
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic analogs of stryphnusin isolated from the marine sponge Stryphnus fortis inhibit acetylcholinesterase with no effect on muscle function or neuromuscular transmission†
Lindon W. K.
Moodie‡
a,
Monika C.
Žužek
b,
Robert
Frangež
b,
Jeanette H.
Andersen
c,
Espen
Hansen
c,
Elisabeth K.
Olsen
c,
Marija
Cergolj
de,
Kristina
Sepčić
d,
Kine Ø.
Hansen
*c and
Johan
Svenson
*af
aDepartment of Chemistry, UiT The Arctic University of Norway, Breivika, N-9037, Tromsø, Norway. E-mail: kine.o.hanssen@uit.no; johan.svenson@sp.se
bInstitute of Preclinical Sciences, Veterinary faculty, University of Ljubljana, Ljubljana, Slovenia
cMarbio, UiT The Arctic University of Norway, Breivika, N-9037, Tromsø, Norway
dDepartment of Biology, Biotechnical Faculty, University of Ljubljana, Ljubljana, Slovenia
eDepartment of Biotechnology, University of Rijeka, Rijeka, Croatia
fDepartment of Chemistry, Materials and Surfaces SP Technical Research Institute of Sweden, Box 857, SE-501 15 Borås, Sweden
First published on 9th November 2016
Abstract
The marine secondary metabolite stryphnusin (1) was isolated from the boreal sponge Stryphnus fortis, collected off the Norwegian coast. Given its resemblance to other natural acetylcholinesterase antagonists, it was evaluated against electric eel acetylcholinesterase and displayed inhibitory activity. A library of twelve synthetic phenethylamine analogs, 2a–7a and 2b–7b, containing tertiary and quaternary amines respectively were synthesized to investigate the individual structural contributions to the activity. Compound 7b was the strongest competitive inhibitor of both acetylcholinesterase and butyrylcholinesterase with IC50 values of 57 and 20 μM, respectively. This inhibitory activity is one order of magnitude higher than the positive control physostigmine, and is comparable with several other marine acetylcholinesterase inhibitors. The physiological effect of compound 7b on muscle function and neuromuscular transmission was studied and revealed a selective mode of action at the investigated concentration. This data is of importance as the interference of therapeutic acetylcholinesterase inhibitors with neuromuscular transmission can be problematic and lead to unwanted side effects. The current findings also provide additional insights into the structure–activity relationship of both natural and synthetic acetylcholinesterase inhibitors.
Introduction
The diverse molecular scaffolds displayed by natural products hold great promise for drug development and have already inspired a number of clinically used drugs.1,2 Historically, most compounds have been isolated from terrestrial sources but, as a result of technological advancements, the last 50 years have seen a rise in the number of isolated marine natural products, with some 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 reported in the scientific literature.2,3 Approximately 500 new marine natural products are reported each year.4 To date, the Food and Drug Administration (FDA) has approved seven drugs of marine origin which illustrates their potential.5 Natural products generally expand into a broader chemical realm than the synthetic libraries screened by the pharmaceutical industry6 and screening success has been shown to increase when including natural-product-like scaffolds.7
000 reported in the scientific literature.2,3 Approximately 500 new marine natural products are reported each year.4 To date, the Food and Drug Administration (FDA) has approved seven drugs of marine origin which illustrates their potential.5 Natural products generally expand into a broader chemical realm than the synthetic libraries screened by the pharmaceutical industry6 and screening success has been shown to increase when including natural-product-like scaffolds.7
Nearly half of the new marine natural products reported are isolated from the Porifera (sponges) taxon which is attributed to a high content of both opportunistic and symbiotic microorganisms.4,8–10 Marine microorganisms are the source of many highly potent natural products which include approved drugs and compounds in clinical trials.1,5 The marine microbes are particularly challenging to cultivate and therefore the collection of larger marine benthic organisms remains highly warranted for the continued discovery of novel compounds of microbial origin.
Our recent studies of Arctic marine organisms have led to the characterization of a range of acetylcholinesterase (AChE) inhibitors.11 During these investigations, we have reported the isolation and AChE-inhibitory properties of the halogenated tyrosine derivatives pulmonarin A and B, isolated from the ascidian Synoicum pulmonaria.12,13 These small, dibrominated compounds displayed AChE inhibition in the pharmaceutically relevant range (Ki = 90 and 20 μM respectively) and represent interesting marine hits for further studies.13
Four brominated indole derivatives have also recently been isolated from the boreal sponge Geodia barretti. A library of 22 synthetic compounds was synthesized in order to establish the structure–activity relationship (SAR) against AChE and the role of indole bromination.14 The most potent natural compounds from that study were the 2,5-diketopiperazines barettin and 8,9-dihydrobarettin which displayed significant inhibition of AChE, (inhibition constants of 29 and 19 μM respectively) and butyrylcholinesterase (BChE; inhibition constants of 14 and 48 μM respectively) via a reversible noncompetitive mechanism.14

The chronic neurodegenerative condition known as Alzheimer's disease (AD) is characterized by progressive degeneration of cholinergic neurons and is the most common cause of dementia.20 AChE (E.C 3.1.1.7) is the key enzyme for termination of neurotransmission in cholinergic pathways via the rapid hydrolysis of the neurotransmitter acetylcholine following its presynaptic release.21,22 Therefore, AChE inhibition is a promising approach for symptomatic treatment of AD.23 Recent studies also indicate that AChE inhibitor binding to the peripheral anionic site of AChE can be beneficial for the inhibition of the amyloid cascade and offer protection of neural cells against free radical induced damage.24 In addition, patients diagnosed with AD show a progressive increase in the activity of the related cholinesterase enzyme, BChE (E.C. 3.1.1.8). This enzyme is found mainly in the blood plasma,25 and serves as a “back-up” when AChE activity is compromised or absent.26 Both enzymes represent relevant therapeutic targets for ameliorating the symptoms of the AD.
