
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical formation of quinone methides from peptides containing modified tyrosine†
Antonija
Husak
a,
Benjamin P.
Noichl
b,
Tatjana
Šumanovac Ramljak
a,
Margareta
Sohora
a,
Đani
Škalamera
a,
Nediljko
Budiša
b and
Nikola
Basarić
*a
aDepartment of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Bijenička cesta 54, 10 000 Zagreb, Croatia. E-mail: nbasaric@irb.hr; Tel: +385 1 4561 141; Fax: +385 1 4680 195
bInstitute for Chemistry, Technical University Berlin, Müller-Breslau-Str. 10, 10623 Berlin, Germany
First published on 27th October 2016
Abstract
We have demonstrated that quinone methide (QM) precursors can be introduced in the peptide structure and used as photoswitchable units for peptide modifications. QM precursor 1 was prepared from protected tyrosine in the Mannich reaction, and further used as a building block in peptide synthesis. Moreover, peptides containing tyrosine can be transformed into a photoactivable QM precursor by the Mannich reaction which can afford monosubstituted derivatives 2 or bis-substituted derivatives 3. Photochemical reactivity of modified tyrosine 1 and dipeptides 2 and 3 was studied by preparative irradiation in CH3OH where photodeamination and photomethanolysis occur. QM precursors incorporated in peptides undergo photomethanolysis with quantum efficiency ΦR = 0.1–0.2, wherein the peptide backbone does not affect their photochemical reactivity. QMs formed from dipeptides were detected by laser flash photolysis (λmax ≈ 400 nm, τ = 100 μs–20 ms) and their reactivity with nucleophiles was studied. Consequently, QM precursors derived from tyrosine can be a part of the peptide backbone which can be transformed into QMs upon electronic excitation, leading to the reactions of peptides with different reagents. This proof of principle showing the ability to photochemically trigger peptide modifications and interactions with other molecules can have numerous applications in organic synthesis, materials science, biology and medicine.
Introduction
Quinone methides (QMs) are reactive intermediates in the chemistry and photochemistry of phenols that have received significant scientific attention over the past twenty years.1,2 Interest in their chemistry has been initiated due to their applications in synthesis,3,4 as well as their biological activity.5–7 To date, the biological effects of QMs have been connected to their reactivity with nucleobases8–10 and DNA.11–14 More importantly, some antineoplastic antibiotics base their action on the metabolic formation of QMs that cross-link DNA.15,16 A significant endeavor in elucidating the reactivity of QMs with DNA has been undertaken by Rokita et al.,1,16 who demonstrated reversible alkylation of DNA by QMs.9 They have shown that QMs act as “immortal” species that are capable of forming thermodynamically most stable DNA cross-adducts.17–19 However, it has also been demonstrated that QMs react with amino acids20,21 and proteins.22 Thus, we have recently shown that the antiproliferative activity of QMs formed in the phototautomerization of hydroxyphenylanthracenes is due to the reaction with intracellular proteins, rather than with DNA.23Formation of QMs often requires harsh conditions such as elevated temperature,24,25 or the use of biologically inacceptable oxidants26 or F−.13 On the contrary, photochemical reactions require mild conditions which allow for the generation of QMs in living cells.7,27 Two main photochemical reactions for the formation of QMs are dehydration of hydroxymethyl substituted phenols28,29 and deamination of aminomethyl30 or ammoniummethylphenols.21,31 Photodeamination can also be initiated by an intramolecular photoinduced electron transfer reaction with naphthalene diimide as a photooxidizing agent.32 Freccero et al. have used photodeamination of Mannich salts for the investigation of biological activity of QMs,33,34 and the ability of QM derivatives to selectively target guanine quadruplexes has been demonstrated.35–37 We have investigated the dependence of the photodeamination efficiency on pH in aqueous media and have shown that this reaction is the most efficient one for zwitterionic prototropic forms at 8.5 < pH < 11.1.38 Furthermore, Freccero et al. focused their study on the structure of QM precursors resulting in high efficiency of deamination or high intersystem crossing yields and population of triplets.39
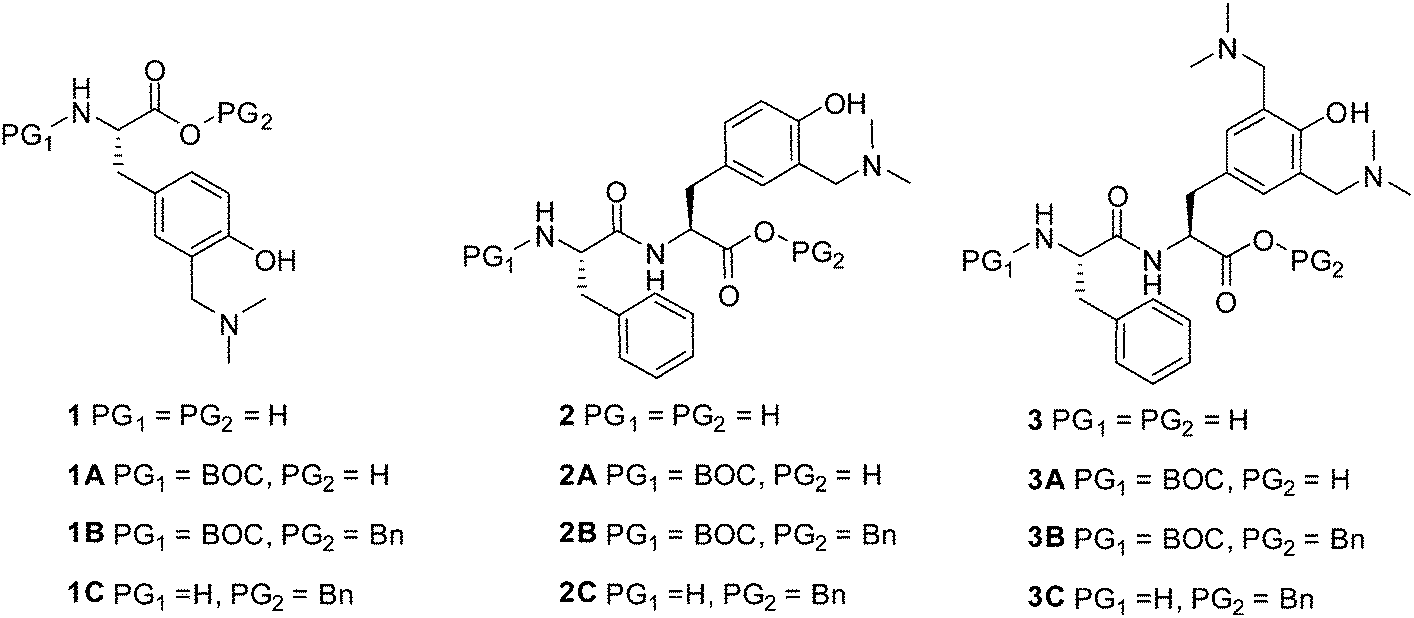
Results and discussion
Synthesis
QM precursor, a derivative of tyrosine 1B was synthesized in the Mannich reaction from BOC-Tyr-OBn and Eschenmoser's salt (Scheme 1), according to the procedure in the literature precedent.38 BOC-Tyr-OBn was prepared according to the usual procedures for the introduction of protective groups.44 Unnatural amino acid 1B was deprotected at the N-site by the use of trifluoroacetic acid (TFA) to yield 1C, or at the C-terminus by hydrogenolysis to furnish 1A. Both deprotections were conducted in high yields without any interference or change of the aminomethylphenol moiety.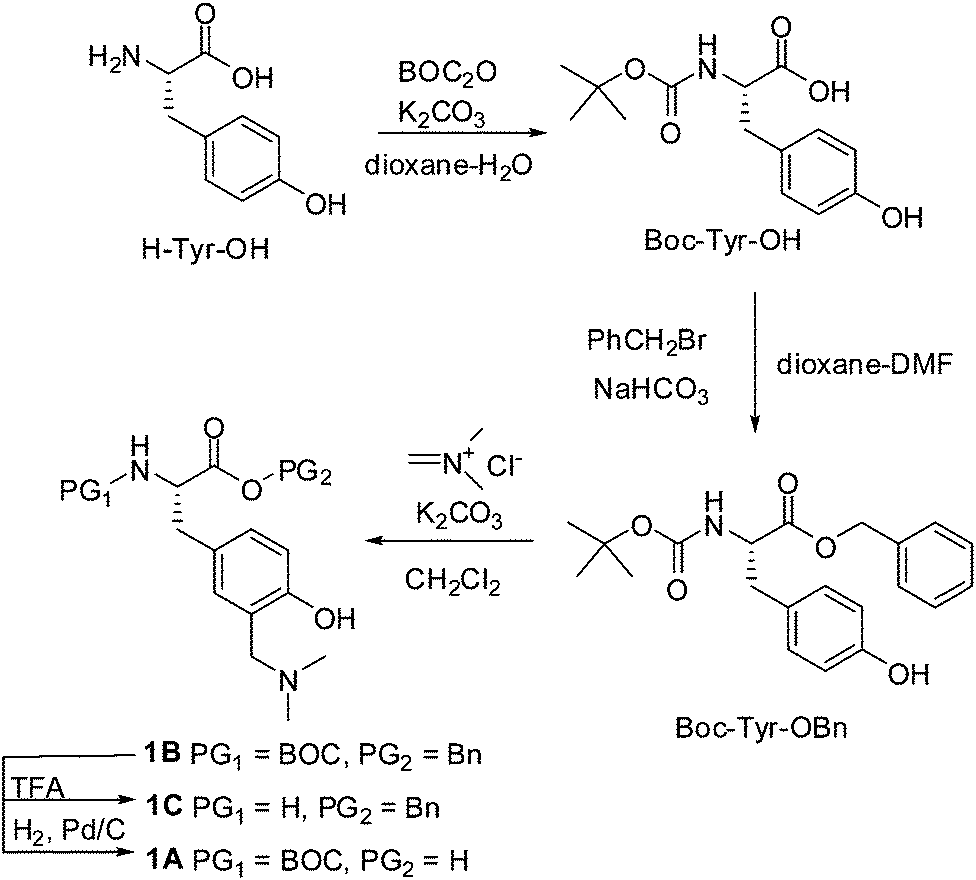 | ||
| Scheme 1 | ||
The synthesis of dipeptides with the incorporated QM precursor was conducted in two ways, by introduction of the functional group in the Mannich reaction after peptide coupling, or by the use of the QM precursor in the peptide coupling (Scheme 2).
