
Chemoenzymatic synthesis of para-nitrophenol (pNP)-tagged α2–8-sialosides and high-throughput substrate specificity studies of α2–8-sialidases†
Nova
Tasnima
,
Hai
Yu
,
Yanhong
Li
,
Abhishek
Santra
and
Xi
Chen
*
Department of Chemistry, University of California, One Shields Avenue, Davis, CA 95616, USA. E-mail: xiichen@ucdavis.edu; Fax: +1 530 752-8995; Tel: +1 530 754-6037
First published on 24th November 2016
Abstract
para-Nitrophenol (pNP)-tagged α2–8-linked sialosides containing different sialic acid forms were chemoenzymatically synthesized using an efficient one-pot three-enzyme α2–8-sialylation system. The resulting compounds allowed high-throughput substrate specificity studies of the α2–8-sialidase activity of a recombinant human cytosolic sialidase hNEU2 and various bacterial sialidases. The sialoside substrate profiles obtained can be used to guide the selection of suitable sialidases for sialylglycan analysis and for cell and tissue surface glycan modification. They can also be used to guide sialidase inhibitor design.
Introduction
Sialic acids are a family of monosaccharides named nonulosonic acids (9-carbon α-keto acids). They are commonly presented as the outermost monosaccharides on the carbohydrate moieties of cell surface glycoproteins and glycolipids in vertebrates and are directly involved in inflammation, immune regulation, cell interactions, bacterial and viral infection, and other important biological and pathological processes.1–3 Variations on the C-4, 5, 7, 8, and 9 positions of sialic acids are common. Among more than 50 naturally occurring sialic acid forms, the most common N-acetylneuraminic acid (Neu5Ac), the non-human sialic acid N-glycolylneuraminic acid (Neu5Gc), and 2-keto-3-deoxy-nonulosonic acid (Kdn) represent three basic forms1–4 that allow further modifications to take place. Many of these modifications occur after the formation of sialyl linkages (so called post-glycosylation modifications or PGMs).5 On the other hand, an azido group has been introduced into various positions of sialic acid to allow metabolic glycan engineering-based chemical and biological investigation of sialoside formation, turnover, and functions in living cells and in animals.6–8 The aspect of how the azido modification affects the functions of sialidases in living cells and animals is less well documented.In addition to the common α2–3- and α2–6-linked sialic acids, the α2–8-sialyl linkage is also well known in vertebrates and in bacteria.9–14 The presentation of sialic acids in nature is regulated by the coordinated and sometimes competing functions of sialyltransferases and sialidases. While the sialyl linkage and substrate specificities of diverse sialyltransferases are well characterized, those of sialidases are less well studied despite the use of sialidases as important analytical tools.1,15 Sialic acid-containing glycoproteins, glycolipids, and oligosaccharides purified from natural sources and some synthetic sialosides have been used to study the substrate specificity of various sialidases.5,16–19 However, the heterogeneous nature of the purified products causes complication for data intepretation. Synthetic biotinylated sialosides20,21 have been used to probe sialidase activities but such a method requires immobolization of the glycans and washing steps before detection. Sialosides with a fluorophore or a chlorophore directly attached to an α-Neu5Ac are commonly used to screen, identify, or even characterize the activities of sialidases. This strategy increases the throughput of the assay, but provides limited information on the linkage preference of sialidases and their substrate specificity toward diverse sialic acid structures.
