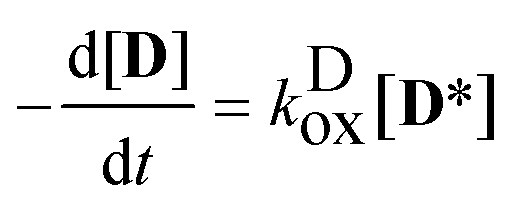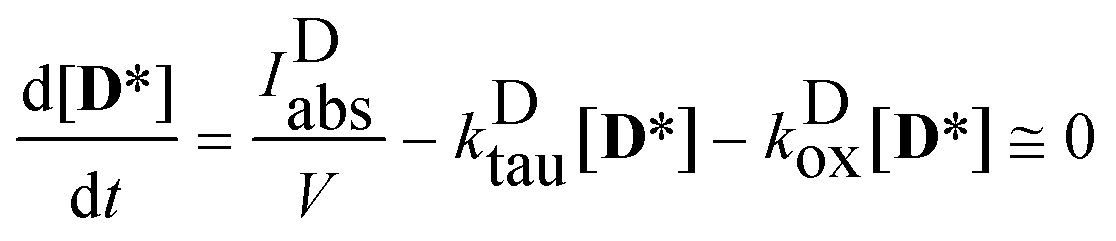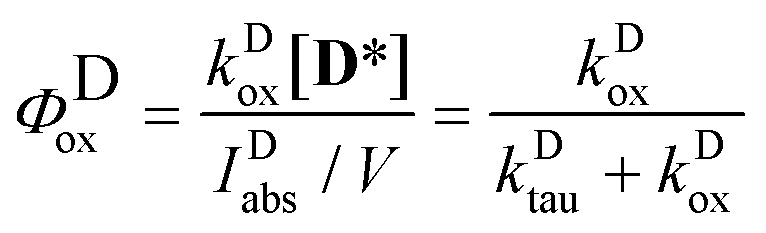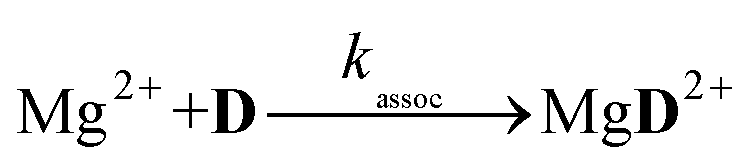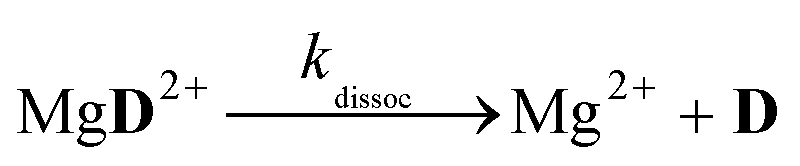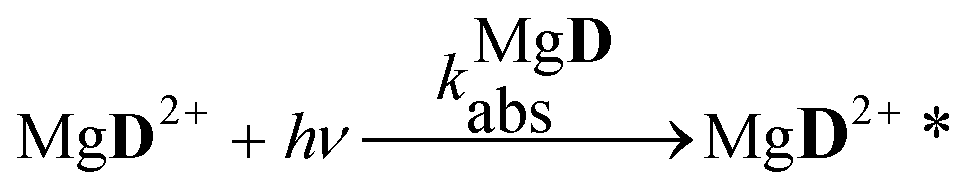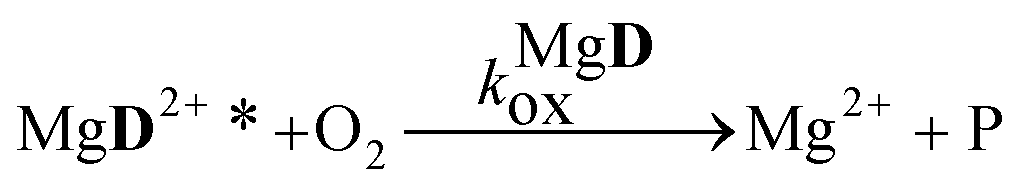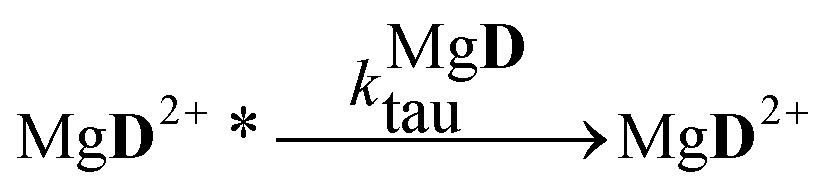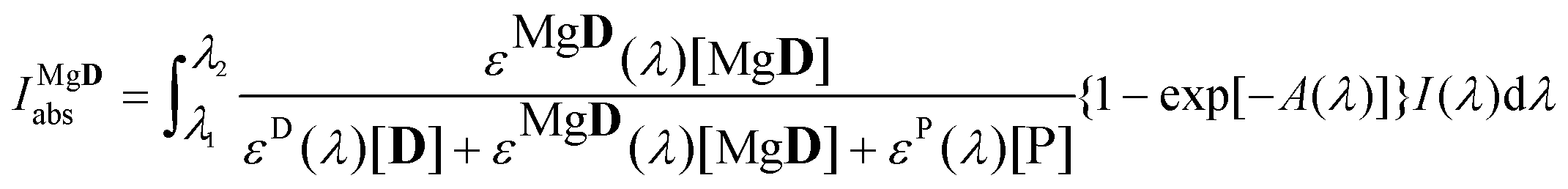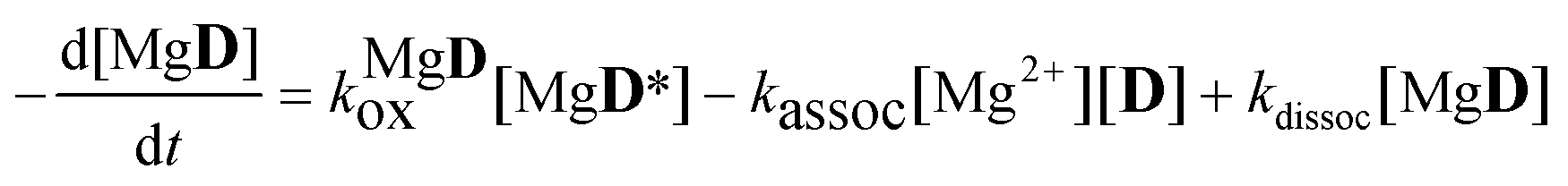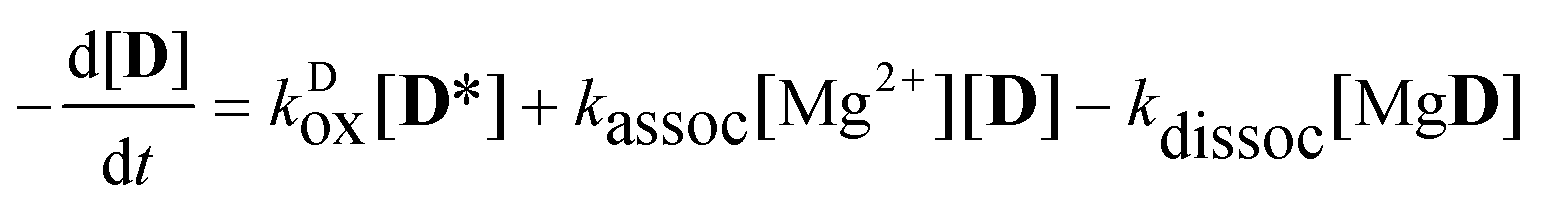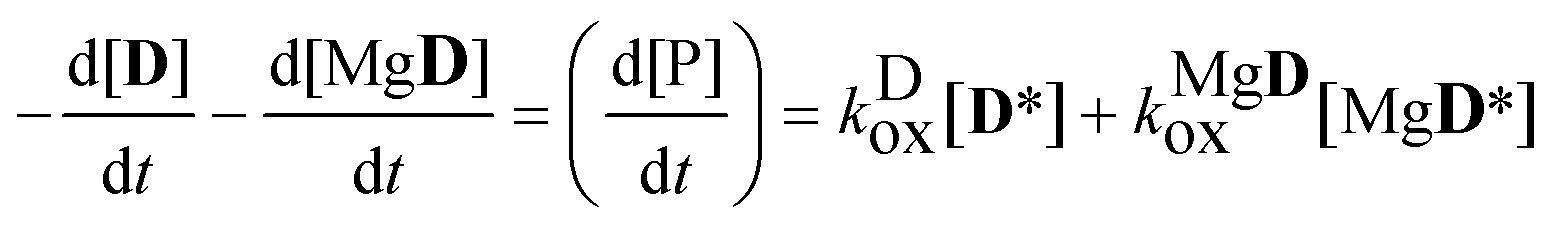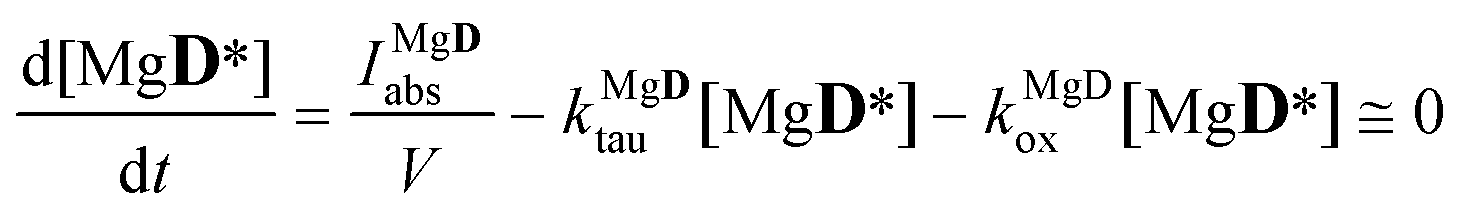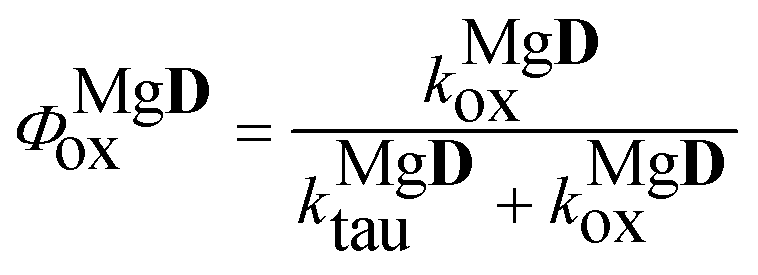The kinetics and mechanism of photooxygenation of 4′-diethylamino-3-hydroxyflavone†
Received
29th September 2015
, Accepted 17th December 2015
First published on 18th December 2015
Abstract
The photolysis reactions of 4′-diethylamino-3-hydroxyflavone (D), a versatile fluorescent probe showing excited-state intramolecular proton transfer (ESIPT), and the magnesium chelate of D (MgD2+) have been studied in acetonitrile solution. Upon UV irradiation both species were oxidized into O-4-diethylaminobenzoyl salicylic acid, differently from the photoreaction of the parent compound 3-hydroxyflavone (3HF) which was described to undergo rearrangement to 3-hydroxy-3-phenyl-indan-1,2-dione. The photooxygenation of the Mg2+ complex was found to be significantly faster than the reaction of the pure dye. As the kinetic analysis of the absorption spectra of samples under irradiation showed, the rate coefficients for the oxygenations of the excited state dye and complex have close values, kox(D*) = 2.4 × 107 min−1, kox(MgD2+*) = 3.9 × 107 min−1; the difference arises from the higher photooxygenation quantum yield of the complex, Φ(MgD2+) = 2.3 × 10−3, than the respective value for the pure dye, Φ(D) = 1.5 × 10−4. The potential energy surface of the photooxygenation of D was calculated assuming a reaction path in which the phototautomer formed from Dvia ESIPT, reacts in its triplet state with triplet molecular oxygen O2, a mechanism similar to that suggested for the photoreaction of the parent 3HF. The moderate values for the transition state energies confirmed the plausibility of the hypothetical mechanism.