The FDA and European Medicines Agency have approved three compounds addressing the cognitive impairment of AD patients: donepezil, rivastigmine and galanthamine.21 The latter two drugs are strongly affiliated with natural products chemistry. Rivastigmine was developed from physostigmine, an alkaloid naturally occurring in the Calabar bean27 while galanthamine was isolated from the bulb of Galanthus woronowii.28,29 All three compounds inhibit AChE in a reversible manner, and interact directly with the active site or adjacent binding pockets.21 Several marine natural products have been shown to display neurological activities although they have yet to reach the market.30 The current state of neurologically active marine natural products was recently reviewed by Sakai and Swanson.31
In the present report, we describe the isolation and evaluation of stryphnusin (1), a brominated marine phenethylamine derivative isolated from the Arctic sponge Stryphnus fortis (Vosmaer 1885). S. fortis is a large, smooth sponge which is found in dense colonies in the northern Atlantic Ocean and is common to the Norwegian coast. S. fortis is known for containing the bioactive secondary metabolite ianthelline which displays both antifouling and cytotoxic bioactivities.32,33 However, the actual primary producer of ianthelline was recently suggested to be the Hexadella dedritifera sponge which commonly grows on S. fortis.34 No attempts were made to search for H. dedritifera in the current S. fortis material. The current study represents an extension of our continued search for novel cholinesterase inhibitors of marine origin. Compound 1 is structurally related to the marine AChE inhibitors isolated from S. pulmonaria and G. baretti and was evaluated for its ability to inhibit AChE. Based on the initial observed inhibitory activity of 1 against electric eel AChE, a library of simplified synthetic analogs were prepared and evaluated. Although the structure of 1 was originally reported in 2000 after isolation from the Caribbean sponge Verongula gigantea,35 and again in 2010, from the Mediterranean phlebobranchiate ascidian Ciona edwardsii,36 only limited bioactivity data has been reported. The effect on BChE, and the physiological effect on neuromuscular transmission and muscle function were evaluated for the most active synthetic analog. The data reported expands the knowledge of marine cholinesterase inhibitors and represents the first study of their effect on muscle function.
Results and discussion
The monobrominated 1 was found in organic phase of the S. fortis extract and was isolated using mass guided preparative HPLC. The compound was identified based on spectroscopic analysis.35 Compound 1 was evaluated as inhibitor of electric eel AChE and was found to exhibit a moderate inhibitory activity (Table 1). Based on the initially observed activity, and in order to supplement our previously obtained structure activity relationship (SAR) data, a range of synthetic analogs were prepared and tested, affording six tertiary (2a–7a) and six quaternary amines (2b–7b) (Scheme 1). The degree and position of phenyl ring bromine, hydroxyl and methoxy substituents was incorporated by consideration of the appropriate phenethylamine starting materials. The tertiary amines 2a–7a were prepared via reductive amination, and further alkylation by methyl iodide yielded the corresponding quaternary amines 2b–7b. The starting materials for compounds 6a37 and 7a38 were prepared using reported methods.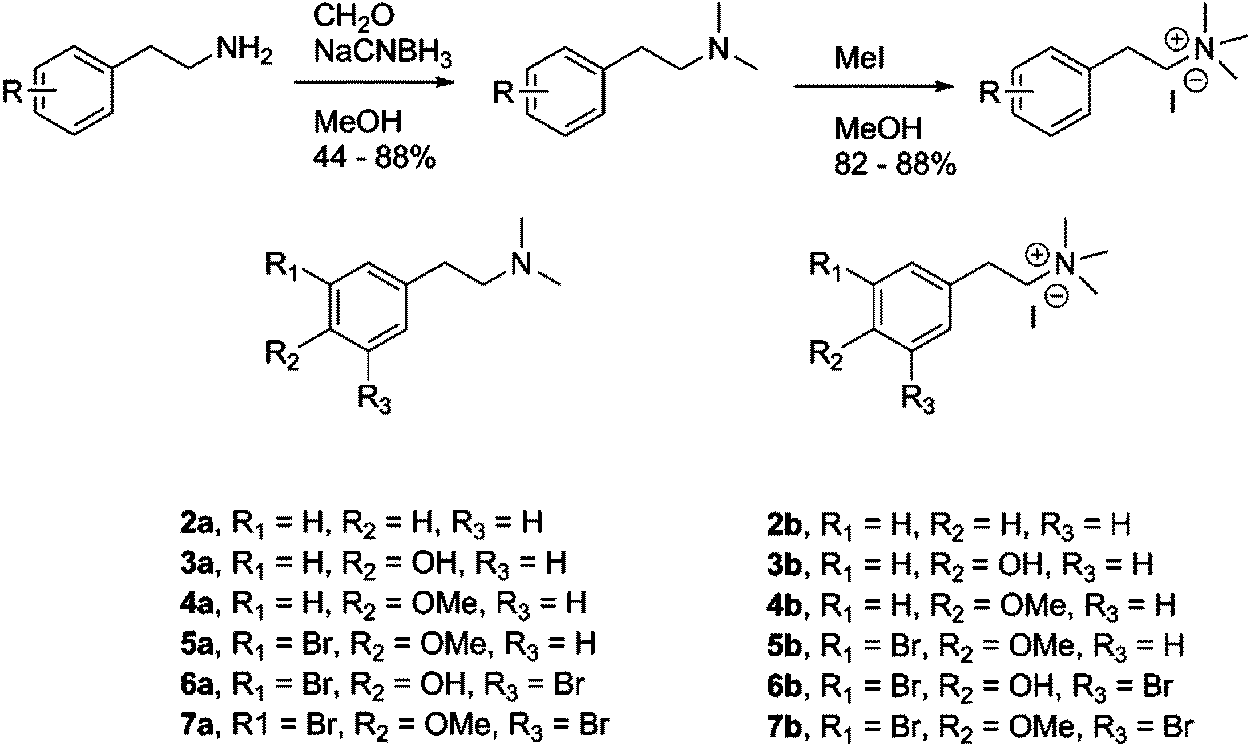 | ||
| Scheme 1 The synthesis of stryphnusin analogs 2a–7a and 2b–7b. | ||
| Compound | IC50a (μM) |
K i (μM) |
|---|---|---|