To mitigate the challenges, we previously constructed a library of para-nitrophenol (pNP)-tagged α2–3- and α2–6-linked sialosides containing diverse sialic acid forms which allowed the investigation of various sialidases for their substrate specificity towards different sialyl linkages and various sialic acid forms in a high-throughput format.6,8,17,18,22,23 The information obtained and the high-throughput screening method established have been used for identifying inhibitors with selectivity towards certain sialidases.7,16 Similar probes and strategies do not exist for elucidating the substrate specificity of α2–8-sialidases in a high-throughput manner. We report herein an efficient synthesis of pNP-tagged α2–8-linked sialosides containing different sialic acid forms using a highly effective one-pot three-enzyme α2–8-sialylation approach. The resulting compounds have been used in the high-throughput substrate specificity studies of the α2–8-sialidase activity of a recombinant human cytosolic sialidase hNEU2 and various bacterial sialidases. The results obtained can be used to guide the selection of suitable sialidases for sialylglycan analysis and for cell and tissue surface glycan modification. They can also be used to guide sialidase inhibitor design.
Results and discussion
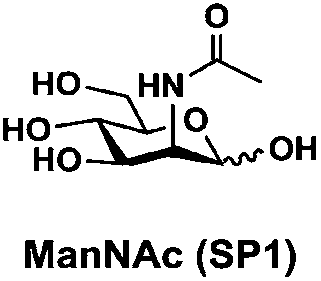
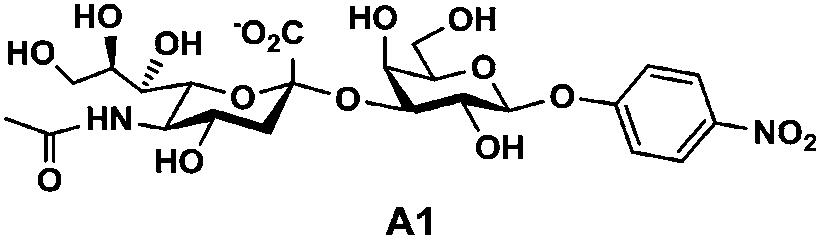
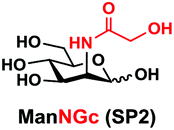
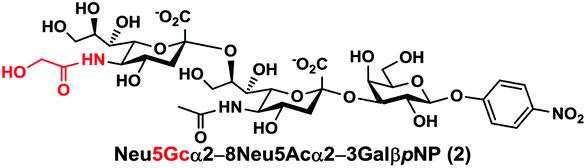
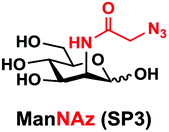
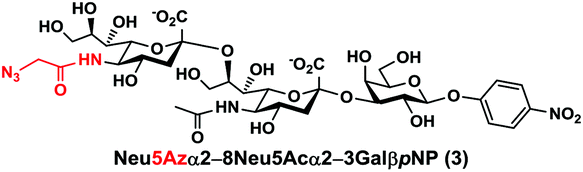
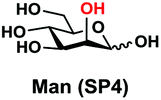
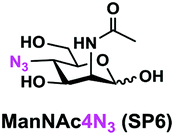
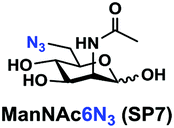
In order to probe the influence of sialic acid structural diversity and modification on the activity of α2–8-specific sialidases, a library of seven pNP-tagged α2–8-sialosides was constructed. To do this, Neu5Acα2–3GalβpNP (A1) was synthesized as reported previously22 in an excellent 90% yield using a one-pot multienzyme (OPME) α2–3-sialylation system24 containing Neisseria meningitidis cytidine 5′-monophosphate (CMP)-sialic acid synthetase (NmCSS)25 and a Pasteurella multocida α2–3-sialyltransferase M144D (PmST1 M144D) mutant.26 It was then used as an acceptor in an efficient OPME chemoenzymatic α2–8-sialylation system27 (Scheme 1) containing Pasteurella multocida sialic acid aldolase (PmNanA),28 NmCSS,25 and Campylobacter jejuni α2–3/8-sialyltransferase (CjCstII)12,29 for synthesizing α2–8-linked sialosides containing Neu5Ac, Neu5Gc, Kdn, N-azidoacetylneuraminic acid (Neu5Az), 5-azido neuraminic acid (Neu5N3), 7-azido-7-deoxy-Neu5Ac (Neu5Ac7N3), or 9-azido-9-deoxy-Neu5Ac (Neu5Ac9N3) from their corresponding six-carbon precursors (SP1–SP7) and Neu5Acα2–3GalβpNP (A1).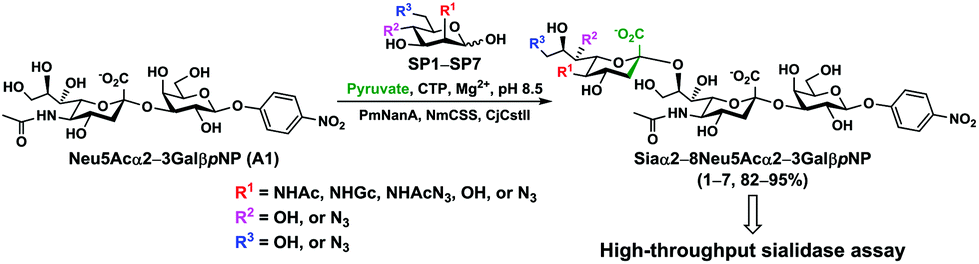 | ||
| Scheme 1 One-pot multienzyme (OPME) chemoenzymatic synthesis of Siaα2–8Neu5Acα2–3GalβpNP (1–7). Enzyme abbreviations: PmNanA, Pasteurella multocida sialic acid aldolase; NmCSS, Neisseria meningitidis CMP-sialic acid synthetase; CjCstII, Campylobacter jejuni α2–3/8-sialyltransferase. | ||
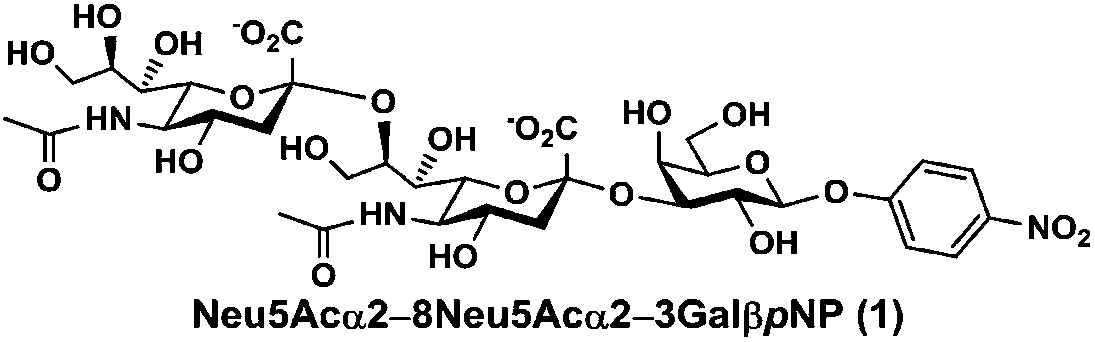
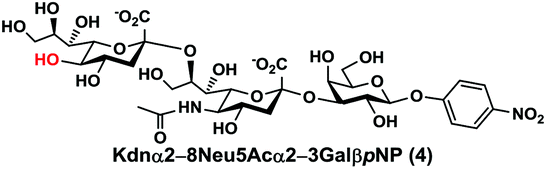
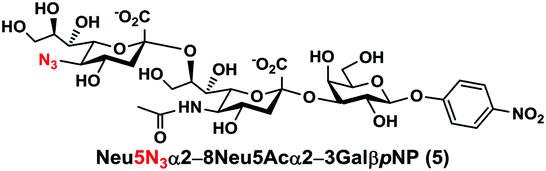
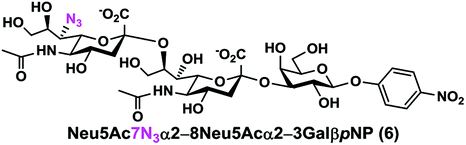
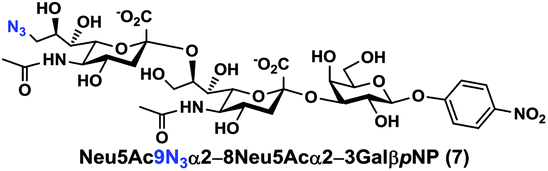
As shown in Table 1, target α2–8-sialosides (1–7) were obtained in excellent 82–95% yields. These include disialylated products containing natural sialic acids form such as Neu5Acα2–8Neu5Acα2–3GalβpNP (1, 85%), Neu5Gcα2–8Neu5Acα2–3GalβpNP (2, 90%), and Kdnα2–8Neu5Acα2–3GalβpNP (4, 82%), as well as those containing non-natural sialic acid forms with an azide at various positions in the sialic acid such as Neu5Azα2–8Neu5Acα2–3GalβpNP (3, 92%), Neu5N3α2–8Neu5Acα2–3GalβpNP (5, 95%), Neu5Ac7N3α2–8Neu5Acα2–3GalβpNP (6, 85%), and Neu5Ac9N3α2–8Neu5Acα2–3GalβpNP (7, 86%).