 | ||
| Scheme 2 | ||
Dipeptide BOC-Phe-Tyr-OH was synthesized from BOC-Phe-OH by the use of N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) activation.45 Protected dipeptide BOC-Phe-Tyr-OBn was transformed into QM precursor dipeptide 2B in the Mannich reaction with Eschenmoser's salt where only one aminomethyl group was introduced. Preparation of the Mannich reagent in situ with an excess of dimethylamine and formaldehyde allowed for the introduction of two aminomethyl groups, yielding 3B. Thus, we have demonstrated that tyrosine in a peptide can be transformed into a QM precursor in the Mannich reaction. However, peptides cannot contain amino acids that react with electrophiles (e.g. tryptophan).
The other synthetic protocol for the preparation of peptide 2B was the coupling of activated BOC-Phe-OH with the N-free site of QM precursor 1C (Scheme 2). Peptide coupling by the use of NHS and EDC activation45 was conducted, giving the desired QM precursor in a moderate yield (57%).
Consequently, we have demonstrated that QM precursor 1C can be used as a building block in peptide synthesis. Attempts to perform activation of the carboxylic group in 1A and couple it with H-Phe-OBn failed by the use of NHS and EDC activation47 or by the N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) and 1-hydroxybenzotriazole (HOBT) activation protocol.46 Nevertheless, dipeptides 2 and 3 can be easily deprotected, and in principle, used in further synthesis of more complex molecules.
The synthesis of more complex peptides with incorporated modified tyrosine was accomplished by a combination of solid and liquid phase peptide synthesis (LPPS & SPPS). Tripeptide 4 was synthesized via LPPS deploying the alternating activation of the C-terminus (EDC·HCl and NHS, Scheme 3) and coupling of the unprotected amino acids Ala-OH and 1 (Scheme 3). For the latter coupling step, the required deprotection of 1B was accomplished in two steps, using H2/Pd in EtOH for the selective removal of the benzyl group, followed by 4 M HCl in dioxane to yield the desired unprotected amino acid 1 (97%, two steps), which was used for the coupling. This approach provides the desired amino acid sequence – Asp–Ala–1– (4) – in a completely protected manner, suitable for the synthesis of longer peptide chains.
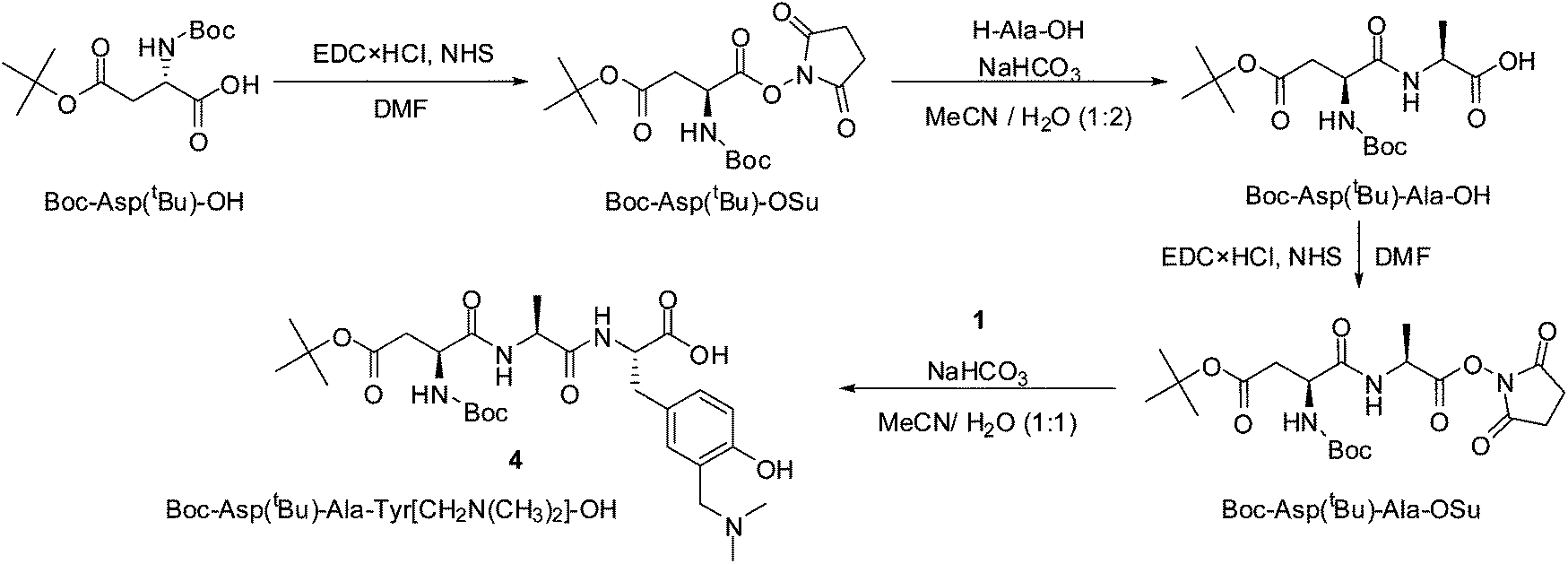 | ||
| Scheme 3 | ||
The synthesis of the corresponding mini-protein was accomplished using SPPS. Therefore, the previously synthesized intermediate 5 on the solid support (see Scheme S1 in the ESI†) and protected tripeptide 4 were utilized. For the intermediate 5 we have chosen the first 17 amino acids of the Trp-Cage analogue TC10b.40 In this way the TC10b analogue was obtained, bearing 1 instead of tyrosine at the position 3. The coupling was performed in the presence of O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) and N,N-diisopropylamine (DIPEA) for 1 hour in N,N-dimethylformamide (DMF), followed by an acid deprotection step, yielding the desired peptide TC10b_3Y1 (Scheme 4). The structure of the peptide was confirmed by MS (ESI-HR: m/z = 1072.53394, calculated for [M + H]+: 1072.53366; Δm/z = 0.2 ppm).
 | ||
| Scheme 4 | ||
Photophysical properties
The spectral properties of QM precursors 1 are anticipated not to differ from those of the parent molecule. However, it is interesting to investigate the spectral properties of dipeptides 2 and 3 that contain two chromophores, phenylalanine, and the modified tyrosine. Their spectral and photophysical properties were investigated in CH3CN (Table 1, for spectra see Fig. S2–S7 in the ESI†).| λ abs/nm (ε/dm3 mol−1 cm−1)a |
λ
em![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b/nm b/nm |
Φ
Fc |
τd/ns |
|
|---|---|---|---|---|
| a Maximum in the absorption spectrum and molar absorption coefficient in the brackets. b Maximum in the emission spectrum. c Approximate quantum yield of fluorescence measured by the use of anisole in cyclohexane (ΦF = 0.29)47 as a reference upon excitation at 260 nm. d Decay of fluorescence at 310 nm measured by time-correlated single photon counting (SPC), pre-exponential factors are given in brackets. | ||||
| 2 | 282 (500) | 305 | ≈0.006 | 0.28 ± 0.05 (0.32) |
| 0.71 ± 0.06 (0.44) | ||||
| 5.0 ± 0.1 (0.24) | ||||
| 3A | 286 (1700) | 315 | ≈0.007 | 0.22 ± 0.04 (0.26) |
| 0.75 ± 0.03 (0.54) | ||||
| 6.0 ± 0.1 (0.12) | ||||
In the absorption spectra of dipeptides 2 and 3A (Fig. S2 and S5 in the ESI†) the typical absorption band at ≈280 nm was observed corresponding to the S0 → S1 absorption of the tyrosine moiety. In the emission spectra recorded in CH3CN (Fig. S3 and S6 in the ESI†), the typical dual fluorescence was observed with one main band at 305–315 nm, and a shoulder at 370 nm for 2, and 430 nm for 3A. This dual fluorescence is due to the emission of phenol and phenolate. Both species are present in equilibrium in S0, but the phenolate is also formed in S1 by excited state intramolecular proton transfer (ESIPT), as seen for the Mannich derivatives of cresol.38 Namely, upon electronic excitation, phenols exhibit enhanced acidity (unsubstituted phenol 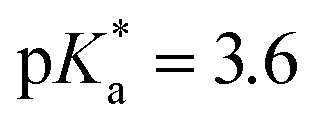 ),48,49 and the methylamine group has a role of a basic site. The appearance of the emission spectra depends on the excitation and emission wavelength, which is expected in a bichromophoric system containing Phe and Tyr. Moreover, due to the acidic and basic functionalities in the molecule, the dipeptides exhibit several prototropic forms. Therefore, fluorescence quantum yield depends on the excitation wavelength, and the decay of fluorescence cannot be fit to single exponential function. Due to the complexity of the chromophoric system, no attempt was made to assign the fluorescence decay components. In Table 1, only an estimate for the fluorescence quantum yield (ΦF) is reported, measured by the use of anisole in cyclohexane (ΦF = 0.29).47 Generally, ΦF values are very low, probably due to the relatively efficient deactivation from S1 by photodeamination giving QMs.38 Addition of H2O to the CH3CN solution until a concentration of 4 M was reached quenched the fluorescence to about 2–3 times, but further additions did not result in additional quenching. Due to very weak fluorescence in the aqueous solution and complexity due to several pH-dependant prototropic forms,38 we constrained our measurements to CH3CN solution only.