| a IC50 is determined as the concentration of the compound inducing 50% inhibition of the enzyme activity. b K i not determined for compounds displaying an IC50 > 250 μM. c Data taken from ref. 13 and 14. | ||
| AChE | ||
| 1 | 232 | 235 |
| 2a | 1675 | n.d. |
| 3a | 1513 | n.d. |
| 4a | 1395 | n.d. |
| 5a | 968 | n.db |
| 6a | 774 | n.d. |
| 7a | 163 | 202 |
| 2b | 1096 | n.d. |
| 3b | 1387 | n.d. |
| 4b | 1287 | n.d. |
| 5b | 293 | n.d. |
| 6b | 444 | n.d. |
| 7b | 57 | 51 |
| Physostigmine | 3 | 4 |
| Pulmonarin Ac | 150 | 90 |
| Pulmonarin Bc | 36 | 20 |
| 6-Bromoconicaminc | 230 | 90 |
| Barettinc | 36 | 29 |
| BChE | ||
| 7b | 20 | n.d. |
| 6-Bromoconicaminc | 14 | 11 |
| Barettinc | 26 | 14 |
The kinetics of the in vitro inhibition of electric eel AChE were assessed by employing the colorimetric assay developed by Ellman39 and the data is presented in Table 1.
The in vitro AChE inhibition of 1 was modest with an IC50 of 232 μM (Fig. 1), which is similar to the recently reported 6-bromoconicamin. The prepared synthetic analogs generally demonstrated weaker inhibition with the exception of 7a and 7b, which displayed IC50s of 163 and 57 μM respectively. Compound 7b was also evaluated as an inhibitor of BChE and displayed an IC50 value of 20 μM as shown in Fig. 1.
 | ||
| Fig. 1 Inhibition of electric eel acetylcholinesterase (solid symbols) and horse serum butyrylcholinesterase (open symbols) by compounds 1 (triangles) and 7b (circles). The IC50 values towards AChE was determined to be 232 μM for 1 and 57 μM for 7b, and the IC50 value for 7b towards BChE was determined to be 20 μM. | ||
For those compounds displaying IC50s < 250 μM the inhibitory constants Ki were also determined using Dixon plot analysis, as shown in Fig. 2 for 1 and 7b. All the examined compounds were shown to be reversible competitive AChE inhibitors, suggesting their binding to the active site of the free enzyme.
 | ||
| Fig. 2 Determination of electric eel AChE inhibition type and the inhibition constants (Ki) for 1 (left graph), and 7b (right graph) by Dixon plot analysis. The concentrations of the substrate acetylthiocholine were 0.125 (●), 0.25 (□), and 0.50 mM (■). Ki towards AChE was determined to be 235 μM for 1 and 51 μM for 7b. | ||
Compound 7b was the most active competitive inhibitor with a Ki of 51 μM in our assays. This was superior to pulmonarin A and 6-bromoconicamin and comparable to pulmonarin B and barettin.13,14 When compared to the natural product 1, it appears that the additional bromination and phenolic methylation are sufficient to increase the inhibitory activity. The reported Ki of the FDA approved AChE inhibitor galanthamine ranges from 2–10 μM (ref. 28) and there is generally a wide concentration range in which AChE inhibitors effectively exerts their mode of action.40 AChE inhibitors also often yield different affinities depending on the enzyme source and experimental setup. The positive control in our study, physostigmine displayed an IC50 of 3 μM which is relatively high yet comparable to other reported IC50 values against electric eel AChE (0.028–6.45 μM).41,42 That implies that the inhibitory activities observed for 7b is near the pharmaceutically relevant concentration range.
While most of the synthetic analogs were not active enough to motivate their detailed Ki analysis, the link between degree of substitution and the inhibitory potency of the compounds was still evident. The quaternary amines also generally displayed a higher inhibitory activity in relation to their tertiary structural counterparts. By dissecting the molecules into individual chemical constituents it was possible to assess both the charged contribution as well as the Connolly solvent excluded volume of the substituted ethylphenyl part of the molecules as presented in Table 2 and Fig. 3.