| Entry | Sia precursor | Acceptor | Sialosides (Siaα2–8Neu5Acα2–3GalβpNP) | Yield |
|---|---|---|---|---|
| A |
|
|
|
85% |
| B |
|
A1 |
|
90% |
| C |
|
A1 |
|
92% |
| D |
|
A1 |
|
82% |
| E |
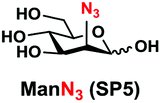
|
A1 |
|
95% |
| F |
|
A1 |
|
85% |
| G |
|
A1 |
|
86% |
α2–8-Sialosides obtained (1–7) were used for the substrate specificity studies of the α2–8-sialidase activity of a recombinant human lysosomal sialidase hNEU230 and various bacterial sialidases in a high-throughput format. In this assay (Scheme 2), each sialoside was incubated with a sialidase of interest as well as excess amounts of an α2–3-sialyl linkage specific sialidase and a β-galactosidase. If the Siaα2–8Neu5Acα2–3GalβpNP (selected from 1–7) were a suitable substrate, the α2–8-sialidase activity of the enzyme would release the terminal sialic acid to produce Neu5Acα2–3GalβpNP (A1). The excess amounts of the α2–3-specific sialidase and β-galactosidase added allow complete release of the terminal Neu5Ac from A1 by the α2–3-sialidase to produce GalβpNP which was further hydrolyzed by the β-galactosidase to produce para-nitrophenol. Without the α2–8-sialidase activity of the sialidase under investigation, no para-nitrophenol would be produced as all enzymes are exo-glycosidases which only catalyze the hydrolysis of terminal monosaccharide residues. At the end of the enzymatic reaction, the pH of the solution was adjusted to higher than 9.5 using CAPS buffer (0.5 M, pH 10.5) to convert para-nitrophenol to para-nitrophenolate which was quantified at A405 nm using a microtiter plate reader with a UV-Vis detector. The assays were carried out in 384-well plates in a high-throughput manner. Streptococcus pneumoniae NanB (SpNanB)31,32 was used as the α2–3-specific sialidase in the assay as it did not show noticeable α2–8-sialidase activity under the reaction conditions (30 min assays) used.
 | ||
| Scheme 2 The high-throughput screening method for substrate specificity studies of α2–8-sialidases. | ||
As shown in Fig. 1, six of the sialidases tested showed α2–8-sialidase activities under the assay conditions described in the Experimental section. While both Neu5Acα2–8Neu5Acα2–3GalβpNP (1) and Neu5Gcα2–8Neu5Acα2–3GalβpNP (2) were able to be cleaved by all six sialidases, other compounds (3–7) could be cleaved by only selected numbers of sialidases. In general, Neu5Ac9N3α2–8Neu5Acα2–3GalβpNP (7) was tolerated by all bacterial sialidases that have α2–8-sialidase activity except for human hNEU2 (Fig. 1A). Neu5Azα2–8Neu5Acα2–3GalβpNP (3) was tolerated by five out of six sialidases including hNEU2 but not by Vibrio cholerae sialidase (Fig. 1B). In comparison, Kdnα2–8Neu5Acα2–3GalβpNP (4) was not a substrate for four out of six sialidases except for CjCstII (Fig. 1C) and SpNanA (Fig. 1D). Neu5N3α2–8Neu5Acα2–3GalβpNP (5) was even more selective and was recognized only by CjCstII (Fig. 1C). Neu5Ac7N3α2–8Neu5Acα2–3GalβpNP (6) was tolerated by four out of six sialidases except for hNEU2 (Fig. 1A) and Vibrio cholerae sialidase (Fig. 1B) which were the least promiscuous ones among the six sialidases with α2–8-sialidase activity.
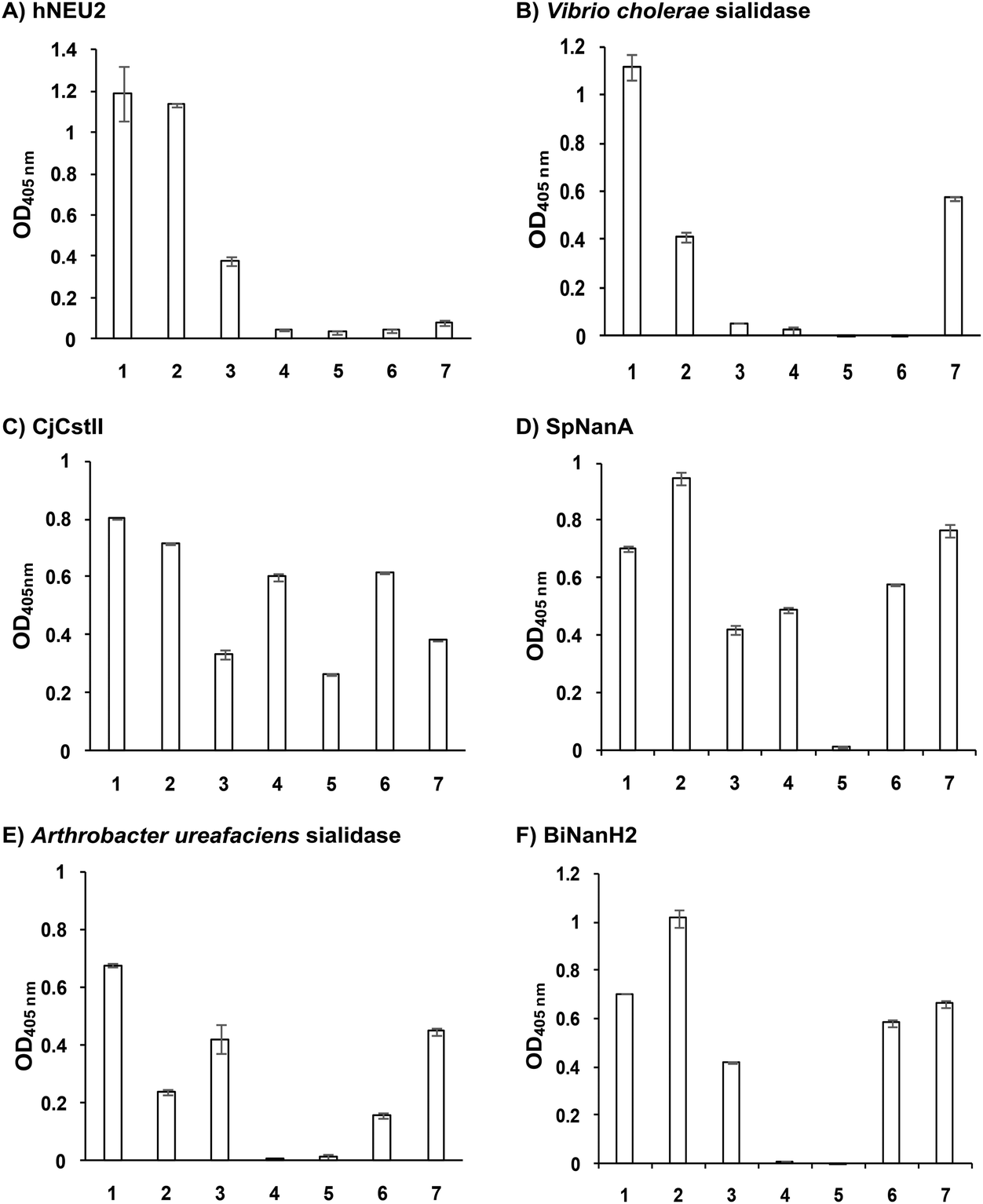 | ||
| Fig. 1 Substrate specificity of the α2–8-sialidase activity of hNEU2 and several bacterial sialidases. Substrates used were Neu5Acα2–8A1 (1), Neu5Gcα2–8A1 (2), Neu5Azα2–8A1 (3), Kdnα2–8A1 (4), Neu5N3α2–8A1 (5), Neu5Ac7N3α2–8A1 (6), and Neu5Ac9N3α2–8A1 (7), where A1 is Neu5Acα2–3GalβpNP. | ||
In addition to sialosides 1 and 2, hNEU2 (Fig. 1A) could tolerate only Neu5Azα2–8Neu5Acα2–3GalβpNP (3) and Vibrio cholerae sialidase (Fig. 1B) could tolerate only Neu5Ac9N3α2–8Neu5Acα2–3GalβpNP (7) as substrates. CjCstII (Fig. 1C) was the most promiscuous α2–8-sialidase among the six and could use all seven sialosides as substrates. Its activity towards three (3, 5, and 7) out of four azido-containing compounds was lower than others. It is quite unique in tolerating Neu5N3α2–8Neu5Acα2–3GalβpNP (5) which was not recognized by any other sialidases in the list. SpNanA (Fig. 1D) was the second most promiscuous one. Both CjCstII and SpNanA could tolerate the Kdn-terminating structure Kdnα2–8Neu5Acα2–3GalβpNP (4). The substrate promiscuities of Arthrobacter ureafaciens sialidase (Fig. 1E) and Bifidobacterium longum subsp. infantis ATCC15697 sialidase 2 (BiNanH2)33 (Fig. 1F) were ranked in the middle. Both could tolerate five out of seven α2–8-sialosides as substrates except for Kdnα2–8Neu5Acα2–3GalβpNP (4) and Neu5N3α2–8Neu5Acα2–3GalβpNP (5).