),48,49 and the methylamine group has a role of a basic site. The appearance of the emission spectra depends on the excitation and emission wavelength, which is expected in a bichromophoric system containing Phe and Tyr. Moreover, due to the acidic and basic functionalities in the molecule, the dipeptides exhibit several prototropic forms. Therefore, fluorescence quantum yield depends on the excitation wavelength, and the decay of fluorescence cannot be fit to single exponential function. Due to the complexity of the chromophoric system, no attempt was made to assign the fluorescence decay components. In Table 1, only an estimate for the fluorescence quantum yield (ΦF) is reported, measured by the use of anisole in cyclohexane (ΦF = 0.29).47 Generally, ΦF values are very low, probably due to the relatively efficient deactivation from S1 by photodeamination giving QMs.38 Addition of H2O to the CH3CN solution until a concentration of 4 M was reached quenched the fluorescence to about 2–3 times, but further additions did not result in additional quenching. Due to very weak fluorescence in the aqueous solution and complexity due to several pH-dependant prototropic forms,38 we constrained our measurements to CH3CN solution only.
Photochemistry
Photodeamination of aminomethylphenols in aqueous CH3OH leads to photomethanolysis products via QM intermediates.32,38,50 To demonstrate that photomethanolysis of modified tyrosine 1 and dipeptides 2 and 3 also takes place, we performed preparative irradiation of 1B, 2B and 3B. Fully protected compounds (bearing BOC and Bn) were chosen for preparative irradiation to make the product isolation easier. The irradiation experiments were conducted by irradiating CH3OH solutions at 300 nm and by analyzing the composition of the solutions by HPLC. After the irradiation, the photoproducts were isolated by preparative TLC and characterized by NMR spectroscopy. Methanolysis of 1B until the conversion of 44% gave methyl ether 6, with the isolated yield of 28% (eqn (1)). | (1) |
Similarly, irradiation of dipeptide 2B until the conversion of 63% was achieved gave cleanly only methyl ether 7 (isolated yield 21%, eqn (2)).
 | (2) |
On the other hand, irradiation of 3B gave a mixture of products 8 and 9 whose ratio depended on the irradiation time (eqn (3)). Thus, after irradiating for 30 min, the ratio of 8:9 was 1:1. After prolonged irradiation until the conversion of 68%, 9 was isolated in 21% yield. Similarly, after irradiation of peptide 3A until the conversion of 40% dimethoxy product 10 was isolated in 5% yield (eqn (4)).
 | (3) |
 | (4) |
The efficiency of the photomethanolysis reaction (ΦR) was measured by the use of a primary actinometer, KI/KIO3Φ254 = 0.74),47,51 and a secondary actinometer, photomethanolysis of 2-hydroxymethylphenol (Φ254 = 0.23).28 The values are compiled in Table 2. The efficiency was measured for all Mannich derivatives 1 to demonstrate that the protective groups on tyrosine do not affect the photomethanolysis reaction. The corresponding photoproducts from 1, 1A and 1C were detected by HPLC, but product isolation was conducted only for the fully protected isomer 1B. Moreover, the ΦR was measured for the dipeptide bearing one or two aminomethyl groups.
The values in Table 2 indicate similar photochemical reaction efficiencies of tyrosine QM precursors 1A–1C, irrespective of the protective groups used. Moreover, QM precursors remain photochemically reactive in dipeptides 2B and 2C where photomethanolysis takes place with similar efficiencies to those of derivatives 1. Dipeptides 2 contain two chromophores, Phe and Tyr both of which can be excited at 254 nm. Similar photomethanolysis efficiency for 2 suggests that energy transfer from Phe to Tyr takes place leading to photoelimination, regardless of which chromophore was initially excited.
Photochemical reactivity was also tested with the protected tripeptide 4 in the presence of benzyl mercaptan. After the irradiation of the reaction solution for 20 min at 254 nm, the completely oxidized product was detected with the conversion of 23%. Only a minor fraction of the products of photoreaction was identified to be the non-oxidized product (see the ESI†). These results indicate that tripeptide also undergoes photodeamination giving QMs that can be trapped with nucleophiles. Irradiation of the mini-protein TC10b_3Y1 was performed in CH3OD where the photomethanolysis reaction is anticipated. After one hour irradiation at 254 nm, MS analysis indicated its conversion to the corresponding methyl ether, which clearly demonstrates that photoreaction of the QM precursor is feasible, and shows this reaction's applicability to protein systems.
Laser flash photolysis (LFP)
To detect QM intermediates in the photochemistry of dipeptides 2 and 3, LFP was used. The samples were excited with a Nd:YAG laser at 266 nm. The measurements were performed in N2 and O2-purged CH3CN solution, where O2 is expected to quench triplets and radicals, but not QMs. Moreover, the spectra and decay kinetics were measured in CH3CN and CH3CN–H2O (1:1) where the difference is expected due to ESPT pathways,52–55 and different reactivities of QMs in aprotic and protic solution (for the transient absorption spectra see Fig. S8–S16 in the ESI†).38,56–60
Dipeptide 2 is not well soluble in CH3CN. However, in neat CH3CN solution a transient was detected with a maximum at 390 nm (Fig. S8 in the ESI†), tentatively assigned to 2-QM based on the comparison with the previously published spectra of o-QM derivatives.38,61 Due to a long lifetime of the transient and its low intensity, decay kinetics in CH3CN was not investigated. Spectra of better quality were obtained in CH3CN–H2O (1:1) solution where the compound is more soluble (Fig. 1). The transient assigned to 2-QM was detected with a maximum at 400 nm. It was formed within a laser pulse and it decayed to the baseline with unimolecular kinetics and lifetime of 20 ± 2 ms (Table 3). To verify its assignment a quenching study was performed with ethanolamine (EtAm) and NaN3, ubiquitous QM quenchers,38,60,62–66 as well as with ethyl vinyl ether (EVE) that reacts with QMs in the Diels–Alder reaction (Fig. S10–S12 in the ESI†).67 The measured quenching constants (Table 3) are in agreement with the expected reactivity of QM with nucleophiles.38