Introduction
3-Hydroxy-flavones (3HFs) have two bands in their fluorescence spectra, one arising from the ‘normal’ (N*) excited molecules, the other from the tautomeric (PT*) species, the latter formed via excited state intramolecular proton transfer (ESIPT) from the hydroxyl to the carbonyl group of the N* molecules.1,2 The positions of the two bands, as well as their intensity ratios are sensitive to the local environment, making many of these compounds attractive ratiometric fluorescent sensors.1 Besides, as was demonstrated by Kasha et al., 3HFs with their normal and tautomeric ground states (N and PT) and N* and PT* excited states can act as four-level laser materials.3
3-Hydroxy-flavones with dialkylamino substituents in their 4′ positions (NR2-3HFs) proved to be particularly successful molecular fluorescent sensors. The intramolecular proton transfer cycle in 4′-diethylamino-3-hydroxyflavone, the title compound of this study, a prototype of these probes is exhibited in Scheme 1. The S0 → S1 excitation of NR2-3HF probes is associated with a strong charge transfer.1,2 As a consequence, the position of their N* band is highly sensitive to the polarity of the environment. Besides, NR2-3HF probes, as hydrogen bond acceptors via their CO groups, confer the additional advantage that the ratio of the fluorescence intensities of their N* and PT* forms characterizes the hydrogen bonding ability of their local environment.4 By exploiting the special spectroscopic features of NR2-3HF probes, detailed information can be obtained on their locations in phospholipid membranes.5–8 Attaching such fluorophores covalently to peptides and proteins, peptide–membrane interactions9,10 and structural changes of proteins11 can be studied. Furthermore, NR2-3HFs may have important future applications as fluorescent sensors for the detection of adenosine triphosphate (ATP), as their excitation spectra are sensitive to ATP,7,12 and this effect is selective over the other nucleotides.13
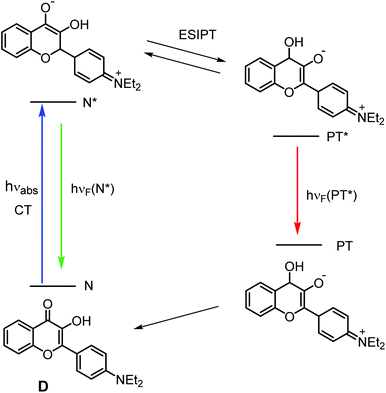 |
| | Scheme 1 Photoinduced intramolecular proton transfer cycle of 4′-diethylamino-3-hydroxyflavone (D). | |
The excitation of 3HFs may be followed by irreversible reactions (oxygenation, and rearrangement),14 the quantum yields of which characterize the photostability of these fluorescent probes, which is a key point when they are applied in analytical and imaging techniques. As no reports on such photoreactions of NR2-3HFs have been found in the literature, we performed this study on the photolysis of 4′-diethylamino-3-hydroxyflavone (D, see Scheme 1), involving the identification of the product(s), the determination of the photophysical and kinetic parameters, and the analysis of the reaction mechanism on the basis of quantum chemical calculations. Our study was extended to the photolysis of the Mg complex, MgD2+, as an example for the photoinduced reaction of a chelate complex of the 4′-diethylamino derivative of 3HF.
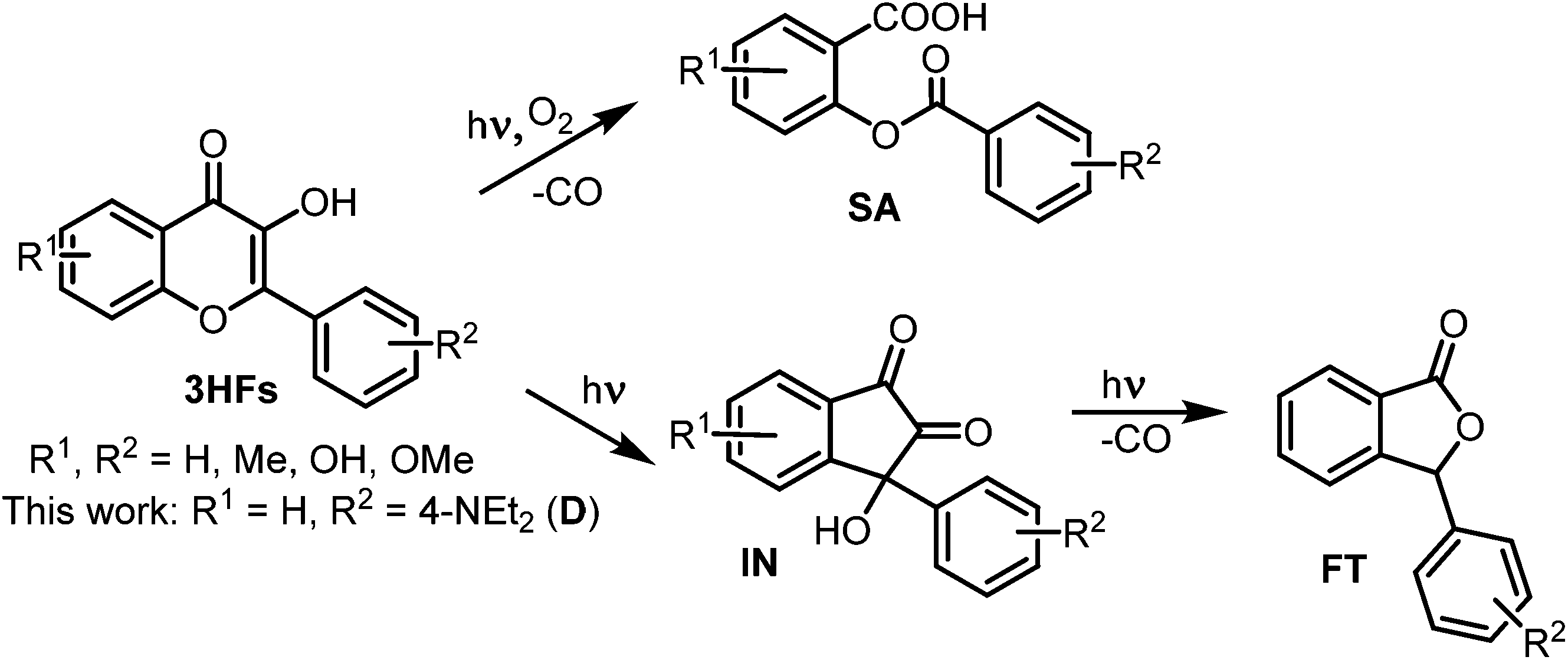
The reactions of other 3HFs, induced by UV irradiation, especially the reactions of the parent compound 3HF, have widely been studied, and basically three kinds of products have been identified (Scheme 2). In the absence of O2, the photolysis of 3HFs resulted in 3-hydroxy-3-aryl-indan-1,2-dione rearrangement products (IN) in apolar, polar aprotic as well as in polar protic solvents.15–18 Some INs lost CO, transforming into 3-aryl-phthalides (FTs).16,17 Under aerated conditions, in apolar solvents 3HFs underwent photooxygenation into O-benzoyl salicylic acid derivatives (SA) via oxidative decarbonylation.19
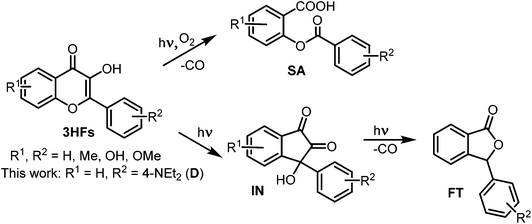 |
| | Scheme 2 Photoreactions of 3-hydroxyflavones. | |
The presence of some metal ions (Mg2+, Ca2+, Hg2+, Zn2+, Pb2+) did not affect the photooxygenation of 3HF in MeOH or in MeOH/H2O mixtures, the same products were obtained – IN or IN + FT as in the neat solvent, whereas other metal ions (Cu2+, Ni2+, Fe3+, Co2+, Al3+) prevented the photoreaction.16,17 Recently, these investigations have been extended to a 3HF–tris(2-picolylamine) ligand pair which forms penta- and hexacoordinated chelates with a series of transition metal ions, and their photooxygenation to SA and degradation to FT products have been observed – the type of reaction depended on the solvent.20,21
The mechanism of the photooxygenation of the parent compound, 3HF, observed in apolar solvents, has been studied in detail. In such solvents the formation of the S1 state phototautomer, 1PT*, from the excited normal molecules, 1N* via ESIPT is a fast process (a transient absorption signal with 35 fs rise time was assigned to 1PT*),22 and the conversion is practically complete, as the less polar 1PT* form is more stable. The singlet phototautomer is converted into the triplet species, 3PT*, with a quantum yield of 0.18 (in n-heptane),23 and it is this 3PT* form of 3HF, which is oxidized by the triplet state oxygen, 3O2.24 The quantum yield of the photooxidation is only ∼0.001.25 For the rate constant of the overall (physical + chemical) quenching of the T1 state dye by 3O2, 3.2 × 109 M−1 s−1 was obtained in n-heptane, a value close to 1/9 of the diffusion controlled rate, which is the theoretical limit for a triplet–triplet energy transfer, i.e. the quenching was dominantly a physical process.23 In the photochemical reaction, an exo-peroxide and an endo-peroxide were reported as intermediates. The exo-peroxide was the photolysis product of 3HF in the O2 matrix, and detected by infrared spectroscopy.26 The endo-peroxide was obtained by photolysing 3HF on a silver electrode, and detected by surface enhanced Raman scattering.27
Materials and methods
Synthesis of D
D was synthesized in two steps, following the general procedure of Smith et al. for the synthesis of flavones.28 First, 2-hydroxy-acetophenone was condensed in ethanol with 4-diethylamino-benzaldehyde to the corresponding chalcone,29 which was oxidized then with H2O2 in alkaline ethanol to obtain D in the form of a yellow solid.30 The product was identified by NMR spectroscopy and HRMS.31
Spectroscopic and photochemical experiments
The acetonitrile solvent was of HPLC grade and purchased from Merck. Mg(ClO4)2 was from Sigma Aldrich, and used without further purification.