The broad tolerance of the sialosides 1–7 as substrates by the α2–8-sialidase activity of CjCstII is reasonable as its α2–8-sialyltransferase activity was used for the synthesis of the probes.
Among the sialidases tested, Clostridium perfringens sialidase22 and Pasteurella multocida multifunctional α2–3-sialyltransferase 1 (PmST1)34 did not show α2–8-sialidase activity under assay conditions (30 minutes assays) described in the Experimental section. Cytidine 5′-monophosphate (CMP, 0.4 mM) was added to the CjCstII and PmST1 reactions as CMP was able to enhance the sialidase activities of these enzymes similar to that reported for the α2–6-sialidase activity of Photobacterium damselae α2–6-sialyltransferase.35
The substrate specificity of the α2–8-sialidase activity of hNEU2 (Fig. 1A) matched very well with its α2–3/6-sialidase activities which recognized α2–3/6-sialosides terminated with Neu5Ac, Neu5Gc, and Neu5Az (previously abbreviated as Neu5AcN3) very well but not those terminated with Neu5Ac7N3, Neu5Ac9N3, Kdn, or Neu5N3 (previously abbreviated as 5N3-Kdn).30,36,37
The substrate specificity of the α2–8-sialidase activity of Vibrio cholerae sialidase (Fig. 1B) matched very well with its α2–3/6-sialidase activities which recognized α2–3/6-sialosides terminated with Neu5Ac, Neu5Gc, and Neu5Ac9N3 very well but not those terminated with Neu5Az, Neu5Ac7N3, Kdn, or Neu5N3.22,36–38
Similarly, the substrate specificity of the α2–8-sialidase activity of Arthrobacter ureafaciens sialidase (Fig. 1E) matched well with its α2–3/6-sialidase activities which recognized α2–3/6-sialosides terminated with Neu5Ac and Neu5Gc (moderately) but not Kdn.22
The substrate specificity of the α2–8-sialidase activity of BiNanH2 (Fig. 1F) matched reasonably well with its α2–3/6-sialidase activities which recognized α2–3/6-sialosides terminated with Neu5Ac and Neu5Az very well but not those terminated with Neu5N3 or Kdn. The tolerance towards Neu5Gc-terminated sialosides differed slightly as Neu5Gcα2–8Neu5Acα2–3GalβpNP (2) was cleaved more efficiently than Neu5Acα2–8Neu5Acα2–3GalβpNP (1) by its α2–8-sialiase activity but Neu5Gcα2–3/6GalβpNP was a much poorer substrate due to its α2–3/6-sialidase activities.33
Conclusions
In conclusion, we report here an efficient synthesis of para-nitrophenol (pNP)-tagged α2–8-linked sialosides containing different sialic acid forms using a highly effective one-pot three-enzyme α2–8-sialylation approach. The resulting compounds have allowed high-throughput substrate specificity studies of the α2–8-sialidase activity of a recombinant human cytosolic sialidase hNEU2 and various bacterial sialidases. The information obtained can be used to guide the selection of suitable sialidases for sialylglycan analysis and for cell and tissue surface glycan modification. It can also be used to guide sialidase inhibitor design.Experimental section
Materials and methods
Chemicals were purchased and used as received. NMR spectra were recorded in the NMR facility of University of California, Davis on a Bruker Avance-800 NMR spectrometer (800 MHz for 1H, 200 MHz for 13C). Chemical shifts are reported in parts per million (ppm) on the δ scale. High resolution (HR) electrospray ionization (ESI) mass spectra were obtained using a Thermo Electron LTQ-Orbitrap Hybrid MS at the Mass Spectrometry Facility in the University of California, Davis. Silica gel 60 Å (230–400 mesh, Sorbent Technologies) was used for flash column chromatography. Thin layer chromatography was performed on silica gel plates (Sorbent Technologies) using anisaldehyde sugar stain for detection. Gel filtration chromatography was performed with a column (100 cm × 2.5 cm) packed with Bio-Gel P-2 Fine resins (Bio-Rad). N-Acetyl-D-mannosamine (ManNAc) and N-acetylneuraminic acid (Neu5Ac) were from Inalco (Italy). Cytidine 5′-triphosphate (CTP) was purchased from Hangzhou Meiya Pharmaceutical Co. Ltd. Aspergillus oryzae β-galactosidase, sodium pyruvate, and cytidine 5′-triphosphate (CMP) were from Sigma (St. Louis, MO). Sialidases from Arthrobacter ureafaciens, Vibrio cholerae, and Clostridium perfringens were purchased from Prozyme (Hayward, CA). Recombinant enzymes hNEU2,30Campylobacter jejuni α2–3/8-sialyltransferase (CjCstII),29Bifidobacterium longum subsp. infantis ATCC15697 sialidase 2 (BiNanH2),33Pasteurella multocida sialic acid aldolase (PmNanA),28Neisseria meningitidis CMP-sialic acid synthetase (NmCSS),25Pasteurella multocida multifunctional α2–3-sialyltransferase 1 (PmST1),34 and PmST1 M144D mutant26 were expressed and purified as described previously.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH:H2O = 5:2:1, by volume). A yield of 90% was achieved. The product NMR spectra matched with those reported previously.22
