
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flexible synthesis of cationic peptide–porphyrin derivatives for light-triggered drug delivery and photodynamic therapy†
R.
Dondi‡
a,
E.
Yaghini‡
b,
K. M.
Tewari
 a,
L.
Wang
ac,
F.
Giuntini§
a,
M.
Loizidou
b,
A. J.
MacRobert
b and
I. M.
Eggleston
*a
a,
L.
Wang
ac,
F.
Giuntini§
a,
M.
Loizidou
b,
A. J.
MacRobert
b and
I. M.
Eggleston
*a
aDepartment of Pharmacy and Pharmacology, University of Bath, Bath BA2 7AY, UK. E-mail: ie203@bath.ac.uk
bUCL Division of Surgery and Interventional Science, University College London, Royal Free Campus, Rowland Hill Street, London NW3 2PF, UK
cSchool of Pharmaceutical Sciences, Shandong University, Jinan, China
First published on 14th November 2016
Abstract
Efficient syntheses of cell-penetrating peptide–porphyrin conjugates are described using a variety of bioconjugation chemistries. This provides a flexible means to convert essentially hydrophobic tetrapyrolle photosensitisers into amphiphilic derivatives which are well-suited for use in light-triggered drug delivery by photochemical internalisation (PCI) and targeted photodynamic therapy (PDT).
Introduction
Photodynamic therapy (PDT) is a light-based therapeutic technique that shows great promise for the selective treatment of a variety of cancers and non-malignant conditions.1 The principle of PDT is that highly localised destruction of tumours, diseased tissue or pathogenic organisms is achieved with light following the administration of a light-activated photosensitising drug or photosensitiser. This generates various cytotoxic reactive oxygen species such as singlet oxygen that can interact with and damage cellular components, leading to cell death. Many photosensitisers have been developed and have received approval or clinical trials for use in the treatment of cancer,2 age-related macular degeneration,3 pre-cancerous conditions such as Barret's oesphagus,4 and oral sterilisation in dental procedures.5 Among these, porphyrins and other tetrapyrrole-related compounds remain the broadest class of compound under study for PDT and other light-based therapies.6 The effectiveness of PDT in a particular application depends upon the wavelength at which the photosensitiser may be activated, and this has led to the development of many modified tetrapyrroles that show enhanced absorption at the red end of the spectrum where absorption by tissue is relatively weak, thus allowing a photodynamic effect at a greater depth in vivo.6,7 Another important consideration in the development of novel photosensitisers is their efficient delivery to the target site, in particular achieving selective delivery of photosensitisers to tumours compared to normal tissues. In this context, in recent years there has been considerable interest in the development of peptide and protein-targeted photosensitisers as a means of improving the pharmacokinetic properties, solubility and tissue specificity of various otherwise hydrophobic derivatives. Conjugation of photosensitisers to antibodies and a range of synthetic peptides has been explored and has been found to provide significant enhancements in both the efficiency and selectivity of cellular uptake of photosensitisers for PDT in a range of cancer models.8 A key innovation in the development of such targeted photosensitisers has been the application of biorthogonal ligation techniques9 which facilitate the efficient attachment of a variety of photosensitisers to unprotected, multifunctional peptides in solution.Cell-penetrating peptides (CPPs) are an intensely researched class of biocompatible carriers which have great potential for targeted drug delivery.10–12 Such molecules, of either natural or synthetic origin typically consist of 8–30 amino acid residues and possess the ability to translocate across biological membranes and transport diverse molecular cargoes, either covalently or non-covalently attached, which would otherwise be poorly internalised. The conjugation of tetrapyrrole-based photosensitisers and other derivatives to CPPs has indeed been shown to provide an attractive means for enhancing photosensitiser delivery for both PDT of cancer13–15 and in antimicrobial PDT applications.16,17 Importantly, CPP-conjugation also offers a means to control the sub-cellular localisation of a particular photosensitiser in eukaryotic systems to potentially maximise therapeutic effect.10,14
We recently demonstrated that the conjugation of a porphyrin derivative to a cationic CPP is an effective way of generating a novel water-soluble amphiphilic photosensitiser suitable for light-triggered drug delivery by photochemical internalisation (PCI).18 PCI is a novel technology for effecting the light-activated intracellular release of biologically active molecules at specific sites in living tissue, often where conventional methods of drug administration prove unsuccessful.19,20 The effectiveness of various macromolecular therapeutic agents can often be severely impaired by their limited ability to reach their intracellular targets due to sequestration in endosomes and lysosomes after uptake by endocytic mechanisms. PCI provides a highly effective means to selectively trigger endosomal escape and intracellular relocalisation of entrapped drugs, by applying the basic principle of the PDT approach. In PCI, a sub-lethal light dose is applied to a photosensitiser that localises in endo/lysosomal membranes, which is sufficient to cause partial rupture of these intracellular organelles (mediated by singlet oxygen). This allows the entrapped drugs to escape but does not compromise the viability of the cells themselves.20 Recent clinical studies have demonstrated both the feasibility and safety of PCI for the treatment of advanced and recurrent solid malignancies.21
A critical requirement in PCI is that the photosensitiser used must possess features that make it localise in the same intracellular vesicles (lysosomes, endosomes) as the administered drug, i.e. they must be lysosomotropic and therefore amphiphilic.20 Many well-known PDT photosensitisers are unsuitable for PCI as they partition non-selectively to other cellular organelles (e.g. mitochondria, Golgi apparatus, endoplasmic reticulum), however as shown in Fig. 1, attachment of an otherwise hydrophobic photosensitiser derivative to a cationic CPP may transform it into an amphiphilic compound that is both water-soluble and amenable to cellular uptake by endocytosis.18 The hydrophobic porphyrin macrocycle then has the potential to localise in the lipid bilayer of the endosomal membranes, for selective oxidative damage,22,23 with the hydrophilic peptide in the fluid phase.18 These properties are ideally suited for PCI (see above).
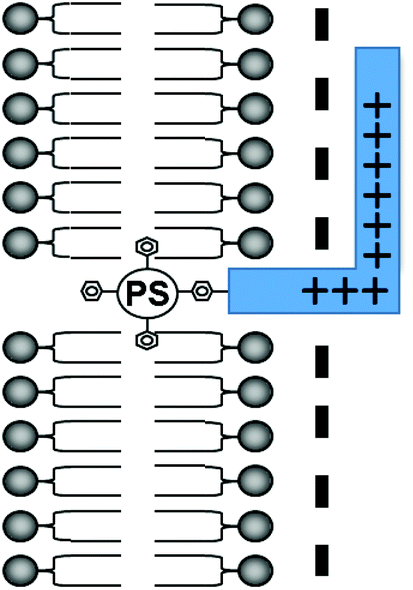 | ||
| Fig. 1 Amphiphilic CPP-targeted photosensitiser. The hydrophobic photosensitiser unit localises in the lipid bilayer of endosomal/lysosomal vesicles while the cationic peptide carrier resides in the fluid phase. | ||
The application of CPP-targeted photosensitisers for PCI and PDT depends on the availability of reliable synthetic protocols to derivatise typical cationic CPPs in a site-specific fashion24 with easily accessible functionalised photosensitisers of maximum clinical potential.25 The aim of this study was to investigate the potential of a number of bioconjugation reactions for the efficient modular preparation of CPP–porphyrin conjugates, as a general strategy to convert classical PDT agents into amphiphilic conjugates suitable for targeted PDT and PCI.