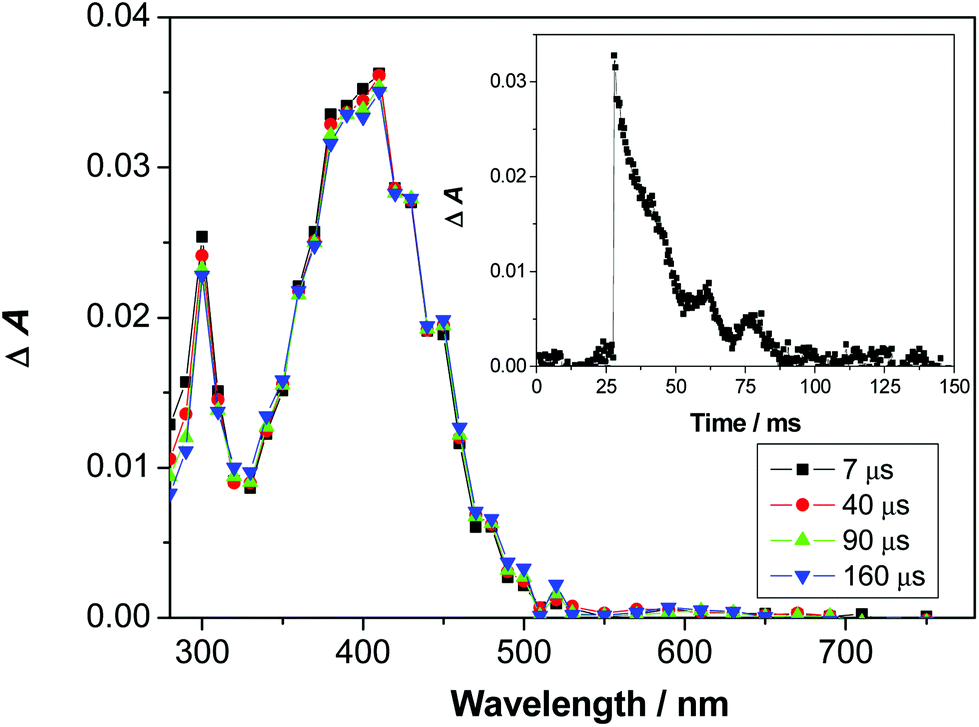 | ||
| Fig. 1 Transient absorption spectra of O2-purged CH3CN–H2O (1:1) solution of 2. Inset: decay of transient absorbance at 400 nm. | ||
| Transient |
τ
CH3CNa/ms |
τ
CH3CN–H2Ob/ms |
k q (EtAm)c/M−1 s−1 | k q (NaN3)d/M−1 s−1 | k q (EVE)e/M−1 s−1 |
|---|---|---|---|---|---|
| a Lifetime in O2-purged CH3CN solution. b Lifetime in O2-purged CH3CN–H2O solution. c Quenching rate constant with ethanolamine (EtAm), measured in CH3CN–H2O. d Quenching rate constant with NaN3, measured in CH3CN–H2O. e Quenching rate constant with ethyl vinyl ether (EVE), measured in CH3CN–H2O. f Measured in CH3CN. | |||||
| 2-QM | — | 20 ± 2 | 2.3 × 105 | 1.1 × 107 | 9.9 × 102 |
| 3A-QM | 0.11 ± 0.01 | 13 ± 3 | 2.3 × 105f 9.9 × 104 |
— | 5.8 × 103 |

Conclusions
We synthesized new QM precursors, tyrosine derivatives 1, and incorporated them in the dipeptides 2. Moreover, we have demonstrated that QM precursors can be made from peptides containing tyrosine in the Mannich reaction, or by using modified tyrosine 1 as a building block in the usual peptide synthesis. Although peptide derivatives have several acidic and basic sites, and their photophysical properties are complex, photochemical reactivity of modified tyrosine and dipeptides in the deamination reaction is not affected by the peptide backbone. Moreover, reactivity of QM-dipeptides that are formed in the photodeamination reactions is not altered by the peptide. Consequently, photochemical reactions of 2, 3 and TC10b_3Y1 show a proof of principle that the QM precursor can be introduced into the peptide and used as a photochemical switch to enable the interaction and alkylation of peptides with other molecules of interest, which can have a tremendous impact in chemistry and biology.Experimental
General
1H and 13C NMR spectra were recorded at 300, 500 or 600 MHz at rt, using TMS as a reference and chemical shifts were reported in ppm. Melting points were determined using Mikroheiztisch apparatus and were not corrected. HRMS were obtained on a MALDI TOF/TOF instrument. ESI-MS were obtained on a LTQ Orbitrap instrument. Irradiation experiments were performed in a reactor equipped with 11 lamps with the output at 300 nm or a reactor equipped with 8 lamps at 254 or 300 nm (1 lamp 8 W). During irradiation, the irradiated solutions were continuously purged with argon and cooled by a tap-water finger-condenser. Solvents for irradiation were of HPLC purity. Chemicals were purchased from the usual commercial sources and were used as received. Solvents for chromatographic separations were used as they were delivered by the supplier (p.a. or HPLC grade) or purified by distillation (CH2Cl2). Preparations of the known compounds, L-BOC-Tyr(OH)-OH,68L-BOC-Tyr(OH)-OBn,44L-BOC-Phe-OSu,45 and L-BOC-Phe-Tyr(OH)-OH,69,70 are given in the ESI.†BOC-Tyr[CH2N(CH3)2]-OBn (1B)
A flask was charged with BOC-Tyr-OBn (1.11 g, 3.0 mmol) and CH2Cl2 (150 mL) was added. To this solution, K2CO3 (0.21 g, 1.5 mmol) and Eschenmoser's salt (0.28 g, 3.0 mmol) were added and the reaction mixture was stirred at rt for 5 days. When the reaction was completed, 0.5 M HCl (100 mL) was added, and the product was extracted with ethyl acetate (3 × 100 mL). The solvent was removed on a rotary evaporator to afford an oily product. The product was purified by column chromatography on aluminium oxide (II–III) using 0 → 5% CH3OH in CH2Cl2 as the eluent to afford the pure product (0.72 g, 56%) in the form of colorless crystals.Colorless crystals, mp 85–87 °C; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.35–7.22 (m, 5H), 6.91 (dd, J = 1.6, 8.0 Hz, 1H), 6.84 (d, J = 1.6 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 5.12 (d, J = 12.0 Hz, 1H), 5.06 (d, J = 12.0 Hz, 1H), 4.37–4.23 (m, 1H), 3.53 (s, 2H), 2.96 (dd, J = 6.4, 13.6 Hz, 1H), 2.84 (dd, J = 8.0, 13.6 Hz, 1H), 2.26 (s, 6H), 1.38 (s, 9H); 13C NMR (CD3OD, 75 MHz) δ/ppm: 173.7 (s, 1C), 157.8 (s, 1C), 157.7 (s, 1C), 137.1 (s, 1C), 131.1 (d, 2C), 130.5 (d, 3C), 129.5 (d, 1C), 129.3 (d, 1C), 128.6 (s, 1C), 123.5 (s, 1C), 116.6 (d, 1C), 80.6 (s, 1C), 67.8 (t, 1C), 62.4 (t, 1C), 56.9 (d, 1C), 44.7 (q, 2C), 37.9 (t, 1C), 28.6 (q, 3C); HRMS (MALDI-TOF) m/z [M + H]+ calcd for C24H32N2O5 429.2389, found 429.2368.
BOC-Tyr[CH2N(CH3)2]-OH (1A)
In a vessel for hydrogenation BOC-Tyr[CH2N(CH3)2]-OBn (1B) (390 mg, 0.9 mmol) was dissolved in anhydrous EtOH (40 mL), and 10% Pd/C (200 mg) was added. The hydrogenation was carried out for 5 h at the pressure of 63 psi. The crude reaction mixture was filtered and the solvent was evaporated on a rotary evaporator. The residue was purified by column chromatography on silica gel using 20 → 100% CH3OH in CH2Cl2 to afford the pure product (274 mg, 89%) in the form of a colorless solid.Colorless solid, mp 133–135 °C; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.10 (s, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 4.16 (s, 2H), 3.34 (s, 2H), 3.03 (dd, J = 5.0, 13.5 Hz, 1H), 2.88 (dd, J = 6.5, 13.5 Hz, 1H), 2.77 (s, 6H), 1.39 (s, 9H); 13C NMR (CD3OD, 75 MHz) δ/ppm: 178.3 (s, 1C), 157.2 (s, 1C), 156.4 (s, 1C), 134.1 (d, 1C), 133.8 (d, 1C), 130.9 (s, 1C), 117.7 (s, 1C), 116.1 (d, 1C), 80.0 (s, 1C), 58.4 (t, 1C), 58.3 (d, 1C), 43.3 (q, 2C), 38.9 (t, 1C), 28.8 (q, 3C); HRMS (MALDI-TOF) m/z [M + H]+ calcd for C17H26N2O5 339.1920, found 339.1911.
TFA·H-Tyr[CH2N(CH3)2·TFA]-OBn (1C)
BOC-Tyr[CH2N(CH3)2]-OBn (1B) (335 mg, 0.8 mmol) was dissolved in CH2Cl2 (3 mL). TFA/CH2Cl2 (1:1, 4 mL) was added and the reaction mixture was stirred at rt for 2 h. The solvent was removed by distillation in a vacuum to afford an oily product quantitatively.
Colorless oil; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.41–7.28 (m, 5H), 7.14–7.09 (m, 2H), 6.86 (d, J = 8.8 Hz, 1H), 5.26 (d, J = 11.5 Hz, 1H), 5.20 (d, J = 11.5 Hz, 1H), 4.31 (dd(t), J = 7.0 Hz), 4.22 (d, J = 12.9 Hz, 1H), 4.13 (d, J = 12.9 Hz, 1H), 3.15 (d, J = 7.5 Hz, 2H), 2.79 (d, J = 7.5 Hz, 6H); 13C NMR (CD3OD, 75 MHz) δ/ppm: 169.9 (s, 1C), 157.5 (s, 1C), 136.2 (s, 1C), 134.4 (d, 1C), 134.0 (d, 1C), 129.9 (d, 1C), 129.8 (d, 2C), 129.7 (d, 2C), 126.6 (s, 1C), 118.0 (s, 1C), 117.0 (d, 1C), 69.3 (t, 1C), 58.2 (t, 1C), 55.2 (d, 1C), 43.2 (q, 2C), 36.5 (t, 1C); HRMS (MALDI-TOF) m/z [M]+ calcd for (C23H26F6N2O7) 556.1644, found 556.1635.