:MeOH:H2O = 5:2:1, by volume). A yield of 90% was achieved. The product NMR spectra matched with those reported previously.22
General procedures. Neu5Acα2–3GalβpNP (A1, 1 eq., 8–10 mM), Man/ManNAc derivatives/ManNGc (1.3–1.5 eq.), CTP (1.3–2.0 eq.), and sodium pyruvate (5 eq.) were dissolved in water in a 50 mL centrifuge tube, the pH of this mixture was adjusted to be neutral. MgCl2 (20 mM) and Tris-HCl buffer (100 mM, pH 8.5) were then added. After adding PmNanA (0.075–3 mg), NmCSS (0.2–3 mg), and CjCstII (0.3–3 mg), water was added to bring the concentration of Neu5Acα2–3GalβpNP to approximately 10 mM. For synthesizing compounds 1 and 4 from ManNAc (SP1) and mannose (SP4) respectively, the reaction mixture was incubated for 2 h at 37 °C. For synthesizing sialoside 6 from ManNAc4N3 (SP6), the reaction mixture was incubated for 2 h at 37 °C and extended for additional 24 hours at room temperature. For synthesizing sialosides 2, 3, 5, and 7 from ManNGc (SP2), ManNAz (SP3), ManN3 (SP5) and ManNAc6N3 (SP7) respectively, the reaction mixtures were gently shaken at room temperature for 14 h. The reaction progress was monitored by liquid chromatography-mass spectrometry (LC-MS) and TLC (EtOAc
:MeOH:H2O = 5:2:1, by volume) analyses. When an optimal yield was achieved, the same volume of cold ethanol was added to the reaction mixture. The mixture was incubated at 4 °C for 30 min and centrifuged. The supernatant was concentrated by rotary evaporation. Compounds 1, 4, and 6 were purified using a BioGel P-2 gel filtration column followed by silica gel column purification (EtOAc:MeOH:H2O = 5:2:1, by volume) and a final BioGel P-2 gel filtration column. Compounds 1 and 4 were then subjected to additional purification using high performance liquid chromatography (HPLC) with a C18 column. Compounds 2, 3, 5, and 7 were purified directly using HPLC with a C18 column.
Neu5Acα2–8Neu5Acα2–3GalβpNP (1). 81 mg, yield 85%; white solid. 1H NMR (800 MHz, D2O) δ 8.27 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.8 Hz, 2H), 5.32 (d, J = 7.2 Hz, 1H), 4.23 (dd, J = 3.2 Hz and 9.6 Hz, 1H), 4.18 (dd, J = 4.0 and 12.0 Hz, 1H), 4.15 (m, 1H), 4.06 (d, J = 1.6 Hz, 1H), 3.94 (t, J = 6.4 Hz, 1H), 3.91–3.57 (m, 15H), 2.75 (dd, J = 4.8 and 12.8 Hz, 1H), 2.71 (dd, J = 4.0 and 12.0 Hz, 1H), 2.06 (s, 3H), 2.02 (s, 3H), 1.77 (t, J = 12.0 Hz, 1H), 1.71 (t, J = 12.8 Hz, 1H). 13C NMR (200 MHz, D2O) δ 174.85, 173.35, 173.24, 161.55, 142.38, 125.95, 116.34, 100.31, 100.07, 99.49, 78.29, 75.31, 75.28, 73.96, 72.53, 71.60, 69.24, 68.72, 68.35, 67.99, 67.83, 66.96, 62.43, 61.43, 60.61, 52.15, 51.61, 40.37, 39.67, 22.20, 21.90. HRMS (ESI) m/z calculated for C34H49N3O24 (M − H) 882.2628, found 882.2604.
Neu5Gcα2–8Neu5Acα2–3GalβpNP (2). 55 mg, yield 90%; white solid. 1H NMR (800 MHz, D2O) δ 8.27 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.8 Hz, 2H), 5.32 (d, J = 8.0 Hz, 1H), 4.23 (dd, J = 2.4 Hz and 9.6 Hz, 1H), 4.18 (dd, J = 4.0 and 12.0 Hz, 1H), 4.16 (m, 1H), 4.12 (s, 2H), 4.06 (d, J = 2.4 Hz, 1H), 3.94 (t, J = 6.4 Hz, 1H), 3.91–3.55 (m, 15H), 2.77 (dd, J = 4.8 and 12.8 Hz, 1H), 2.72 (dd, J = 4.8 and 12.8 Hz, 1H), 2.06 (s, 3H), 1.77 (t, J = 12.8 Hz, 1H), 1.74 (t, J = 12.0 Hz, 1H). 13C NMR (200 MHz, D2O) δ 175.61, 174.85, 173.35, 173.27, 161.56, 142.38, 125.95, 116.34, 100.33, 100.08, 99.49, 78.28, 75.31, 75.28, 73.96, 72.25, 71.66, 69.23, 68.72, 68.09, 67.92, 67.83, 66.96, 62.39, 62.07, 61.43, 60.84, 60.62, 52.15, 51.31, 40.42, 39.67, 22.20. HRMS (ESI) m/z calculated for C34H49N3O25 (M − H) 898.2577, found 898.2561.
Neu5Azα2–8Neu5Acα2–3GalβpNP (3). 51 mg, yield 92%; white solid. 1H NMR (800 MHz, D2O) δ 8.27 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 9.6 Hz, 2H), 5.32 (d, J = 8 Hz, 1H), 4.23 (dd, J = 3.2 and 10.4 Hz, 1H), 4.18 (dd, J = 4 and 12 Hz, 1H), 4.15 (m, 1H), 4.05 (s, 2H), 3.94–3.66 (m, 17H), 2.76 (dd, J = 4 and 12 Hz, 1H), 2.71 (dd, J = 4 and 12.8 Hz, 1H), 2.06 (s, 3H), 1.76 (t, J = 12 Hz, 1H), and 1.72 (t, J = 12 Hz, 1H). 13C NMR (200 MHz, D2O): δ 174.85, 173.35, 173.24, 170.99, 161.56, 142.38, 125.95, 116.34, 100.33, 100.07, 99.49, 78.29, 75.31, 75.29, 73.96, 72.20, 71.68, 69.22, 68.72, 68.20, 67.94, 67.83, 66.96, 62.41, 61.42, 61.16, 60.62, 52.15, 51.78, 51.69, 40.39, 39.67, 22.20. HRMS (ESI) m/z calculated for C34H48N6O24 (M − H) 923.2642, found 923.2629.