Results and discussion
Photosensitiser synthesis
The preparation of the ligatable photosensitisers used in this study is outlined in Scheme 1. As in previous studies by us, meso-tetrakistetraphenylporphyrin 1 provided the template for these compounds, and was transformed to the mono-amino derivative 2 by a modification of the method of Luguya et al.26 Treatment of 1 with sodium nitrite and trifluoroacetic acid under the reported conditions (3 min contact time) gave a mixture of mono- and dinitrated porphyrins that were reduced directly with sodium borohydride and palladium on charcoal. 2 was then easily separated from disubstituted products and unchanged 1 in reproducible yields of around 30% over the two steps. The porphyrin scaffold was then elaborated with several functionalities that would permit biorthogonal ligation with a suitably modified CPP component, via acylation with a suitable carboxylic acid component using either EDC/HOBt or PyBOP activation.27 Compounds 3–8 were obtained in satisfactory yields via either activation protocol, and in the cases of 3 and 8 were used directly with their complementary ligatable CPP partners (see below). The azido and alkynyl derivatives 5–7 were further converted into the corresponding zinc complexes by treatment with Zn(OAc)2 in DCM in excellent yields, ready for use in copper-catalysed ligation reactions.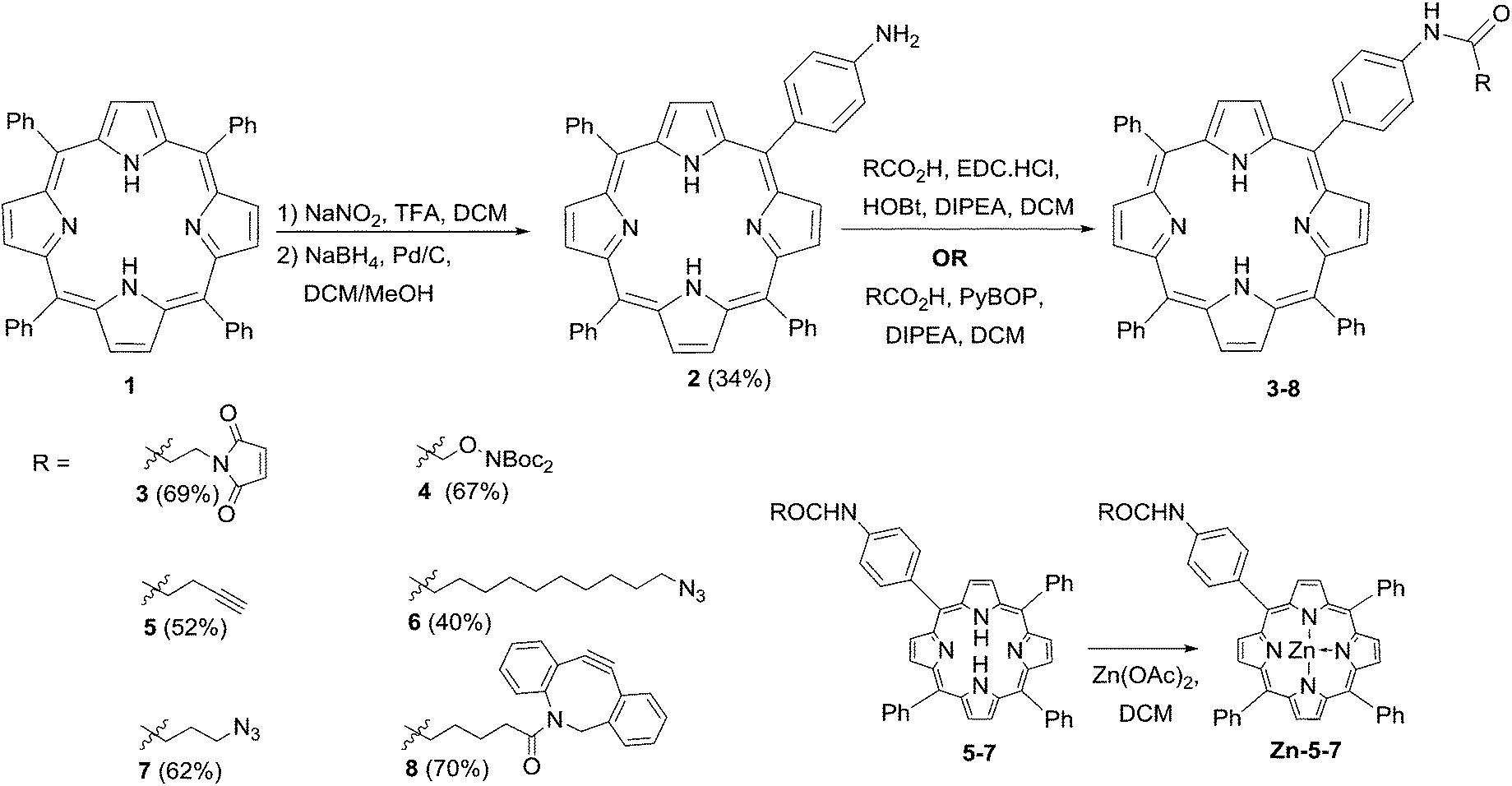 | ||
| Scheme 1 Synthesis of ligatable porphyrin derivatives. | ||
Peptide synthesis
All peptides were synthesised by 9-fluorenylmethoxycarbonyl (Fmoc) peptide synthesis on Rink amide resin, using PyBOP activation. Peptides 10–14 were based on the decapeptide CPP sequence derived from the transcriptional activator (Tat) protein from human immunodeficiency virus 1 (HIV-1), commonly known as HIV-1 Tat(48–57),28 which provided the template for N-terminally ligatable derivatives in this study. In each case, the final peptide was functionalised on-resin to introduce a complementary reactive group to permit one of the following ligation chemistries with the appropriate porphyrin derivative. Peptides 10–12 were obtained via straightforward acylation of the resin-bound peptide with either Fmoc-L-Cys-OH (for 10), azidoundecanoic acid (for 11), or pentynoic acid (for 12), using HATU activation.27 For peptide 13, an N-terminal azido function was introduced by direct diazo transfer upon the resin-bound Tat sequence using the Stick reagent (imidazole-1-sulfonyl azide hydrochloride),29 with completion of the reaction being assessed by the Kaiser test,30 as for the on-resin acylations to generate 10–12. In the case of the keto-functionalised derivative 14, acylation of the resin-bound peptide could only be satisfactorily achieved using the preformed succinimido ester of pyruvic acid.31 Other activation methods (DIC, PyBOP, HATU) failed to give desired product, presumably due to the tendency of keto acids such as pyruvate to undergo Claisen-type condensations under strong carboxyl activation conditions.32 The C-terminally ligatable peptides 15–18 were obtained directly by standard solid phase peptide synthesis. Cys-containing peptide 15, was originally reported by Santra et al.33 and is also based upon the HIV-1 Tat(48–57) sequence. It provided the template for our prototype CPP-targeted photosensitiser for PCI and applications in antimicrobial PDT.17,19 Azidopeptide 16 was derived from 15, by replacement of the terminal Cys residue with ε-azidolysine34 and insertion of a Gly residue at the C-terminus itself, to facilitate loading of the Rink amide resin. Azidopeptides 17 and 18 were obtained in an analogous fashion by respectively the replacement of one Lys residue in the sequence of pVEC, a CPP derived from murine vascular endothelial-cadherin protein, or a fragment of penetratin, a CPP derived from the Antennapedia homeoprotein.12 The final N-terminally functionalised CPPs 11–14 were obtained in yields of 35–58%, and the C-terminally functionalised derivatives 15–18 in yields of 59–67% after cleavage from the resin and side chain deprotection with TFA/TIS/H2O or TFA/TIS/H2O/EDT, for Cys-containing peptides 10 and 15 (Scheme 2).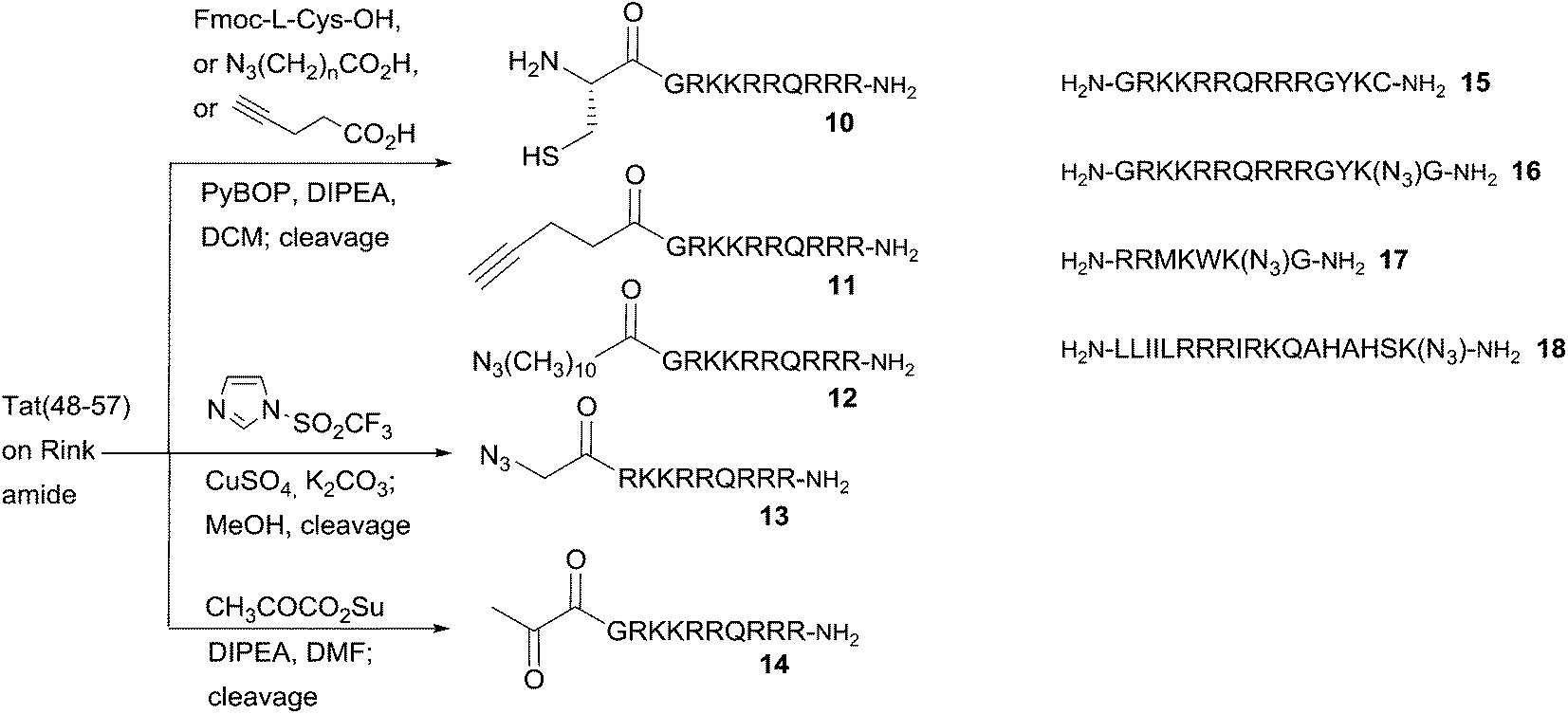 | ||
| Scheme 2 Preparation of N- and C-terminally ligatable CPP derivatives. | ||
Ligation reactions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17,37 in DMSO as shown in Scheme 3. This furnished the desired CPP conjugates 19 and 20 in excellent yields, with no significant difference in the efficiency of N-terminal or C-terminal attachment. This is in contrast to a report by Kitagishi et al.37 who observed that an analogous ligation to the N-terminus of an octaarginine derivative proceeded only in very low yield. Indeed, using an alternative polar ligation at the N-terminus of the Tat-CPP unit via keto peptide 14 also proved to be highly effective. In this case, the Boc-protected porphyrin 4 was first treated with TFA in DCM to generate aminoxyderivative 9, which isolated as the free base in 94% yield. The complementary fully deprotected peptide 14 was then treated with a four-fold excess of 9 in 0.1% TFA/DMSO to give the desired oxime conjugate in 75% yield following purification by semi-preparative HPLC. DMSO proved to be the solvent of choice for these ligations as a compromise between the otherwise mutually exclusive solubilities of the highly hydrophobic porphyrins and the unprotected peptides, since preliminary studies with water/PEG systems were found to give very sluggish reactions and variable yields. Notwithstanding this, it was typically possible to greatly simplify the isolation of 19–21 and similar conjugates by precipitation of the crude products or removal of the reaction solvent by isolation on a solid-phase extraction cartridge prior to semi-preparative HPLC.