BOC-Phe-Tyr[CH2N(CH3)2]-OBn (2B)
A flask was charged with TFA·H-Tyr[CH2N(CH3)2·TFA]-OBn (1C) (594 mg, 1.8 mmol), NaHCO3 (605 mg, 7.2 mmol) and THF–H2O (1:1, 24 mL). To the mixture, a solution of succinimide-activated phenylalanine (732 mg, 2.0 mmol) in THF (14 mL) was added dropwise, and the reaction mixture was stirred at rt for 2 days. THF was removed on a rotary evaporator, and the reaction mixture was acidified with 0.5 M HCl to pH 2, and the product was extracted with ethyl acetate (3 × 30 mL). The organic layer was washed with water and dried over anhydrous Na2SO4. After filtration and evaporation of the solvent, the product was purified by column chromatography on silica gel using 0 → 5% CH3OH in CH2Cl2 as the eluent to afford the pure product (749 mg, 57%) in a form of a colorless solid.
Colorless solid; mp 92–94 °C; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.37–7.27 (m, 5H), 7.25–7.15 (m, 5H), 7.11 (s, 1H), 7.08 (d, J = 8.1 Hz, 1H), 6.78 (d, J = 8.1 Hz, 1H), 5.12 (s, 2H), 4.68 (ddd(t), J = 7.0 Hz, 1H), 4.30–4.22 (m, 1H), 4.13 (s, 2H), 3.09 (dd, J = 6.0, 14.0 Hz, 1H), 3.02–2.89 (m, 2H), 2.75 (s, 6H), 2.75–2.65 (m, 1H), 1.34 (s, 9H); 13C NMR (CD3OD, 75 MHz) δ/ppm: 174.3 (s, 1C), 172.3 (s, 1C), 157.5 (s, 1C), 156.7 (s, 1C), 138.5 (s, 1C), 137.0 (s, 1C), 134.1 (d), 133.9 (d), 134.0 (d), 133.0 (d), 129.6 (d), 129.5 (d), 129.4 (d), 129.4 (s), 129.3 (d), 129.2 (d), 127.7 (d), 127.5 (d), 118.2 (s, 1C), 116.4 (d, 1C), 80.6 (s, 1C), 68.0 (t, 1C), 58.3 (t, 1C), 57.1 (d, 1C), 55.3 (d, 1C), 43.2 (q, 2C), 39.3 (t, 1C), 37.1 (t, 1C), 28.6 (q, 3C); HRMS (MALDI-TOF) m/z [M + H]+ calcd for C33H41N3O6 576.3074, found 576.3087.
BOC-Phe-Tyr[CH2N(CH3)2]-OBn (2B)
A flask was charged with BOC-Phe-Tyr-OBn (146 mg, 0.28 mmol) dissolved in CH2Cl2 (20 mL). To the reaction mixture, K2CO3 (20 mg, 0.14 mmol) and Eschenmoser's salt (26 mg, 0.28 mmol) were added and the reaction was stirred at rt for 5 days. When the reaction was completed, 0.3 M HCl (50 mL) was added, and the product was extracted with ethyl acetate (3 × 50 mL). The solvent was removed on a rotary evaporator to afford an oily substance which was purified by column chromatography on silica gel using 5 → 10% CH3OH in CH2Cl2 as the eluent to afford the pure product (50 mg, 31%) in a form of a colorless solid.BOC-Phe-Tyr[CH2N(CH3)2]-OH (2A)
In a vessel for hydrogenation BOC-Phe-Tyr[CH2N(CH3)2]-OBn (2B) (263 mg, 0.4 mmol) was dissolved in anhydrous EtOH (30 mL), and 10% Pd/C (100 mg) was added. Hydrogenation was carried out for 6 h at the pressure of 63 psi. The crude reaction mixture was filtered and the solvent was removed on a rotary evaporator to afford the pure product (221 mg, 99%).Yellowish oil; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.29–7.13 (m, 7H), 6.84 (d, J = 8.0 Hz, 1H), 4.63–4.56 (m, 1H), 4.32–4.18 (m, 3H), 3.15 (dd, J = 5.0, 14.0 Hz, 1H), 3.04 (dd, J = 5.0, 14.0 Hz, 1H), 2.99–2.88 (m, 1H), 2.83 (s, 6H), 2.78–2.69 (m, 1H), 1.35 (s, 9H); 13C NMR (CD3OD, 150 MHz) δ/ppm: 174.6 (s, 1C), 174.0 (s, 1C), 157.6 (s, 1C), 156.5 (s, 1C), 138.6 (s, 1C), 134.3 (d, 1C), 134.1 (d, 1C), 130.3 (d, 2C), 130.0 (s, 1C), 129.4 (d, 2C), 127.7 (d, 1C), 117.4 (s, 1C), 116.4 (d, 1C), 80.7 (s, 1C), 58.3 (t, 1C), 57.4 (d, 1C), 55.5 (d, 1C), 43.3 (q, 2C), 39.2 (t, 1C), 37.8 (t, 1C), 28.7 (q, 3C), one singlet was not seen; HRMS (MALDI-TOF) m/z [M + H]+ calcd for C26H35N3O6 486.2604, found 486.2627.
BOC-Phe-Tyr[CH2N(CH3)2]2-OH (3A)
To a solution of BOC-Phe-Tyr-OH (200 mg, 0.5 mmol) in 40% aqueous dimethylamine (1 mL, 7.8 mmol), formalin (37% aq, 0.5 mL, 6.4 mmol) was added dropwise and the temperature was maintained below 30 °C (overall time was 30 min). The reaction mixture was stirred at rt for 1 h, and then at the temperature of 90–95 °C for 2 h. Then, sodium chloride was added and stirring was continued for 20 min. The resulting oil was subjected to column chromatography on aluminum oxide (act. V) using 5 → 10% CH3OH in CH2Cl2 as the eluent to afford the pure product (21 mg, 8%) in the form of a colorless solid.Colorless solid; mp 124–126 °C; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.30–7.15 (m, 5H), 7.04 (s, 2H), 4.39 (ddd(t), J = 5.0 Hz, 1H), 4.27–4.19 (m, 1H), 4.00 (d, J = 13.6 Hz, 2H), 3.95 (d, J = 13.6 Hz, 2H), 3.15–2.95 (m, 3H), 2.80–2.65 (m, 1H), 2.59 (s, 12H), 1.35 (s, 9H); 13C NMR (CD3OD, 150 MHz) δ/ppm: 177.2 (s, 1C), 173.3 (s, 1C), 158.6 (s, 1C), 157.5 (s, 1C), 138.8 (s, 1C), 133.3 (d, 1C), 130.3 (d, 2C), 129.8 (s, 1C), 129.4 (d, 2C), 127.7 (d, 2C), 120.2 (s, 2C), 80.4 (s, 1C), 60.4 (t, 2C), 57.7 (d, 1C), 57.4 (d, 1C), 43.8 (q, 4C), 39.3 (t, 1C), 38.2 (t, 1C), 28.7 (q, 3C); HRMS (MALDI-TOF) m/z [M + H]+ calcd for C29H42N4O6 543.3183, found 543.3177.
BOC-Phe-Tyr[CH2N(CH3)2]2-OBn (3B)
Prepared from BOC-Phe-Tyr-OBn (500 mg, 1.0 mmol), dimethylamine (40% aq, 2 mL, 3.0 mmol), and formalin (37% aq, 1 mL, 2.5 mmol), according to the procedure for 3A. The reaction after purification afforded the product (68 mg, 11%) in the form of colorless oil.Colorless oil; 1H NMR (CD3OD, 600 MHz) δ/ppm: 7.36–7.27 (m, 5H), 7.25–7.21 (m, 2H), 7.20–7.15 (m, 3H), 6.87 (s, 2H), 5.10 (s, 2H), 4.68 (ddd(t), J = 7.0 Hz, 1H), 4.31–4.24 (m, 1H), 3.55 (d, J = 13.0 Hz, 2H), 3.51 (d, J = 13.0 Hz, 2H), 3.06–2.97 (m, 2H), 2.93 (dd, J = 7.5, 13.5 Hz, 1H), 2.73–2.67 (m, 1H), 2.26 (s, 12H), 1.34 (s, 9H); 13C NMR (CD3OD, 150 MHz) δ/ppm: 174.1 (s, 1C), 174.0 (s, 1C), 157.2 (s, 1C), 157.1 (s, 1C), 138.5 (s, 1C), 136.9 (s, 1C), 131.6 (d, 2C), 130.4 (d, 2C), 129.6 (d, 1C), 129.4 (d, 2C), 128.2 (d, 1C), 127.9 (d, 1C), 127.8 (s, 1C), 127.7 (d, 2C), 123.4 (s, 2C), 80.6 (s, 1C), 67.9 (t, 1C), 60.7 (t, 2C), 57.2 (d, 1C), 55.2 (d, 1C), 44.7 (q, 4C), 39.4 (t, 1C), 37.7 (t, 1C), 28.7 (q, 3C); HRMS (MALDI-TOF) m/z [M + H]+ calcd for C36H48N4O6 633.3652, found 633.3623.