Kdnα2–8Neu5Acα2–3GalβpNP (4). 70 mg, yield 82%; white solid. 1H NMR (800 MHz, D2O) δ 8.27 (d, J = 9.6 Hz, 2H), 7.26 (d, J = 8.8 Hz, 2H), 5.32 (d, J = 7.2 Hz, 1H), 4.22 (dd, J = 3.2 Hz and 9.6 Hz, 1H), 4.17 (dd, J = 4.0 and 12.8 Hz, 1H), 4.17 (m, 1H), 4.13 (s, 1H), 4.06 (d, J = 2.4 Hz, 1H), 3.94 (t, J = 6.4 Hz, 1H), 3.9–3.55 (m, 15H), 2.71–2.67 (m, 1H), 2.06 (s, 3H), 1.77 (t, J = 12.0 Hz, 1H), 1.67 (t, J = 12.0 Hz, 1H). 13C NMR (200 MHz, D2O) δ 174.86, 173.45, 173.35, 161.56, 142.38, 125.95, 116.34, 100.33, 100.09, 99.49, 78.21, 75.31, 75.28, 73.96, 73.53, 71.90, 70.25, 69.69, 69.24, 68.71, 67.83, 67.64, 66.98, 62.50, 61.41, 60.61, 52.15, 39.93, 39.64, 22.20. HRMS (ESI) m/z calculated for C32H46N2O24 (M − H) 841.2362, found 841.2347.
Neu5N3α2–8Neu5Acα2–3GalβpNP (5). 56 mg, yield 95%; white solid. 1H NMR (800 MHz, D2O) δ 8.27 (d, J = 9.6 Hz, 2H), 7.26 (d, J = 9.6 Hz, 2H), 5.31 (d, J = 8 Hz, 1H), 4.23 (dd, J = 3.2 and 10.4 Hz, 1H), 4.16 (dd, J = 4 Hz and 12 Hz, 1H), 4.13 (m, 1H), 4.06 (d, J = 3.2 Hz, 1H), 3.95–3.44 (m, 16H), 2.73 (dd, J = 4.8 and 12.8 Hz, 1H), 2.70 (dd, J = 4.8 and 12 Hz, 1H), 2.06 (s, 3H), 1.77 (t, J = 12.8 Hz, 1H), 1.72 (t, J = 12 Hz, 1H). 13C NMR (200 MHz, D2O) δ 174.86, 173.40, 173.10, 161.56, 142.38, 125.96, 116.34, 100.40, 100.13, 99.50, 78.20, 75.29, 75.27, 73.96, 72.51, 71.82, 69.45, 69.17, 68.71, 68.31, 67.82, 67.05, 62.54, 62.40, 61.40, 60.61, 52.14, 40.12, 39.59, 22.19. HRMS (ESI) m/z calculated for C32H45N5O23 (M − H) 866.2427, found 866.2410.
Neu5Ac7N3α2–8Neu5Acα2–3GalβpNP (6). 71 mg, yield 85%; white solid. 1H NMR (800 MHz, D2O) δ 8.27 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 9.6 Hz, 2H), 5.31 (d, J = 8 Hz, 1H), 4.21 (dd, J = 3.2 and 10.4 Hz, 1H), 4.16 (dd, J = 4.0 and 12 Hz, 1H), 4.12 (m, 1H), 4.05 (d, J = 2.4 Hz, 1H), 4.04–3.47 (m, 16H), 2.74–2.70 (m, 2H), 2.06 (s, 3H), 2.02 (s, 3H), 1.77–1.72 (m, 2H). 13C NMR (200 MHz, D2O) δ 174.82, 174.28, 173.32, 173.02, 161.55, 142.38, 125.95, 116.34, 100.50, 100.06, 99.50, 78.37, 75.31, 75.28, 73.97, 71.66, 70.52, 69.33, 68.70, 68.34, 67.77, 66.93, 62.29, 61.37, 60.61, 60.56, 52.33, 52.14, 40.11, 39.67, 22.18, 22.02. HRMS (ESI) m/z calculated for C34H48N6O23Mass calculated (M − H) 907.2693, found 907.2669.
Neu5Ac9N3α2–8Neu5Acα2–3GalβpNP (7). 41 mg, yield 86%; white solid. 1H NMR (800 MHz, D2O) δ 8.27 (d, J = 9.6 Hz, 2H), 7.26 (d, J = 8.8 Hz, 2H), 5.31 (d, J = 8 Hz, 1H), 4.22 (dd, J = 3.2 and 9.6 Hz, 1H), 4.17 (dd, J = 3.2 and 12 Hz, 1H), 4.11 (m, 1H), 4.06 (d, J = 2.4 Hz, 1H), 4.01 (m, 1H), 3.94–3.48 (m, 15H), 2.74 (dd, J = 4 and 12 Hz, 1H), 2.71 (dd, J = 4.8 and 12.8 Hz, 1H), 2.06 (s, 3H), 2.02 (s, 3H), 1.75 (t, J = 12.8 Hz, 1H) and 1.70 (t, J = 12 Hz, 1H). 13C NMR (200 MHz, D2O) δ 174.83, 174.80, 173.33, 173.21, 161.56, 142.38, 125.95, 116.35, 100.41, 100.04, 99.51, 78.37, 75.29, 73.93, 72.35, 70.16, 69.24, 68.71, 68.54, 68.35, 67.83, 66.93, 61.41, 60.62, 52.84, 52.15, 51.60, 40.37, 39.70, 22.20, 21.92. HRMS (ESI) m/z calculated for C34H48N6O23 Mass calculated (M − H) 907.2693, found 907.2679.
Cloning of SpNanA and SpNanB
The catalytic domain of SpNanA (318–792 aa, NCBI Reference Sequence: NP_359129.1) was cloned as an N-His6-tagged fusion protein in the pET15b vector. The truncated SpNanB (30–697 aa, NCBI Reference Sequence: NP_359124.1) was cloned as a C-His6-tagged fusion protein in the pET22b(+) vector. Genomic DNA of Streptococcus pneumoniae R6 was used as a template for polymerase chain reactions (PCR). The primers used for SpNanA were: forward primer 5′-GATC![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) CCTGAAGGAGCGGCTTTAAC-3′ (NdeI restriction site is underlined) and reverse primer 5′-CGCTTAATCTTTGCTCAAAAAGTCC-3′ (BamHI restriction site is underlined). The primers used for SpNanB were: forward primer 5′-GATCGAATGAATTAAACTATGGTCAACT-3′ (BamHI restriction site is underlined) and reverse primer 5′-CGCTTTTGTTAAATCATTAATTTCCAAA-3′ (XhoI restriction site is underlined). PCR was performed in a reaction mixture (50 μL) containing genomic DNA (1 μg), forward and reverse primers (1 μM each), 10 × Herculase buffer (5 μL), dNTP mixture (1 mM), and 5 U (1 μL) of Herculase-enhanced DNA polymerase. The reaction mixture was subjected to 30 cycles of amplification with an annealing temperature of 52 °C. The resulting PCR product was purified and digested with the corresponding restriction enzymes. The purified and digested PCR product was ligated with the predigested pET15b or pET22b(+) vector and transformed into electrocompetent E. coli DH5α cells. Selected clones were grown for minipreps. Positive clones were characterized by restriction mapping and DNA sequencing performed by Davis Sequencing Facility at the University of California-Davis.