17,37 in DMSO as shown in Scheme 3. This furnished the desired CPP conjugates 19 and 20 in excellent yields, with no significant difference in the efficiency of N-terminal or C-terminal attachment. This is in contrast to a report by Kitagishi et al.37 who observed that an analogous ligation to the N-terminus of an octaarginine derivative proceeded only in very low yield. Indeed, using an alternative polar ligation at the N-terminus of the Tat-CPP unit via keto peptide 14 also proved to be highly effective. In this case, the Boc-protected porphyrin 4 was first treated with TFA in DCM to generate aminoxyderivative 9, which isolated as the free base in 94% yield. The complementary fully deprotected peptide 14 was then treated with a four-fold excess of 9 in 0.1% TFA/DMSO to give the desired oxime conjugate in 75% yield following purification by semi-preparative HPLC. DMSO proved to be the solvent of choice for these ligations as a compromise between the otherwise mutually exclusive solubilities of the highly hydrophobic porphyrins and the unprotected peptides, since preliminary studies with water/PEG systems were found to give very sluggish reactions and variable yields. Notwithstanding this, it was typically possible to greatly simplify the isolation of 19–21 and similar conjugates by precipitation of the crude products or removal of the reaction solvent by isolation on a solid-phase extraction cartridge prior to semi-preparative HPLC.
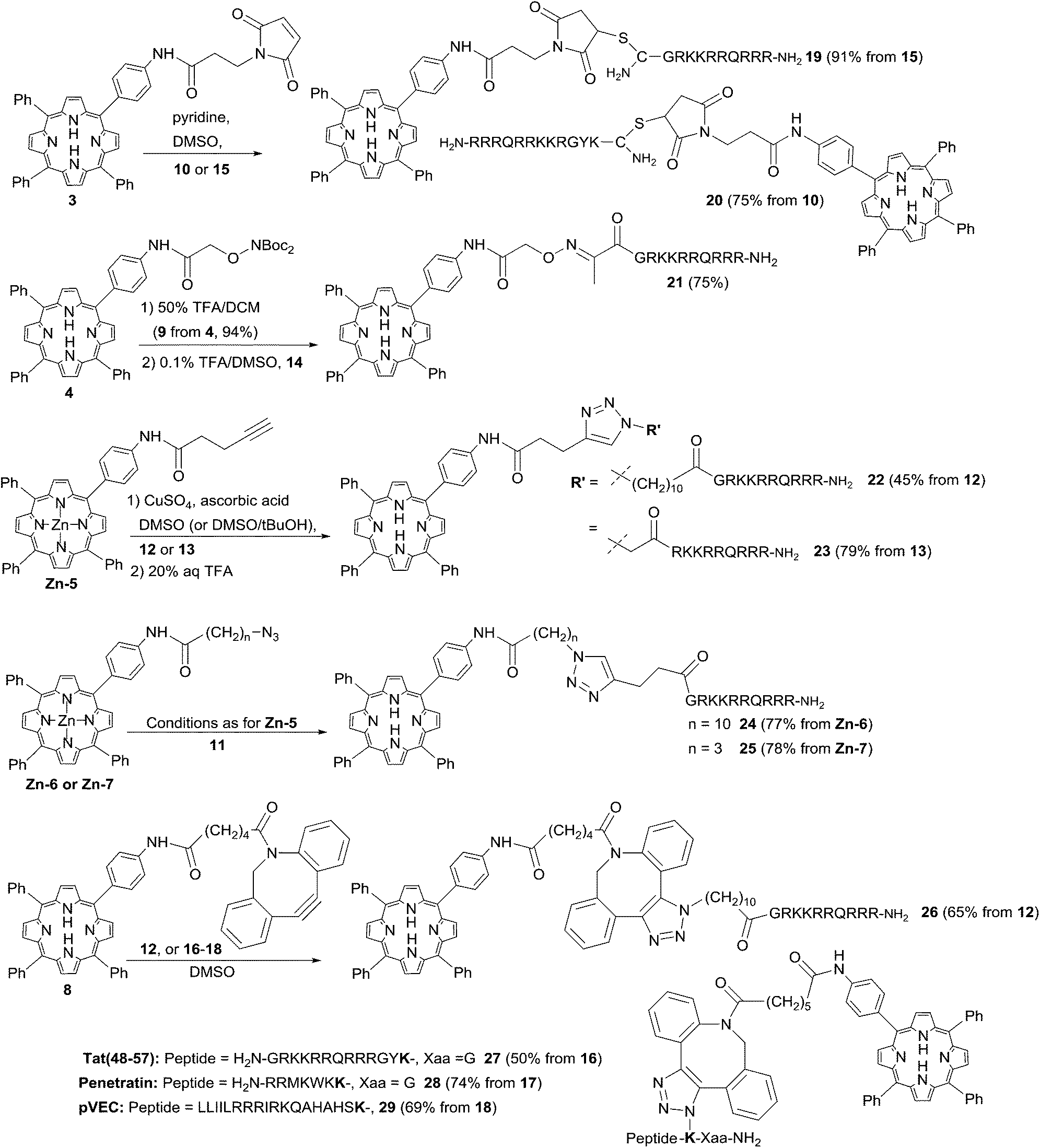 | ||
| Scheme 3 Bioconjugations of functionalised porphyrins to CPP derivatives. The azidolysine-derived residue in conjugates 27–29 is highlighted in bold. Conjugates 27 and 28 contain a spacer residue (Gly) between the azidolysine and the C-terminus. | ||
Recent advances in biorthogonal ligation chemistry44–46 have yielded a range of novel variants on the original triazole approach in which the use of a strained alkyne component removes the need for copper catalysis, and this has indeed recently been shown to be suitable for generating specific porphyrin–antibody conjugates.36 We chose to investigate SPAAC ligations for the effective synthesis of both N- and C-terminally linked CPP–porphyrin conjugates, and as described above, porphyrin derivative 8 bearing a strained dibenzocyclooctyne (DBCO) function could be readily prepared by acylation of 2 with the commercially available carboxylic acid derivative. Owing to the chemical reactivity of the DBCO unit, the preferred sense of combination was with the azido function in the peptide component as detailed above, either through N-terminal acylation with a suitable azidoalkanoic acid (Tat peptide 12), or by incorporation of a C-terminally located azidolysine (peptides 16–18). Reaction of 2 eq. of DBCO porphyrin 8 with the N-terminal azido Tat peptide 12 proceeded smoothly in DMSO to give the expected conjugate 26 in 65% yield as an inseparable mixture of triazole regioisomers.44 Again there was no significant loss of efficiency upon switching from an N- to C-terminal ligation: combination of 8 with the relevant C-terminally ligatable Tat peptide 16 also proceeded effectively to give conjugate 27 in 50% yield after HPLC isolation. When 8 was combined with the penetratin and pVEC derivatives 17 and 18, once again the desired C-terminally ligated conjugates 28 and 29 were obtained with high efficiency irrespective of the length of the peptide component and without requiring a significant excess of 8 (see Scheme 3).