BOC-Asp(tBu)-Ala-1-OH (4)
To a solution of the activated Boc-Asp(tBu)-Ala-OSu (62.7 mg, 137.0 μmol) in CH3CN (500 μL), a solution of NaHCO3 (34.5 mg, 3.0 mmol, 10.0 eq.) and 1·HCl (37.7 mg, 137.0 μmol, 1.0 eq.) in CH3CN–H2O (1:2, 1.5 mL) was added. The reaction mixture was stirred for 48 h at rt, before it was diluted with H2O (2 mL) and the pH was adjusted to 4.0 with 1 M HCl. The aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic phases were washed with 1 M HCl (2 mL) and brine (10 mL), dried over Na2SO4 and filtered. The solvent was removed under reduced pressure to afford the desired tripeptide 4 (54.5 mg 67%).
1H NMR (CD3CN, 500 MHz) δ/ppm: 7.23–7.22 (m, 1NH), 7.16 (s, 1H), 7.11 (d, J = 6.4 Hz, 1H), 7.02–7.00 (m, 1NH), 6.90 (d, J = 8.2 Hz, 1H), 5.90–5.89 (m, 1NH), 4.54 (q, J = 6.7 Hz, 1H), 4.34–4.33 (m, 1H), 4.27 (t, J = 7.0 Hz, 1H), 4.21 (s, 2H), 3.07 (dd, J = 5.3, 14.0 Hz, 1H), 2.95 (dd, J = 6.9, 14.0 Hz, 1H), 2.77 (s, 6H), 2.68 (dd, J = 5.4, 16.3 Hz, 1H), 2.54 (dd, J = 7.7, 16.3 Hz, 1H), 1.41 (s, 18H), 1.24 (d, J = 7.0 Hz, 3H); 13C NMR (CD3CN, 126 MHz) δ/ppm: 173.0 (s, 1C), 172.9 (s, 1C), 172.3 (s, 1C), 171.1 (s, 1C), 155.9 (s, 1C), 134.2 (s, 1C), 133.5 (s, 1C), 129.4 (s, 1C), 129.3 (s, 1C), 117.4 (s, 1C), 116.8 (s, 1C), 82.0 (s, 1C), 80.6 (s, 1C), 57.3 (s, 1C), 54.6 (s, 1C), 52.4 (s, 1C), 49.9 (s, 1C), 43.4 (s, 2C), 38.2 (s, 1C), 36.6 (s, 1C), 28.5 (s, 3C), 28.2 (s, 3C), 18.1 (s, 1C); ESI-MS [M + H]+ calcd for C28H45N4O9 581.31811, found 581.31805.
General SPPS procedure
All peptides were synthesized using 2-CT resin via standard Fmoc solid phase peptide synthesis with TBTU and DIPEA as coupling reagents. Manual coupling of canonical amino acids was performed for 40 min per coupling, with 2.5 equivalents of Fmoc-amino acid and 4.0 equiv. of DIPEA and TBTU. Tripeptide coupling to the resin was performed for 50 min, with 1.0 equiv. of the tripeptide and 3.0 equiv. of DIPEA and TBTU. Deprotection of the Fmoc-group was performed with a 20% piperidine solution in DMF for 2 × 15 min. The peptides were deprotected and cleaved from the resin under acidic conditions for 2 h [84%TFA/4% H2O/4% phenol/4% triisopropylsilane/4% 2,2′-(ethylenedioxy)diethanethiol]. The peptides were then precipitated in cold Et2O, the supernatant was then decanted and the precipitate was dried under reduced pressure. The purification was achieved using reversed phase HPLC on a 1260 Infinity system (Agilent Technologies) using a C18 preparative column (21.2 × 250 mm, 10 μm, Agilent Technologies) with gradients of System B (CH3CN, 0.1% TFA) in System A (H2O, 0.1% TFA). The peptides were characterized by electrospray ionization (ESI) mass spectrometry in positive ion mode on an LTQ Orbitrap XL mass spectrometer. A Grace Grom-Sil-120-ODS-4-HE (length 50 mm, ID 2 mm, 3 μm) column was used employing a linear gradient of 20–100% over 10 minutes at 0.3 mL min−1. The solvent system used was A (0.1% formic acid in H2O) and B (0.1% formic acid in CH3CN).TC10b_3Y1
The peptide was prepared according to the general procedure from 4 and 17-peptide intermediate bound to the resin. The coupling conditions were: TBTU, DIPEA, DMF, 50 min. The cleavage from the resin was achieved under acidic conditions.ESI-MS: (M + H)+: 2144.06005.
m/z = 1072.53381 (M + 2H)2+, calcd for (M + 2H)2+: 1072.53366.
m/z = 715.35858 (M + 3H)3+, calcd for (M + 3H)3+: 715.35820.
m/z = 536.77075 (M + 4H)4+, calcd for (M + 4H)4+: 536.77047.
Irradiation experiments, general
A quartz vessel was filled with a solution of the tyrosine derivative or peptide (0.1 mmol) in CH3OH (50 or 100 mL). The solution was purged with Ar (30 min) and irradiated using a Rayonet reactor at 300 nm with 11 lamps over 30–90 min (1 lamp, 8 W). During irradiation, the solution was continuously purged with Ar and cooled with a finger condenser. The composition of the irradiated solution was analyzed by HPLC. After the irradiation, the solvent was removed on a rotary evaporator and the residue was purified by chromatography.Irradiation of 1B
Irradiation of 1B (54 mg, 0.12 mmol) in CH3OH (100 mL) for 40 min after purification by TLC using 10% CH3OH in CH2Cl2 as the eluent afforded the pure product 6 (14 mg, 28%).Colorless oil; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.38–7.26 (m, 6H), 6.86 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (s, 1H), 5.17 (d, J = 12.6 Hz, 1H), 5.08 (d, J = 12.6 Hz, 1H), 4.96 (d, J = 7.6 Hz, 1H), 4.60–4.45 (m, 3H), 3.40 (s, 3H), 3.05–2.89 (m, 2H), 1.41 (s, 9H); 13C NMR (CD3OD, 150 MHz) δ/ppm: 171.8 (s, 1C), 155.5 (s, 1C), 155.1 (s, 1C), 135.3 (s, 1C), 130.3 (d, 1C), 128.9 (d, 2C), 128.6 (d, 1C), 128.5 (d, 2C), 127.9 (s, 1C), 127.0 (s, 1C), 122.0 (d, 1C), 116.6 (d, 1C), 79.9 (s, 1C), 74.0 (t, 1C), 67.0 (t, 1C), 58.2 (q, 1C), 54.6 (d, 1C), 37.4 (t, 1C), 28.7 (q, 3C); HRMS (MALDI-TOF) m/z [M]+ calcd for C23H29NO6 415.2073, found 415.2094.
Irradiation of 2B
Irradiation of 2B (62 mg, 0.11 mmol) in CH3OH (100 mL) for 30 min after purification by TLC using 10% CH3OH in CH2Cl2 as the eluent afforded the pure product 7 (13 mg, 21%).Colorless oil; 1H NMR (CD3OD, 600 MHz) δ/ppm: 7.38–7.33 (m, 3H), 7.30–7.24 (m, 5H), 7.24–7.18 (m, 1H), 7.17 (d, J = 7.0 Hz, 2H), 6.70–6.65 (m, 2H), 6.59 (s, 1H), 6.28 (br. s), 5.10 (d, J = 12.0 Hz, 1H), 5.07 (d, J = 12.0 Hz, 1H), 4.97 (br. s), 4.77–4.73 (m, 1H), 4.49 (d, J = 12.5 Hz, 1H), 4.46 (d, J = 12.5 Hz, 1H), 4.32 (br. s), 3.38 (s, 3H), 3.02 (d, J = 6 Hz, 2H), 2.94 (d, J = 6 Hz, 2H), 1.34 (s, 9H); 13C NMR (CD3OD, 150 MHz) δ/ppm: 170.8 (s), 170.7 (s), 155.2 (s), 136.5 (s), 135.0 (s), 130.2 (d), 129.4 (d), 129.0 (d), 128.7 (d), 128.6 (s), 128.54 (d), 128.51 (d), 126.9 (d), 126.6 (s), 122.1 (s), 116.5 (d), 80.2 (s), 73.8 (t), 67.1 (t), 58.2 (q), 55.7 (d), 53.4 (d), 38.3 (t), 37.0 (t), 28.2 (q), one singlet was not seen; HRMS (MALDI-TOF) m/z [M + K]+ calcd for C32H38N2O7 601.2316, found 601.2328.