The absorption spectra were recorded on an Agilent 8453 diode array UV-Vis spectrometer. The fluorescence spectra and the fluorescence decay curves were recorded with an FLS920 Edinburgh Instruments combined steady state and fluorescence lifetime spectrometer. Two pulsed diode lasers – an EPL-375 (emitting at 378 nm, pulse width ∼90 ps) and an EPL-470 (emitting at 473 nm, pulse width ∼100 ps) were used as light sources when the decay curves were recorded. NMR spectra were recorded on a Bruker Avance DRX-500 spectrometer. The mass spectra were obtained using a Waters Q-TOF Premier mass spectrometer (Manchester, UK) in positive electrospray mode.
In the photolysis experiments, the samples were irradiated by using an Osram XBO OFR 150 W xenon arc lamp, placing a BG3/4g band-pass filter in front of the lamp, with a transmission range of ∼300–420 nm. The photon flux incident on the samples was determined by chemical actinometry, via measuring the yield of photoconversion of o-nitrobenzaldehyde into o-nitrosobenzoic acid.32,33
The optical spectroscopic and photochemical experiments were carried out at 25 °C, in aerated solutions.
Quantum chemical calculations
The theoretical calculations were carried out using the Gaussian09 program package34,35 at the density functional theory (DFT) and time-dependent DFT (TD-DFT)36–38 levels. We employed the M06-2X functional39 with the 6-31++G** basis set for geometry optimizations and for vibrational frequency calculations as well. The solvent effects were described by the integral equation formalism polarizable continuum model (IEF-PCM) with acetonitrile as the solvent. The excited-state energy minimizations were started from the corresponding optimized ground state geometries. The transition state (TS) searches were executed in two steps. First, we used the quadratic synchronous transit (QST3)40 method with a smaller 6-31G* basis set using the optimized structures of the reactants and products. Secondly, a TS optimization was carried out using the Berny algorithm with the 6-31++G** basis set, recomputing the Hessian in each geometry optimization cycle.
Results and discussion
Optical spectra
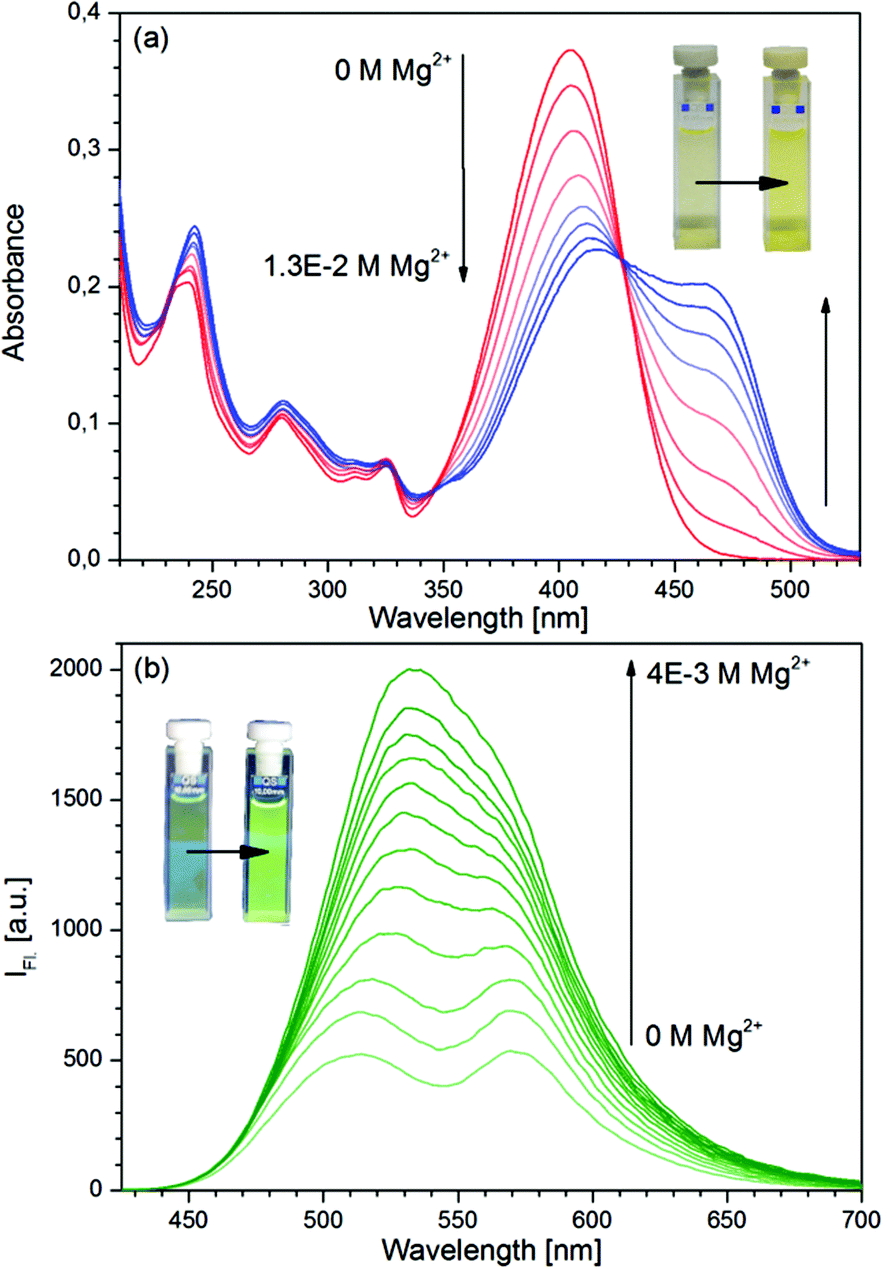
The absorption and fluorescence spectra of D in pure solvent and in the presence of Mg(ClO4)2 in various concentrations are shown in Fig. 1. The fluorescence band at 510 nm belongs to the N*, the band at 575 nm to the PT* form of the dye.41 The large shifts of the absorption and N* fluorescence bands and the disappearance of the PT* band are typical of the Mg complexes of 3HFs: the parent compound 3HF and the dimethyl analogue of D, 4′-NMe2-3HF were reported to show similar spectral changes in the presence of Mg2+ ions.42,43 In contrast, the addition of Mg2+ ions to the solutions of the respective 3-methoxyflavones, did not induce such changes in the spectra. By a simple explanation, the hydroxyl protons of 3HFs dissociated and MgL+ chelates were formed (L = ligand). Recent combined conductometric, spectroscopic and theoretical studies have concluded that the parent compound 3HF forms a complex [MgClO4(3HF)]+, i.e. an ion pair of the salt is bound by the ligand, in which the intramolecular CO⋯HO hydrogen bond is ruptured, but the proton is not dissociated.44,45
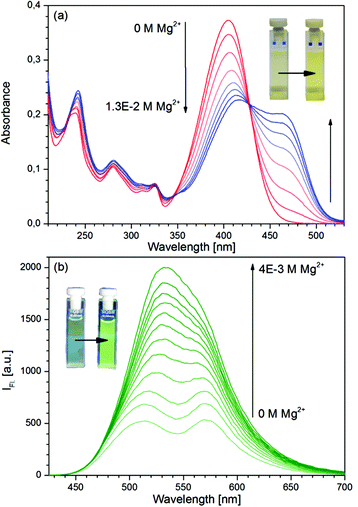 |
| | Fig. 1 (a) Absorption and (b) fluorescence (λex = 427 nm) spectra of D at various Mg(ClO4)2 concentrations. [D]0 = 10−5 M. | |
A least-squares fitting of the absorbance data confirmed that ligand D and Mg2+ ions react in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, and yielded a value of KS = 110 M−1 for the stability constant. For comparison, the stability constants of the other alkali earth metal complexes of D have also been determined, and significantly lower values have been obtained (see Table S1 in the ESI†).
:1 ratio, and yielded a value of KS = 110 M−1 for the stability constant. For comparison, the stability constants of the other alkali earth metal complexes of D have also been determined, and significantly lower values have been obtained (see Table S1 in the ESI†).