Uptake and subcellular localisation of CPP conjugates
The efficient uptake and endolysosomal localisation of the amphiphilic CPP-photosensitiser conjugates was determined by microscopy in MC28 rat fibrosarcoma cells. All the N- and C-terminally linked conjugates studied showed the expected sub-cellular localisation predicted for their overall amphiphilic character (see Fig. 1) and which would render them potentially suitable photosensitisers for PCI applications. Fig. 2 shows typical images demonstrating the efficient uptake of C-linked derivative 20. This parallels the behaviour of 20 previously observed in the clinically important HN5 head and neck cancer cell line, wherein greatly enhanced cellular uptake of the porphyrin moiety is provided by the function of the CPP to which it is conjugated.18 Endolysosomal localisation of the conjugate was confirmed by colocalisation of the porphyrin fluorescence with Lysotracker Green as visualised by the yellow colour in the merged image in Fig. 2C. All the conjugates were highly water soluble, unlike the parent porphyrin 1 and functionalised but non-ligated porphyrins 2–8.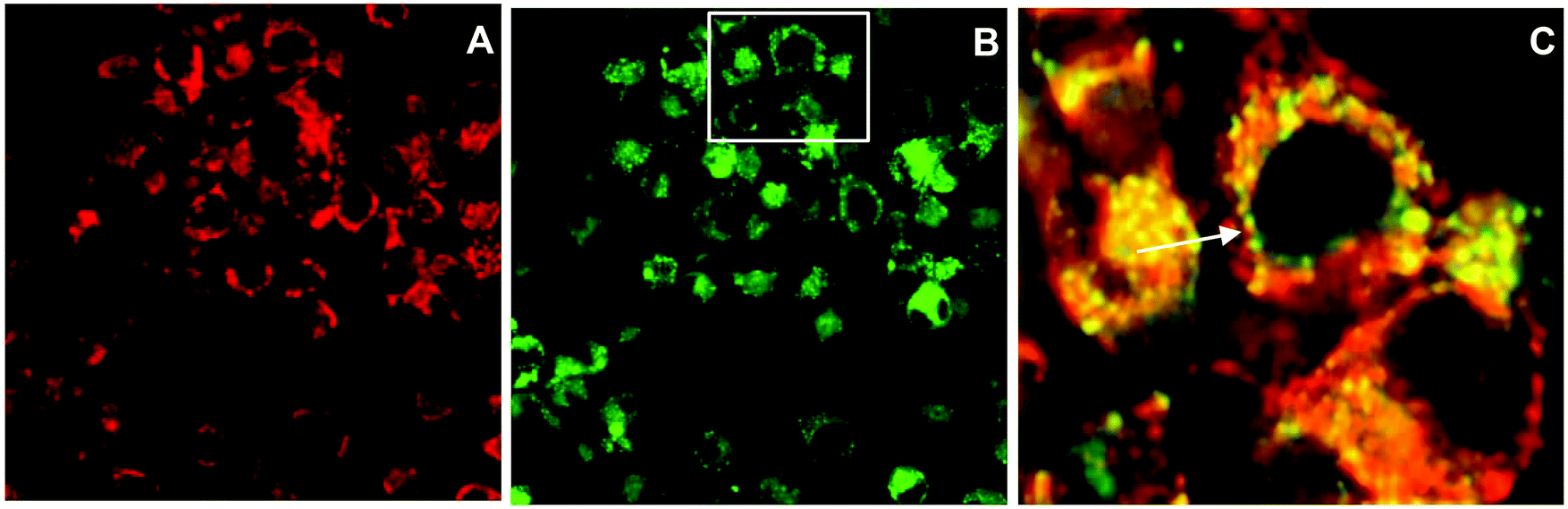 | ||
| Fig. 2 Cellular uptake and colocalisation of conjugate 20 with LysoTracker Green in MC28 cells using laser scanning confocal microscopy. Cells were incubated with 20 (2.5 μM) for 24 h. LysoTracker Green (100 nM) was applied to cells 30 min before imaging. A: 20 alone, B: LysoTracker Green, C: merged A and B inset figure highlighting the overlap of fluorescence from 20 and LysoTracker Green. Scale bar: 20 μm. | ||
Phototoxicity
The localisation of CPP-targeted porphyrin derivatives in lysosomal compartments should result in a highly efficient targeted PDT effect, if as desired, the photosensitiser is localised in the membrane of these vesicles where singlet oxygen generated can effectively photo-oxidise unsaturated lipid materials.20 The photoxicities of selected N- and C-linked conjugates were therefore examined in monolayer culture in MC28 and MCF-7 human breast cancer cells. A significant photo-induced reduction in cell viability was observed with both N- and C-terminally linked maleoyl conjugates 19 and 20 in MC28 cells, which was enhanced with increased photosensitiser concentration and dose of blue light. Comparison of 19 and 20 however revealed little difference in performance as a result of switching the photosensitiser location within the peptide carrier (Fig. 3).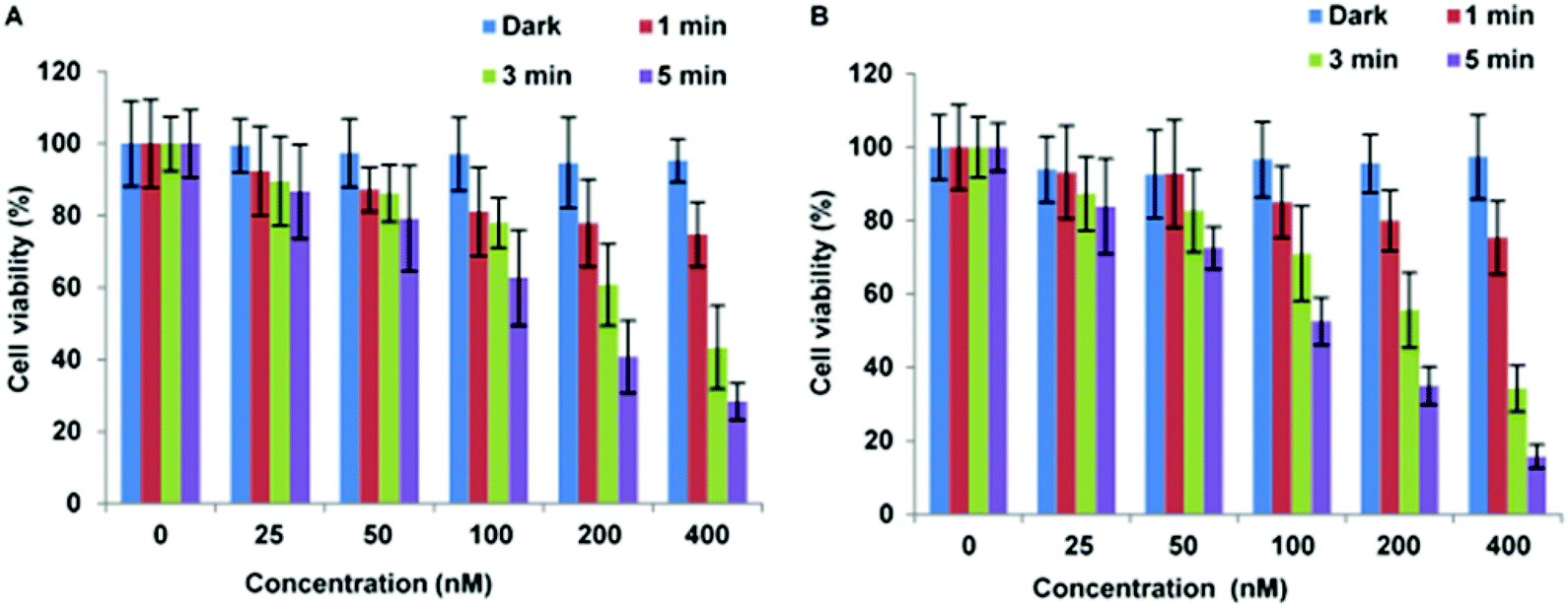 | ||
| Fig. 3 PDT effect of 19 (A) and 20 (B) in MC28 cells. Cells were incubated with Tat–porphyrin conjugates at various concentrations and were illuminated with a blue lamp for up to 5 min. MTT assay was carried out 48 h after light exposure. Data are presented as mean value ± standard deviation (SD) of three independent experiments. | ||
Importantly, Fig. 3 shows that control experiments in the dark for both conjugates resulted in no chemical toxicity at concentrations that were significantly greater than those typically employed for in vitro PCI experiments (see below). This confirmed that the observed reduction in cell viability was light-induced rather than being associated with any membrane-disrupting effects due to the CPP moiety. The triazole conjugates 24 and 26 also displayed a concentration and light dose-dependent increase in the PDT effect (Fig. 4), again without dark toxicity under the experimental conditions. Table 1 shows the porphyrin dose required to induce 50% toxicity (LD50) after 5 min illumination for the four conjugates 19, 20, 24, and 26. These values show that while the PDT performance is indeed similar for 19 and 20 (N- vs. C-terminal porphyrin conjugation), connecting the hydrophobic porphyrin and the polycationic hydrophilic peptide components via the more extended triazole-based linker of 26 seems to be highly beneficial. Notwithstanding this observation, the spectroscopic properties of the porphyrin unit did not appear to be affected by peptide ligation for these or any of the conjugates (see ESI†).