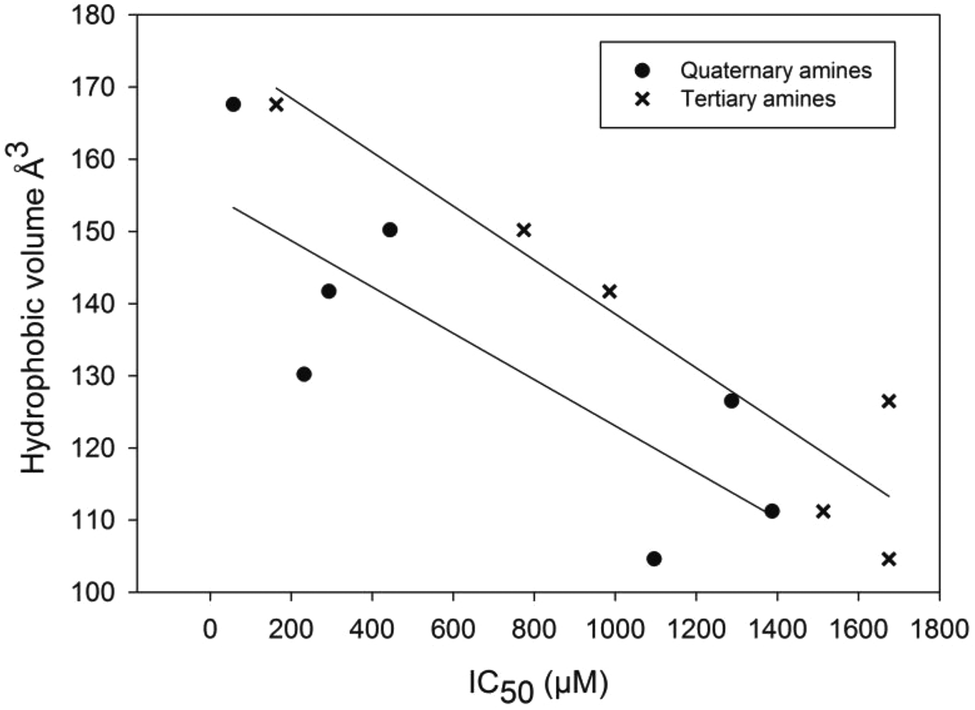 | ||
| Fig. 3 Correlation plot illustration the influence of the hydrophobic volume on the inhibitory effect of the different types of analogs. -×- are the tertiary amines, 2a–7a and -•- represent the quaternary amines 1, 2b–7b. | ||
| Compound | Solvent excluded volumea (Å3) | logPa |
IC50 tertiary (“a”) (μM) | IC50 quaternary (“b”) (μM) |
|---|---|---|---|---|
| a Calculated using ChemBio3D Ultra 14.0 disregarding the substituted and ionized nitrogen atom, hence only the contribution from the substituted ethylphenyl moiety. | ||||
| 1 | 130.2 | 3.38 | n.a. | 232 |
| 2 | 104.6 | 2.94 | 1675 | 1096 |
| 3 | 111.2 | 2.81 | 1513 | 1387 |
| 4 | 126.5 | 2.55 | 1395 | 1287 |
| 5 | 141.7 | 3.64 | 968 | 293 |
| 6 | 150.2 | 4.21 | 774 | 444 |
| 7 | 167.6 | 4.47 | 163 | 57 |
The correlation between the bulk of the molecules and their inhibitory activity is clear and this trend is also evident when examining the influence of logP on activity. The quaternary amines generally displayed higher inhibitory activities in comparison to their tertiary amine counterparts. Although all molecules of the current study can be regarded as basic in a physiological context, it is obvious that the constant positive charge of the quaternary compounds 1 and 2b–7b is beneficial. This is not surprising given that this functionality is chemically analogous with the natural substrate of AChE.43 Isolated 1 and its methoxy analogue 5b, demonstrate similar activities (232 and 293 μM, respectively), suggesting that these compounds do not engage in any crucial hydrogen bond formation with groups within the active site. It is of interest though, that the dibromomethoxy 7b (57 μM) is significantly more active than its phenolic counterpart 6b (444 μM). Considering the data as a whole, we propose that compounds containing large, hydrophobic substituents on the phenyl group, in addition to the quaternary amine, display the most effective inhibitory behavior. These findings contrast our recent study of bromotryptamines where no obvious trend between the hydrophobicity and AChE inhibition was seen.14
The cytotoxicity of the compounds was also evaluated employing human MRC-5 fibroblast. None the included compounds displayed any significant toxic effects at concentrations up to 150 μM (data not included). Compound 1 has previously been tested against rat PC 12 cells and shown to display no toxicity at 10 μg mL−1.36 The use of AChE inhibitors can have several drawbacks including unwanted muscle contraction and neuromuscular transmission.44,45 Given that 7b was the most potent molecule from our initial studies, an extensive physiological evaluation of its effects on muscle contraction employing isolated mouse hemidiaphragm was conducted.
The effects of 7b at the concentration which significantly reduced the AChE activity by 20% (20 μM) on both nerve evoked and directly elicited single twitch and tetanic contractions in isolated mouse hemidiaphragm preparation were studied. AChE inhibition in hippocampus CA1, CA3 and striatum produced by ethanol extract from Ptychopetalum olacoides was shown to be 33%, 20% and 17%, respectively, and these levels of inhibition significantly improved cognitive abilities in old rats.46 In line with this, we performed the experiments on muscle function and neuromuscular transmission employing a 20 μM concentration. Reversible AChE inhibitor neostigmine methylsulfate (3 μM) (Sigma-Aldrich, USA) was employed as a positive control. At this concentration neostigmine inhibits AChE in mouse diaphragm muscle by 96%.47 In the muscle contraction experiment neostigmine induced characteristic facilitation of neuromuscular transmission associated with anticholinesterase treatment followed by a decrease in indirectly elicited muscle twitches. Neostigmine produced the complete block of tetanic contractions evoked by repetitive nerve stimulation. 7b appeared to have no effect on directly and indirectly evoked muscle twitch amplitude and the amplitude of directly and indirectly evoked tetanic muscle contraction (Fig. 4 and 5). The potential extended effects of 7b on indirectly evoked muscle twitch amplitude were also investigated by incubating 7b for 60 min with the mouse hemidiaphragm preparation (Fig. 6). Compound 7b behaved in a similar fashion to the negative control (methanol) in the time-course study and appeared to have little effect on muscle contraction, an advantageous property for AChE drugs.