The fluorescence decay data of the pure dye and its Mg complex are presented in Table 1. The decay curves of the dye were taken at an emission wavelength on the rising edge of the N* band (470 nm), on the falling edge of the PT* band (630 nm) and at some intermediate wavelengths (510 nm is shown in Table 1). Biexponential curves were obtained, with a dominant ∼380 ps slower component, which practically did not change with the wavelength, indicating quasi-equilibrium between the two excited state tautomers. Femtosecond fluorescence studies proved that the forward and reverse ESIPT reactions in D and related molecules are much faster than the deactivations of the N* and PT* forms.41,46
Table 1 Fluorescence decay time constants of D and MgD2+
| |
λ
ex (nm) |
λ
em (nm) |
τ
1 (ps) (rel. amplitude) |
τ
2 (ps) (rel. amplitude) |
|
D
|
378 |
470 |
65 (9%) |
376 (91%) |
|
|
510 |
97 (7%) |
383 (93%) |
|
|
630 |
|
376 (100%) |
| |
| MgD2+ |
473 |
535 |
3570 (100%) |
|
The decay of the Mg complex was studied in a solution containing ∼52% of the dye in the complexed state (the initial concentrations were [D]0 = 2.5×10−6 M and [Mg(ClO4)2]0 = 10−2 M). The longer lifetime of the excited state complex, 3570 ps can be associated with the rigidity of the chelate structure and the lack of proton transfer as the deactivation channel.43
Product of photolysis
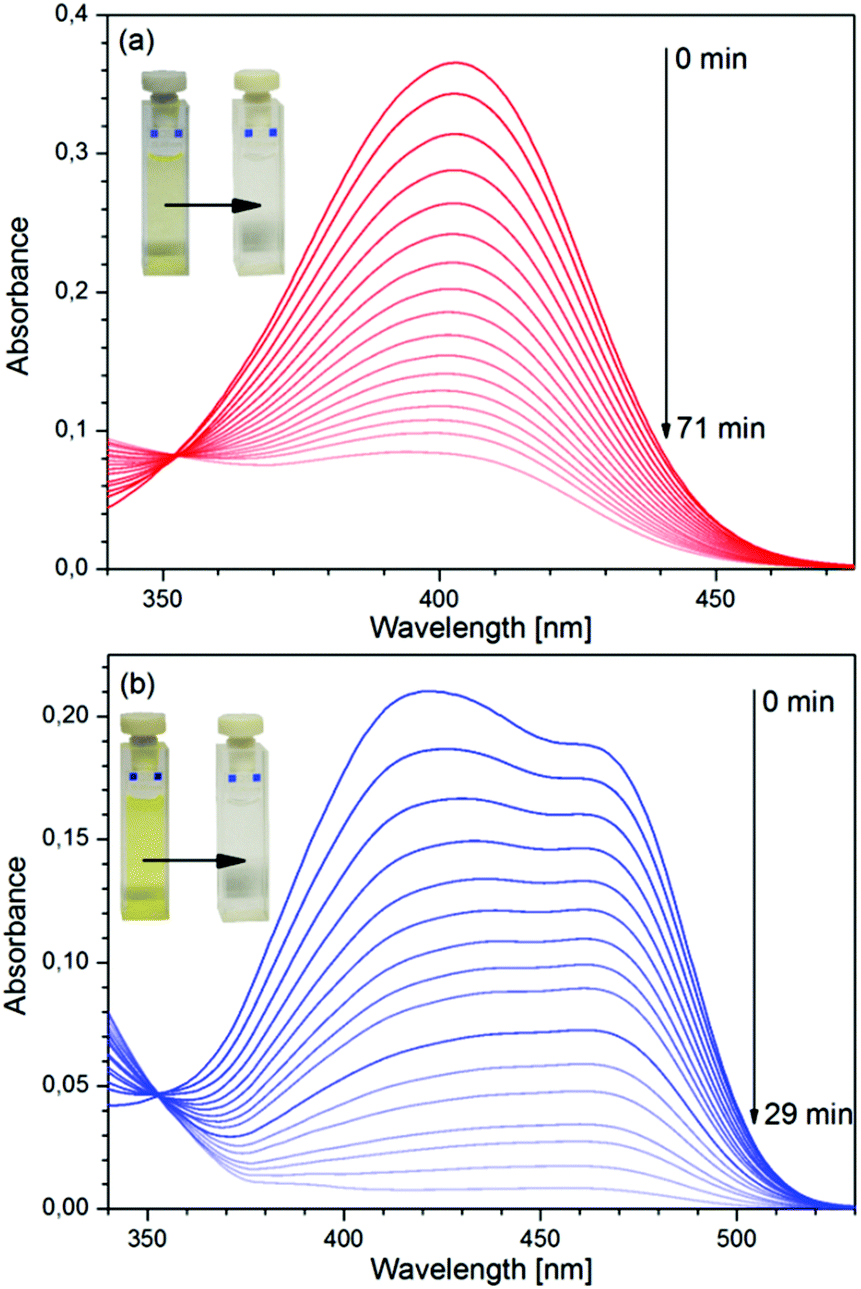
The variation of the absorption spectrum of the aerated solution of the dye upon irradiation by UV light, measured in a 1 cm cell is shown in Fig. 2a. Like in the case of the parent compound, 3HF,3 photoinduced reaction was observed only in aerated solutions of D, and the samples unaerated by nitrogen bubbling were insensitive to UV light. The spectra obtained by irradiating a D–Mg(ClO4)2 mixture (aerated), in which ∼52% of D was present in the complexed form, are exhibited in Fig. 2b.
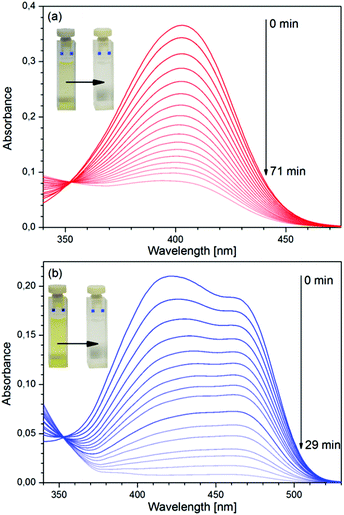 |
| | Fig. 2 Variation of the absorption spectra of solutions of D under irradiation by UV light. (a) Pure D, (b) D–Mg(ClO4)2 mixture. Initial concentrations: (a) [D]0 = 10−5 M, (b) [D]0 = 5 × 10−5 M, [Mg(ClO4)2]0 = 10−2 M. | |
A larger amount of the product, required for measuring the NMR spectra was obtained then by irradiating a 5 × 10−4 M solution of D in a vessel for 24 h, using the same band-pass filter and UV lamp, achieving a ∼70% conversion by the UV-VIS spectrum. The product was isolated by column chromatography (Merck silica gel, toluene–methanol 9:1).
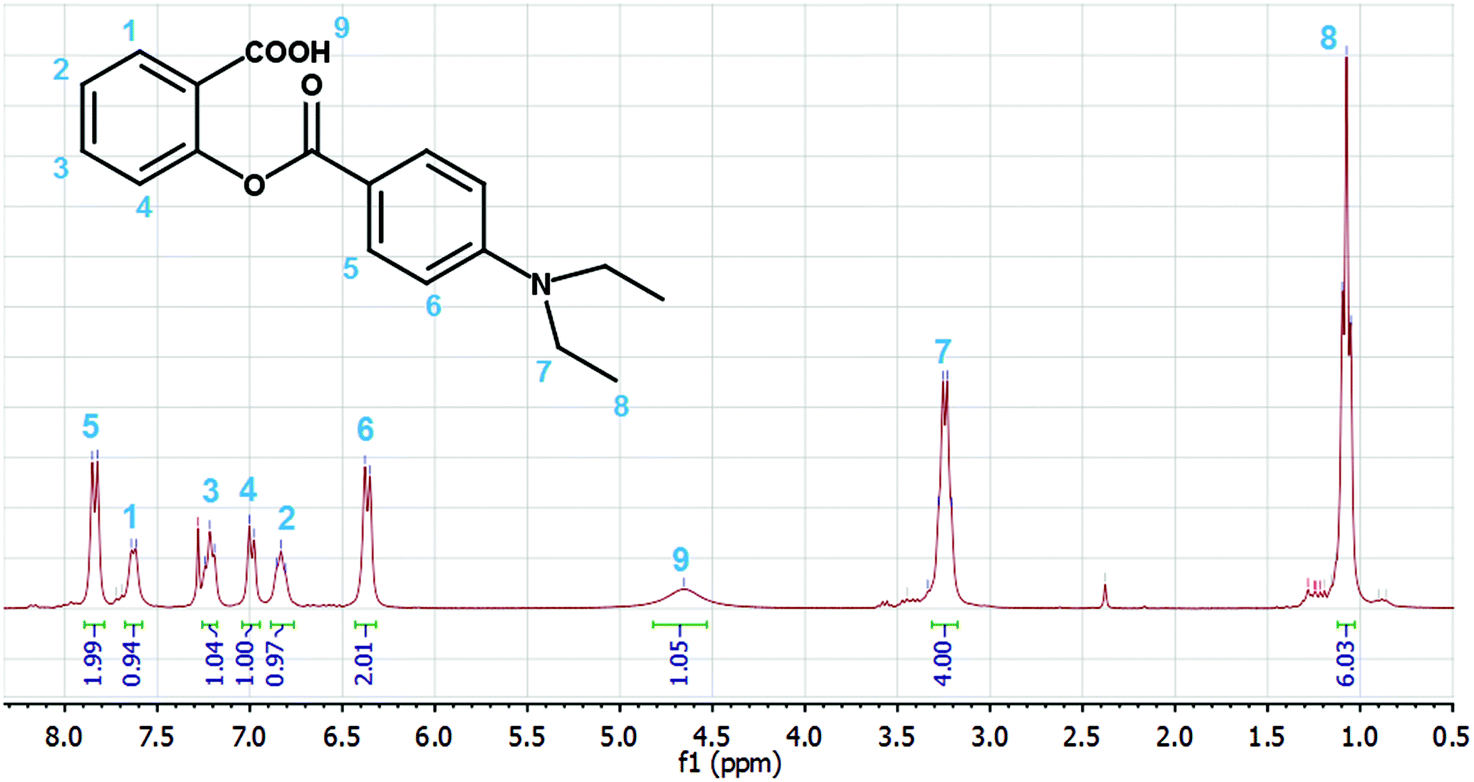
The 1H NMR spectrum of the product (Fig. 3) is consistent with the salicylic acid derivative SA (see Scheme 2) and not with the 1,2-indanedione IN. The position of the doublet of the H5 protons (7.82 ppm) indicates the connection of an electron acceptor CO group, while the relatively low values of the H2 (6.83 ppm) and H4 (6.99 ppm) chemical shifts are indicative of an electron donating group (ester oxygen).