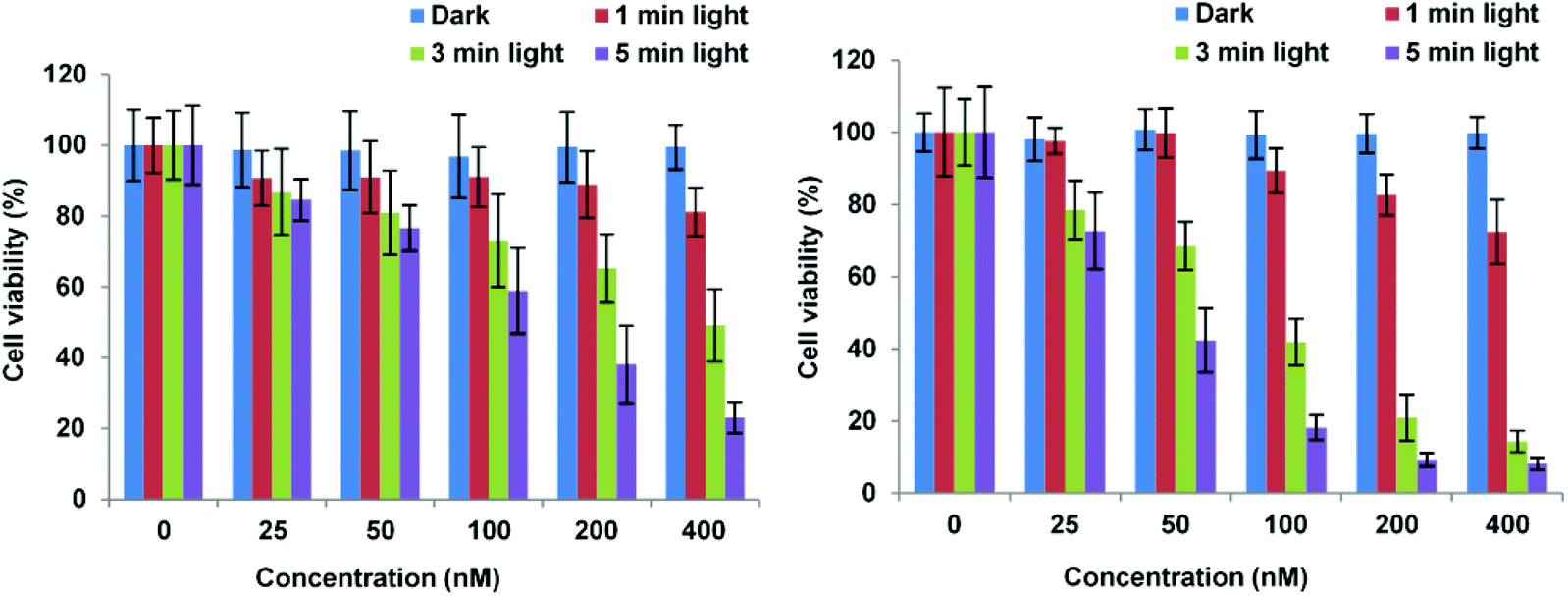 | ||
| Fig. 4 PDT effect of 24 (A) and 26 (B) in MC28 cells. Cells were incubated with Tat–porphyrin conjugates at various concentrations and were illuminated with a blue lamp for up to 5 min. MTT assay was carried out 48 h after light exposure. Data are presented as mean value ± SD of three independent experiments. | ||
| Compound | LD50 PDT (nM) | LD50 dark (μM) |
|---|---|---|
| 19 | 114 ± 8 | ≫1 |
| 20 | 94 ± 6 | ≫1 |
| 24 | 108 ± 6 | ≫1 |
| 26 | 37 ± 2 | ≫1 |
Finally, the C-terminally linked triazole conjugate 29 exemplifying SPAAC ligation was evaluated in MCF7 cells. Once again, an effective concentration and light dose-dependent reduction in cell viability was observed (Fig. 5), with LD50 = 449 nM for 7 min irradiation. As expected, no dark toxicity was shown when the cells were incubated with 29 at various concentrations up to 1 μM.
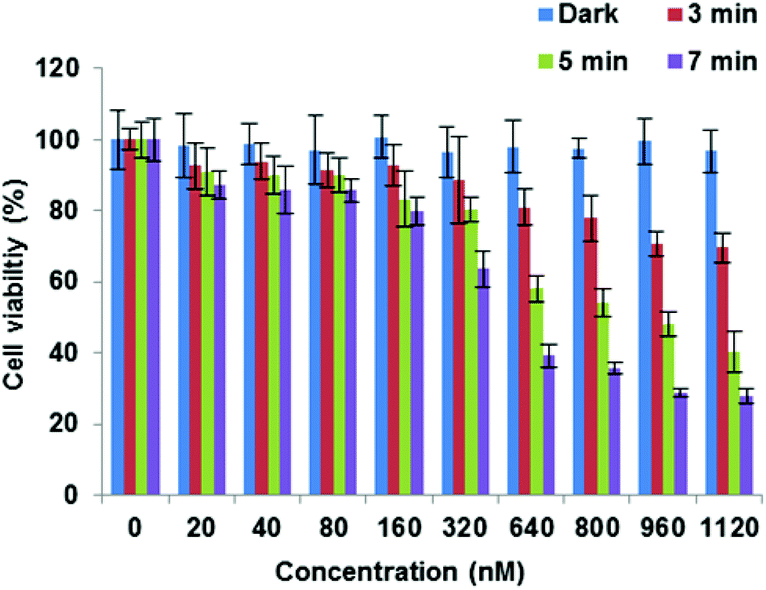 | ||
| Fig. 5 PDT effect of 29 in MCF-7 cells. Cells were incubated with photosensitiser at various concentrations and were illuminated with a blue lamp for up to 7 min. The MTT assay was carried out 48 h after light exposure. Data are presented as mean value ± SD of three independent experiments. | ||
Light-triggered drug delivery
Two of the N-terminally linked Tat conjugates, 19 and 26 were studied further for their ability to promote light-induced relocalisation and endosomal escape of a co-administered protein toxin. Saporin is a 30 kDa ribosome inactivating protein, which has been widely used in neuronal research and as a model compound for PCI studies. Upon cellular uptake, it is ordinarily entrapped within lysosomes, thus severely limiting its toxicity. PDT treatment and PCI of saporin with 19 and 26 resulted in almost the same level of cytotoxic response for each compound (Fig. 6). This data shows that for the PCI experiments the effect of combining either conjugate with saporin is synergistic rather than additive. Indeed, with both conjugates a significant reduction in cell viability was observed in PCI-treated cells versus PDT-treated cells (p < 0.0001). For example at 5 min illumination in Fig. 6B for 26, PCI resulted in a four-fold reduction in viability compared to PDT. PCI efficacy is defined as the ratio of the viability measured using PDT over the PCI viability, and since the reduction in viability using saporin alone is small (approximately 10% in either case) this ratio gives a measure of the enhancement in saporin cytotoxicity induced by PCI.47 In the absence of effective light-triggered endolysosomal disruption and saporin release, the PDT:PCI ratio would be near unity. The ratios observed here of 3.3 for 19 and 4.0 for 26 showing that effective PCI is induced and to similar extents in both conjugates, notwithstanding the difference in the CPP-photosensitiser linkage.