Irradiation of 3B
Irradiation of 3B (28 mg, 0.04 mmol) in CH3OH (50 mL) for 30 min after purification by TLC using 10% CH3OH in CH2Cl2 as the eluent afforded the pure product 9 (5 mg, 21%).Colorless oil; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.38–7.33 (m, 5H), 7.31–7.27 (m, 2H), 7.25–7.21 (m, 2H), 7.21–7.15 (m, 5H), 6.71 (s, 2H), 5.11–5.08 (s, 2H), 4.81–4.73 (m, 1H), 4.46 (s, 4H), 4.39–4.28 (m, 1H), 3.42–3.36 m, 6H), 3.13–2.88 (m, 4H), 1.37 (s, 9H); 13C NMR (CD3OD, 150 MHz) δ/ppm: 170.8 (s), 153.1 (s), 136.8 (s), 135.1 (s), 129.4 (d), 129.1 (d), 128.64 (d), 128.60 (d), 128.5 (d), 128.4 (d), 126.9 (d), 126.4 (s), 123.6 (s), 80.5 (s), 71.6 (t), 67.1 (t), 58.3 (q), 53.5 (d), 53.4 (d), 38.2 (t), 37.1 (t), 28.7 (q), two singlets were not seen; HRMS (MALDI-TOF) m/z [M + K]+ calcd for C34H42N2O8 645.2578, found 645.2570.
Irradiation of 3A
Irradiation of 3A (59 mg, 0.11 mmol) in CH3OH (100 mL) for 75 min after purification by TLC using 10% CH3OH in CH2Cl2 as the eluent afforded the pure product 10 (5 mg, 9%).Colorless oil; 1H NMR (CD3OD, 300 MHz) δ/ppm: 7.30–7.15 (m, 5H), 7.04 (s, 2H), 6.94–6.84 (m, 1H), 4.68–4.57 (m, 1H), 4.54–4.50 (m, 4H), 4.31–4.24 (m, 1H), 3.38 (s, 6H), 3.19–2.98 (m, 2H), 2.76–2.63 (m, 2H), 1.33 (s, 9H); 13C NMR (CD3OD, 150 MHz) δ/ppm: 174.0 (s, 1C), 157.5 (s, 1C), 154.0 (s, 1C), 138.7 (s, 1C), 131.2 (d, 1C), 130.3 (d, 2C), 129.4 (d, 2C), 129.0 (s, 1C), 127.6 (d, 1C), 125.4 (s, 2C), 80.7 (s, 1C), 72.0 (t, 2C), 58.5 (q, 2C), 57.5 (d, 1C), 55.3 (d, 1C), 39.2 (t, 1C), 37.6 (t, 1C), 28.6 (q, 3C), one singlet was not seen; HRMS (MALDI-TOF) m/z [M + K]+ calcd for C27H36N2O8 555.2109, found 555.2083.
Quantum yield of methanolysis
Quantum yields for photomethanolysis reactions were determined by using two actinometers simultaneously: KI/KIO3 (Φ254 = 0.74),47,51 and photomethanolysis of 2-hydroxymethylphenol (Φ254 = 0.23),28 as recently described by us.71 The measurement was performed in eight quartz cells of the same dimensions that were irradiated from the front side only. Solutions of tyrosines or peptide derivatives in CH3OH, and actinometers were freshly prepared and their concentrations were adjusted to have absorbances of 0.4–0.8 at 254 nm. After adjustment of the concentrations and measurement of the corresponding UV-vis spectra, the solutions were purged with a stream of N2 (20 min), and then sealed with a cap. The cells were placed in a holder which ensured an equal distance of all samples from the lamp and were irradiated at the same time in the reactor with 1 lamp at 254 nm for 1–15 min. Before and after the irradiation, the samples were taken from the cells by the use of a syringe and analyzed by HPLC to determine photochemical conversions. The conversion did not exceed 30% to avoid a change of the absorbance, or filtering of the light by the product. From the conversion of actinometers irradiance was calculated. Similar values were obtained for both actinometers. The mean value of two measurements was reported. All equations for the calculation of quantum yields are given in the ESI.†Steady-state and time-resolved fluorescence measurements
The steady-state measurements were performed on a PTI QM40 fluorometer at 20 °C. Excitation slits were set to a bandpass of 3 nm, and emission slits to a bandpass of 2 nm. The spectra were corrected for the fluctuations in lamp intensity and transmission of optics. The samples were dissolved in CH3CN, or CH3CN–H2O (1:1) and the concentrations were adjusted to absorbances less than 0.1 at the excitation wavelengths of 250, 260 or 270 nm. Fluorescence quantum yields were determined by comparison of the integral of the emission bands with the one of anisole in cyclohexane (Φf = 0.29)47 upon excitation at 260 nm. The quantum yields were calculated according to eqn (S1) in the ESI.† Fluorescence decays, collected over 1023 time channels, were obtained on a single photon counter using a light emitting diode for excitation at 260 nm. The instrument response functions, using LUDOX as the scatterer, were recorded at the same wavelengths as the excitation wavelength and had a half width of ≈0.2 ns. Emission decays for samples in CH3CN solutions were recorded at 310 nm. The counts in the peak channel were 1 × 103. The time increment per channel was 0.020 ns. The obtained histograms were fit as sums of exponentials using Gaussian-weighted non-linear least-squares fitting based on Marquardt–Levenberg minimization implemented in the software package from the instrument. The fitting parameters (decay times and pre-exponential factors) were determined by minimizing the reduced chi-square χ2 and graphical methods were used to judge the quality of the fit that included plots of the weighted residuals vs. channel number.
Laser flash photolysis (LFP)
All LFP studies were performed on a system at the University of Victoria72 using as an excitation source a pulsed Nd:YAG laser at 266 nm (<20 mJ per pulse), with a pulse width of 10 ns. Static cells (7 mm × 7 mm) were used and the solutions were purged with nitrogen or oxygen for 30 min prior to perform the measurements. Absorbances at 266 nm were ∼0.3–0.5. For the collection of decays on long time scales, a modification of the setup was used, wherein the probing light beam from the Xe-lamp was not pulsed.73Acknowledgements
These materials are based on work financed by the ESF project HR.3.2.01-0254 and the Croatian Science Foundation (HRZZ grant IP-2014-09-6312). The authors thank the Department of Chemistry at the University of Victoria, Canada and Prof. P. Wan and C. Bohne for the support and the use of LFP facilities.Notes and references
- Quinone Methides, ed. S. E. Rokita, Wiley, Hoboken, 2009 Search PubMed.
- M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury, RSC Adv., 2014, 4, 55924–55959 RSC.
- R. W. Van De Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367–5405 CrossRef CAS.
- T. P. Pathak and M. S. Sigman, J. Org. Chem., 2011, 76, 9210–9215 CrossRef CAS PubMed.
- M. Frecero, Mini-Rev. Org. Chem., 2004, 1, 403–415 CrossRef.
- P. Wang, Y. Song, L. Zhang, H. He and X. Zhou, Curr. Med. Chem., 2005, 12, 2893–2913 CrossRef CAS PubMed.
- N. Basarić, K. Mlinarić-Majerski and M. Kralj, Curr. Org. Chem., 2014, 18, 3–18 CrossRef.
- S. E. Rokita, J. Yang, P. Pande and W. A. Greenberg, J. Org. Chem., 1997, 62, 3010–3012 CrossRef CAS PubMed.
- W. F. Veldhuyzen, A. J. Shallop, R. A. Jones and S. E. Rokita, J. Am. Chem. Soc., 2001, 123, 11126–11132 CrossRef CAS PubMed.
- E. E. Weinert, K. N. Frankenfield and S. E. Rokita, Chem. Res. Toxicol., 2005, 18, 1364–1370 CrossRef CAS PubMed.
- M. Chatterjee and S. E. Rokita, J. Am. Chem. Soc., 1994, 116, 1690–1697 CrossRef CAS.
- Q. Zeng and S. E. Rokita, J. Org. Chem., 1996, 61, 9080–9081 CrossRef CAS.
- P. Pande, J. Shearer, J. Yang, W. A. Greenberg and S. E. Rokita, J. Am. Chem. Soc., 1999, 121, 6773–6779 CrossRef CAS.
- W. F. Veldhuyzen, P. Pande and S. E. Rokita, J. Am. Chem. Soc., 2003, 125, 14005–14013 CrossRef CAS PubMed.
- S. N. Richter, S. Maggi, S. Colloredo Mels, M. Palumbo and M. Freccero, J. Am. Chem. Soc., 2004, 126, 13973–13979 CrossRef CAS PubMed.
- H. Wang, Curr. Org. Chem., 2014, 18, 44–60 CrossRef CAS.
- H. Wang, M. S. Wahi and S. E. Rokita, Angew. Chem., Int. Ed., 2008, 47, 1291–1293 CrossRef CAS PubMed.
- H. Wang and S. E. Rokita, Angew. Chem., Int. Ed., 2010, 49, 5957–5960 CrossRef CAS PubMed.
- C. S. Rossiter, E. Modica, D. Kumar and S. E. Rokita, Chem. Commun., 2011, 47, 1476–1478 RSC.
- P. G. McCracken, J. L. Bolton and G. R. J. Thatcher, J. Org. Chem., 1997, 62, 1820–1825 CrossRef CAS.
- E. Modica, R. Zanaletti, M. Freccero and M. Mella, J. Org. Chem., 2001, 66, 41–52 CrossRef CAS PubMed.
- S. Arumugam, J. Guo, N. E. Mbua, F. Friscourt, N. Lin, E. Nekongo, G.-J. Boons and V. V. Popik, Chem. Sci., 2014, 5, 1591–1598 RSC.