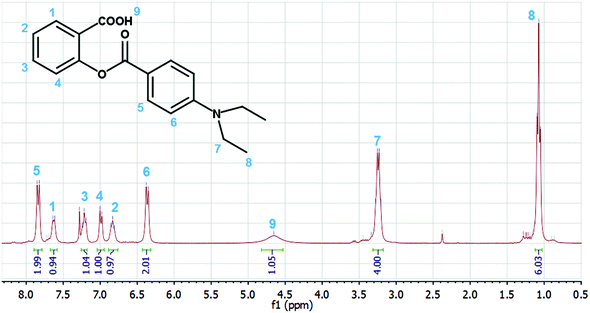 |
| | Fig. 3
1H-NMR spectrum of the photolysis product of D. | |
The HRMS measurement (see Fig. S1 in the ESI†) confirmed this structure, as the measured mass [M + H]+ = 314.1396 showed good agreement (1.9 ppm) with the calculated mass for the salicylic acid ester SA (C18H19NO4, MH+ = 314.1392), whereas the rearrangement product IN (C19H19NO3, MH+ = 310.1443) would have had a mass, identical to that of D.
The irradiation of a mixture with MgClO4, with [D]0 = 5 × 10−4 M, and [Mg(ClO4)2]0 = 7 × 10−3 M initial concentrations, led also to SA, as proved by the 1H NMR spectrum of the product.
Kinetic analysis
The aim of the analysis has been to compare the photooxidation rates and quantum yields of the dye and its Mg complex. The photooxidation of the pure dye has been followed by recording the absorption spectra of the sample at 2 minute intervals for an hour. The instantaneous concentrations of the dye and the photooxidation product were obtained by resolving the spectra into the contributions of D and the product, P. The photoinduced processes in the solution of the pure dye were described by the set of equations:| | 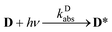 | (1) |
| | 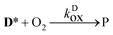 | (2) |
| | 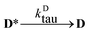 | (3) |
The rate of process (1), the excitation of the dye, expressed in mol photons L−1 s−1, could be obtained as the fraction of the intensity of the incident light, absorbed by the dye, IDabs, related to the volume of the sample:
| | 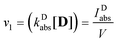 | (4) |
where
| | 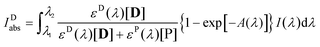 | (5) |
which is the extension of a formula derived for the absorption of monochromatic light by a single component of a multi-absorber solution,
47 to polychromatic light. In
eqn (5)εD(
λ) and
εP(
λ) are the absorption coefficients of the dye and the product, respectively, at wavelength
λ,
A(
λ) is the absorption spectrum of the sample, and
I(
λ) is the spectrum of the irradiating light. (
I(
λ) was derived from the spectra of the xenon arc lamp and the transmission filter applied, it is related to the intensity of the incident light,
I0, determined by actinometry, as
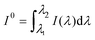
.) The photooxidation of the dye, reaction
(2), was treated as a first order reaction, because of the large excess of dissolved O
2 in the samples. (The reaction has been conducted under air-equilibrated conditions, with intensive stirring. The concentration of O
2 is [O
2]
airsatd = 2.42 × 10
−3 M in air-saturated acetonitrile,
48 two orders higher than the total concentration of
D, a fraction of which are the excited state
D* molecules in reaction
(2).) The rate coefficient of the oxidation was calculated from the (numerical) derivatives of the [
D] −
t function:
| | 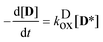 | (6) |
obtaining the concentration of the excited state dye from
| | 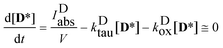 | (7) |
i.e. applying the quasi steady-state approximation for this species.
The quantum yield of the photooxidation of the dye was obtained then from kDox and kDtau as
| | 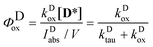 | (8) |
When Mg2+ has been added to the reaction mixture in great excess (so that over 50% of D was complexed at the start of the reaction), the overall rate of photooxidation has increased considerably. Absorption spectra of the sample were recorded at 1 minute intervals for 15 minutes. The concentrations of the dye, the complex and the product, the latter identical as in the photolysis of the pure dye, were obtained by resolving the spectra of the reaction mixtures into the spectra of the three absorbing components, taking the known equilibrium constant for the complexation into consideration, i.e. ensuring that [MgD2+]/([D]·[Mg2+]) is constant all the time.
The reaction schemes (1)–(3) have been amended by the reactions:
| |  | (9) |
| | 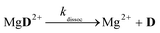 | (10) |
| | 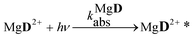 | (11) |
| | 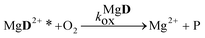 | (12) |
| | 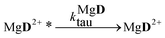 | (13) |
The amount of light absorbed by MgD2+ was calculated according to the following equation:
| | 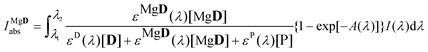 | (14) |
The rate coefficient of the oxidation of the excited complex was obtained then from the sum of the numerical derivatives of the [D] − t and [MgD] − t functions:
| | 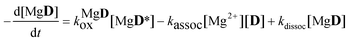 | (15) |
| | 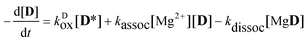 | (16) |
hence
| | 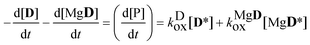 | (17) |
where
kDox was known from the previous experiment. The concentrations of the excited state species were obtained using the steady-state approximations
(7) and
| | 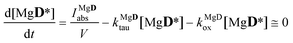 | (18) |
Finally, the quantum yield of the photooxidation of the complex was obtained as
| | 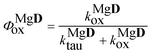 | (19) |
The values obtained for the photooxidation rate coefficients and quantum yields are collected in Table 2. For D a lower Φox value was obtained than Φox ∼ 0.001, reported for the parent compound, 3HF.25 The rate coefficients for the oxidations of the excited state dye and Mg complex have close values. The higher rate of the photoreaction in the presence of the Mg salt, observable through the change in the absorption spectrum, is associated with the higher quantum yield for the oxidation of the complex.
Table 2 Rate coefficients and quantum yields for photooxygenation
| Reactant |
k
ox/min−1 |
Φ
ox
|
|
D
|
2.4 × 107 |
1.5 × 10−4 |
| MgD2+ |
3.9 × 107 |
2.3 × 10−3 |
Reaction mechanism
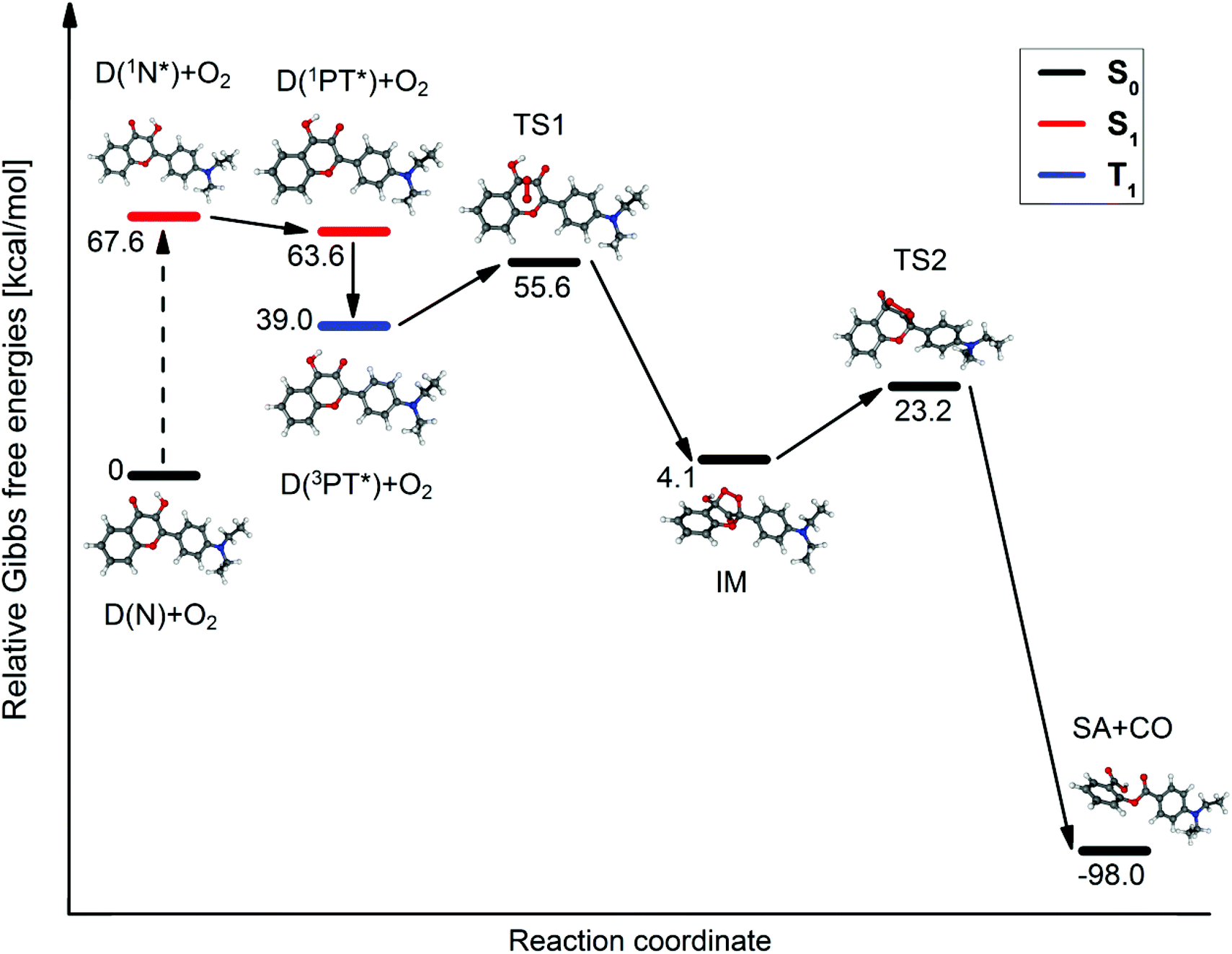
The transition states and intermediates of the photooxidation of D were characterized with the help of potential energy surface (PES) calculations. A reaction path analogous to the experimentally established steps of the photooxidation of the parent compound, 3HF,23–27 was considered. The results are illustrated in Fig. 4. The reaction takes place with the PT* form, since the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O(δ−) and CH–OH(δ+) functional groups as nucleophilic/electrophilic centers at the given positions in the tautomeric species (see Scheme 1), promote the addition of O2.24 The first steps are the S0 → S1 excitation of the N form, and the subsequent conversion of 1N* to its tautomer, 1PT*. We did not search for the TS of the ESIPT reaction on the S1 PES since this phototautomerization is well documented in the literature.49,50 Since the triplet O2 molecule most probably attacks triplet species for spin rules,23,24 we suppose that 1PT* undergoes inter-system crossing to give 3PT*. The reaction of 3PT* with 3O2 leads to an S0 state endo-peroxide intermediate (IM in Fig. 4), which decomposes – on the S0 surface – into the final product (an SA derivative, see Scheme 2) and CO. Although the approaches (deficiencies of the TD-DFT method, relatively small basis set, and continuum solvation model) introduced in the calculations allow only for qualitative results, the reasonable values for the Gibbs free energies of the intermediate, IM and transition states, TS1, TS2 confirm the plausibility of this hypothesized reaction mechanism.