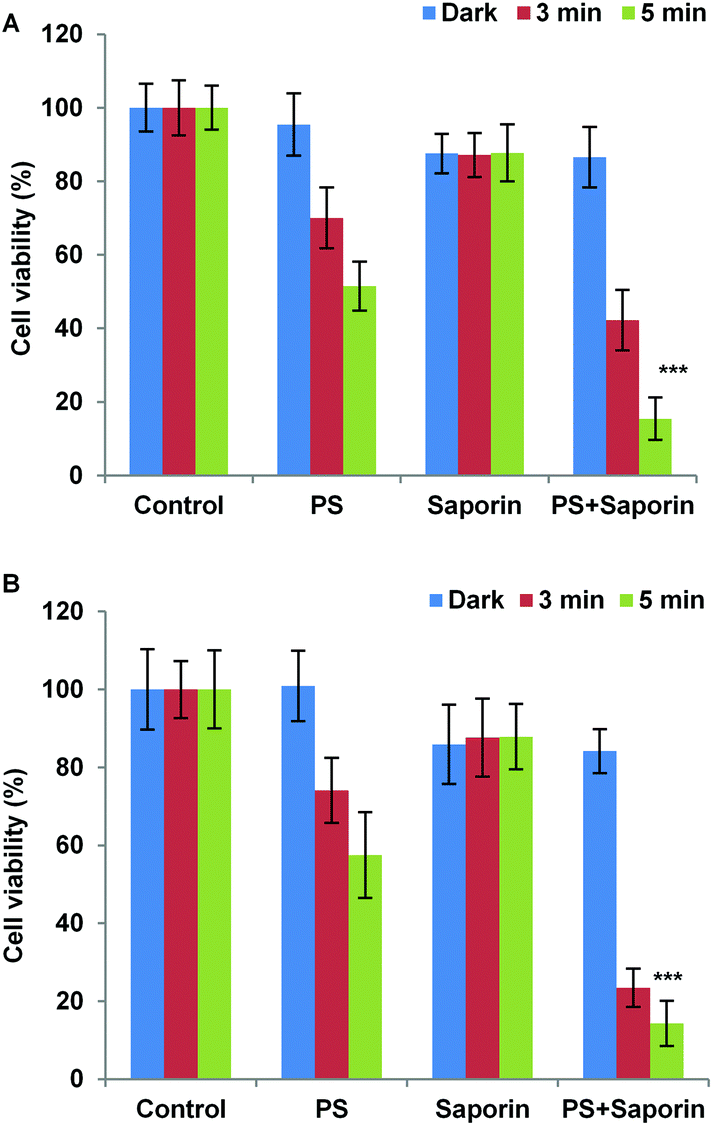 | ||
| Fig. 6 Light-induced cytotoxic response for 19 (A) and 26 (B) in MC28 cells, showing PDT (without saporin) and PCI (with saporin) effects. Cells were incubated with photosensitiser (50 nM) with or without saporin (20 nM) for 18 h. Irradiation was carried out for 3 and 5 min with a blue lamp. MTT assay was carried 72 h after irradiation. The experiments were repeated 3 times, and representative data are shown. Error bars = SD. Statistically significant difference between PCIconjugate19-saporinversus PDTconjugate19 (p < 0.0001) is indicated by ***. Likewise, statistically significant difference between PCIconjugate26-saporinversus PDTconjugate26 (p < 0.0001) is indicated by ***. | ||
Experimental
General information
Chemical reagents were purchased from Sigma-Aldrich, Fluka, Acros, Novabiochem, and Bachem. Peptide grade DMF was purchased from Rathburn Chemicals. Anhydrous DCM was obtained by distillation over calcium hydride. Dowex-1 basic anion exchange resin 100–200 mesh (hydroxide form) was purchased from Sigma and was activated by overnight soaking in 2 M aq. NaOH; the beads were then washed with deionised water and with MeOH. All other solvents were purchased from Fisher Scientific and used as received. Analytical TLC was performed using silica gel 60 F254 pre-coated on aluminium sheets (Merck). Column chromatography was performed on silica gel 60 (35–70 micron) from Sigma-Aldrich. Melting points were recorded on an Electrothermal IA9200 melting point apparatus in open capillaries, and are quoted uncorrected. UV spectra were recorded on a Perkin-Elmer Lambda 19 uv/vis spectrophotometer. Fluorescence spectra were recorded on a Cary Eclipse fluorimeter. 1H and 13C NMR were recorded using a Varian Mercury-VX spectrometer at 400 MHz (1H) and 100 MHz (13C) or a Bruker Avance III 500 at 500 MHz (1H) and 125 MHz (13C). Chemical shift values are given in ppm (δ). J values are given in Hz. Analytical RP-HPLC was performed on a Dionex Ultimate 3000 system (Dionex, UK), with a VWD-3400 variable wavelength detector, and a RF-2000 fluorescence detector. Analyses were performed at 35 ± 0.1 °C on a Gemini 5 μ C18 110A column, (150 × 4.6 mm – Phenomenex, UK), equipped with a Security Guard C18 (ODS) 4 × 3.0 mm ID guard column (Phenomenex, UK), at a flow rate of 1 mL min−1. Mobile phase A was 0.1% aq. TFA, mobile phase B was 0.1% TFA in MeCN. (Method 1: 0.0–10.0 min 0–95% B, 10.0–15.0 min 95% B, 15.0–15.1 min at 95–5% B, 15.1–18.0 min 5% B. Method 2: 0.0–10.0 min 0–95% B, 10.0–20.0 min 95% B, 20.0–20.1 min at 95–5% B, 20.1–23.0 min 5% B). Preparative RP-HPLC was performed on a Dionex HPLC system equipped with a Phenomenex Gemini 5 μ C18 (250 × 10 mm) column at a flow rate of 2.5 mL min−1. Solid phase extractions were performed with Supelco Discovery DSC-18 cartridges (2 g). High resolution mass spectrometry was performed using a Bruker MicroTOF autospec ESI mass spectrometer.5-(4-Aminophenyl)-10,15,20-triphenylporphyrin (2)
The method of Luguya et al.26 was modified as follows. Nitration: a solution of meso-tetrakistetraphenylporphyrin 1 (1.60 g, 2.60 mmol) in TFA (80 mL) in an air-open round-bottom flask was treated at 18 °C with NaNO2 (321 mg, 4.64 mmol). After 3 min, the reaction was quenched with H2O (40 mL), then transferred to a separatory funnel and further diluted with H2O (80 mL) and DCM (160 mL). After separation, the aqueous phase was extracted with DCM (2 × 150 mL). The organic phases were combined, washed with saturated aq. NaHCO3 (2 × 175 mL) and brine (175 mL), then dried over MgSO4. The combined organic phases were filtered and evaporated to give a residue (1.89 g), which was used directly in the next step. Reduction: a solution of the preceding crude mixture of unreacted 1 and mono- and dinitrated products in DCM (640 mL) and MeOH (160 mL) in an air-open round-bottom flask was treated with 5% Pd/C (320 mg). NaBH4 (241 mg, 63.7 mmol) was added in small portions with stirring during 10 min, then the mixture was stirred for an additional 15 min. The reaction was quenched by the addition of H2O (150 mL), and the resulting suspension was transferred to a separatory funnel. The organic phase was separated, washed with brine (100 mL), dried over MgSO4, filtered and evaporated. The crude product was purified by column chromatography, eluting first with toluene to remove unreacted 1 (589 mg, 37% recovery), then 0–10% EtOAc in DCM to elute 2 (564 mg, 34% from 1) and then diaminoporphyrins (368 mg, 22% from 1). The spectroscopic data (1H NMR, 13C NMR) were indistinguishable from the literature.26Preparation of ligatable porphyrins (3)–(8)
Compounds 3 and 5–7 were prepared according to the general procedure detailed below.General procedure: a stirred solution of amino derivative 2 (0.08 mmol) in anhydrous DCM (2.5 mL) was treated with the corresponding carboxylic acid (0.16 mmol), followed by EDC (0.16 mmol), HOBt (0.16 mmol) and DIPEA (0.47 mmol). The solution was protected from light and stirred under nitrogen at room temperature overnight. The reaction mixture was diluted with DCM, washed with 0.1 M HCl, saturated aq. NaHCO3, and brine. The organic phase was dried (MgSO4), filtered, and the solvent was evaporated. The crude product was purified by column chromatography (0–20% EtOAc/DCM gradient).
5-[4-(2-Aminoxy)-acetylamidophenyl]-10,15,20-triphenylporphyrin (9)
A solution of 4 (20.0 mg, 0.02 mmol) in 50% TFA/DCM (2 mL) was stirred at room temperature for 30 min, then it was treated with activated Dowex-1 resin (hydroxide form) until the acid was neutralised. The resin was filtered off, and the solvent was evaporated. Filtration through a pad of silica gel (eluent: DCM) followed by recrystallization (DCM/MeOH), gave 9 as a dark purple solid (14 mg, 94%); mp 266–269 °C; UV-Vis (CHCl3), nm (%): 418 (100), 515 (6.6), 548 (4.9), 590 (4.0), 646 (3.6); 1H-NMR (400 MHz, CDCl3) δ 8.86–8.77 (m, 8H), 8.41 (s, 1H), 8.20–8.12 (m, 8H), 7.95–7.89 (m, 2H), 7.76–7.68 (m, 9H), 5.85 (s, 2H), 4.40 (s, 2H) −2.79 (s, 2H); 13C-NMR (100 MHz, CDCl3) δ 168.1, 165.4, 142.1, 138.4, 136.9, 135.1, 134.5, 131.1, 129.7, 127.7, 126.7, 120.2, 119.4, 118.1, 118.0, 75.3; HRMS [found (ESI+) 703.2791 [M + H]+, C46H34N6O2 requires 703.2821].Preparation of porphyrin Zn complexes
Peptide synthesis
All peptides were assembled according to the Fmoc strategy, on Rink Amide MBHA Resin (Novabiochem, 200–400 mesh, 0.60 mmol g−1 loading) using an Activo P11 automated synthesiser fitted with a reactor heating jacket. The synthesis was performed on 250 mg of resin (0.15 mmol scale). Removal of the Fmoc group from the resin was performed with 20% piperidine/DMF (2.5 mL, 4 × 3 min at 60 °C). The first residue attachment for peptides starting with a Gly, Arg, or azidolysinyl residue was performed manually at room temperature using 5 min preactivation then 1 h coupling with the amino acid (4 eq.), DIC (4 eq.) and DIPEA (6 eq.). This was followed by an acetylation step (Ac2O/DIPEA/DMF = 1/1/8, 2.5 mL, 1 × 10 min). For peptide 15 with C-terminal Cys, a low-racemisation protocol by Angell et al. was employed,48 utilising a 1 h coupling at room temperature without preactivation with Fmoc-Cys(Trt)-OH (4 eq.), HATU (4 eq.), HOBt (4 eq.) and collidine (4 eq.). Subsequent Fmoc deprotection steps were performed using 25% piperidine/DMF (3 mL, 1 × 5 min, 1 × 10 min), with the chain elongation steps being performed at 60 °C for 35 min using 3 eq. of each Fmoc-protected amino acid (Fmoc-Arg(Pmc)-OH, Fmoc-Gln(Trt)-OH), Fmoc-Lys(Boc)-OH, or Fmoc-Gly-OH), PyBOP (3 eq.), and DIPEA (6 eq.) in DMF (4.3 mL) (35 min,). The Fmoc deprotection step was omitted after the coupling of the final residue. The peptide resin was washed thoroughly with DMF, DCM, MeOH and Et2O, and dried in vacuo.(10)–(12): derivatisation was carried with the appropriate acid (3 eq.), HATU (2.9 eq.), and DIPEA (6 eq.) in DMF (2 mL) for 1 h. Completion of the acylation was confirmed by the Kaiser test.