CCTGAAGGAGCGGCTTTAAC-3′ (NdeI restriction site is underlined) and reverse primer 5′-CGCTTAATCTTTGCTCAAAAAGTCC-3′ (BamHI restriction site is underlined). The primers used for SpNanB were: forward primer 5′-GATCGAATGAATTAAACTATGGTCAACT-3′ (BamHI restriction site is underlined) and reverse primer 5′-CGCTTTTGTTAAATCATTAATTTCCAAA-3′ (XhoI restriction site is underlined). PCR was performed in a reaction mixture (50 μL) containing genomic DNA (1 μg), forward and reverse primers (1 μM each), 10 × Herculase buffer (5 μL), dNTP mixture (1 mM), and 5 U (1 μL) of Herculase-enhanced DNA polymerase. The reaction mixture was subjected to 30 cycles of amplification with an annealing temperature of 52 °C. The resulting PCR product was purified and digested with the corresponding restriction enzymes. The purified and digested PCR product was ligated with the predigested pET15b or pET22b(+) vector and transformed into electrocompetent E. coli DH5α cells. Selected clones were grown for minipreps. Positive clones were characterized by restriction mapping and DNA sequencing performed by Davis Sequencing Facility at the University of California-Davis.
Expression and purification of SpNanA and SpNanB
E. coli BL21 (DE3) cells containing a recombinant plasmid in the pET15b or pET22b(+) vector was cultured in LB-rich medium (10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl) supplemented with 100 μg mL−1 ampicillin. The cells were grown until the OD600nm of the culture reached 0.8–1.0. Isopropyl-1-thio-β-D-galactopyranoside (IPTG, 0.1 mM) was added to induce the overexpression of the target protein. The culture was incubated at 20 °C for 20 h with shaking at 250 rpm in a C25KC incubator shaker (New Brunswick Scientific, Edison, NJ). The cells were then harvested by centrifuging at 4 °C, 4000 rpm for 2 h. The cell pellet from 1 L culture was resuspended in 20 mL of lysis buffer (100 mM Tris-HCl, pH 8.0, containing 0.1% Triton X-100). The suspension was sonicated (amplitude at 68% for big tip, 3 s pulse on and 2 s pulse off for 80 cycles) and centrifuged at 12000g for 15 min at 4 °C. The lysate (supernatant) was collected and added to a Ni2+-NTA affinity column that was pre-equilibrated with 6 column volumes of binding buffer (50 mM Tris-HCl buffer, pH 7.5, 0.5 M NaCl, 5 mM imidazole). The column was washed with 10 column volumes of binding buffer (50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 5 mM imidazole), 10 column volumes of washing buffer (50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 20 mM imidazole) followed by 8 volumes of elute buffer (50 mM Tris-HCl, pH 7.5, 0.5 NaCl, 200 mM imidazole). The fractions containing the purified enzyme were collected, dialyzed against Tris-HCl buffer (20 mM, pH 7.5) containing 10% glycerol, and stored at 4 °C for sialidase assays.
Sialidase assays
Sialidase assays (20 μL for each reaction) were carried out in duplicate at 37 °C for 30 min in a 384-well plate (Fisher Scientific, Chicago, IL) in a reaction mixture containing Siaα2–8Neu5Acα2–3GalβpNP (selected from 1–7 for each sample, 0.3 mM), SpNanB (452 ng, used as an α2–3-sialidase), and Aspergillus oryzae β-galactosidase (12 μg, 126 mU). The amounts of SpNanB and β-galactosidase required to completely hydrolyze Neu5Acα2–3GalβpNP (0.3 mM) to release pNP within the time frame of the assay were predetermined and confirmed by control assays with Neu5Acα2–3GalβpNP (0.3 mM) in the absence of the sialidase to be tested. The reactions were stopped by adding 40 μL of N-cyclohexyl-3-aminopropane sulfonic acid (CAPS) buffer (0.5 M, pH 10.5). The amount of the para-nitrophenolate formed was determined by measuring the A405nm of the reaction mixtures using a microtiter plate reader.In order to ensure that the concentrations of the sialosides were consistent, the stock solutions of the sialosides were quantified using capillary electrophoresis (CE) equipped with a photodiode array (PDA) detector as described previously.36 The assay conditions for bacterial sialidases were: hNEU2 (51.4 μg) in MES buffer (100 mM, pH 5.0); Vibrio cholerae sialidase (2 mU) in sodium acetate buffer (50 mM, pH 5.5) containing CaCl2 (4 mM) and bovine serum albumin (0.1 mg mL−1); CjCstII (115.2 μg) in MES buffer (100 mM, pH 6.0) containing CMP (0.4 mM); SpNanA (450 ng) in sodium acetate buffer (100 mM, pH 6.0); A. ureafaciens sialidase (3 mU) in sodium acetate buffer (100 mM, pH 5.5); BiNanH2 (11.4 μg) in sodium acetate buffer (100 mM, pH 5.0); Clostridium perfringens sialidase (60 mU) in MES buffer (100 mM, pH 5.0); PmST1 (36 μg) in sodium acetate buffer (100 mM, pH 5.5) containing CMP (0.4 mM).
Acknowledgements
This work was supported by US NIH grants R01HD065122 and U01GM120419. The Bruker Avance-800 NMR spectrometer was funded by NSF grant DBIO-722538. We would like to thank Prof. David A. Mills from the University of California-Davis in providing the plasmid containing BiNanH2. H. Y., Y. L., and X. C. are co-founders of Glycohub, Inc., a company focusing on the development of carbohydrate-based reagents, diagnostics, and therapeutics. Glycohub, Inc. played no role in the design, execution, interpretation, or publication of this study.Notes and references
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS PubMed.
- T. Angata and A. Varki, Chem. Rev., 2002, 102, 439–469 CrossRef CAS PubMed.
- R. Schauer, Glycoconjugate J., 2000, 17, 485–499 CrossRef CAS PubMed.
- H. Yu, S. Huang, H. Chokhawala, M. Sun, H. Zheng and X. Chen, Angew. Chem., Int. Ed., 2006, 45, 3938–3944 CrossRef CAS PubMed.