C–O(δ−) and CH–OH(δ+) functional groups as nucleophilic/electrophilic centers at the given positions in the tautomeric species (see Scheme 1), promote the addition of O2.24 The first steps are the S0 → S1 excitation of the N form, and the subsequent conversion of 1N* to its tautomer, 1PT*. We did not search for the TS of the ESIPT reaction on the S1 PES since this phototautomerization is well documented in the literature.49,50 Since the triplet O2 molecule most probably attacks triplet species for spin rules,23,24 we suppose that 1PT* undergoes inter-system crossing to give 3PT*. The reaction of 3PT* with 3O2 leads to an S0 state endo-peroxide intermediate (IM in Fig. 4), which decomposes – on the S0 surface – into the final product (an SA derivative, see Scheme 2) and CO. Although the approaches (deficiencies of the TD-DFT method, relatively small basis set, and continuum solvation model) introduced in the calculations allow only for qualitative results, the reasonable values for the Gibbs free energies of the intermediate, IM and transition states, TS1, TS2 confirm the plausibility of this hypothesized reaction mechanism.
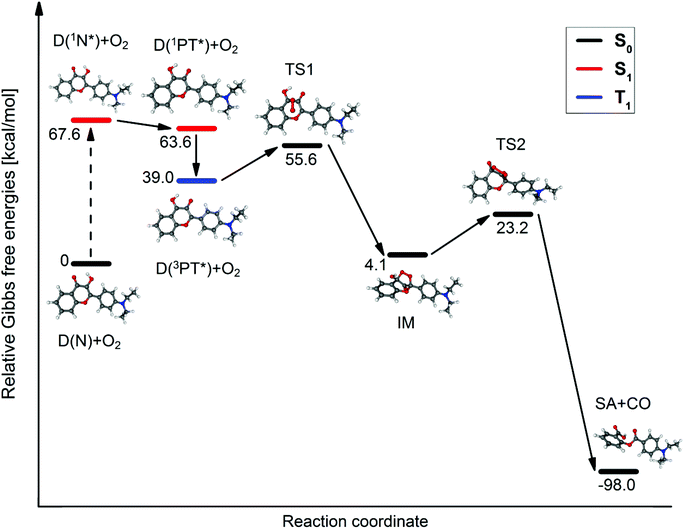 |
| | Fig. 4 The proposed reaction path of the photooxygenation. | |
The parent compound, 3HF was reported to show a ground-state solute–solvent proton transfer in DMSO, a hydrogen bond acceptor solvent.51 Such anion formation would obviously affect the reactivity of our derivative toward molecular O2. The absorption spectrum of D in neat acetonitrile (Fig. S2 in the ESI†), however, does not indicate the occurrence of anionic species. On the other hand, the spectrum of the MgD2+ complex shows similarity to the spectrum of the anionic form of the dye, yet, its photolysis results in the same oxidation product, as the photolysis of the free dye. It is difficult to assess the reaction mechanism for the oxygenation of complexed D, but we note that the anionic form of 3HF was found to exhibit significantly increased reactivity toward 1O2, related to the uncharged species.24
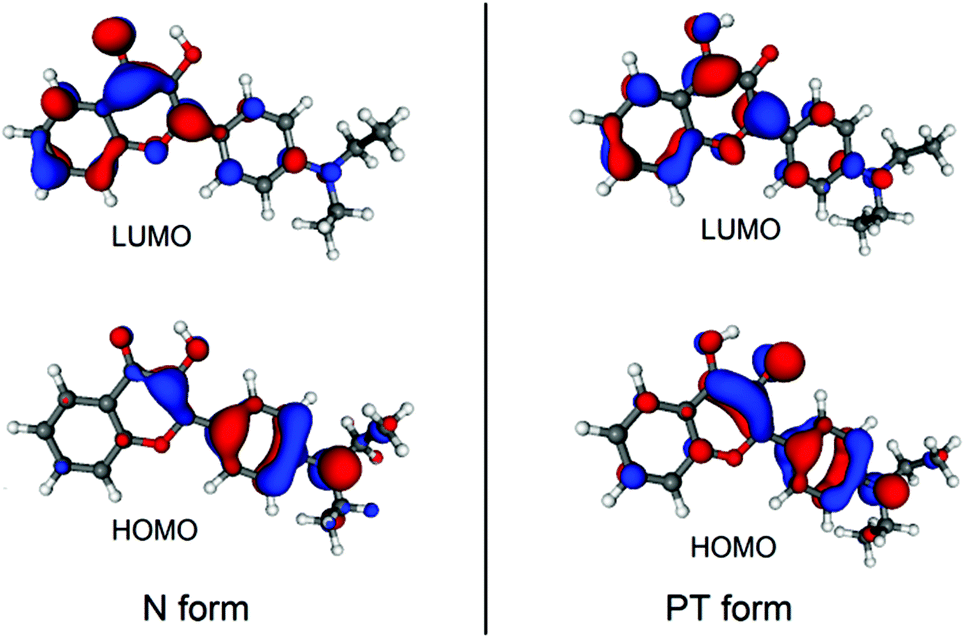
To understand why the oxygenation is preferred in the excited state, it is instructive to inspect the frontier molecular orbitals (MOs) of D in its N and T forms, which are visualized in Fig. 5. As can be seen, the lowest unoccupied molecular orbitals (LUMOs) are localized with opposite phases on the carbons where the oxygen molecule binds, unlike the highest occupied molecular orbitals (HOMOs). Thus, considering the electronic structure of the oxygen molecule, the electronic excitation to the S1 state (with the subsequent inter-system crossing) facilitates the formation of the T–O2 complex.
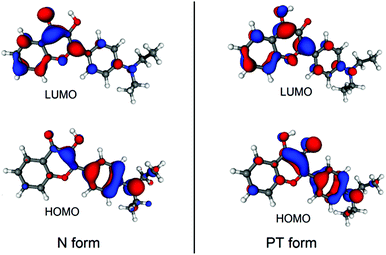 |
| | Fig. 5 Calculated molecular orbitals of the N and PT forms of D. | |
Conclusions
In studies on the photolysis of 3-hydroxyflavone and its substituted derivatives, two types of reactions had been observed: photooxygenation leading to the respective O-benzoyl salicylic acid and rearrangement into the respective 3-hydroxy-3-aryl-indane-1,2-dione. The type of reaction depended on the substituents, the solvent and the aeration/deaeration of the sample. In our present work, it has been found that 4′-diethylamino-3-hydroxyflavone (D) which is a versatile fluorescent probe, undergoes the former reaction (photooxygenation) in acetonitrile. Our theoretical calculations on the S0, S1, and T1 PESs of the system proved that the photooxygenation reaction is a possible reaction route. The excited molecule, after tautomerization, undergoes inter-system crossing, and the triplet-state species reacts with the oxygen molecule. Our study has been extended to the Mg chelate of D (MgD2+), the photolysis of which resulted in the same product as that of the pure dye. Analyzing the temporal change of the absorption spectra of the reaction mixtures and with the help of actinometric measurements, the quantum yields for the photoreactions of D and its Mg chelate and the rate coefficients for the oxidations of the excited state species (D* and MgD2+*) have been determined, striving for obtaining data, which are, in principle independent of the intensity of the irradiating light. It can be hoped that these results will be utilized in the development of further 3-hydroxychromone based and other fluorescent sensors, in particular in the evaluation of their photostability.
Acknowledgements
This work was financially supported by the Hungarian Scientific Research Fund (Grant K108752) and the National Office for Research and Innovation (Grant KMOP-1.1.2-07/1-2008-0002). The authors are grateful to Dr András Simon for the NMR experiments.
References
-
S. Protti and A. Mezzetti, Any colour you like. Excited state and ground state proton transfer in flavonols and applications, in Specialist Periodical Report: Photochemistry, ed. A. Albani, Royal Society of Chemistry, Cambridge, 2012, vol. 40, pp. 295–322 Search PubMed.
- A. P. Demchenko, K. C. Tang and P. T. Chou, Excited-state proton coupled charge transfer modulated by molecular structure and media polarization, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
- P. Chou, D. McMorrow, T. J. Aartsma and M. Kasha, The Proton-Transfer Laser. Gain Spectrum and Amplification of Spontaneous Emission of 3-Hydroxyflavone, J. Phys. Chem., 1984, 88, 4596–4599 CrossRef CAS.