 | ||
| Fig. 4 Effects of 7b on contractions in isolated mouse hemidiaphragm preparation. (A) Representative control tracing. Arrow (MeOH) indicates superfusion of methanol solution in 0.35 v/v% final concentration. (B) ‘Positive control’ with AChE inhibitor neostigmine (3 μM). (C) 7b (20 μM). N—denotes nerve evoked muscle contraction; D—denotes directly elicited muscle contraction; Tn—denotes nerve evoked tetanic contraction; Td—denotes directly elicited tetanic contraction; W—wash out. | ||
 | ||
| Fig. 5 The time-course effects of 7b on indirectly evoked muscle twitch of isolated mouse hemidiaphragm preparation. Each point represents the mean value ± SEM obtained from 2–3 different nerve muscle preparations. Graphs including the positive control neostigmine can be found in the ESI.† | ||
 | ||
| Fig. 6 Effects of 7b on both, nerve evoked (A) and directly elicited tetanic (B) contractions of isolated mouse hemidiaphragm preparation. Note that 7b (20 μM) have no effect on the amplitude of directly and indirectly evoked tetanic muscle contraction. Graphs including the positive control neostigmine can be found in the ESI.† | ||
Inhibition of AChE in the neuromuscular junction is associated with the inability to sustain a tetanic contraction produced by the repetitive high frequency stimulation of the motor nerve.48 The effect of 7b on the maximal amplitude of nerve evoked tetanic contraction was thus also established. 7b did not influence the tetanic contractions and tetanic fade was only seen for the positive control neostigmine in our studies (Fig. S11 in ESI†).
Finally the depolarization effect of 7b on the skeletal muscle end plate potentials (EPPs) was investigated. For the EPPs experiments the mouse hemidiaphragm preparations were pretreated for 30 min with a 2 μM solution of conotoxin GIIIB, and all experiments were further performed in the presence of 2 μM conotoxin GIIIB to record full sized EPPs and to prevent muscle twitches. Compound 7b displayed no prominent activity towards the EPPs and induced no changes in evoked neurotransmitter release (A) or EPPs half decay (B) after both 30 and 60 minutes exposure of the neuromuscular preparation to the compound (Fig. 7).
 | ||
| Fig. 7 Effects of 7b on EPPs amplitude and EPPs half decay. Mouse hemidiaphragm preparations were pretreated for 30 min with 2 μM conotoxin GIIIB, and all experiments were performed in the presence of 2 μM conotoxin GIIIB to record full sized EPPs and to prevent muscle twitches. Each point represents the mean value ± SEM obtained from 8–12 muscle fibers of each from 2–3 different nerve muscle preparations. Evoked neurotransmitter release (A) and EPPs half decay (B) were determined after 30 and 60 min. | ||
From the neuromuscular data we can conclude that 7b, our most potent analog of stryphnusin, inhibits AChE without also inflicting unwanted collateral physiological responses in the neuromuscular system. Of the prepared compounds, only compound 7b was investigated due to its relatively high inhibitory activity and its structural similarity with both natural and synthetic phenethylamine analogs. Intrigued by this selectivity, and to further investigate the generality of these findings, an analogous synthetic compound, a monobrominated tryptamine from our recent study (compound “9a” in ref. 14) was also included.14 The brominated tryptamine behaved in a very similar fashion (see ESI Fig. S8–S13† for comparison with 7b and neostigmine) in the neuromuscular experiments. This illustrates that both these types of compounds exert their AChE inhibition without side effects on muscular transmission. To the best our knowledge, this is the first reported neuromuscular investigation of these types of small, halogenated AChE inhibitors. Several natural AChE inhibitors such as the bufotenins and related compounds display a similar size, degree of substitution and distribution of functionalities and these results illustrate that this structural motif can be used to generate small selective reversible AChE inhibitors.