- R. Drzeniek, Curr. Top. Microbiol. Immunol., 1972, 59, 35–74 CAS.
- E. Saxon, S. J. Luchansky, H. C. Hang, C. Yu, S. C. Lee and C. R. Bertozzi, J. Am. Chem. Soc., 2002, 124, 14893–14902 CrossRef CAS PubMed.
- R. Xie, L. Dong, Y. Du, Y. Zhu, R. Hua, C. Zhang and X. Chen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5173–5178 CrossRef CAS PubMed.
- J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS PubMed.
- R. Isomura, K. Kitajima and C. Sato, J. Biol. Chem., 2011, 286, 21535–21545 CrossRef CAS PubMed.
- S. Inoue and M. Iwasaki, Biochem. Biophys. Res. Commun., 1978, 83, 1018–1023 CrossRef CAS PubMed.
- L. K. Mahal, N. W. Charter, K. Angata, M. Fukuda, D. E. Koshland, Jr. and C. R. Bertozzi, Science, 2001, 294, 380–381 CrossRef CAS PubMed.
- H. Yu, J. Cheng, L. Ding, Z. Khedri, Y. Chen, S. Chin, K. Lau, V. K. Tiwari and X. Chen, J. Am. Chem. Soc., 2009, 131, 18467–18477 CrossRef CAS PubMed.
- M. Fukuda, M. Lauffenburger, H. Sasaki, M. E. Rogers and A. Dell, J. Biol. Chem., 1987, 262, 11952–11957 CAS.
- M. Gilbert, M. F. Karwaski, S. Bernatchez, N. M. Young, E. Taboada, J. Michniewicz, A. M. Cunningham and W. W. Wakarchuk, J. Biol. Chem., 2002, 277, 327–337 CrossRef CAS PubMed.
- R. P. Estrella, J. M. Whitelock, R. H. Roubin, N. H. Packer and N. G. Karlsson, Methods Mol. Biol., 2009, 534, 171–192 CAS.
- A. P. Corfield, H. Higa, J. C. Paulson and R. Schauer, Biochim. Biophys. Acta, 1983, 744, 121–126 CrossRef CAS.
- H. Kodama, L. G. Baum and J. C. Paulson, Carbohydr. Res., 1991, 218, 111–119 CrossRef CAS PubMed.
- A. Frisch and E. F. Neufeld, Anal. Biochem., 1979, 95, 222–227 CrossRef CAS PubMed.
- Y. Uchida, Y. Tsukada and T. Sugimori, J. Biochem., 1979, 86, 1573–1585 CAS.
- J. E. McCombs, C. Zou, R. B. Parker, C. W. Cairo and J. J. Kohler, ACS Chem. Biol., 2016, 11, 185–192 CrossRef CAS PubMed.
- H. A. Chokhawala, S. Huang, K. Lau, H. Yu, J. Cheng, V. Thon, N. Hurtado-Ziola, J. A. Guerrero, A. Varki and X. Chen, ACS Chem. Biol., 2008, 3, 567–576 CrossRef CAS PubMed.
- H. A. Chokhawala, H. Yu and X. Chen, ChemBioChem, 2007, 8, 194–201 CrossRef CAS PubMed.
- Y. Li, H. Cao, N. Dao, Z. Luo, H. Yu, Y. Chen, Z. Xing, N. Baumgarth, C. Cardona and X. Chen, Virology, 2011, 415, 12–19 CrossRef CAS PubMed.
- H. Yu, H. A. Chokhawala, S. Huang and X. Chen, Nat. Protoc., 2006, 1, 2485–2492 CrossRef CAS PubMed.
- H. Yu, H. Yu, R. Karpel and X. Chen, Bioorg. Med. Chem., 2004, 12, 6427–6435 CrossRef CAS PubMed.
- G. Sugiarto, K. Lau, J. Qu, Y. Li, S. Lim, S. Mu, J. B. Ames, A. J. Fisher and X. Chen, ACS Chem. Biol., 2012, 7, 1232–1240 CrossRef CAS PubMed.
- H. Yu and X. Chen, Org. Biomol. Chem., 2016, 14, 2809–2818 CAS.
- Y. Li, H. Yu, H. Cao, K. Lau, S. Muthana, V. K. Tiwari, B. Son and X. Chen, Appl. Microbiol. Biotechnol., 2008, 79, 963–970 CrossRef CAS PubMed.
- J. Cheng, H. Yu, K. Lau, S. Huang, H. A. Chokhawala, Y. Li, V. K. Tiwari and X. Chen, Glycobiology, 2008, 18, 686–697 CrossRef CAS PubMed.
- Y. Li, H. Cao, H. Yu, Y. Chen, K. Lau, J. Qu, V. Thon, G. Sugiarto and X. Chen, Mol. BioSyst., 2011, 7, 1060–1072 RSC.
- G. Xu, J. A. Potter, R. J. Russell, M. R. Oggioni, P. W. Andrew and G. L. Taylor, J. Mol. Biol., 2008, 384, 436–449 CrossRef CAS PubMed.
- G. Xu, M. J. Kiefel, J. C. Wilson, P. W. Andrew, M. R. Oggioni and G. L. Taylor, J. Am. Chem. Soc., 2011, 133, 1718–1721 CrossRef CAS PubMed.
- D. A. Sela, Y. Li, L. Lerno, S. Wu, A. M. Marcobal, J. B. German, X. Chen, C. B. Lebrilla and D. A. Mills, J. Biol. Chem., 2011, 286, 11909–11918 CrossRef CAS PubMed.
- H. Yu, H. Chokhawala, R. Karpel, H. Yu, B. Wu, J. Zhang, Y. Zhang, Q. Jia and X. Chen, J. Am. Chem. Soc., 2005, 127, 17618–17619 CrossRef CAS PubMed.
- J. Cheng, S. Huang, H. Yu, Y. Li, K. Lau and X. Chen, Glycobiology, 2010, 20, 260–268 CrossRef CAS PubMed.
- H. Cao, Y. Li, K. Lau, S. Muthana, H. Yu, J. Cheng, H. A. Chokhawala, G. Sugiarto, L. Zhang and X. Chen, Org. Biomol. Chem., 2009, 7, 5137–5145 CAS.
- Z. Khedri, M. M. Muthana, Y. Li, S. M. Muthana, H. Yu, H. Cao and X. Chen, Chem. Commun., 2012, 48, 3357–3359 RSC.
- Z. Khedri, Y. Li, S. Muthana, M. M. Muthana, C. W. Hsiao, H. Yu and X. Chen, Carbohydr. Res., 2014, 389, 100–111 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for products. See DOI: 10.1039/c6ob02240e |
| This journal is © The Royal Society of Chemistry 2017 |