- M. Kralj, L. Uzelac, Y.-H. Wang, P. Wan, M. Tireli, K. Mlinarić-Majerski, I. Piantanida and N. Basarić, Photochem. Photobiol. Sci., 2015, 14, 1082–1092 CAS.
- G. G.-H. Qiao, K. Lenghaus, D. H. Solomon, A. Reisinger, I. Bytheway and C. Wentrup, J. Org. Chem., 1998, 63, 9806–9811 CrossRef CAS.
- E. Dorrestijn, M. Kranenburg, M. V. Ciriano and P. Mulder, J. Org. Chem., 1999, 64, 3012–3018 CrossRef CAS PubMed.
- D. A. Bolon, J. Org. Chem., 1970, 35, 3666–3670 CrossRef CAS.
- C. Percivalle, F. Doria and M. Freccero, Curr. Org. Chem., 2014, 18, 19–43 CrossRef CAS.
- L. Diao, C. Yang and P. Wan, J. Am. Chem. Soc., 1995, 117, 5369–5370 CrossRef CAS.
- B. Baker, L. Diao and P. Wan, J. Photochem. Photobiol., A, 1997, 104, 91–96 CrossRef.
- K. Nakatani, N. Higashida and I. Saito, Tetrahedron Lett., 1997, 38, 5005–5008 CrossRef CAS.
- S. Colloredo-Mels, F. Doria, D. Verga and M. Freccero, J. Org. Chem., 2006, 71, 3889–3895 CrossRef CAS PubMed.
- M. Di Antonio, F. Doria, M. Mella, D. Merli, A. Profumo and M. Freccero, J. Org. Chem., 2007, 72, 8354–8360 CrossRef CAS PubMed.
- D. Verga, M. Nadai, F. Doria, C. Percivalle, M. Di Antonio, M. Palumbo, S. N. Richter and M. Freccero, J. Am. Chem. Soc., 2010, 132, 14625–14637 CrossRef CAS PubMed.
- F. Doria, S. N. Richter, M. Nadai, S. Colloredo-Mels, M. Mella, M. Palumbo and M. Freccero, J. Med. Chem., 2007, 50, 6570–6579 CrossRef CAS PubMed.
- M. Nadai, F. Doria, M. Di Antonio, G. Sattin, L. Germani, C. Percivalle, M. Palumbo, S. N. Richter and M. Freccero, Biochimie, 2011, 93, 1328–1340 CrossRef CAS PubMed.
- F. Doria, M. Nadai, M. Folini, M. Di Antonio, L. Germani, C. Percivalle, C. Sissi, N. Zaffaroni, S. Alcaro, A. Artese, S. N. Richter and M. Freccero, Org. Biomol. Chem., 2012, 10, 2798–2806 CAS.
- F. Doria, M. Nadai, M. Folini, M. Scalabrin, L. Germani, G. Sattin, M. Mella, M. Palumbo, N. Zaffaroni, D. Fabris, M. Freccero and S. N. Richter, Chem. – Eur. J., 2013, 19, 78–81 CrossRef CAS PubMed.
- Đ. Škalamera, C. Bohne, S. Landgraf and N. Basarić, J. Org. Chem., 2015, 80, 10817–10828 CrossRef PubMed.
- F. Doria, A. Lena, R. Bargiggia and M. Freccero, J. Org. Chem., 2016, 81, 3665–3673 CrossRef CAS PubMed.
- B. Barua, J. C. Lin, W. D. Williams, P. Kummler, J. W. Neidigh and N. H. Andersen, Protein Eng., Des. Sel., 2008, 21, 171–185 CrossRef CAS PubMed.
- J. W. Neidigh, R. M. Fesinmeyer and N. H. Andersen, Nat. Struct. Biol., 2002, 9, 425–430 CrossRef CAS PubMed.
- L. Qiu, S. A. Pabit, A. E. Roitberg and S. J. Hagen, J. Am. Chem. Soc., 2002, 124, 12952–12953 CrossRef CAS PubMed.
- B. P. Noichl, P. M. Durkin and N. Budiša, Biopolymers, 2015, 104, 585–600 CrossRef CAS PubMed.
- Synthesis of Peptides and Peptidomimetics, ed. A. Felix, L. Moroder and C. Toniolo, Thieme, Stuttgart, 2004, vol. 22 Search PubMed.
- B. Siddique and J. Duhamel, Langmuir, 2011, 27, 6639–6650 CrossRef CAS PubMed.
- V. Dourtoglov and B. Gross, Synthesis, 1984, 572–575 CrossRef.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, in Handbook of Photochemistry, CRC Taylor and Francis, Boca Raton, 2006 Search PubMed.
- J. F. Ireland and P. A. H. Wyatt, Adv. Phys. Org. Chem., 1976, 12, 131–221 CrossRef CAS.
- L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol., A, 1993, 75, 1–20 CrossRef CAS.
- E. E. Weinert, R. Dondi, S. Colloredo-Melz, K. N. Frankenfield, C. H. Mitchell, M. Freccero and S. E. Rokita, J. Am. Chem. Soc., 2006, 128, 11940–11947 CrossRef CAS PubMed.
- S. Goldstein and J. Rabani, J. Photochem. Photobiol., 2008, 193, 50–55 CrossRef CAS.
- J. Lee, G. W. Robinson, S. P. Webb, L. A. Philips and J. H. Clark, J. Am. Chem. Soc., 1986, 108, 6538–6542 CrossRef CAS.
- G. W. Robinson, J. Phys. Chem., 1991, 95, 10386–10391 CrossRef CAS.
- L. M. Tolbert and J. E. Haubrich, J. Am. Chem. Soc., 1994, 116, 10593–10600 CrossRef CAS.
- K. M. Solntsev, D. Huppert, N. Agmon and L. M. Tolbert, J. Phys. Chem. A, 2000, 104, 4658–4669 CrossRef CAS.
- Y. Chiang, A. J. Kresge and Y. Zhu, J. Am. Chem. Soc., 2000, 122, 9854–9855 CrossRef CAS.
- Y. Chiang, A. J. Kresge and Y. Zhu, J. Am. Chem. Soc., 2001, 123, 8089–8094 CrossRef CAS PubMed.
- Y. Chiang, A. J. Kresge and Y. Zhu, J. Am. Chem. Soc., 2002, 124, 717–722 CrossRef CAS PubMed.
- M. M. Toteva and J. P. Richard, Adv. Phys. Org. Chem., 2011, 45, 39–91 CrossRef CAS PubMed.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 11892–11899 CrossRef CAS PubMed.
- N. Basarić, I. Žabčić, K. Mlinarić-Majerski and P. Wan, J. Org. Chem., 2010, 75, 102–116 CrossRef PubMed.
- D. Brousmiche, M. Xu, M. Lukeman and P. Wan, J. Am. Chem. Soc., 2003, 125, 12961–12970 CrossRef CAS PubMed.
- M. Xu, M. Lukeman and P. Wan, Photochem. Photobiol., 2006, 82, 50–56 CrossRef CAS PubMed.
- N. Basarić, N. Cindro, D. Bobinac, K. Mlinarić-Majerski, L. Uzelac, M. Kralj and P. Wan, Photochem. Photobiol. Sci., 2011, 10, 1910–1925 Search PubMed.
- N. Basarić, N. Cindro, D. Bobinac, K. Mlinarić-Majerski, L. Uzelac, M. Kralj and P. Wan, Photochem. Photobiol. Sci., 2012, 11, 381–396 Search PubMed.
- J. Veljković, L. Uzelac, K. Molčanov, K. Mlinarić-Majerski, M. Kralj, P. Wan and N. Basarić, J. Org. Chem., 2012, 77, 4596–4610 CrossRef PubMed.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2011, 133, 5573–5579 CrossRef CAS PubMed.
- O. Keller, W. E. Keller, G. van Look and G. Wersin, Org. Synth., 1990, 7, 70–78 Search PubMed.
- J. R. Luly, N. Yi, J. Soderquist, H. Stein, J. Cohen, T. J. Perun and J. J. Plattner, J. Med. Chem., 1987, 30, 1609–1616 CrossRef CAS PubMed.
- S. Liaqat, S. S. Panda, A. Rauf, A. O. Al-Youbi and A. Katritzky, Synthesis, 2014, 67–72 Search PubMed.
- Đ. Škalamera, K. Mlinarić-Majerski, I. Martin-Kleiner, M. Kralj, P. Wan and N. Basarić, J. Org. Chem., 2014, 79, 4390–4397 CrossRef PubMed.
- Y. Liao and C. Bohne, J. Phys. Chem., 1996, 100, 734–743 CrossRef CAS.
- R. H. Mitchell, C. Bohne, Y. Wang, S. Bandyopadhyay and C. B. Wozniak, J. Org. Chem., 2006, 71, 327–336 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures for the preparation of known precursors, fluorescence spectra of 2 and 3, LFP data, and 1H and 13C NMR spectra. See DOI: 10.1039/c6ob02191c |
| This journal is © The Royal Society of Chemistry 2016 |