Conclusions
The marine natural product stryphnusin (1) was isolated from an organic extract of the marine sponge S. fortis. 1 shares structural features with known AChE inhibitors and the natural substrate and was therefore evaluated for inhibitory activity against electric eel AChE, displaying moderate inhibitory properties. In order to identify analogues with greater activity and to develop a pharmacophore model, a library of 12 compounds was synthesized. The majority of the synthetic compounds were less active than the natural product but 7b, which contained an additional bromine and methyl functionality, displayed inhibitory properties comparable to several other marine AChE inhibitors. SAR analysis of the library highlighted that both phenyl ring substituents contribute to steric bulk and hydrophobicity, and that analogues bearing a quaternary amine improved activity, thus providing new insights into the structure–activity relationship of AChE inhibitors. Our most promising compound, 7b, and a structurally related tryptamine, were employed in neuromuscular transmission studies and showed no significant effect; a desirable property when developing therapeutic AChE inhibitors.Experimental
General experimental procedures
The preparatory HPLC system used to isolate 1 consisted of a 600 pump, a 2996 Photodiode Array UV detector, a 3100 Mass Detector, and a 2767 sample manager (Waters, Milford, MA, USA). NMR spectra were acquired on either a Varian VNMRS 600 MHz or a Varian 7000e 400 MHz spectrometer. Carbon resonances were either acquired directly or derived from gHMBC experiments and the chemical shifts were referenced to the residual solvent peaks. HRMS was recorded on an LTQ Orbitrap XL Hybrid Fourier Transform mass spectrometer from Thermo Scientific and the Thermo Scientific Accela HPLC-LTQ Ion Trap-Orbitrap Discovery system was used to determine accurate mass of the synthetic compounds. Infrared spectra were recorded on an Avatar 320 FT-IR spectrometer from Nicolet. Solvents, reagents and compound 2a were acquired from commercial sources and used without further purification. The starting materials for compounds 6a37 and 7a38 were prepared using reported methods. Spectroscopic data is included for novel compounds, or those lacking characterization in the literature. For the neuromuscular investigation adult male Balb/C mice were used.Isolation and characterization of 1
Specimens of S. fortis were collected northwest off Spitsbergen (79°33′N, 8°53′E) at 333 m depth using an Agassiz dredge trawl in September 2007. The sample was stored at −23 °C until use. A subsample is kept at the Norwegian National Marine Biobank (Marbank, reference number M10037), UiT The Arctic University of Norway, Tromsø. Frozen sponge material (2.0 kg) was extracted as previously described yielding 34.62 g of organic extract.33 The organic extract (2 g) was partitioned between n-hexane (150 mL) and 90% MeOH (100 mL). The 90% MeOH fraction was dried under vacuum and further purified by mass guided prep-HPLC using a XTerra RP18 HPLC column employing a linear gradient from 5 to 10% acetonitrile in ultra-pure water (both containing 0.1% formic acid) at a flow rate of 6 mL min−1 over 13 min resulting in the isolation of 1. The structure of 1 was confirmed using MS, 1D and 2D NMR (COSY) spectroscopic techniques and comparison with literature data.35General procedure for reductive amination
The spectral data of compounds 3a (55%, 0.38 mmol),494a (88%, 0.52 mmol),50 and 6a (73%, 0.1 mmol)51 were consistent with previous reports.
General procedure for quaternary amine formation
The spectral data of compounds 6b (82%, 0.04 mmol)52 was consistent with those reported. Compounds 2b,533b,544b55 and 7b56 have been reported but lack full characterization data.
Cholinesterase inhibition assay
Cholinesterase activity was measured by Ellman's method, using acetylthiocholine chloride (0.125, 0.25, and 0.5 mM, respectively) as a substrate in 100 mM potassium phosphate buffer pH 7.4 at 25 °C, and electric eel AChE, or horse serum BChE as enzyme sources (Sigma, final concentration in the test 0.0075 U mL−1). Hydrolysis of acetylthiocholine chloride was followed on a VIS microplate reader (Dynex Technologies, USA) at 405 nm. AChE or BChE inhibition was monitored for 5 minutes at 20 °C for each compound (prepared from a 2 mg mL−1 stock in methanol and then progressively diluted in 100 mM potassium phosphate buffer pH 7.4). The positive control (physostigmine, Sigma) was prepared in ethanol (at a 10 mM final concentration) and progressively diluted in the same buffer. The effect of the pure methanol or ethanol on enzyme inhibition was also checked, and all readings were corrected for their appropriate blanks. Every measurement was repeated at least three times.Cytotoxicity testing
The potential cytotoxicity of compounds 1–7b was evaluated against human MRC-5 normal lung fibroblasts, using the tetrazolium based (MTS) CellTiter 96® Aqueous One Solution Cell Proliferation Assay. Percent cell survival was calculated by comparing exposed cells to untreated cells and cells treated with Triton X-100 (0.01%), as previously described.33Muscle contraction experiments
Mice were sacrificed by cervical dislocation, followed by immediate exsanguination. The diaphragm with corresponding phrenic nerves was dissected and used.Hemidiaphragm was tightly pinned to the Rhodorsil coated organ bath containing oxygenated standard Krebs-Ringer solution composed of (in mM): 154 NaCl, 2 CaCl2, 5 KCl, 1 MgCl2, 5 HEPES and 11 D-glucose, pH 7.4, at 22–24 °C. The tendinous side of the hemidiaphragm was attached with a steel hook via silk thread to an isometric force displacement transducer FT 03 (Grass instruments, West Warwick, RI, USA). Nerve-evoked single isometric twitches were recorded as follows: the motor nerve of isolated neuromuscular preparation was stimulated with a square pulse S-48 stimulator (Grass instruments, West Warwick, RI, USA) via a suction electrode with pulses of 0.1 ms duration, 0.1 Hz stimulation rate and with the supramaximal voltage of 8–10 V. Directly evoked single isometric twitches were evoked by stimulating hemidiaphragm preparation with a platinum electrode assembly placed along the organ bath with pulses of 0.1 ms in duration, with a 0.1 Hz stimulation rate and with the supramaximal voltage of 60–80 V. Directly or nerve-evoked tetanic muscle contraction recordings were obtained by stimulating the hemidiaphragm with train of pulses (1000 ms duration at 80 Hz). Each hemidiaphragm preparation was then left to equilibrate for 20 min to achieve stable resting tension before beginning the experiments. Electrical signals were amplified by a P122 strain gage amplifier (Grass instruments, West Warwick, RI, USA) and then digitized at a sampling rate of 1 kHz using a data acquisition system (Digidata 1440A; Molecular Devices, Sunnyvale, CA, USA).57 The effect of 7b on the neuromuscular hemidiaphragm preparation was measured for 60 min.