(14): derivatisation was carried out using pyruvic acid succinimido ester49 (2 eq.) and DIPEA (2 eq.) in DMF (2 mL) overnight. Completion of the acylation was confirmed as previously.
Following the acylation step, the resin beads were filtered off under vacuum, and washed thoroughly with DMF, DCM, MeOH, and Et2O, and dried in vacuo. For the cleavage and deprotection of the ligatable peptides, the acylated peptidyl resin was swollen in DCM for 20 min, then it was treated with TFA/TIS/H2O (95/2.5/2.5) for 4 h. Cysteine-containing peptide (10) was instead treated with TFA/TIS/H2O/EDT (95/2.5/2.5/1) for 3 h. The resin beads were filtered off, washed with TFA, and the combined filtrates were evaporated to a small volume then anhydrous Et2O was added. The resulting precipitate was collected by centrifugation and was washed twice more with Et2O. The precipitated material was dissolved in 1% aq. TFA, filtered using a 0.2 μm syringe filter and the resulting solution was directly purified by semi-preparative HPLC. The purified peptides were then freeze-dried.
(10) (24 mg, 40%); HPLC: tR (Method 1): 4.07 min; HRMS [found (ESI+) 749.9608 [M + 2H]2+, C58H117N33O12S2 requires 749.9635].
(11) (26 mg, 58%); HPLC: tR (Method 1): 3.76 min; HRMS [found (ESI+) 738.4662 [M + 2H]2+, C60H116N32O12 requires 738.4720].
(12) (24 mg, 55%); HPLC: tR (Method 1): 6.53 min; HRMS [found (ESI+) 803.0359 [M + 2H]2+, C66H131N35O12 requires 803.0353].
(14) (16 mg, 35%); HPLC: tR (Method 1): 3.91 min; HRMS [found (ESI+) 489.3067 [M + 3H]3+, C58H115N32O13 requires 489.3102].
(15) (20 mg, 33%); HPLC: tR (Method 1): 3.90 min; compound was previously described.9
(16) (54 mg, 67%). HPLC tR (Method 1) 7.61 min; [Found (ESI+) 609.7098 [M + 3H]3+, C74H138N39O16 requires 609.7056].
(17) (33 mg, 65%). HPLC tR (Method 1) 5.06 min; [Found (ES+) 557.8337 [M + 2H]2+, C48H85N21O8S requires 557.8330].
(18) (40 mg, 59%). HPLC tR (Method 1) 5.43 min; [Found (ESI+) 745.8039 [M + 3H]3+, C98H179N40O20 requires 745.8076].
Ligation chemistry
(23): a solution of azidopeptide 13 (4.0 mg, 1.7 μmol) in DMSO (500 μL) was treated with alkynyl-porphyrin (Zn-4) (6.0 mg, 7.7 μmol), CuSO4·5H2O (1 mg, 8 μmol) and ascorbic acid (2.5 mg, 14 μmol). The mixture was allowed to stand at room temperature, shielded from light for 1.5 h, then it was applied to a solid phase extraction cartridge. The cartridge was washed with 0.1% aq. TFA, then 0.1% TFA in H2O/MeCN (80/20), and then the desired conjugate was eluted with 0.1% TFA in H2O/MeCN, (50/50). The eluate obtained was evaporated to dryness, then the residue was dissolved in 20% aq. TFA and the resulting solution allowed to stand at room temperature until HPLC analysis indicated complete decomplexation of the porphyrin moiety. The solution was then evaporated to a small volume and the desired conjugate was isolated by semi-preparative HPLC and freeze-drying, to give 16 as a dark green solid (4.1 mg, 79%). HPLC: tR (Method 2): 6.83 min; UV-Vis (0.1% aq. TFA), nm (%): 438 (100), 657 (15.4); Fluorescence λmax. (0.1% aq. TFA, λexc. = 420 nm) 685 nm; HRMS [found (ESI+) 1066.5903 [M + 2H]2+. C104H145N39O12 requires 1066.5975].
(24): A solution of alkynyl-Tat peptide (11) (1.0 mg, 0.42 μmol) in DMSO (200 μL), tBuOH (30 μL) and H2O (30 μL) was treated with azidoundecanoyl-porphyrin (Zn-6) (1.0 mg, 2.6 μmol), CuSO4·5H2O (6.2 μL of a 1 M solution in H2O, 6.2 μmol) and sodium ascorbate (10 mg, 50 μmol). The mixture was allowed to stand at room temperature, shielded from light for 3 h, then was diluted with 5.0% aq. TFA, left to stand at room temperature for 30 min and directly purified by semi-preparative HPLC. The purified conjugate was freeze-dried, to give 24 as a dark green solid (3.1 mg, 77%). HPLC: tR (Method 2): 7.61 min; UV-Vis (0.1% aq. TFA), nm (%): 437 (100), 651 (15.2); Fluorescence λmax. (0.1% aq. TFA, λexc. = 420 nm), 684 nm; HRMS [found (ESI+) 772.4495 [M + 3H]3+, C115H167N40O132 requires 772.4549].
(25): A solution of alkynyl-Tat peptide 11 (6.0 mg, 8.1 μmol) in DMSO (1.2 mL), tBuOH (180 μL) and H2O (180 μL) was treated with azidobutanoyl-porphyrin (Zn-7) (6.0 mg, 2.6 μmol), CuSO4·5H2O (37.4 μL of a 1 M solution in H2O, 37.4 μmol) and sodium ascorbate (60 mg, 300 μmol). The mixture was allowed to stand at room temperature, shielded from light for 3 h, then was diluted with 5.0% aq. TFA, left to stand at room temperature for 30 min and directly purified by semi-preparative HPLC. The purified conjugate was freeze-dried, to give 25 as a dark green solid (6.1 mg, 78%). HPLC: tR (Method 2): 7.12 min; UV-Vis (0.1% aq. TFA), nm (%): 438 (100), 648 (15.9); Fluorescence λmax. (0.1% aq. TFA, λexc. = 420 nm), 684 nm; HRMS [found (ESI+) 554.8321 [M + 4H]4+, C108H150N40O13 requires 554.8149].
(28): As for 27, but with azidopeptide 17 (4.8 mg, 2.6 μmol) and DBCO-porphyrin 8 (2.6 mg, 2.8 μmol). Purification by semi-preparative HPLC gave 28 as a dark green solid (6.9 mg, 74%). HPLC: tR (Method 2): 7.90 min; UV-vis (0.1% aq. TFA), nm (%): 433 (100), 652 (10); Fluorescence λmax. (0.1% aq. TFA, λexc. = 420 nm), 684 nm; HRMS [found (ESI+) 697.6903 [M + 3H]3+, C115H132N30O8S requires 697.6850].
(29): As for 27, but with azidopeptide 18 (10.0 mg, 3.3 μmol) and DBCO-porphyrin 8 (3.1 mg, 3.3 μmol) in DMSO (500 μL). Purification by semi-preparative HPLC gave 29 as a dark green solid (9.0 mg, 69%). HPLC: tR (Method 1): 7.61 min; UV-Vis (MeOH), nm (%): 415 (100), 513 (12.96), 547 (9.50), 590 (8.23), 646 (7.81); Fluorescence λmax. (MeOH, λexc. = 420 nm) nm (%) 653 (100), 715 (34); HRMS [found (ESI+) 795.4614 [M + 4H]4+, C163H228N46O22 requires 795.4529].
Cell lines and cultivation
MC28 cells, a methylcholanthrene-induced fibrosarcoma cell line, were grown in DMEM supplemented with 10% FCS. Human breast cancer cells (MCF-7) were grown in DMEM-F12 medium containing 10% FCS. Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. Unless otherwise stated materials for the cell studies were purchased from Sigma-Aldrich Gillingham, UK.Statistical analysis
Data were analysed using two-tailed Students T-test with appropriate testing post hoc using Prism 6 software. The dose required to reduce viability by 50% (LD50) was also calculated using Graphpad Prism. Error bars from the mean show ±standard deviation (SD). Values of P < 0.05 were considered to be significant.Conclusions
Bioconjugation strategies offer a highly efficient and flexible means to generate conjugates between hydrophobic porphyrins and polyfunctional CPPs under very mild conditions. These amphiphilic photosensitisers with regiospecific attachment of a tetrapyrrole component within the peptide backbone are effective agents for both targeted PDT and PCI. As exemplified in this study, strain-promoted azide–alkyne ligations offer an ideal way to repurpose simple porphyrin derivatives for PCI by attachment to a variety of CPPs. This approach opens the way for adapting clinically relevant photosensitisers with the most attractive spectroscopic properties that are currently used in PDT for minimally invasive therapies in conjunction with selected targeted CPPs.Acknowledgements
This work was supported by BBSRC grants BB/D0127831 and BB/J009164/1 (IME) and BB0113291 and BB/J009318/1 (AJM) at Bath and UCL respectively. We thank the University of Bath Faculty of Science for a Fee Waiver Scholarship (KMT) and a Bath-Shandong Pharmacy Vacation Scholarship (LW). We thank Dr S. Pascu for assistance with the fluorescence analyses. We are also grateful to Dr C. Walsh for assistance with the statistical analysis.Notes and references
- T. J. Dougherty, J. Clin. Laser Med. Surgery, 2002, 20, 3–7 Search PubMed.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- P. K. Kaiser, D. S. Boyer, A. F. Cruess, J. S. Slakter, S. Pilz, A. Weisberger and D. S. Grp, Ophthalmology, 2012, 119, 1001–1010 CrossRef PubMed.