- A. S. Klymchenko and A. P. Demchenko, Multiparametric probing of intermolecular interactions with fluorescent dye exhibiting excited state intramolecular proton transfer, Phys. Chem. Chem. Phys., 2003, 5, 461–468 RSC.
- V. V. Shynkar, A. S. Klymchenko, Y. Mély, G. Duportail and V. G. Pivovarenko, Anion Formation of 4′-(Dimethylamino)-3-hydroxyflavone in Phosphatidylglycerol Vesicles Induced by HEPES Buffer: A Steady-State and Time-Resolved Fluorescence Investigation, J. Phys. Chem. B, 2004, 108, 18750–18755 CrossRef CAS.
- A. S. Klymchenko, G. Duportail, A. P. Demchenko and Y. Mély, Bimodal Distribution and Fluorescence Response of Environment-Sensitive Probes in Lipid Bilayers, Biophys. J., 2004, 86, 2929–2941 CrossRef CAS PubMed.
- G. M'Baye, O. V. Martyloga, G. Duportail and V. G. Pivovarenko, 3-Hydroxy-4′-[di(2-hydroxymethyl)amino]flavone as a new step in search of an ideal membrane ratiometric fluorescent probe, J. Photochem. Photobiol., A, 2006, 184, 113–124 CrossRef.
- D. Ghosh, S. Batuta, S. Das, N. A. Begum and D. Mandal, Proton Transfer Dynamics of N,N’-Dimethylamino-3-hydroxyflavone Observed in Hydrogen-Bonding Solvents and Aqueous Micelles, J. Phys. Chem. B, 2015, 119, 5650–5661 CrossRef CAS PubMed.
- V.Y. Postupalenko, V.V. Shvadchak, G. Duportail, V.G. Pivovarenko, A.S. Klymchenko and Y. Mély, Monitoring membrane binding and insertion of peptides by two-color fluorescent label, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 424–432 CrossRef CAS PubMed.
- V. Y. Postupalenko, O. M. Zamotaiev, V. V. Shvadchak, A. V. Strizhak, V. G. Pivovarenko, A. S. Klymchenko and Y. Mely, Dual-Fluorescence L-Amino Acid Reports Insertion and Orientation of Melittin Peptide in Cell Membranes, Bioconjugate Chem., 2013, 24, 1998–2007 CrossRef CAS PubMed.
- S. V. Avilov, C. Bode, F. G. Tolgyesi, A. S. Klymchenko, J. Fidy and A. P. Demchenko, Heat perturbation of bovine eye lens alpha-crystallin probed by covalently attached ratiometric fluorescent dye 4′-diethylamino-3-hydroxyflavone, Biopolymers, 2005, 78, 340–348 CrossRef CAS PubMed.
- D. A. Yushchenko, O. B. Vadzyuk, S. O. Kosterin, G. Duportail, Y. Mély and V. G. Pivovarenko, Sensing of adenosine-5′-triphosphate anion in aqueous solutions and mitochondria by a fluorescent 3-hydroxyfklavone dye, Anal. Biochem., 2007, 369, 218–225 CrossRef CAS PubMed.
- V. G. Pivorenko, O. B. Vadzyuk and S. O. Kosterin, Fluorometric Detection of Adenosine Triphosphate with 3-Hydroxy-(4′-dimethylamino)flavone in Aqueous Solutions, J. Fluorescence, 2006, 16, 9–15 CrossRef PubMed.
- M. Sisa, S. L. Bonnet, D. Ferreira and J. H. Van der Westhuizen, Photochemistry of Flavonoids, Molecules, 2010, 15, 5196–5245 CrossRef CAS PubMed.
- T. Matsuura, T. Takemoto and R. Nakashima, Photoinduced reactions – LXXI. Photorearrangement of 3-hydroxyflavones to 3-aryl-3-hydroxy-1,2-indandiones, Tetrahedron, 1973, 29, 3337–3340 CrossRef CAS.
- I. Yokoe, K. Higuchi, Y. Shirataki and M. Komatsu, Photochemistry of Flavonoids. III. Photorearrangement of Flavonols, Chem. Pharm. Bull., 1981, 29, 894–898 CrossRef CAS.
- S. Protti, A. Mezzetti, C. Lapouge and J.-P. Cornard, Photochemistry of metal complexes of 3-hydroxyflavone: towards a better understanding of the influence of solar light on the metal–soil organic matter interactions, Photochem. Photobiol. Sci., 2008, 7, 109–119 CAS.
- S. Protti and A. Mezzetti, Solvent effects on the photophysics and photoreactivity of 3-hydroxyflavone: A combined spectroscopic and kinetic study, J. Mol. Liq., 2015, 205, 110–114 CrossRef CAS.
- P.-T. Chou and M. L. Martinez, Photooxygenation of 3-hydroxyflavone and molecular design of the radiation-hard scintillator based on the excited-state proton transfer, Radiat. Phys. Chem., 1993, 41, 373–378 CrossRef CAS.
- K. Grubel, A. R. Marts, S. M. Greer, D. L. Tierney, C. J. Allpress, S. N. Anderson, B. J. Laughlin, R. C. Smith, A. M. Arif and L. M. Berreau, Photoinitiated Dioxygenase-Type Reactivity of Open-Shell 3d Divalent Metal Flavonolato Complexes, Eur. J. Inorg. Chem., 2012, 4750–4757 CrossRef CAS.
- K. Grubel, S. L. Saraf, S. N. Anderson, B. J. Laughlin, R. C. Smith, A. M. Arif and L. M. Berreau, Synthesis, characterization, and photoinduced CO-release reactivity of a Pb(II) flavonolate complex: Comparisons to Group 12 analogs, Inorg. Chim. Acta, 2013, 407, 91–97 CrossRef CAS.
- S. Ameer-Beg, S. M. Ormson, R. G. Brown, P. Matousek, M. Towrie, E. T. J. Nibbering, P. Foggi and F. V. R. Neuwahl, Ultrafast measurements of excited state intramolecular proton transfer (ESIPT) in room temperature solutions of 3-hydroxyflavone and derivatives, J. Phys. Chem. A, 2001, 105, 3709–3718 CrossRef CAS.
- W. E. Brewer, S. L. Studer, M. Standiford and P.-T. Chou, Dynamics of the Triplet State and the Reverse Proton Transfer of 3-Hydroxyfiavone, J. Phys. Chem., 1989, 93, 6088–6094 CrossRef CAS.
- S. L. Studer, W. E. Brewer, M. L. Martinez and P.-T. Chou, Time-Resolved Study of the Photooxygenation of 3-Hydroxyflavone, J. Am. Chem. Soc., 1989, 111, 7643–7644 CrossRef CAS.
- M. L. Martinez, S. L. Studer and P.-T. Chou, Direct Evidence of the Triplet-State Origin of the Slow Reverse Proton Transfer Reaction of 3-Hydroxyflavone, J. Am. Chem. Soc., 1990, 112, 2427–2429 CrossRef CAS.
- P.-T. Chou, S. L. Studer, M. L. Martinez, E. Orton and M. Young, Photooxygenation of 3-hydroxyflavone in a 12 K O2 matrix, Photochem. Photobiol., 1991, 53, 587–593 CrossRef CAS.
- M. Wang, T. Spataru, J. R. Lombardi and R. L. Birke, Time Resolved Surface Enhanced Raman Scattering Studies of 3-Hydroxyflavone on a Ag Electrode, J. Phys. Chem. C, 2007, 111, 3044–3052 CAS.
- M. A. Smith, R. M. Neumann and R. A. Webb, A modification of the Algar-Flynn-Oyamada preparation of flavonols, J. Heterocycl. Chem., 1968, 5, 425–426 CrossRef CAS.
- S. Park and H.-J. Kim, Highly activated Michael acceptor by an intramolecular hydrogen bond as a fluorescence turn-on probe for cyanide, Chem. Commun., 2010, 46, 9197–9199 RSC.
- S. M. Ormson, R. G. Brown, F. Vollmer and W. Rettig, Switching between charge-transfer and proton-transfer emission in the excited-state of a substituted 3-hydroxyflavone, J. Photochem. Photobiol., A, 1994, 81, 65–72 CrossRef CAS.
- V. G. Pivovarenko, L. Jozwiak and J. Błazejowski, 2,8-Bis[4-(diethylamino)phenyl]-3,7-dihydroxy-4H,6H-pyrano[3,2-g]chromene-4,6-dione - A new liquid-phase-sensitive fluorescent probe utilising intramolecular one- or two-proton transfer phenomena, Eur. J. Org. Chem., 2002, 3979–3985 CrossRef CAS.
- K. L. Willett and R. A. Hites, Chemical Actinometry: Using o-Nitrobenzaldehyde to Measure Light Intensity in Photochemical Experiments, J. Chem. Educ., 2000, 77, 900–902 CrossRef CAS.
- E. S. Galbavy, K. Ram and C. Anastasio, 2-Nitrobenzaldehyde as a chemical actinometer for solution and ice photochemistry, J. Photochem. Photobiol., A, 2010, 209, 186–192 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
-
J. B. Foresman and A. E. Frisch, Exploring Chemistry With Electronic Structure Methods, Gaussian, Pittsburgh, 1996 Search PubMed.
- P. Hohenberg and W. Kohn, Inhomogenous Electron Gas, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- F. Furche and D. Rappoport, Density functional methods for excited states: Equilibrium structure and electronic spectra, Theor. Comput. Chem., 2005, 16, 93–128 CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- C. Y. Peng and H. B. Schlegel, Combining synchronous transit and quasi-newton methods to find transition-states, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- P.-T. Chou, S.-C. Pu, Y.-M. Cheng, W.-S. Yu, Y.-C. Yu, F.-T. Hung and W.-P. Hu, Femtosecond Dynamics on Excited-State Proton/Charge-Transfer Reaction in 4′-N,N-Diethylamino-3-hydroxyflavone. The Role of Dipolar Vectors in Constructing a Rational Mechanism, J. Phys. Chem. A, 2005, 109, 3777–3787 CrossRef CAS PubMed.