Recordings of end plate potentials (EPPs)
The experiments were performed at 22–24 °C on oxygenated mouse hemidiaphragm preparations, pretreated for 30 min with 2 μM μ-conotoxin GIIIB, an inhibitor of muscle sodium channels, to record full-sized endplate potentials without contracting the muscle. The resting membrane potentials and endplate potentials (EPPs) were recorded from endplate regions in superficial muscle fibres using intracellular borosilicate microelectrodes filled with 3 M KCl and pulled with a P-97 Flaming/Brown microelectrode puller (Sutter Instruments, Novato, CA, USA). Microelectrodes with resistance from 10–20 MΩ were used. Recordings were performed before, 30 and 60 min after application of 7b, and 15 min after washing-out the 7b. EPPs were evoked by stimulating the phrenic nerve with supramaximal square pulses of 0.1 ms duration and with a frequency of 1 Hz. EPP and MEPP recordings were digitized using Digidata 1440A and the pClamp 10 software. Data were analyzed using the pClamp-Clampfit 10 program. Amplitudes of EPPs were normalized to a membrane potential of −70 mV using the formula formula Vc = V0 × (−70)/E, where Vc is the normalized amplitude of EPPs, V0 is the recorded amplitude and E is the resting membrane potential.Data analysis and statistics
Data were statistically analysed using SigmaPlot for Windows 11.0 (Systat Software Inc., Germany). The results are presented as the mean ± SEM. Data were firstly tested for normality (Shapiro–Wilk) and equal variance for assignment to parametric or non-parametric analysis. For the statistical analysis of the data, a two-tailed Student t-test was used and P value ≤0.05 was considered to be statistically significant.Acknowledgements
This work was partly supported with grants from the Norwegian research council (ES508288) and JS and LM are grateful for the support. The Slovenian authors wish to thank the Slovenian research agency for financial support (Grant P1-0207 and P4-0053), and the ERASMUS Student mobility programme for financial support of MC.Notes and references
- W. H. Gerwick and B. S. Moore, Chem. Biol., 2012, 19, 85–98 CrossRef CAS PubMed.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- T. F. Molinski, D. S. Dalisay, S. L. Lievens and J. P. Saludes, Nat. Rev. Drug Discovery, 2009, 8, 69–85 CrossRef CAS PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2015, 32, 116–211 RSC.
- W. H. Gerwick and A. M. Fenner, Microb. Ecol., 2013, 65, 800–806 CrossRef CAS PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- J. Hert, J. J. Irwin, C. Laggner, M. J. Keiser and B. K. Shoichet, Nat. Chem. Biol., 2009, 5, 479–483 CrossRef CAS PubMed.
- M. C. Leal, J. Puga, J. Serôdio, N. C. Gomes and R. Calado, PLoS One, 2012, 7, 1–15 Search PubMed.
- G. Steinert, S. Whitfield, M. W. Taylor, C. Thoms and P. J. Schupp, Mar. Biotechnol., 2014, 16, 594–603 CrossRef CAS PubMed.
- U. Hentschel, J. Piel, S. M. Degnan and M. W. Taylor, Nat. Rev. Microbiol., 2012, 10, 641–654 CrossRef CAS PubMed.
- J. Svenson, Phytochem. Rev., 2013, 12, 567–578 CrossRef CAS PubMed.
- M. Tadesse, M. B. Strøm, J. Svenson, M. Jaspars, B. F. Milne, V. Tørfoss, J. H. Andersen, E. Hansen, K. Stensvåg and T. Haug, Org. Lett., 2010, 12, 4752–4755 CrossRef CAS PubMed.
- M. Tadesse, J. Svenson, K. Sepčić, L. Trembleau, M. Engqvist, J. H. Andersen, M. Jaspars, K. Stensvåg and T. Haug, J. Nat. Prod., 2014, 77, 364–369 CrossRef CAS PubMed.
- E. K. Olsen, E. Hansen, L. W. K. Moodie, J. Isaksson, K. Sepčić, M. Cergolj, J. Svenson and J. H. Andersen, Org. Biomol. Chem., 2016, 14, 1629–1640 CAS.
- J. Harley-Mason and A. Jackson, J. Chem. Soc., 1954, 1165–1171 RSC.
- S. Bhattacharya and A. Sanyal, Indian J. Physiol. Pharmacol., 1971, 15, 133–134 CAS.
- N. Lysek, E. Rachor and T. Lindel, Z. Naturforsch., 2002, 57, 1056–1061 CAS.
- L. Peters, G. M. König, H. Terlau and A. D. Wright, J. Nat. Prod., 2002, 65, 1633–1637 CrossRef CAS.
- I. E. Kasheverov, I. V. Shelukhina, D. S. Kudryavtsev, T. N. Makarieva, E. N. Spirova, A. G. Guzii, V. A. Stonik and V. I. Tsetlin, Mar. Drugs, 2015, 13, 1255–1266 CrossRef CAS PubMed.
- A. V. Terry and J. J. Buccafusco, J. Pharmacol. Exp. Ther., 2003, 306, 821–827 CrossRef CAS PubMed.
- M. B. Colovic, D. Z. Krstic, T. D. Lazarevic-Pasti, A. M. Bondzic and V. M. Vasic, Curr. Neuropharmacol., 2013, 11, 315–335 CrossRef CAS PubMed.
- V. N. Talesa, Mech. Ageing Dev., 2001, 122, 1961–1969 CrossRef CAS PubMed.
- D. Munoz-Torrero, Curr. Med. Chem., 2008, 15, 2433–2455 CrossRef CAS PubMed.
- N. Tabet, Age Ageing, 2006, 35, 336–338 CrossRef CAS PubMed.
- L. Pezzementi and A. Chatonnet, Chem.-Biol. Interact., 2010, 187, 27–33 CrossRef CAS PubMed.
- B. Li, J. A. Stribley, A. Ticu, W. Xie, L. M. Schopfer, P. Hammond, S. Brimijoin, S. H. Hinrichs and O. Lockridge, J. Neurochem., 2000, 75, 1320–1331 CrossRef CAS PubMed.
- D. J. Triggle, J. M. Mitchell and J. Filler, CNS Drug Rev., 1998, 4, 87–136 CrossRef CAS.
- H. Geerts, P. O. Guillaumat, C. Grantham, W. Bode, K. Anciaux and S. Sachak, Brain Res., 2005, 1033, 186–193 CrossRef CAS PubMed.
- M. Heinrich and H. L. Teoh, J. Ethnopharmacol., 2004, 92, 147–162 CrossRef CAS PubMed.
- D. Kudryavtsev, T. Makarieva, N. Utkina, E. Santalova, E. Kryukova, C. Methfessel, V. Tsetlin, V. Stonik and I. Kasheverov, Mar. Drugs, 2014, 12, 1859–1875 CrossRef PubMed.
- R. Sakai and G. T. Swanson, Nat. Prod. Rep., 2014, 31, 273–309 RSC.
- K. Ø. Hanssen, G. Cervin, R. Trepos, J. Petitbois, T. Haug, E. Hansen, J. H. Andersen, H. Pavia, C. Hellio and J. Svenson, Mar. Biotechnol., 2014, 16, 684–694 CrossRef CAS PubMed.
- K. Ø. Hanssen, J. H. Andersen, T. Stiberg, R. A. Engh, J. Svenson, A.-M. Genevière and E. Hansen, Anticancer Res., 2012, 32, 4287–4297 CAS.
- P. Cárdenas, J. Chem. Ecol., 2016, 42, 339–347 CrossRef PubMed.
- P. Ciminiello, C. Dell'Aversano, E. Fattorusso, S. Magno and M. Pansini, J. Nat. Prod., 2000, 63, 263–266 CrossRef CAS.
- A. Aiello, E. Fattorusso, C. Imperatore, M. Menna and W. E. Müller, Mar. Drugs, 2010, 8, 285–291 CrossRef CAS PubMed.
- E. García-Egido, J. Paz, B. Iglesias and L. Muñoz, Org. Biomol. Chem., 2009, 7, 3991–3999 Search PubMed.
- R. M. Van Wagoner, J. Jompa, A. Tahir and C. M. Ireland, J. Nat. Prod., 1999, 62, 794–797 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- K. Hostettmann, A. Borloz, A. Urbain and A. Marston, Curr. Org. Chem., 2006, 10, 825–847 CrossRef CAS.
- M. R. Loizzo, R. Tundis, F. Menichini and F. Menichini, Curr. Med. Chem., 2008, 15, 1209–1228 CrossRef CAS PubMed.
- F. Menichini, R. Tundis, M. R. Loizzo, M. Bonesi, M. Marrelli, G. A. Statti, F. Menichini and F. Conforti, Fitoterapia, 2009, 80, 297–300 CrossRef CAS PubMed.
- M. Pohanka, Biomed. Pap., 2011, 155, 219–229 CrossRef CAS PubMed.
- J. Morrison, Br. J. Pharmacol., 1977, 60, 45–53 CrossRef CAS PubMed.
- A. L. Clark and F. Hobbiger, Br. J. Pharmacol., 1983, 78, 239–246 CrossRef CAS PubMed.
- M. Figueiró, J. Ilha, D. Pochmann, L. Porciúncula, L. Xavier, M. Achaval, D. Nunes and E. Elisabetsky, Phytomedicine, 2010, 17, 956–962 CrossRef PubMed.
- J. Minic, A. Chatonnet, E. Krejci and J. Molgó, Br. J. Pharmacol., 2003, 138, 177–187 CrossRef CAS PubMed.
- C. Chang, S. Hong and J. L. Ko, Br. J. Pharmacol., 1986, 87, 757–762 CrossRef CAS PubMed.
- A. Küçükosmanoǧlu Bahçeevli, S. Kurucu, U. Kolak, G. Topçu, E. Adou and D. G. I. Kingston, J. Nat. Prod., 2005, 68, 956–958 CrossRef PubMed.
- M. Saravanan, K. S. Kumar, P. P. Reddy and B. Satyanarayana, Synth. Commun., 2010, 40, 1880–1886 CrossRef CAS.
- H. Kigoshi, K. Kanematsu, K. Yokota and D. Uemura, Tetrahedron, 2000, 56, 9063–9070 CrossRef CAS.
- X. Fu and F. J. Schmitz, J. Nat. Prod., 1999, 62, 1072–1073 CrossRef CAS PubMed.
- H. Decker and P. Becker, Ber. Dtsch. Chem. Ges., 1912, 45, 2404–2409 CrossRef.
- G. Barger, J. Chem. Soc., 1909, 95, 2193–2197 RSC.
- K. W. Rosenmund, Ber. Dtsch. Chem. Ges., 1910, 43, 306–313 CrossRef CAS.
- E. Leete, R. M. Bowman and M. F. Manuel, Phytochemistry, 1971, 10, 3029–3033 CrossRef CAS.
- M. Grandič, R. Aráoz, J. Molgó, T. Turk, K. Sepčić, E. Benoit and R. Frangež, Toxicol. Appl. Pharmacol., 2012, 265, 221–228 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for synthetic compounds not previously reported and additional neuromuscular experiments. See DOI: 10.1039/c6ob02120d |
| ‡ Present address: Department of Chemistry, University of Umeå, SE-901 87, Umeå, Sweden. |
| This journal is © The Royal Society of Chemistry 2016 |