- B. J. Qumseya, W. David and H. C. Wolfsen, Clin. Endosc., 2013, 46, 30–37 CrossRef PubMed.
- T. Maisch, Lasers Med. Sci., 2007, 22, 83–91 CrossRef PubMed.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS PubMed.
- R. G. W. Jinadasa, Z. Zhou, M. G. H. Vicente and K. M. Smith, Org. Biomol. Chem., 2016, 14, 1049–1064 CAS.
- P. M. R. Pereira, B. Korsak, B. Sarmento, R. J. Schneider, R. Fernandes and J. P. C. Tome, Org. Biomol. Chem., 2015, 13, 2518–2529 Search PubMed; K. Liu, R. Xing, Q. Zou, G. Ma, H. Möhwald and X. Yan, Angew. Chem., Int. Ed., 2016, 55, 3036–3039 CrossRef CAS PubMed; R. Xing, K. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, Adv. Mater., 2016, 28, 3669–3676 CrossRef PubMed.
- F. Giuntini, C. M. A. Alonso and R. W. Boyle, Photochem. Photobiol. Sci., 2011, 10, 759–791 CAS.
- K. M. Stewart, K. L. Horton and S. O. Kelley, Org. Biomol. Chem., 2008, 6, 2242–2255 CAS.
- S. R. Jean, D. V. Tulumello, S. P. Wisnovsky, E. K. Lei, M. P. Pereira and S. O. Kelley, ACS Chem. Biol., 2014, 9, 323–333 CrossRef PubMed.
- M. Zahid and P. D. Robbins, Molecules, 2015, 20, 13055–13070 CrossRef CAS PubMed.
- M. Sibrian-Vazquez, T. J. Jensen, R. P. Hammer and M. G. H. Vicente, J. Med. Chem., 2006, 49, 1364–1372 CrossRef CAS PubMed.
- M. Sibrian-Vazquez, T. J. Jensen and M. G. H. Vicente, J. Med. Chem., 2008, 51, 2915–2923 CrossRef CAS PubMed.
- M. Sibrian-Vazquez, X. Hu, T. J. Jensen and M. G. H. Vicente, J. Porphyrins Phthalocyanines, 2012, 16, 603–615 CrossRef CAS.
- F. Moret, M. Gobbo and E. Reddi, Photochem. Photobiol. Sci., 2015, 14, 1238–1250 CAS.
- L. Bourré, F. Giuntini, I. M. Eggleston, C. A. Mosse, A. J. MacRobert and M. Wilson, Photochem. Photobiol. Sci., 2010, 9, 1613–1620 Search PubMed.
- J. T. W. Wang, F. Giuntini, I. M. Eggleston, S. G. Bown and A. J. MacRobert, J. Controlled Release, 2012, 157, 305–313 CrossRef CAS PubMed.
- K. Berg, P. K. Selbo, L. Prasmickaite, T. E. Tjelle, K. Sandvig, D. Moan, G. Gaudernack, O. Fodstad, S. Kjolsrud, H. Anholt, G. H. Rodal, S. K. Rodal and A. Hogset, Cancer Res., 1999, 59, 1180–1183 CAS.
- A. M. d. P. Bayona, C. M. Moore, M. Loizidou, A. J. MacRobert and J. H. Woodhams, Int. J. Cancer, 2016, 138, 1049–1057 CrossRef PubMed.
- A. A. Sultan, W. Jerjes, K. Berg, A. Hogset, C. A. Mosse, R. Hamdoudi, Z. Hamdoon, C. Simeon, D. Carnell, M. Forster and C. Hopper, Lancet Oncol., 2016, 17, 1217–1229 CrossRef CAS PubMed.
- G. A. Johnson, E. A. Ellis, H. Kim, N. Muthukrishnan, T. Snavely and J.-P. Pellois, PLoS One, 2014, 9, e91220 Search PubMed.
- I. Meerovich, N. Muthukrishnan, G. A. Johnson, A. Erazo-Oliveras and J.-P. Pellois, Biochim. Biophys Acta, Gen. Subj., 2014, 1840, 507–515 CrossRef CAS PubMed.
- N. Muthukrishnan, S. Donovan and J.-P. Pellois, Photochem. Photobiol., 2014, 90, 1034–1042 CAS.
- T. Ohtsuki, S. Miki, S. Kobayashi, T. Haraguchi, E. Nakata, K. Hirakawa, K. Sumita, K. Watanabe and S. Okazaki, Sci. Rep., 2015, 5, 18577 CrossRef CAS PubMed.
- R. Luguya, L. Jaquinod, F. R. Fronczek, A. G. H. Vicente and K. M. Smith, Tetrahedron, 2004, 60, 2757–2763 CrossRef CAS.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- A. Ziegler, P. Nervi, M. Durrenberger and J. Seelig, Biochemistry, 2005, 44, 138–148 CrossRef CAS PubMed.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS PubMed.
- E. Kaiser, R. I. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS PubMed.
- H. F. Gaertner, F. Cerini, A. Kamath, A.-F. Rochat, C.-A. Siegrist, L. Menin and O. Hartley, Bioconjugate Chem., 2011, 22, 1103–1114 CrossRef CAS PubMed.
- O. Melnyk, J. A. Fehrentz, J. Martinez and H. Gras-Masse, Biopolymers, 2000, 55, 165–186 CrossRef CAS PubMed.
- S. Santra, H. Yang, J. T. Stanley, P. H. Holloway, B. M. Moudgil, G. Walter and R. A. Mericle, Chem. Commun., 2005, 3144–3146 RSC.
- T. J. Sminia and D. S. Pedersen, Synlett, 2012, 2643–2646 CAS.
- C. M. A. Alonso, A. Palumbo, A. J. Bullous, F. Pretto, D. Neri and R. W. Boyle, Bioconjugate Chem., 2010, 21, 302–313 CrossRef CAS PubMed.
- A. Maruani, H. Savoie, F. Bryden, S. Caddick, R. Boyle and V. Chudasama, Chem. Commun., 2015, 51, 15304–15307 RSC.
- H. Kitagishi, S. Hatada, T. Itakura, Y. Maki, Y. Maeda and K. Kano, Org. Biomol. Chem., 2013, 11, 3203–3211 CAS.
- S. Acherar, L. Colombeau, C. Frochot and R. Vanderesse, Curr. Med. Chem., 2015, 22, 3217–3254 CrossRef CAS PubMed.
- Y. Ikawa, H. Harada, M. Toganoh and H. Furuta, Bioorg. Med. Chem. Lett., 2009, 19, 2448–2452 CrossRef CAS PubMed.
- M. Thandu, V. Rapozzi, L. Xodo, F. Albericio, C. Comuzzi and S. Cavalli, ChemPlusChem, 2014, 79, 90–98 CrossRef CAS.
- F. Dumoulin and V. Ahsen, J. Porphyrins Phthalocyanines, 2011, 15, 481–504 CrossRef CAS.
- A. Leonidova, V. Pierroz, L. A. Adams, N. Barlow, S. Ferrari, B. Graham and G. Gasser, ACS Med. Chem. Lett., 2014, 5, 809–814 CrossRef CAS PubMed.
- J. Jiang, M. Taniguchi and J. S. Lindsey, New J. Chem., 2015, 39, 4534–4550 RSC.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- M. King and A. Wagner, Bioconjugate Chem., 2014, 25, 825–839 CrossRef CAS PubMed.
- J. A. Wagner, D. Mercadante, I. Nikic, E. A. Lemke and F. Graeter, Chem. – Eur. J., 2015, 21, 12431–12435 CrossRef CAS PubMed.
- B. Bull-Hansen, Y. Cao, K. Berg, E. Skarpen, M. G. Rosenblum and A. Weyergang, J. Controlled Release, 2014, 182, 58–66 CrossRef CAS PubMed.
- Y. M. Angell, J. Alsina, F. Albericio and G. Barany, J. Pept. Res., 2002, 60, 292–299 CrossRef CAS PubMed.
- H. F. Gaertner, F. Cerini, A. Kamath, A.-F. Rochat, C.-A. Siegrist, L. Menin and O. Hartley, Bioconjugate Chem., 2011, 22, 1103–1114 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C spectra, HPLC traces, UV and fluorescence spectra. See DOI: 10.1039/c6ob02135b |
| ‡ These authors contributed equally. |
| § Current address: School of Pharmacy, Liverpool John Moores University, Byrom Street, Liverpool L3 3AF, UK. |
| This journal is © The Royal Society of Chemistry 2016 |