- A. D. Roshal, A. V. Grigorovich, A. O. Doroshenko, V. G. Pivorenko and A. P. Demchenko, Flavonols and Crown-Flavonols as Metal Cation Chelators. The Different Nature of Ba2+ and Mg2+ Complexes, J. Phys. Chem. A, 1998, 102, 5907–5914 CrossRef CAS.
- A. D. Roshal, A. V. Grigorovich, A. O. Doroshenko, V. G. Pivovarenko and A. P. Demchenko, Flavonols as metal-ion chelators: complex formation with Mg2+ and Ba2+ cations in the excited state, J. Photochem. Photobiol., A, 1999, 127, 89–100 CrossRef CAS.
- V. N. Agieienko, Y. V. Kolesnik and O. N. Kalugin, Structure, solvation, and dynamics of Mg2+, Ca2+, Sr2+ and Ba2+ complexes with 3-hydroxyflavone, and perchlorate anion in acetonitrile medium: A molecular dynamics simulation study, J. Chem. Phys., 2014, 140, 194501 CrossRef PubMed.
- V. N. Agieienko and O. N. Kalugin, Complexation of Ni(ClO4)2 and Mg(ClO4)2 with 3-Hydroxyflavone in Acetonitrile Medium: Conductometric, Spectroscopic, and Quantum Chemical Investigation, J. Phys. Chem. B, 2014, 118, 12251–12262 CrossRef CAS PubMed.
- V. V. Shynkar, Y. Mély, G. Duportail, E. Piémont, A. S. Klymchenko and A. P. Demchenko, Picosecond time-resolved fluorescence studies are consistent with reversible excited-state intramolecular proton transfer in 4′ -(dialkylamino)-3-hydroxyflavones, J. Phys. Chem. A, 2003, 107, 9522–9529 CrossRef CAS.
- R. C. Michaelson and L. F. Loucks, Expressions for the Light Absorbed by a Single Component of a Multi-Absorber Solution, J. Chem. Educ., 1975, 52, 652–653 CrossRef CAS.
- C. Franco and J. Olmsted, Photochemical determination of the solubility of oxygen in various media, Talanta, 1990, 37, 905–909 CrossRef CAS PubMed.
- R. Casadesús, O. Vendrell, M. Moreno, J. M. Lluch and K. Morokuma, On the intramolecular proton transfer of 3-hydroxyflavone in the first singlet excited state: A theoretical study, Chem. Phys., 2006, 325, 243–250 CrossRef.
- C.-C. Lin, C.-L. Chen, M.-W. Chung, Y.-J. Chen and P.-T. Chou, Effects of Multibranching on 3-Hydroxyflavone-Based Chromophores and the Excited-State Intramolecular Proton Transfer Dynamics, J. Phys. Chem. A, 2010, 114, 10412–10420 CrossRef CAS PubMed.
- S. Protti, A. Mezzetti, J.-P. Cornard, C. Lapouge and M. Fagnoni, Hydrogen bonding properties of DMSO in ground-state formation and optical spectra of 3-hydroxyflavone anion, Chem. Phys. Lett., 2008, 467, 88–93 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5pp00358j |
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 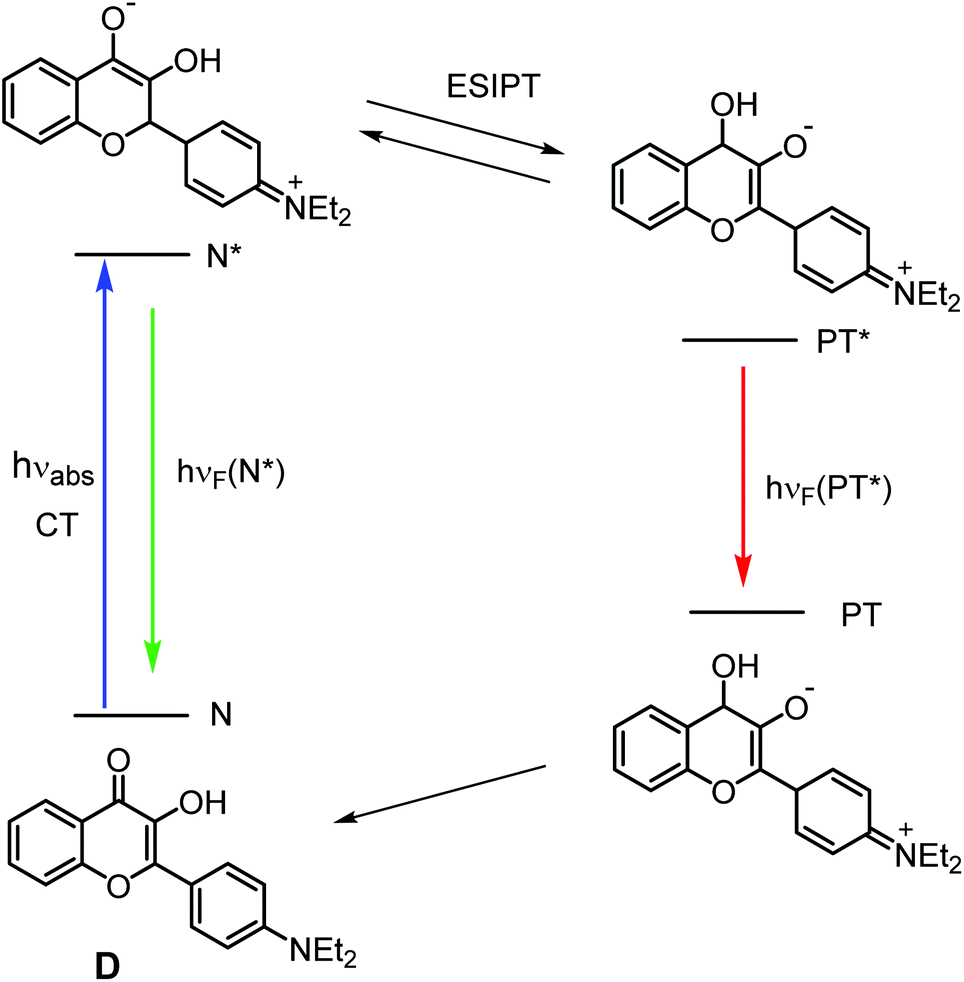
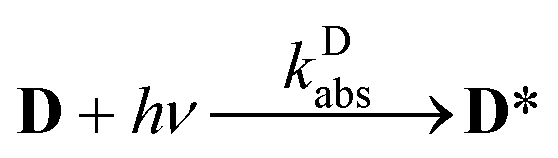
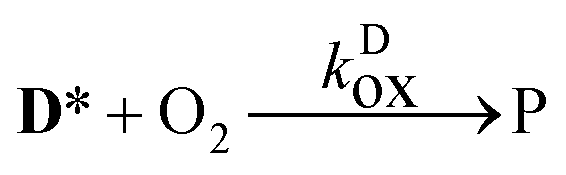
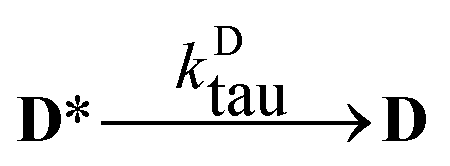
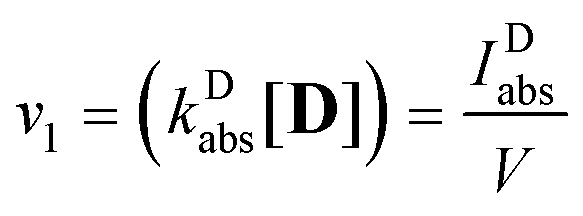
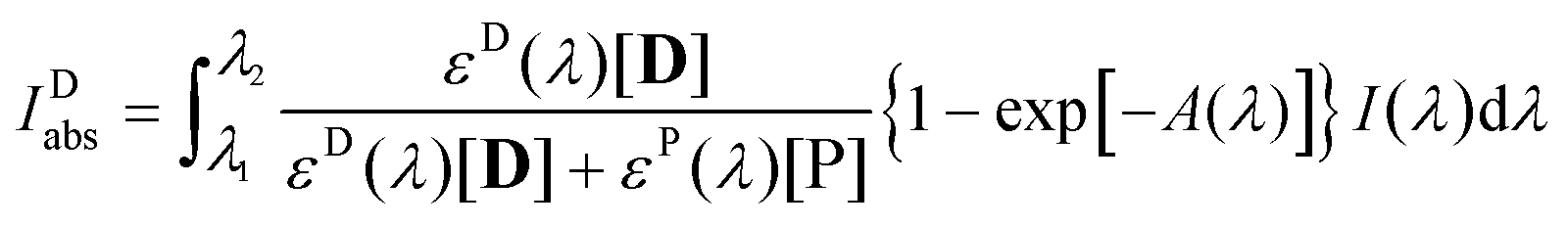
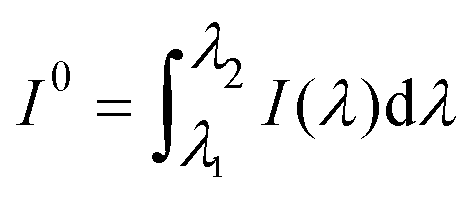 .) The photooxidation of the dye, reaction (2), was treated as a first order reaction, because of the large excess of dissolved O2 in the samples. (The reaction has been conducted under air-equilibrated conditions, with intensive stirring. The concentration of O2 is [O2]airsatd = 2.42 × 10−3 M in air-saturated acetonitrile,48 two orders higher than the total concentration of D, a fraction of which are the excited state D* molecules in reaction (2).) The rate coefficient of the oxidation was calculated from the (numerical) derivatives of the [D] − t function:
.) The photooxidation of the dye, reaction (2), was treated as a first order reaction, because of the large excess of dissolved O2 in the samples. (The reaction has been conducted under air-equilibrated conditions, with intensive stirring. The concentration of O2 is [O2]airsatd = 2.42 × 10−3 M in air-saturated acetonitrile,48 two orders higher than the total concentration of D, a fraction of which are the excited state D* molecules in reaction (2).) The rate coefficient of the oxidation was calculated from the (numerical) derivatives of the [D] − t function: