kim and Y. Kurasawa, J. Heterocycl. Chem., 1999, 36, 321–332 CrossRef CAS.
- S. Fustero, A. Simón-Fuentes and J. F. Sanz-Cervera, Org. Prep. Proced. Int., 2009, 41, 253–290 CrossRef CAS.
- D. L. Reger, J. R. Gardinier, P. J. Pellechia, M. D. Smith and K. J. Brown, Inorg. Chem., 2003, 42, 7635–7643 CrossRef CAS PubMed.
- Y. B. Borozdina, V. Kamm, F. Laquai and M. Baumgarten, J. Mater. Chem., 2012, 22, 13260–13267 RSC.
- A. Miniewicz, K. Palewska, L. Sznitko and J. Lipinski, J. Phys. Chem. A, 2011, 115, 10689–10697 CrossRef CAS PubMed.
- H. Lee, M. Y. Berezin, R. Tang, N. Zhegalova and S. Achilefu, Photochem. Photobiol., 2013, 89, 326–331 CrossRef CAS PubMed.
- Z. Yang, K. Zhang, F. Gong, S. Li, J. Chen, J. S. Ma, L. N. Sobenina, A. I. Mikhaleva, B. A. Trofimov and G. Yang, J. Photochem. Photobiol., A, 2011, 217, 29–34 CrossRef CAS.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2009, 110, 624–655 CrossRef PubMed.
- T. Asaumi, N. Chatani, T. Matsuo, F. Kakiuchi and S. Murai, J. Org. Chem., 2003, 68, 7538–7540 CrossRef CAS PubMed.
- N. Umeda, K. Hirano, T. Satoh, N. Shibata, H. Sato and M. Miura, J. Org. Chem., 2010, 76, 13–24 CrossRef PubMed.
- P. M. Liu and C. G. Frost, Org. Lett., 2013, 15, 5862–5865 CrossRef CAS PubMed.
- H. Zhao, Y. Shang and W. Su, Org. Lett., 2013, 15, 5106–5109 CrossRef CAS PubMed.
- R. Flamholc, D. Plażuk, J. Zakrzewski, R. Métivier, K. Nakatani, A. Makal and K. Woźniak, RSC Adv., 2014, 4, 31594–31601 RSC.
- P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 3075–3078 RSC.
- O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- M. Piotrowicz and J. Zakrzewski, Organometallics, 2013, 32, 5709–5712 CrossRef CAS.
- M. Piotrowicz, J. Zakrzewski, A. Makal, J. Bak, M. Malińska and K. Woźniak, J. Organomet. Chem., 2011, 696, 3499–3506 CrossRef CAS.
- M. Piotrowicz, J. Zakrzewski, R. Métivier, A. Brosseau, A. Makal and K. Woźniak, J. Org. Chem., 2015, 80, 2573–2581 CrossRef CAS PubMed.
- H. E. Eastman, C. Jamieson and A. J. B. Watson, Aldrichimica Acta, 2015, 48, 51–55 Search PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- W. Weigel, W. Rettig, M. Dekhtyar, C. Modrakowski, M. Beinhoff and A. D. Schlüter, J. Phys. Chem. A, 2003, 107, 5941–5947 CrossRef CAS.
- A. D'Aléo, E. Cecchetto, L. De Cola and R. M. Williams, Sensors, 2009, 9, 3604–3626 CrossRef PubMed.
- S. Leroy-Ihez, C. Belin, A. D'Aléo, R. M. Williams, L. De Cola and F. Fages, Supramol. Chem., 2003, 15, 627–637 CrossRef.
- S. Leroy, T. Soujanya and F. Fages, Tetrahedron Lett., 2001, 42, 1665–1667 CrossRef CAS.
- A. D'Aléo, A. Karapetyan, M. Giorgi and F. Fages, Tetrahedron, 2015, 71, 2255–2259 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1054146 and 1054147. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6pp00009f |
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |