
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA radical addition/cyclization of diverse ethers to 2-isocyanobiaryls under mildly basic aqueous conditions†
Cintia
Anton-Torrecillas
,
Diego
Felipe-Blanco
and
Jose C.
Gonzalez-Gomez
*
Departamento de Química Orgánica, Facultad de Ciencias and Instituto de Síntesis Orgánica (ISO), Universidad de Alicante, Apdo. 99, 03080 Alicante, Spain. E-mail: josecarlos.gonzalez@ua.es
First published on 25th October 2016
Abstract
Mildly basic aqueous conditions facilitated the tert-butyl peroxybenzoate (TBPB) mediated dehydrogenative addition of a range of ethers, including acetals, to diverse substituted 2-isocyanobiaryls. Mechanistic studies suggest that this radical cascade is an example of base promoted homolytic aromatic substitution (BHAS).
Introduction
Phenanthridine is the scaffold of many naturally occurring products1 that exhibit a wide range of bioactivities, such as antitumor,2 antituberculosis,3 antimicrobial,4 antiviral,5 acaricidal,6 and fungicidal activities.7 Some phenanthridine derivatives have also found application in biological chemistry as dyes for cytofluorometric assays8 or even in materials science due to their optoelectronic properties.9 Recently, the assembly of the phenanthridine framework has been elegantly achieved with a cascade that involves the addition of radicals to somophilic 2-isocyanobiphenyls, followed by intramolecular homolytic aromatic substitution.10,11 In this context, and given the progress made in the Cα–H oxidation of ethers with peroxides to form radicals,12 the oxidative addition of ethers to 2-isocyanobiphenyls has been successfully achieved using different conditions.13 This transformation requires a dual selective C–H bond functionalization, and constitutes a powerful tool to increase molecular complexity with high atom-economy, where only hydrogen is lost (Scheme 1). Moreover, ethers are very important raw materials and they are also common substructures of natural and synthetic bioactive products.14 Consequently, the development of methods that make use of relatively unreactive ethers to build complex molecules, is highly demanded by organic and medicinal chemists. | ||
| Scheme 1 Oxidative addition of 1,4-dioxane to 2-isocyanobiphenyls: a radical cascade. | ||
It is worth noting that 1,4-dioxane is the model substrate for C–H bond functionalization of ethers in most of the reported studies.13 This symmetric ether presents all eight C–H bonds adjacent to an oxygen atom, which stabilizes the radical by hyperconjugation with the non-bonding electrons. Moreover, theoretical studies suggest that the release of ring strain upon radical formation is greater in 1,4-dioxane than in other cyclic ethers such as tetrahydrofuran.15 As a matter of fact, when TBPB was used as the oxidant in the insertion of 2-isocyanobiaryls with ethers, only 1,4-dioxane was successful and THF failed.13a Notably, when benzoyl peroxide was used in the same transformation, another three ethers could be inserted with moderate results (40–45%).13b To the best of our knowledge, the greatest number of ethers tolerated in this reaction (seven examples) has been possible with the use of DTBP as the oxidant, FeCl3 as the catalyst and DBU as the cocatalyst.13c With these precedents we decided to develop a new protocol for this reaction aimed at accommodating a range of ethers, free of transition-metal catalysts and using economical and environmentally friendly conditions (Scheme 2).
 | ||
| Scheme 2 Previous results and our reaction conditions for THF. | ||
Results and discussion
To explore new reaction conditions we selected 2-isocyanobiphenyl (1a) and THF (2a) as model substrates. In a control experiment we performed the reaction using BPO as the oxidant at 100 °C and we obtained 35% yield of the desired product 3aa, a similar result to the one obtained by the Cheng group,13b together with byproduct 4a (Table 1, entry 1). Surprisingly, the addition of K2CO3 and H2O completely inhibited the formation of 3aa (entry 2). Prompted by the excellent results in oxidative transformations achieved with Bu4NI as the catalyst and TBHP,16 we tried this combination, but 3aa was obtained in low yield (entry 3). The use of DTBP or (NH4)2S2O8 in combination with Bu4NI was also unsuccessful in this transformation (entries 4–6). It is reported that TBPB fails to afford product 3aa in this reaction and that only compound 4a is obtained.13a To our surprise, the inclusion of H2O in this reaction mixture afforded product 3aa, although in low yield (entry 7). We were pleased to find that the same oxidant, but in the presence of K2CO3 and H2O, allowed full conversion of 1a to obtain compound 3aa in good yield (entry 10). While the addition of Bu4NI had no important impact on the reaction, the amount of base and the control of the reaction temperature were crucial to obtain good and reliable results (entries 8–11). Similar results were obtained with Na2HPO4, and lower yields were obtained for the desired product when non-nucleophilic organic bases were used in substoichiometric amounts (entries 12–14).|
|
||||
|---|---|---|---|---|
| Entry | Oxidant (mol%)/additive (mol%) | Base (mol%) | Conv.a (%) | (3aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a)b :4a)b |
| a By GC of the crude reaction mixture. b Yield calculated by 1H-NMR of the crude reaction mixture using durene as the internal standard. c Formation of other products is observed by GC. d The conversion was 90% at 80 °C and 0% at 60 °C. BPO = benzoyl peroxide. DTBP = di-tert-butylperoxide. TBPB = tert-butylperoxybenzoate. DABCO = 1,4-diazabicyclo[2.2.2]octane. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||||
| 1c | BPO (120) | None | 100 | 35:22 |
| 2 | BPO (120) | K2CO3 (150) | 37 | — |
| 3 | TBHP (300)/Bu4NI (20) | K2CO3 (150) | 45 | 20:5 |
| 4 | DTBP (300)/Bu4NI (20) | None | 0 | — |
| 5 | DTBP (300)/Bu4NI (20) | K2CO3 (150) | 0 | — |
| 6 | (NH4)2S2O8 (220) | K2CO3 (150) | 0 | — |
| 7 | TBPB (220) | None | 100 | 12:4c |
| 8 | TBPB (120)/Bu4NI (20) | K2CO3 (150) | 55 | — |
| 9 | TBPB (220) | K2CO3 (100) | 70 | — |
| 10 | TBPB (220) | K 2 CO 3 (150) | 100 |
77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :23 :23
|
| 11 | TBPB (220)/Bu4NI (50) | K2CO3 (150) | 100 | 75:25 |
| 12 | TBPB (220) | Na2HPO4 (150) | 90 | 75:25 |
| 13c | TBPB (220) | DABCO (50) | 100 | 18:0 |
| 14c | TBPB (220) | DBU (50) | 100 | 40:0 |

Having found very simple optimized conditions for the model reaction, we evaluated its scope using different 2-isocyanobiaryls and THF (Scheme 3). We first examined substrates with substituents on the upper aromatic ring. Methyl-substituted products 3ba and 3ca were obtained in good to moderate yields, without observing any benzylic oxidation. During the formation of 3ca, some compound 3aa was also observed (GC-MS), likely by ipso-homolytic aromatic substitution, and its isolated yield was affected due to difficulties in the chromatographic purification. A more electron-donating group such as methoxy was compatible with this protocol, affording compound 3da in reasonably good yield, as well as the corresponding byproduct 4d (detected by GC-MS and 1H-NMR). When 2-(2-isocyanophenyl)-naphthalene was used, compound 3ea was the only regioisomer isolated in moderate yield, despite the steric hindrance between the ether moiety and the extra aromatic ring.17 Electron-withdrawing groups, such as acetyl, trifluoromethyl, chloro and fluoro, were suitable substituents in the upper aromatic ring, furnishing products (3fa, 3ga, 3ha and 3ia) in good isolated yields. Interestingly, when meta-substituted substrates 1j and 1k were used, both possible regioisomers were isolated after column chromatography. While the ratio of 3ja/3′ja was 1:1, for 1k the major product was the most crowded regioisomer (3ka).18 We also examined isocyanobiaryls substituted in the lower aromatic ring by electron-donating and electron-withdrawing groups, obtaining uniformly moderate yields in all cases (3la–3oa).
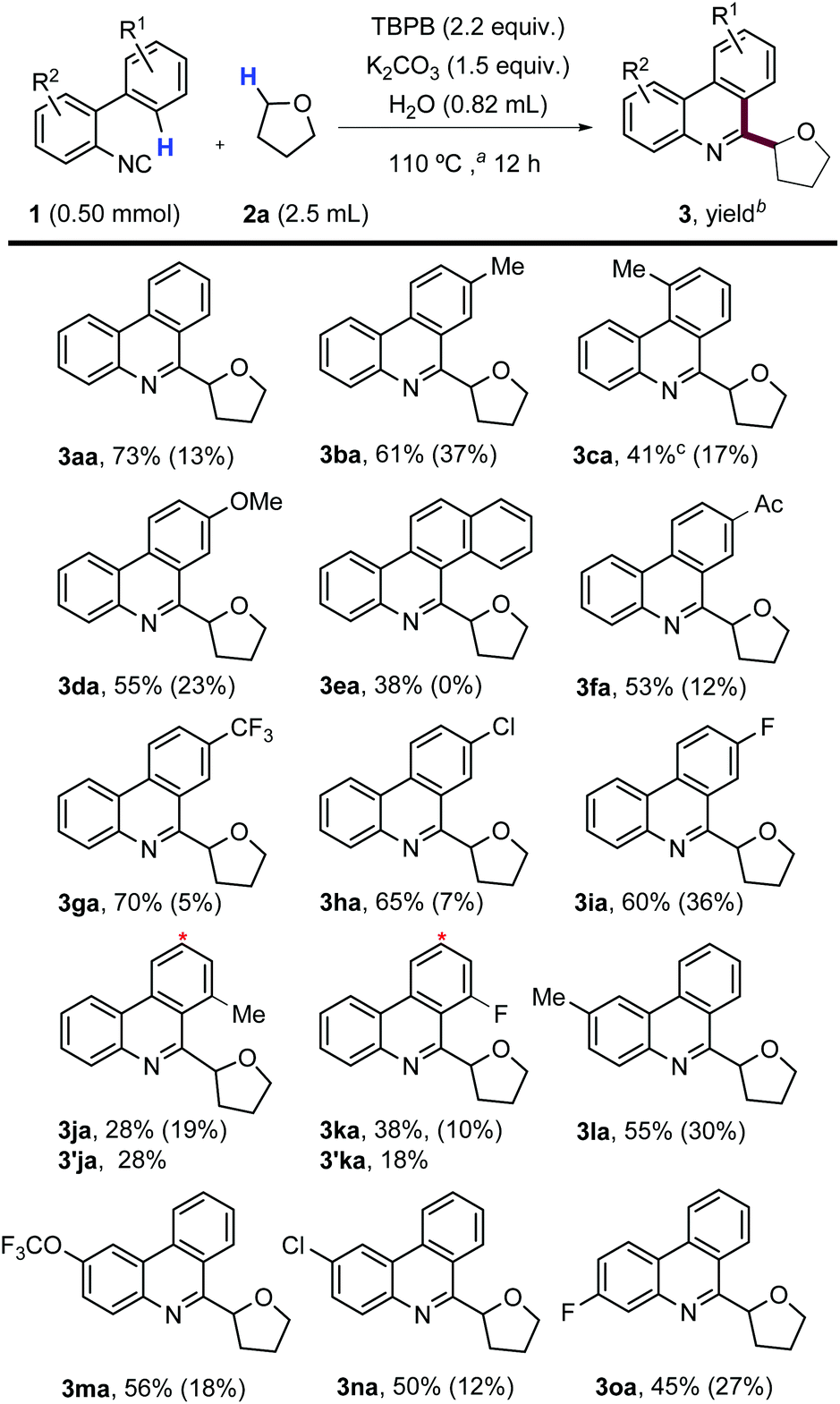 | ||
| Scheme 3 Scope of 2-isocyanobiaryls with THF. aMeasured temperature of the sand bath. bYields after purification of isolated products and in parentheses are yields of 6-H phenanthridines 4 estimated by GC-MS of crude reaction mixtures. cProduct 3aa was also formed and purification of 3ca was more difficult. *The other regioisomer isolated (3′). | ||
We further explore the performance of different ethers using the optimized procedure (Scheme 4). 1,4-Dioxane (2b) and THP (2c) were also suitable for this protocol, obtaining products 3ab and 3ac in moderate yields after purification, as well as compound 4a as the byproduct. With THP, minor amounts of regioisomers were formed (see the ESI† for GC-MS of isomers), complicating the isolation of 3ac in a pure form. Acyclic ethers such as i-Pr2O (2d), n-Bu2O (2e) and t-BuOMe (2g) afforded the corresponding products in good to excellent yields, with linear n-Bu2O providing the best yield. 2-MeTHF (2f) was also examined, affording the more substituted compound 3af as the major product and regioisomer 3′af as an inseparable trans/cis mixture. Surprisingly, when Bn2O (2h) was examined, the expected product (3ah) was obtained in only 17% yield and the addition of a benzyl radical took place preferentially (3′ah). It seems reasonable that after H-abstraction from Bn2O, a β-fragmentation gives rise to the benzyl radical, which is finally added to 1a. The addition of phthalan (2i) was also possible, but compound 3ai was obtained in lower yield. Importantly, acetals 2j and 2k led, respectively, to products 3aj and 3ak in synthetically useful yields. These compounds could be easily hydrolyzed to obtain the corresponding 6-formylphenanthridine and the acetal moiety has the potential to be transformed into other functionalities.19
 | ||
| Scheme 4 Scope of ethers. aMeasured temperature of the sand bath. bYields after purification of isolated products and in parentheses are yields of 4a. | ||
Investigation of the reaction mechanism
To gain insight into the reaction mechanism we performed some experiments. As shown in Scheme 5a, the use of TEMPO as a radical scavenger in our model reaction, under standard conditions, completely inhibited the formation of 3aa and the coupling product 5 was detected by MS. Since compounds 4 are consistent byproducts of the radical cyclization studied here, we were intrigued about their formation and we have not found any experimental evidence of this in the literature.20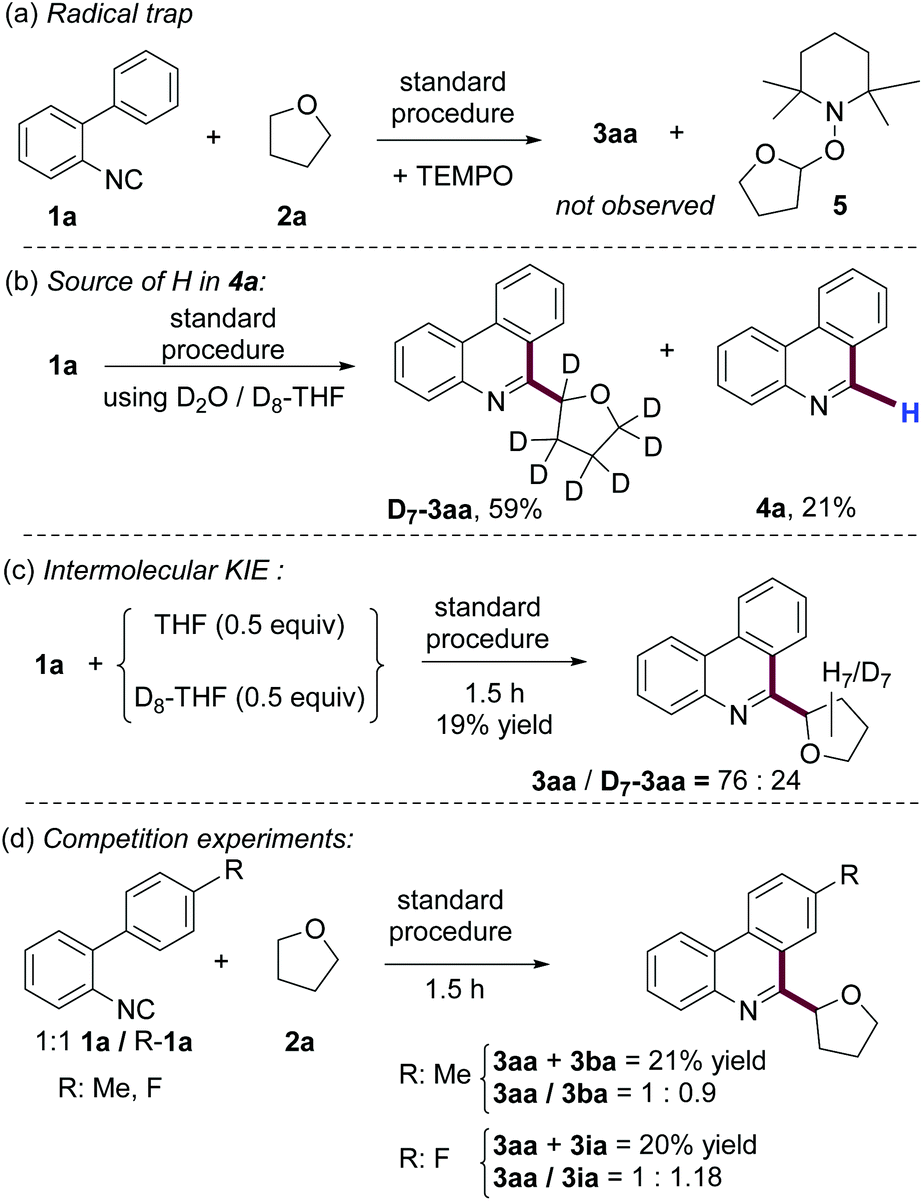 | ||
| Scheme 5 Mechanistic investigations. | ||
When the reaction was conducted using D2O and D8-THF (Scheme 5b), the expected product D7-3aa was obtained, accompanied by phenanthridine 4a. Importantly, the byproduct of this reaction is not deuterated, indicating that neither THF nor H2O is the source of hydrogen for the formation of this phenanthridine. We also studied the intermolecular kinetic isotopic effect (KIE) by competition experiments of equimolar amounts of THF and D8-THF with 1a at the initial stage of the reaction (Scheme 5c). The large KIE obtained (kH/kD = 3.16) suggests that the cleavage of the C(sp3)–H bond is involved in the rate determining step of this reaction. In addition, we also performed competition experiments at the initial state of the reaction, using equimolar quantities of 1a and 2-isocyanobiphenyls substituted in position 4′ by methyl or fluoro groups. The results shown in Scheme 4d suggested that electron-withdrawing substituents in the upper aromatic ring accelerate the reaction. Since this class of group stabilizes the LUMO of the upper aromatic ring, this result is also consistent with a radical cyclization pathway.21
An important feature of this protocol is the crucial role of K2CO3 and H2O in a successful transformation (Table 1, entries 6 and 9). Since the seminal contribution of Studer and Curran,22 several known reactions have been recognized as “base promoted homolytic substitutions” (BHAS) and many new transformations have been developed with this concept.11 Based on literature precedents and our own results, we believe that this reaction is an example of BHAS. A possible reaction mechanism is depicted in Scheme 6. Firstly, homolysis of TBPB takes place upon heating. From the two generated radicals, it is reported that t-BuO˙ abstracts a hydrogen atom from THF faster than Bz˙·to generate the α-furanyl radical.23 Addition of this intermediate to isonitrile 1a, followed by intramolecular addition to the upper aromatic ring, is well documented in a number of examples.11 DFT studies performed by the Studer group indicate that cyclohexadienyl radicals, such as I, are extremely strong acids (pKa ∼ −15 in H2O).24 Therefore, this radical intermediate can be deprotonated under mild basic aqueous conditions (e.g. K2CO3 in H2O) to generate a radical anion II. This species is a potent reductant that can easily transfer an electron to radical Bz˙ or to oxidant TBPB, while generating product 3aa. In addition, intermediate I can transfer a hydrogen atom to 1a, which after a similar BHAS process leads to the formation of byproduct 4a. This explanation is consistent with our observation that neither THF nor H2O was the hydrogen source of 4a.25
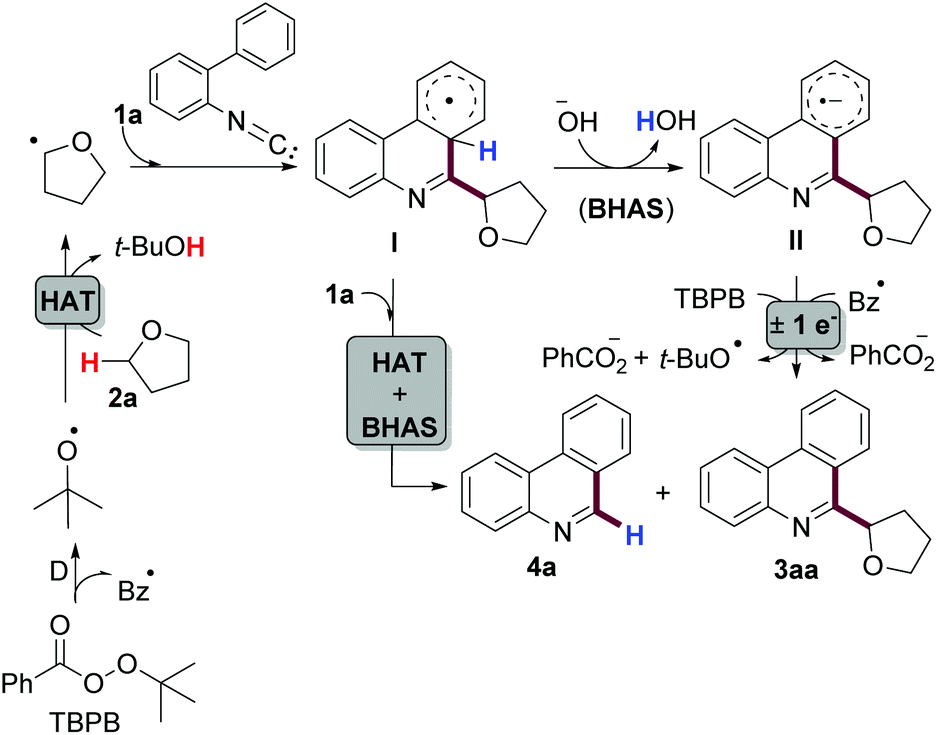 | ||
| Scheme 6 Plausible mechanism. | ||
Conclusions
In conclusion, we have demonstrated that TBPB can be used under mildly basic aqueous conditions to promote the radical addition of a range of ethers to 2-isocyanobiaryls followed by cyclization. Mechanistic investigations suggest that this transformation is an example of BHAS and a plausible explanation is provided for the formation of byproduct 4. Some salient features of the developed methodology are: (a) neither transition metals nor any expensive additives are needed; (b) the wastes generated are harmless inorganic salts and can be easily removed by aqueous workup; (c) the reaction medium is constituted by the reactant ethers and water.Experimental section
General remarks
All starting isonitriles were prepared according to a reported method.10b TLC was performed on silica gel 60 F254, using aluminium plates and visualized by exposure to ultraviolet light. Flash chromatography was carried out on hand-packed columns of silica gel 60 (230–400 mesh). Infrared (IR) spectra were recorded with a spectrophotometer equipped with an ATR component; wavenumbers are given in cm−1. LRMS were obtained using a mass spectrometer coupled with a gas chromatographer (GC); the mobile phase was helium (2 mL min−1); HP-1 column of 12 m was used; temperature program starts at 80 °C for 3 min, then up to 270 °C at a rate of 20 °C min−1, and 17.5 min at 270 °C. HRMS analyses were carried out using Electron Impact (EI) mode at 70 eV by Q-TOF. 1H NMR spectra were recorded at 300 or 400 MHz for 1H-NMR and 75 or 100 MHz for 13C-NMR, using CDCl3 as the solvent and TMS as an internal standard (0.00 ppm). The data are being reported as (s = singlet, d = doublet, t = triplet, m = multiplet or unresolved, br s = broad signal, coupling constant(s) in Hz, integration). 13C-NMR spectra were recorded with 1H-decoupling at 100 MHz and referenced to CDCl3 at 77.16 ppm. DEPT-135 experiments were performed to assign CH, CH2 and CH3.General procedure for the synthesis of compounds 3
Into a pressure tube were added K2CO3 (103 mg, 0.75 mmol) and H2O (0.82 mL). Then, to it were added sequentially the 2-isocyanobiaryl derivative 1 (0.50 mmol), ether 2 (2.5 mL) and tert-butyl peroxibenzoate (TBPB) (213 μL, 1.10 mmol). The reaction mixture was stirred under an Ar atmosphere for 12–14 h at 110 °C (sand bath temperature). Upon full conversion of the isonitrile (TLC or GC), a saturated solution of NaHCO3 (5 mL) and EtOAc (10 mL) was added to the reaction mixture. After phase separation, the aqueous phase was extracted with EtOAc (3 × 15 mL) and the combined organic layers were washed with brine (5 mL), dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel to give the desired product.:1) as a yellow pale solid (91 mg, 0.36 mmol, 73%): Rf 0.23 (95:5 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.63 (dd, J = 8.2, 1.3 Hz, 1H), 8.55 (dd, J = 8.0, 1.5 Hz, 1H), 8.45 (dd, J = 8.7, 1.0 Hz, 1H), 8.19 (dd, J = 7.9, 1.6 Hz, 1H), 7.83 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.75–7.61 (m, 3H), 5.78 (t, J = 6.9 Hz, 1H), 4.26–4.15 (m, 1H), 4.13–4.01 (m, 1H), 2.82–2.65 (m, 1H), 2.50–2.35 (m, 1H), 2.30–2.03 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 159.4 (C), 143.4 (C), 133.4 (C), 130.6 (CH), 130.4 (CH), 128.6 (CH), 127.3 (CH), 127.0 (CH), 126.6 (CH), 124.9 (C), 124.2 (C), 122.5 (CH), 122.0 (CH), 79.7 (CH), 69.1 (CH2), 30.1 (CH2), 26.1 (CH2) ppm; IR ν 3073, 2981, 2881, 1583, 1444, 1300, 1057, 727 cm−1; LRMS (EI) m/z (%) = 249 (M+, 5), 220 (15), 206 (100), 193 (48), 178 (15).
:7) as a yellow pale solid (80 mg, 0.31 mmol, 61%): Rf 0.20 (93:7 hexane/EtOAc); 1H-NMR (400 MHz, CDCl3) δ 8.54–8.48 (m, 2H), 8.20 (br s, 1H), 8.16 (dd, J = 8.1, 1.1 Hz, 1H), 7.71–7.59 (m, 3H), 5.79–5.74 (m, 1H), 4.23–4.15 (m, 1H), 4.10–4.03 (m, 1H), 2.83–2.72 (m, 1H), 2.60 (s, 3H), 2.46–2.35 (m, 1H), 2.27–2.17 (m, 1H), 2.17–2.06 (m, 1H) ppm; 13C-NMR (101 MHz, CDCl3) δ 159.0 (C), 143.1 (C), 137.2 (C), 132.1 (CH), 131.2 (C), 130.5 (CH), 128.1 (CH), 126.9 (CH), 126.0 (CH), 125.1 (C), 124.3 (C), 122.3 (CH), 121.8 (CH), 79.5 (CH), 69.1 (CH2), 30.0 (CH2), 26.1 (CH2), 22.1 (CH3) ppm; IR ν 2955, 2867, 1577, 1460, 1052, 760 cm−1; LRMS (EI) m/z (%) = 263 (M+, 6), 234 (10), 220 (100), 207 (57); HRMS (EI) m/z calcd for C18H17NO 263.1310, found 263.1307.
:5–9:1) as a yellow pale solid (54 mg, 0.20 mmol, 41%): Rf 0.20 (95:5 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.77 (br d, J = 8.2 Hz, 1H), 8.35 (dd, J = 8.1, 1.6 Hz, 1H), 8.23 (dd, J = 8.1, 1.5 Hz, 1H), 7.70 (ddd, J = 8.2, 7.0, 1.4 Hz, 1H), 7.67–7.55 (m, 3H), 5.81–5.75 (t, J = 6.8 Hz, 1H), 4.25–4.15 (m, 1H), 4.10–4.01 (m, 1H), 3.11 (s, 3H), 2.84–2.67 (m, 1H), 2.48–2.32 (m, 1H), 2.27–2.00 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 159.6 (C), 144.5 (C), 135.6 (C), 134.6 (CH), 132.9 (C), 130.9 (CH), 127.8 (CH), 126.7 (CH), 126.5 (CH), 126.4 (C), 126.1 (CH), 125.6 (C), 124.9 (CH), 79.7 (CH), 69.1 (CH2), 30.1 (CH2), 27.1 (CH2), 26.1 (CH3) ppm; IR ν 3069, 2968, 2871, 1587, 1437, 1054, 760 cm−1; LRMS (EI) m/z (%) = 263 (M+, 8), 234 (18), 220 (100), 207 (59), 204 (21), 165 (18); HRMS (EI) m/z calcd for C18H17NO 263.1310, found 263.1306.
:5–75:25) as a white solid (78 mg, 0.28 mmol, 55%): Rf 0.20 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.53 (d, J = 9.1 Hz, 1H), 8.47–8.41 (m, 1H), 8.17–8.12 (m, 1H), 7.82 (d, J = 2.6 Hz, 1H), 7.68–7.56 (m, 2H), 7.44 (dd, J = 9.1, 2.6 Hz, 1H), 5.69 (t, J = 6.9 Hz, 1H), 4.24–4.12 (m, 1H), 4.12–4.01 (m, 1H), 3.98 (s, 3H), 2.87–2.71 (m, 1H), 2.47–2.32 (m, 1H), 2.28–2.05 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 158.6 (C), 158.4 (C), 142.5 (C), 130.5 (CH), 127.8 (C), 127.6 (CH), 127.0 (CH), 126.3 (C), 124.3 (C), 124.1(CH), 121.5 (CH), 120.8 (CH), 107.1 (CH), 80.1 (CH), 69.1 (CH2), 55.6 (CH3), 29.8 (CH2), 26.2 (CH2) ppm; IR ν 3000, 2964, 2858, 1571, 1059, 754 cm−1; LRMS (EI) m/z (%) = 279 (M+, 12), 250 (8), 236 (100), 223 (44), 207 (35); HRMS (EI) m/z calcd for C18H17NO2 279.1259, found 279.1261.
:2–95:5) as a white solid (57 mg, 0.19 mmol, 38%): Rf 0.35 (96:4 hexane/EtOAc); 1H-NMR (400 MHz, CDCl3) δ 9.19 (H-1, d, J = 8.5 Hz, 1H), 8.57 (H-12 + H-4, dd, J = 8.7, 5.2 Hz, 2H), 8.23 (H-7, dd, J = 8.2, 1.1 Hz, 1H), 8.10 (H-11, d, J = 8.9 Hz, 1H), 7.98 (H-10, d, J = 7.9 Hz, 1H), 7.78–7.71 (H-2 + H-8, m, 2H), 7.68–7.63 (H-3 + H-9, m, 2H), 5.91 (H-2′, t, J = 6.4 Hz, 1H), 4.47–4.38 (H-5′, m, 1H), 4.20–4.13 (H-5′, m, 1H), 2.99–2.86 (H-3′, m, 1H), 2.59–2.37 (H-4′, m, 1H), 2.27–2.07 (H-3′ + H-4′, m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 157.7 (C), 144.0 (C), 133.8 (C), 133.2 (C), 131.9(C), 130.1 (CH), 129.9 (C), 128.8 (CH), 128.7 (CH), 128.4 (CH), 127.2 (CH), 126.9 (CH), 126.7 (CH), 123.7(C), 123.0 (C), 122.5 (CH), 120.2 (CH), 80.5 (CH), 69.4 (CH2), 31.3 (CH2), 26.8 (CH2) ppm; IR ν 3019, 2961, 2937, 1561, 1465, 1350, 1051, 750 cm−1; LRMS (EI) m/z (%) = 299 (M+, 14), 256 (100), 242 (28), 227 (19); HRMS (EI) m/z calcd for C21H17NO 299.1310, found 299.1304.
:2) as a yellow pale solid (77 mg, 0.27 mmol, 53%): Rf 0.20 (8:2 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 9.10 (br s, 1H), 8.65 (d, J = 8.7 Hz, 1H), 8.54 (d, J = 8.2 Hz, 1H), 8.35 (dd, 2H), 8.19 (dd, J = 8.1, 1.2 Hz, 1H), 7.77 (ddd, J = 8.3, 7.0, 1.4 Hz, 1H), 7.67 (ddd, J = 8.3, 7.1, 1.4 Hz, 1H), 5.77 (t, J = 7.0 Hz, 1H), 4.22–4.13 (m, 1H), 4.13–4.04 (m, 1H), 2.88–2.79 (m, 1H), 2.77 (s, 3H), 2.52–2.37 (m, 1H), 2.31–2.10 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 197.5 (C), 159.8 (C), 144.2 (C), 136.5 (C), 135.3 (C), 130.7 (C), 129.8 (CH), 128.8 (CH), 128.2 (CH), 127.4 (CH), 124.5 (C), 123.5 (C), 122.9 (CH), 122.6 (CH), 79.9 (CH), 69.2 (CH2), 29.9 (CH2), 26.9 (CH2), 26.2 (CH3) ppm; IR ν 2977, 2921, 2856, 1683, 1615, 1251, 1052, 760 cm−1; LRMS (EI) m/z (%) = 291 (M+, 5), 263 (20), 248 (100), 235 (65), 207 (17); HRMS (EI) m/z calcd for C19H17NO2 291.1259, found 291.1265.
:5–9:1) as a yellow pale solid (111 mg, 0.35 mmol, 70%): Rf 0.25 (95:5 hexane/EtOAc); 1H-NMR (400 MHz, CDCl3) δ 8.81 (s, 1H), 8.72 (d, J = 8.6 Hz, 1H), 8.54 (d, J = 8.1 Hz, 1H), 8.20 (d, J = 8.1 Hz, 1H), 8.00 (d, J = 8.6 Hz, 1H), 7.78 (t, J = 7.4 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 5.72 (t, J = 6.9 Hz, 1H), 4.19–4.12 (m, 1H), 4.12–4.03 (m, 1H), 2.88–2.75 (m, 1H), 2.48–2.35 (m, 1H), 2.30–2.08 (m, 2H); 13C-NMR (101 MHz, CDCl3) δ 159.2 (C), 144.0 (C), 135.6 (C), 130.7 (CH), 129.8 (CH), 129.1(CH), 129.0 (C-8, d, 2JC–F = 32.4 Hz), 127.6 (CH), 126.3 (CH, q, 3JC–F = 3.3 Hz), 124.5 (CH, q, 3JC–F = 4.3 Hz), 124.4 (CH), 124.25 (d, 1JC–F = 272.2 Hz), 123.5 (CH), 123.3 (C), 122.4 (CH), 80.0 (CH), 69.2 (CH2), 29.8 (CH2), 26.2 (CH2) ppm; IR ν 2966, 2874, 1722, 1625, 1173, 1122, 763 cm−1; LRMS (EI) m/z (%) = 317 (M+, 4), 288 (8), 274 (100), 261 (73), 247 ([(M + 1)+ − THF], 14), 226 (12); HRMS (EI) m/z calcd for C14H8F3N, 247.0609, found 247.0612.
:5) as an orange pale solid (92 mg, 0.32 mmol, 65%): Rf 0.25 (95:5 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.53 (d, J = 8.9 Hz, 1H), 8.46 (dd, J = 7.5, 1.8 Hz, 2H), 8.16 (dd, J = 8.1, 1.1 Hz, 1H), 7.78–7.68 (m, 2H), 7.64 (ddd, J = 8.4, 7.1, 1.5 Hz, 1H), 5.65 (t, J = 6.9 Hz, 1H), 4.20–4.11 (m, 1H), 4.11–4.01 (m, 1H), 2.87–2.71 (m, 1H), 2.47–2.30 (m, 1H), 2.29–2.00 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 158.3 (C), 143.3 (C), 133.3 (C), 131.8 (C), 130.9 (CH), 130.7 (CH), 128.9 (CH), 127.4 (CH), 126.2 (CH), 126.0 (C), 124.2 (CH), 123.6 (C), 121.9 (CH), 79.8 (CH), 69.1 (CH2), 29.7 (CH2), 26.1 (CH2) ppm; IR ν 3063, 2939, 2839, 1617, 1460, 1220, 1038, 759 cm−1; LRMS (EI) m/z (%) = 283 (M+, 8), 254 (15), 240 ([M+ − C2H3O], 100), 217 (12), 207 (65), 177 (39); HRMS (EI) m/z calcd for C15H11ClN 240.0580, found 240.0586.
:5–8:2) as a yellow pale solid (80 mg, 0.30 mmol, 60%): Rf 0.21 (95:5 hexane/EtOAc); 1H-NMR (400 MHz, CDCl3) δ 8.60 (H-10, dd, JH–F = 9.1, 5.4 Hz, 1H), 8.46 (dd, J = 8.1, 1.1 Hz, 1H), 8.17 (dd, J = 8.1, 1.2 Hz, 1H), 8.12 (H-7, dd, JH–F = 10.2, 2.6 Hz, 1H), 7.70 (ddd, J = 8.2, 7.0, 1.5 Hz, 1H), 7.64 (ddd, J = 8.3, 7.0, 1.5 Hz, 1H), 7.56 (H-9, ddd, JH–F = 9.1, 8.0, 2.7 Hz, 1H), 5.63 (H-2′, t, J = 6.9 Hz, 1H), 4.20–4.13 (H-5′, m, 1H), 4.09–4.02 (H-5′, m, 1H), 2.84–2.71 (m, 1H), 2.44–2.34 (m, 1H), 2.27–2.06 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 161.33 (C-8, d, 1JC–F = 248.0 Hz), 158.53 (C, d, 4JC–F = 4.3 Hz), 142.94 (C), 130.62 (CH), 130.11 (C), 128.51 (CH), 127.43 (CH), 126.24 (C-6a, d, 3JC–F = 7.8 Hz), 124.93 (C-10, d, 3JC–F = 8.5 Hz), 123.82 (C), 121.76 (CH), 119.64 (CH, d, 2JC–F = 23.9 Hz), 111.59 (CH, d, 2JC–F = 21.8 Hz), 80.08 (CH), 69.14 (CH2), 29.79 (CH2), 26.08 (CH2); IR ν 2958, 2873, 1773, 1697, 1480, 1196, 1054,759 cm−1; LRMS (EI) m/z (%) = 267.1 (M+, 5), 238 (12), 224 (100), 211 (59), 197 (19), 169 (11); HRMS (EI) m/z calcd for C17H14FNO 267.1059, found 267.1042.
:2–95:5) as a yellow pale solid (38 mg, 0.14 mmol, 28%): Rf 0.16 (98:2 hexane/EtOAc); 1H-NMR (400 MHz, CDCl3) δ 8.53 (d, J = 8.3 Hz, 1H), 8.51 (d, J = 8.3 Hz, 1H), 8.12 (dd, J = 8.1, 1.4 Hz, 1H), 7.71–7.57 (m, 3H), 7.49 (d, J = 7.2 Hz, 1H), 5.97 (dd, J = 6.9, 5.2 Hz, 1H), 4.20–4.12 (m, 1H), 4.04–3.95 (m, 1H), 3.09 (s, 3H), 2.89–2.80 (m, 1H), 2.32–2.12 (m, 2H), 2.11–1.96 (m, 1H) ppm; 13C-NMR (101 MHz, CDCl3) δ 159.4 (C), 142.5 (C), 136.5 (C), 134.9 (C), 132.0 (CH), 130.2 (CH), 129.6 (CH), 128.4 (CH), 126.9 (CH), 125.4 (C), 124.4 (C), 122.2 (CH), 120.9 (CH), 80.3 (CH), 68.9(CH2), 30.3 (CH2), 25.8 (CH2), 25.2 (CH3) ppm; IR ν 2959, 2874, 1573, 1449, 1286, 1048, 749 cm−1; LRMS (EI) m/z (%) = 263 (M+, 8), 248 (6), 234 (13), 220 (100), 207 (13), 193 (19); HRMS (EI) m/z calcd for C18H17NO 263.1310, found 263.1312.
:5–100% EtOH), isomer 3′ja was isolated as a yellow pale solid (37 mg, 0.14 mmol, 28%): Rf 0.19 (95:5 hexane/EtOAc); 1H-NMR (400 MHz, CDCl3) δ 8.52 (dd, J = 8.1, 1.5 Hz, 1H), 8.40 (H-10, s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.16 (dd, J = 8.1, 1.0 Hz, 1H), 7.68 (ddd, J = 8.2, 7.1, 1.4 Hz, 1H), 7.60 (ddd, J = 8.3, 7.1, 1.4 Hz, 1H), 7.50 (dd, J = 8.4, 1.4 Hz, 1H), 5.73 (t, J = 6.9 Hz, 1H), 4.23–4.16 (m, 1H), 4.11–4.00 (m, 1H), 2.76–2.65 (m, 1H), 2.62 (s, 3H), 2.46–2.32 (m, 1H), 2.27–2.04 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 159.3 (C), 143.6 (C), 140.7 (C), 133.5 (C), 130.5 (CH), 129.0 (CH), 128.4 (CH), 126.7 (CH), 126.4 (CH), 124.1 (C), 123.0 (C), 122.1 (CH), 121.9 (CH), 79.8 (CH), 69.1 (CH2), 30.2 (CH2), 26.1 (CH2), 22.3 (CH3) ppm; IR ν 2875, 2868, 1618, 1460, 1053, 760; LRMS (EI) m/z (%) = 263 (M+, 5), 234 (13), 220 (100), 207 (51), 192 (23); HRMS (EI) m/z calcd for C18H17NO 263.1310, found 263.1312.
:7) as a white solid (51 mg, 0.19 mmol, 38%): Rf 0.19 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.49 (d, J = 8.2 Hz, 1H), 8.45 (d, J = 8.4 Hz, 1H), 8.22 (dd, J = 8.2, 1.2 Hz, 1H), 7.82–7.70 (m, 2H), 7.64 (ddd, J = 8.3, 7.1, 1.4 Hz, 1H), 7.36 (ddd, J = 12.5, 7.9, 1.0 Hz, 1H), 6.02 (dt, J = 8.2, 4.3 Hz, 1H), 4.43–4.32 (m, 1H), 4.17–4.05 (m, 1H), 2.60–2.45 (m, 1H), 2.40–2.23 (m, 1H), 2.13–1.96 (m, 1H) ppm; 13C-NMR (75 MHz, CDCl3) δ 160.2 (C-7, d, 1JC–F = 255.2 Hz), 158.8 (C, d, 3JC–F = 7.9 Hz), 143.3 (CH), 136.0 (C, d, 4JC–F = 4.4 Hz), 131.0 (C-9, d, 3JC–F = 9.8 Hz), 130.8 (CH), 129.3 (CH), 127.3 (CH), 122.7 (C, d, 5JC–F = 2.7 Hz), 122.3 (CH), 118.7 (C-10, d, 4JC–F = 4.0 Hz), 114.2 (CH), 114.1 (CH), 114.0 (C-6a, d, 2JC–F = 24.6 Hz), 81.9 (C-2′, d, 4JC–F = 14 Hz), 69.3 (CH2), 31.7 (CH2), 25.1 (CH2) ppm; IR ν 2970, 2870, 1581, 1451, 1240, 757 cm−1; LRMS (EI) m/z (%) = 267 (M+, 2), 224 (100), 211 (64), 197 (19), 169 (9); HRMS (EI) m/z calcd for C17H14FNO 267.1059, found 267.1050.
:5), isomer 3′ka was isolated as a white solid (24 mg, 0.09 mmol, 18%): Rf 0.27 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.52 (H-7, dd, J = 9.1, 5.8 Hz, 1H), 8.41 (H-1, dd, J = 8.2, 1.1 Hz, 1H), 8.22 (H-10, dd, J = 10.5, 2.6 Hz, 1H), 8.17 (H-4, dd, J = 8.3, 1.2 Hz, 1H), 7.74 (ddd, J = 8.3, 7.0, 1.5 Hz, 1H), 7.64 (ddd, J = 8.3, 7.0, 1.4 Hz, 1H), 7.42 (H-8, ddd, J = 9.1, 8.2, 2.6 Hz, 1H), 5.70 (H-2′, t, J = 6.9 Hz, 1H), 4.21–4.11 (H-5′, m, 1H), 4.10–4.01 (H-5′, m, 1H), 2.88–2.71 (m, 1H), 2.47–2.31 (m, 1H), 2.29–2.05 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 163.70 (C-9, d, 1JC–F = 251.5 Hz); 158.7 (C), 143.6 (C), 135.9 (C-10a, d, 3JC–F = 9.4 Hz), 130.6 (CH), 129.8 (C-7, d, 3JC–F = 9.3 Hz), 129.3 (CH), 127.1 (CH), 123.8 (C-6a, d, 4JC–F = 4.1 Hz), 122.2 (CH), 122.0 (C-6, d, 5JC–F = 2 Hz), 116.3 (C-10/C-8, d, 2JC–F = 23.7 Hz), 107.5 (C-8/C-10, d, 2JC–F = 22.1 Hz), 80.0 (CH), 69.1 (CH2), 29.8 (CH2), 26.1 (CH2) ppm; IR ν 2962, 2877, 1619, 1496, 1195, 1052, 760 cm−1; LRMS (EI) m/z (%) = 267 (M+, 3), 238 (13), 224 (100), 211 (55), 197 (17), 169 (9); HRMS (EI) m/z calcd for C17H14FNO 267.1059, found 267.1054.
:6–93:7) as a yellow pale solid (43 mg, 0.16 mmol, 55%): Rf 0.17 (93:7 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.61 (d, J = 8.3 Hz, 1H), 8.42 (dd, J = 8.3, 0.7 Hz, 1H), 8.31 (s, 1H), 8.07 (d, J = 8.3 Hz, 1H), 7.79 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.66 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.53 (dd, J = 8.3, 1.6 Hz, 1H), 5.74 (t, J = 6.9 Hz, 1H), 4.25–4.14 (m, 1H), 4.11–4.00 (m, 1H), 2.83–2.66 (m, 1H), 2.61 (s, 3H), 2.48–2.33 (m, 1H), 2.28–2.02 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 158.3 (C), 141.7 (C), 136.8 (C), 133.1 (C), 130.3 (CH), 130.3 (CH), 130.1(CH), 127.1 (CH), 126.5 (CH), 125.0 (C), 124.0 (C), 122.4 (CH), 121.6 (CH), 79.8 (CH), 69.1 (CH2), 30.1 (CH2), 26.1 (CH2), 22.1 (CH3) ppm; IR ν 2971, 2923, 2867, 1582, 1496, 1295, 1051, 822, 729 cm−1; LRMS (EI) m/z (%) = 263 (M+, 6), 234 (12), 220 (100), 207 (47), 192 (15), 165 (12); HRMS (EI) m/z calcd for C18H17NO 263.1310, found 263.1312.
:6) as a red solid (84 mg, 0.25 mmol, 55%): Rf 0.20 (93:7 hexane/EtOAc); 1H-NMR (400 MHz, CDCl3) δ 8.55 (d, J = 8.3 Hz, 1H), 8.48 (d, J = 7.9 Hz, 1H), 8.34 (d, J = 1.7 Hz, 1H), 8.21 (d, J = 8.9 Hz, 1H), 7.87 (ddd, J = 8.4, 7.1, 1.3 Hz, 1H), 7.75 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.57 (ddd, J = 8.9, 2.6, 1.1 Hz, 1H), 5.77 (t, J = 6.9 Hz, 1H), 4.23–4.15 (m, 1H), 4.12–4.04 (m, 1H), 2.81–2.65 (m, 1H), 2.50–2.35 (m, 1H), 2.31–2.06 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 160.1 (C), 147.7 (C), 141.7 (C), 132.8 (C), 132.5 (CH), 130.7 (CH), 128.2 (CH), 126.8 (CH), 125.1 (C-2, d, 3JC–F = 10.5 Hz), 122.6 (CH), 121.9 (CH), 120.8 (q, 1JC–F = 257.5 Hz), 113.7 (CH), 79.6 (CH), 69.2 (CH2), 30.1 (CH2), 26.1 (CH2) ppm; IR ν 2964, 2860, 1619, 1587, 1492, 1254, 1214 cm−1; LRMS (EI) m/z (%) = 333 (M+, 5), 304 (24), 290 (100), 277 (51), 263 (8); HRMS (EI) m/z calcd for C18H14F3NO2 333.0977, found 333.0950.
:6–93:7) as a white solid (42 mg, 0.15 mmol, 50%): Rf 0.20 (93:7 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.53 (d, J = 8.2 Hz, 1H), 8.48 (d, J = 2.1 Hz, 1H), 8.44 (d, J = 8.1 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 7.87–7.79 (m, 1H), 7.71 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.64 (dd, J = 8.7, 2.3 Hz, 1H), 5.74 (t, J = 6.9 Hz, 1H), 4.24–4.14 (m, 1H), 4.11–4.01 (m, 1H), 2.79–2.65 (m, 1H), 2.49–2.34 (m, 1H), 2.28–2.03 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 159.7 (C), 141.8 (C), 132.9 (C), 132.4 (C), 132.0 (CH), 130.7 (CH), 129.1 (CH), 128.0 (CH), 126.7 (CH), 125.3 (C), 125.1 (C), 122.5 (CH), 121.7 (CH), 79.6 (CH), 69.2 (CH2), 30.0 (CH2), 26.1(CH2) ppm; IR ν 2967, 2869, 1584, 1495, 1255, 1054, 822, 767 cm−1; LRMS (EI) m/z (%) = 283 (M+, 4), 254 (16), 240 ([M+ − C2H3O], 100), 227 (51), 213 (13) 177 (29); HRMS (EI) m/z calcd for C15H11ClN 240.0580, found 240.0576.
:4) as a brown oil (36 mg, 0.13 mmol, 45%): Rf 0.20 (93:7 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.54 (d, J = 8.3 Hz, 1H), 8.49 (H-1, dd, J = 9.1, 5.9 Hz, 1H), 8.43 (d, J = 8.2 Hz, 1H), 7.87–7.78 (m, 2H), 7.67 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.38 (H-2, ddd, J = 8.9, 8.1, 2.7 Hz, 1H), 5.76 (t, J = 6.9 Hz, 1H), 4.24–4.14 (m, 1H), 4.11–4.01 (m, 1H), 2.78–2.64 (m, 1H), 2.48–2.34 (m, 1H), 2.29–2.03 (m, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 162.7 (C-3, d, 1JC–F = 247.6 Hz), 160.9 (C), 144.7 (C-4a, d, 3JC–F = 11.9 Hz), 133.2 (C), 130.8 (CH), 127.2 (CH), 126.7 (CH), 124.5 (C, d, 4JC–F = 1.0 Hz), 123.9 (C-1, d, 3JC–F = 9.5 Hz), 122.3 (CH), 120.9 (d, JC–F = 2.1 Hz), 116.0 (CH, d, 2JC–F = 23.7 Hz), 115.0 (CH, d, 2JC–F = 20.5 Hz), 79.5 (CH), 69.2 (CH2), 30.1 (CH2), 26.1(CH2) ppm; IR ν 2972, 2871, 1618, 1580, 1484, 1459, 1053, 764 cm−1; LRMS (EI) m/z (%) = 267 (M+, 5), 238 (15), 224 (100), 211 (55), 196 (17); HRMS (EI) m/z calcd for C17H14FNO 267.1059, found 267.1048.
:15–75:25) as a white solid (69 mg, 0.26 mmol, 52%): Rf 0.25 (8:2 hexane/EtOAc). 1H-NMR (300 MHz, CDCl3) δ 8.66 (d, J = 8.3 Hz, 1H), 8.60–8.52 (m, 1H), 8.48–8.39 (m, 1H), 8.26–8.15 (m, 1H), 7.90–7.80 (m, 1H), 7.78–7.63 (m, 3H), 5.49 (p, J = 6.3 Hz, 1H), 4.34–4.29 (m, 2H), 4.21–4.06 (m, 2H), 3.99–3.88 (m, 2H) ppm; 13C-NMR (75 MHz, CDCl3) δ 156.2 (C), 143.3 (C), 133.3 (C), 130.6 (CH), 130.6 (CH), 128.7 (CH), 127.5 (CH), 127.4 (CH), 126.2 (CH), 124.6 (C), 124.1 (C), 122.6 (CH), 122.0 (CH), 76.3 (CH), 70.2 (CH2), 67.9 (CH2), 66.7 (CH2) ppm; IR ν 2965, 2856, 1114, 10856, 912, 759 cm−1; LRMS (EI) m/z (%) = 265 (M+, 4), 206 (100), 179 (24), 151 (12), 102 (5).
:2–9:1) as a white solid (57 mg, 0.22 mmol, 43%): Rf 0.20 (95:5 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.62 (ddd, J = 8.2, 1.3, 0.6 Hz, 1H), 8.56–8.50 (m, 2H), 8.25–8.19 (m, 1H), 7.80 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.74–7.66 (m, 2H), 7.65–7.57 (m, 1H), 5.20 (dd, J = 11.1, 2.2 Hz, 1H), 4.34–4.24 (m, 1H), 3.81 (td, J = 11.6, 2.4 Hz, 1H), 2.37–2.19 (m, 1H), 2.15–2.00 (m, 2H), 1.96–1.77 (m, 2H), 1.74–1.63 (m, 1H) ppm; 13C-NMR (75 MHz, CDCl3) δ 159.6 (C), 143.4 (C), 133.5 (C), 130.5 (CH), 130.3 (CH), 128.6 (CH), 127.1 (CH), 127.0 (CH), 126.8 (CH), 124.5 (C), 124.1 (C), 122.4 (CH), 121.9 (CH), 80.7 (CH), 69.6 (CH2), 30.6 (CH2), 26.1 (CH2), 24.0 (CH2) ppm; IR ν 3075, 2930, 2839, 1083, 1039, 754, 723 cm−1; LRMS (EI) m/z (%) = 262 (M+ − 1, 1), 235 (20), 206 (100), 193 (12).
:2) as a brown pale oil (79 mg, 0.28 mmol, 57%): Rf 0.29 (100% hexane); 1H-NMR (300 MHz, CDCl3) δ 9.25 (ddd, J = 8.5, 1.4, 0.6 Hz, 1H), 8.70–8.64 (m, 1H), 8.57 (dd, J = 8.0, 1.6 Hz, 1H), 8.17–8.12 (m, 1H), 7.81 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.77–7.61 (m, 3H), 3.75 (p, J = 6.1 Hz, 1H), 1.92 (s, 6H), 1.02 (d, J = 6.1 Hz, 6H) ppm; 13C-NMR (101 MHz, CDCl3) δ 163.7 (C), 142.9 (C), 133.9 (C), 130.6 (CH), 130.3 (CH), 129.9 (CH), 128.5 (CH), 127.0 (CH), 126.1 (CH), 124.5 (C), 124.1 (C), 122.3 (CH), 121.9 (CH), 81.8 (C), 67.0 (CH), 29.1 (CH3), 24.8 (CH3); IR ν 2983, 2933, 1578, 1379, 1162, 1110, 996, 760, 730 cm−1; LRMS (EI) m/z (%) = 264 (M+, 0.2), 236 (18), 221 ([M+ − C5H7O], 100), 204 (34), 179 (54), 150 (18); HRMS (EI) m/z calcd for C16H14N 220.1126, found 220.1128.
:1–98:2) as a yellow oil (123 mg, 0.40 mmol, 80%): Rf 0.65 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.94–8.89 (m, 1H), 8.69–8.64 (m, 1H), 8.57 (dd, J = 8.0, 1.6 Hz, 1H), 8.20–8.15 (m, 1H), 7.84 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.77–7.62 (m, 3H), 4.98 (dd, J = 8.8, 5.4 Hz, 1H), 3.52–3.33 (m, 2H), 2.28–2.13 (m, 1H), 2.01–1.87 (m, 1H), 1.76–1.46 (m, 4H), 1.40–1.23 (m, 4H), 0.95 (t, J = 7.4 Hz, 3H), 0.83 (t, J = 7.3 Hz, 3H) ppm; 13C-NMR (101 MHz, CDCl3) δ 161.9 (C), 143.4 (C), 133.5 (C), 130.5 (CH), 130.2 (CH), 128.7 (CH), 127.0 (CH), 124.5 (C), 124.1 (C), 122.4 (CH), 122.0 (CH), 86.5 (CH), 69.5 (CH2), 38.3 (CH2), 32.2 (CH2), 20.0 (CH2), 19.5 (CH2), 14.1 (CH3), 14.0 (CH3) ppm; IR ν 2957, 2932, 2870, 1759, 1459, 1092, 727 cm−1; LRMS (EI) m/z (%) = 265 (M+ − C3H6, 12), 250 (9), 235 (37), 206 (100), 151 (9).
:1–9:1) as a colorless oil (43 mg, 0.16 mmol, 33%): Rf 0.75 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 9.14 (d, J = 8.4 Hz, 1H), 8.63 (d, J = 8.3 Hz, 1H), 8.53 (dd, J = 8.1, 1.5 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.78 (ddd, J = 8.1, 6.6, 1.2 Hz, 1H), 7.72–7.58 (m, 3H), 4.14–4.03 (m, 1H), 3.80–3.69 (m, 1H), 3.66–3.56 (m, 1H), 2.10–1.88 (m, 3H), 1.83 (s, 3H) ppm; 13C-NMR (75 MHz, CDCl3) δ 163.1, 142.9, 134.0, 130.4, 130.0, 129.3, 128.4, 126.9, 126.6, 124.4, 124.1, 122.4, 121.9, 88.7, 68.1, 37.4, 28.2, 25.2 ppm; IR ν 3065, 2970, 2930, 2872, 1570, 1097, 758 cm−1; LRMS (EI) m/z (%) = 263 (M+, 18), 235 (48), 207 (86), 179 (55), 85 (100); HRMS (EI) calcd for C18H17NO 263.1310, found 263.1308.
:1–9:1), isomer 3′af was isolated as a white solid (26 mg, 0.10 mmol, 21%) [trans/cis mixture in a 58:42 ratio (1H-NMR)]): Rf 0.75 (9:1 hexane/EtOAc); 1H-NMR (400 MHz, CDCl3) δ 8.62 (d, J = 8.3 Hz, 1.10H), 8.53 (d, J = 8.2 Hz, 1.51H), 8.46 (d, J = 7.9 Hz, 0.6H), 8.21–8.15 (m, 1H), 7.81 (ddt, J = 8.3, 7.0, 1.3 Hz, 1.09H), 7.75–7.59 (m, 3.58H), 5.91 (t, J = 6.9 Hz, 0.58H), 5.69 (t, J = 7.1 Hz, 0.42H), 4.53–4.43 (m, 0.64H), 4.37–4.27 (m, 0.47H), 2.91–2.73 (m, 1.06H), 2.52–2.42 (m, 0.62H), 2.42–2.27 (m, 1.02H), 2.27–2.14 (m, 0.44H), 1.87–1.67 (m, 1.5H), 1.39 (dd, J = 6.1, 3.3 Hz, 3H) ppm; 13C-NMR (75 MHz, CDCl3) δ 159.9 (C), 159.2 (C), 143.34 (C), 143.28 (C), 133.43 (C), 133.42 (C), 130.5 (CH), 130.4 (CH), 128.6 (CH), 128.6 (CH), 127.3 (CH), 127.3 (CH), 127.0 (CH), 126.9 (CH), 126.7 (CH), 125.1 (C), 124.9 (C), 124.3 (C), 124.2 (C), 122.5 (CH), 122.4 (CH), 122.0 (CH), 80.6 (CH), 79.2 (CH), 76.1 (CH2), 33.9 (CH2), 33.2 (CH2), 30.6 (CH2), 30.0 (CH2), 21.6 (CH3), 21.4 (CH3) ppm; IR ν 3074, 2971, 2929, 2877, 2861, 1583, 1444, 1073, 725 cm−1; LRMS (EI) m/z (%) = 263 (M+, 11), 220 (72), 208 (100), 179 (58); HRMS (EI): calcd for C18H17NO 263.1310, found 263.1301.
:5–93:2) as a yellow solid (53 mg, 0.20 mmol, 40%): Rf 0.24 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.61 (d, J = 8.3 Hz, 1H), 8.53 (td, J = 8.1, 1.3 Hz, 2H), 8.17 (dd, J = 7.8, 1.4 Hz, 1H), 7.87–7.79 (m, 1H), 7.75–7.61 (m, 3H), 5.09 (s, 2H), 1.40 (s, 9H) ppm; 13C-NMR (75 MHz, CDCl3) δ 158.7 (C), 143.6 (C), 133.4 (C), 130.6 (CH), 130.2 (CH), 128.6 (CH), 127.6 (CH), 127.3 (CH), 127.1 (CH), 125.7 (C), 124.5 (C), 122.2 (CH), 122.0, 74.7 (CH), 66.9 (CH2), 27.9 (3CH3) ppm; IR ν 2974, 2932, 1719, 1364, 1145, 760 cm−1; LRMS (EI) m/z (%) = 265 ([(M + 1)+ − CH3], 1), 235 (37), 208 (100), 192 (48), 180 (51), 165 (25).
:1–98:2) as a yellow solid (32 mg, 0.08 mmol, 17%): Rf 0.5 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.65–8.55 (m, 3H), 8.30–8.25 (m, 1H), 7.81–7.66 (m, 3H), 7.57–7.43 (m, 3H), 7.39–7.25 (m, 7H), 7.24–7.19 (m, 1H), 6.27 (s, 1H), 4.73 (d, J = 11.7 Hz, 1H), 4.64 (d, J = 11.7 Hz, 1H) ppm; 13C-NMR (101 MHz, CDCl3) δ 159.9 (C), 143.4 (C), 140.8 (C), 138.3 (C), 133.9 (C), 130.5 (CH), 130.5 (CH), 128.8 (CH), 128.4 (CH), 128.3 (CH), 128.0 (CH), 127.7 (CH), 127.4 (CH), 127.1 (CH), 126.3 (CH), 124.5 (C), 124.3 (C), 122.3 (CH), 122.1 (CH), 86.5 (CH), 71.7 (CH2) ppm; IR ν 3060, 3029, 2915, 2850, 1572, 1449, 1067, 722 cm−1. LRMS (EI-DIP) m/z (%) = 284 (M+ − C7H7, 52), 269 (100), 268 (52), 178 (16), 91 (32); HRMS (EI) calcd for C27H21NO 375.1623, found 375.1605.
:1–98:2), compound 3′ah was isolated as a yellow solid (62 mg, 0.23 mmol, 46%): Rf 0.5 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.56 (br d, J = 8.3 Hz, 1H), 8.51 (dd, J = 8.2, 1.4 Hz, 1H), 8.18 (ddd, J = 9.7, 8.1, 1.3 Hz, 2H), 7.72 (ddd, J = 8.2, 7.0, 1.4 Hz, 2H), 7.61 (ddd, J = 8.3, 7.0, 1.4 Hz, 1H), 7.53 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.33–7.28 (m, 2H), 7.26–7.10 (m, 4H), 4.74 (s, 2H) ppm; 13C-NMR (101 MHz, CDCl3) δ 160.2 (C), 143.8 (C), 139.2 (C), 133.3 (C), 130.4 (CH), 129.9 (CH), 128.7 (CH), 128.6 (2CH), 127.4 (CH), 127.1 (CH), 126.7 (CH), 126.4 (CH), 125.4 (C), 124.0 (C), 122.5 (CH), 122.0 (CH), 43.2 (CH2) ppm; IR ν 3062, 3025, 1684, 1581, 1362, 723 cm−1. LRMS (EI-DIP) m/z (%) = 269 (M+, 41), 268 (M+ − 1, 100), 254(6), 134 (11), 57 (6).
:1–98: 2) as a yellow crystalline solid (52 mg, 0.17 mmol, 35%): Rf 0.4 (9:1 hexane/EtOAc); mp 120–121 °C (3:1 EtOAc/MeOH); 1H-NMR (400 MHz, CDCl3) δ 8.65 (br d, J = 8.3 Hz, 1H), 8.56 (dd, J = 8.1, 1.4 Hz, 1H), 8.25 (br d, J = 8.3 Hz, 1H), 8.17 (dd, J = 8.0, 1.2 Hz, 1H), 7.80 (s, 1H), 7.72 (ddd, J = 8.2, 7.0, 1.6 Hz, 1H), 7.66 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.59 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.41–7.37 (m, 1H), 7.35–7.30 (m, 1H), 7.23–7.13 (m, 2H), 6.97 (t, J = 2.5 Hz, 1H), 5.59 (dd, J = 12.3, 2.7 Hz, 1H), 5.42 (dd, J = 12.3, 2.0 Hz, 1H) ppm; 13C-NMR (101 MHz, CDCl3) δ 158.8 (C), 143.4 (C), 140.9 (C), 139.2 (C), 133.9 (C), 130.7 (CH), 130.4 (CH), 128.7 (CH), 128.0 (CH), 127.5 (CH), 127.3 (CH), 127.3 (CH), 126.6 (CH), 124.6 (C), 124.4 (C), 122.8 (CH), 122.6 (CH), 122.0 (CH), 121.3(CH), 87.8 (CH), 73.8 (CH2) ppm; IR ν 3074, 2851, 1572, 1027, 720 cm−1. LRMS (EI) m/z (%) = 297 (M+, 2), 268 (100), 251 (3), 119 (12); HRMS (EI) calcd for C21H15NO 297.1154, found 297.1141.
:2) as a white solid (96 mg, 0.26 mmol, 52%): Rf 0.2 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 9.10–9.04 (m, 1H), 8.64 (d, J = 8.3 Hz, 1H), 8.60–8.55 (m, 1H), 8.23–8.18 (m, 1H), 7.90–7.83 (m, 1H), 7.79–7.67 (m, 1H), 6.48 (s, 1H), 5.50 (q, J = 6.5 Hz, 4H) ppm; 13C-NMR (75 MHz, CDCl3) δ 153.6 (C), 142.8 (C), 134.0 (C), 131.0 (CH), 130.6 (CH), 128.9 (CH), 128.3 (CH), 128.1 (CH), 127.4 (CH), 125.0 (C), 123.7 (C), 122.2 (2CH), 106.1(CH), 94.2 (2CH2) ppm; IR ν 3037, 2884, 1418, 1199, 1100, 1047, 944, 750, 723 cm−1; LRMS (EI) m/z (%) = 267 (M+, 1), 208 (67), 179 (100), 151 (31); HRMS (EI) calcd for C16H13NO3 267.0895, found 267.0879.
:1–98:2) as a white crystalline solid (90 mg, 0.3 mmol, 60%): Rf 0.49 (9:1 hexane/EtOAc); mp 105–107 °C (MeOH); 1H-NMR (300 MHz, CDCl3) δ 8.64 (br d, J = 8.3 Hz, 1H), 8.56 (dd, J = 8.0, 1.4 Hz, 1H), 8.30–8.21 (m, 2H), 7.82 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H), 7.79–7.66 (m, 2H), 7.60 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.43 (s, 1H), 7.04–6.91 (m, 4H) ppm; 13C-NMR (75 MHz, CDCl3) δ 152.5 (C), 147.5 (2C), 142.9 (C), 133.9 (C), 130.9 (2CH), 129.0 (CH), 128.4 (CH), 127.7 (CH), 125.9 (CH), 125.0 (C), 123.8 (C), 122.6 (CH), 122.3 (2CH), 122.1 (CH), 112.2 (CH), 109.4 (2CH) ppm; IR ν 3078, 2909, 1479, 1338, 1229, 724 cm−1; LRMS (EI) m/z (%) = 299 (M+, 2), 270 (100), 241 (9), 178 (7); HRMS (EI) calcd for C20H13NO2 299.0946, found 299.0949.
:1), it was isolated as a white solid. Rf 0.25 (9:1 hexane/EtOAc); 1H-NMR (300 MHz, CDCl3) δ 9.28 (s, 1H), 8.63–8.54 (m, 1H), 8.20 (dd, J = 8.1, 1.3 Hz, 1H), 8.03 (br d, J = 7.9 Hz, 1H), 7.84 (ddd, J = 8.4, 7.1, 1.4 Hz, 1H), 7.78–7.63 (m, 2H) ppm; 13C-NMR (75 MHz, CDCl3) δ 153.7 (CH), 144.6 (C), 132.7 (C), 131.1 (CH), 130.3 (CH), 128.9 (CH), 128.8 (CH), 127.6 (CH), 127.2 (CH), 126.5 (C), 124.2 (C), 122.3 (CH), 122.0 (CH) ppm; IR ν 2924, 2851, 1457, 1245, 890, 745 cm−1; LRMS (EI) m/z (%) = 179 (M+, 100), 151 (13), 76(100), 179 (8).
Acknowledgements
We thank the Ministerio de Economia y Competitividad (CTQ2015-66624-P) and the University of Alicante (VIGROB-173) for financial support. C. A.-T. thanks the ISO for a grant.Notes and references
- Examples can be found in: O. B. Abdel-Halim, T. Morikawa, S. Ando, H. Matsuda and M. Yoshikawa, J. Nat. Prod., 2004, 67, 1119 CrossRef CAS PubMed.
- (a) Y. L. Janin, A. Croisy, J.-F. Riou and E. Bisagni, J. Med. Chem., 1993, 36, 3686 CrossRef CAS PubMed; (b) S. D. Fang, L. K. Wang and S. M. Hecht, J. Org. Chem., 1993, 58, 5025 CrossRef CAS; (c) T. Nakanishi, M. Suzuki, A. Saimoto and T. Kabasawa, J. Nat. Prod., 1999, 62, 864 CrossRef CAS PubMed; (d) I. Slaninova, K. Pencikova, J. Urbanova, J. Slanina and E. Taborska, Phytochem. Rev., 2014, 13, 51 CrossRef CAS.
- T. Ishikawa, Med. Res. Rev., 2001, 21, 61 CrossRef CAS PubMed.
- (a) X. Yang, Y. Yao, Y. Qin, Z. Hou, R. Yang, F. Miao and L. Zhou, Chem. Pharm. Bull., 2013, 61, 731 CrossRef CAS PubMed; (b) J. Hu, X. Shi, J. Chen, X. Mao, L. Zhu, L. Yu and J. Shi, Food Chem., 2014, 148, 437 CrossRef CAS PubMed.
- E. Vanquelef, M. Amoros, J. Boustie, M. A. Lynch, R. D. Waigh and O. Duval, J. Enzyme Inhib. Med. Chem., 2004, 19, 481 CrossRef CAS PubMed.
- F. Miao, X.-J. Yang, Y.-N. Ma, F. Zheng, X.-P. Song and L. Zhou, Chem. Pharm. Bull., 2012, 60, 1508 CAS.
- M. Zhang, L. Liu, H. Xiao, T. Zhao, L. Yang and X. Xu, J. Heterocycl. Chem., 2016, 53, 234 CrossRef CAS.
- H. Lecoeur, Exp. Cell Res., 2002, 277, 1 CrossRef CAS PubMed.
- (a) N. Stevens, N. O'Connor, H. Vishwasrao, D. Samaroo, E. Kandel, D. L. Akins, C. M. Drain and N. J. Turro, J. Am. Chem. Soc., 2008, 130, 7182 CrossRef CAS PubMed; (b) Y. Chen, W. Huang, C. Li and Z. Bo, Macromolecules, 2010, 43, 10216 CrossRef CAS.
- For pioneer studies, see: (a) D. Nanni, P. Pareschi, C. Rizzoli, P. Sagarabotto and A. Tundo, Tetrahedron, 2010, 51, 9045 CrossRef; (b) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed.
- For a recent comprehensive review, see: B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505 RSC.
- For some selected examples, see: (a) T. Yoshimitsu, Y. Arano and H. Nagaoka, J. Org. Chem., 2005, 70, 2342 CrossRef CAS PubMed; (b) L. Chen, E. Shi, Z. Liu, S. Chen, W. Wei, H. Li, K. Xu and X. Wan, Chem. – Eur. J., 2011, 17, 4085 CrossRef CAS PubMed; (c) G. S. Kumar, B. Pieber, K. R. Reddy and C. O. Kappe, Chem. – Eur. J., 2012, 18, 6124 CrossRef CAS PubMed; (d) Z.-Q. Liu, L. Zhao, X. Shang and Z. Cui, Org. Lett., 2012, 14, 3218 CrossRef CAS PubMed; (e) S.-R. Guo, Y.-Q. Yuan and J.-N. Xiang, Org. Lett., 2013, 15, 4654 CrossRef CAS PubMed; (f) D. Liu, C. Liu, H. Li and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 4453 CrossRef CAS PubMed.
- (a) J.-J. Cao, T.-H. Zhu, S.-Y. Wang, Z.-Y. Gu, X. Wang and S.-J. Ji, Chem. Commun., 2014, 50, 6439 RSC; (b) L. Wang, W. Sha, Q. Dai, X. Feng, W. Wu, H. Peng, B. Chen and J. Cheng, Org. Lett., 2014, 16, 2088 CrossRef CAS PubMed; (c) C. Pan, J. Han and Z. ChengJian, Sci. China: Chem., 2014, 57, 1172 CrossRef CAS; (d) During the preparation of the manuscript, the following paper was published: Y. Xu, Y. Chen, W. Li, Q. Xie and L. Shao, J. Org. Chem., 2016, 81, 8426 CrossRef CAS PubMed.
- For reviews on the synthesis of oxacyclic natural products, see: (a) E. Kang and E. Lee, Chem. Rev., 2005, 105, 4348 CrossRef CAS PubMed; (b) T. Nakata, Chem. Rev., 2005, 105, 4314 CrossRef CAS PubMed.
- F. Agapito, B. J. Costa Cabral and J. A. Martinho Simoes, J. Mol. Struct.: THEOCHEM, 2005, 719, 109 CrossRef CAS.
- For a recent review, see: X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807 CAS.
- A similar result was observed by the group of Ji (ref. 13a) with 1,4-dioxane and TBPB as the oxidant.
- This regiochemistry was observed by the group of Chen, using 1,4-dioxane and BPO as the oxidant. A plausible explanation is given in ref. 13b.
- For selected examples, see: (a) T. Akindele, Y. Yamamoto, M. Maekawa, H. Umeki, K.-I. Yamada and K. Tomioka, Org. Lett., 2006, 8, 5729 CrossRef CAS PubMed; (b) I. Hayakawa, H. Watanabe and H. Kigoshi, Tetrahedron, 2008, 64, 5873 CrossRef CAS.
- It is worth noting that compound 4a is significantly formed with TBPB in the absence of base (ref. 13a and checked by us in Table 1, entry 7); or with BPO in neutral media (ref. 13b and checked by us in Table 1, entry 1).
- S. Z. Zard, Radical Reactions in Organic Synthesis, Oxford University Press, 2003, pp. 26–30 Search PubMed.
- (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018 CrossRef CAS PubMed; (b) For a recent excellent review, see: A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed.
- The following rate constants are reported for α-hydrogen abstraction of THF: k (t-BuO˙) ∼ 8.3 × 106vs. k (PhCOO˙) ∼ 2.5 × 103 M−1 s−1. See: J. Fossy, D. Lefort and J. Sorba, Free Radicals in Organic Chemistry, John Wiley & Sons, 1995, p. 290 Search PubMed.
- B. Zhang, C. Mück-Lichtenfeld, C. D. Daniliue and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792 CrossRef CAS PubMed.
- An intramolecular electrophilic aromatic substitution might also be considered as a plausible pathway for the formation of byproduct 4a. Due to the extremely high acidity of cyclohexadienyl radical I, a proton transfer to the isonitrile moiety of 1a would form a cationic intermediate that could undergo Friedel–Crafts-type cyclization to give 4a.
- Z. Liang, L. Ju, Y. Xie, L. Huang and Y. Zhang, Chem. – Eur. J., 2012, 18, 15816 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for mechanistic studies and copies of NMR spectra. See DOI: 10.1039/c6ob02103d |
| This journal is © The Royal Society of Chemistry 2016 |