
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemarkable acceleration of template-directed photodimerisation of 9-phenylethynylanthracene derivatives assisted by complementary salt bridge formation†
Junki
Tanabe
,
Daisuke
Taura
,
Naoki
Ousaka
and
Eiji
Yashima
*
Department of Molecular Design and Engineering, Graduate School of Engineering, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: yashima@apchem.nagoya-u.ac.jp
First published on 12th October 2016
Abstract
The photoirradiation of 9-phenylethynylanthracene in degassed chloroform and benzene afforded not only a [4 + 2]-anti Diels–Alder addition dimer, but also a [4 + 4]-anti-dimer as a minor product for the first time as revealed by single-crystal X-ray analysis, while the anthracene residue was quantitatively oxidised in undegassed dilute chloroform, giving the corresponding endoperoxides. The photochemical reactions of carboxylic acid monomers bearing a 9-phenylethynylanthracene unit at one and both ends were further investigated in the presence and absence of the complementary amidine dimer as the template. It was found that a similar photooxidation reaction of the monomers was significantly suppressed in the presence of the template even in undegassed chloroform. In addition, the template-directed photodimerisation of the mono- and di-9-phenylethynylanthracene-bound monomers was remarkably accelerated 30- or 61-fold in the degassed chloroform, giving the [4 + 2]-anti- and [4 + 4]-anti-dimers as major and minor products, respectively, whereas the di-9-phenylethynylanthracene-bound monomer was preferentially photo-polymerised in the absence of the template.
Introduction
Photochemical reactions often promote the formation of products inaccessible by thermal reactions, leading to one of the key reactions in the state-of-the-art organic synthesis.1 However, it remains difficult to control the selectivity and specificity during the photochemical transformations in homogeneous solutions in a predictable way, thus producing a mixture of regioisomers along with stereoisomers. Therefore, the template-directed photoreaction has been developed not only to improve the efficiency, but also to control the regioselectivity and/or enantioselectivity.2Among a variety of photoreactions, the photodimerisation of anthracene and its derivatives is one of the most well-known photochemical reactions and has been extensively investigated.3 The photodimers are connected by two covalent bonds resulting from the [4 + 4] cycloaddition and revert to anthracenes thermally or under UV irradiation by using light below 300 nm. Taking advantage of this feature, the anthracene skeleton has been successfully applied to two-dimensional polymers,4 supramolecular polymers,5 reversible cross-linking reactions,6 photochemical molecular switches7 and photoinduced shape-changeable materials.8 Another important synthetic feature during the anthracene photodimerisation is that substituted anthracenes at specific positions afford regio- and/or stereoisomers.3 For instance, the irradiation of 9-substituted anthracenes gives two regioisomeric dimers, the [4 + 4]-anti and -syn photodimers (Fig. 1a), and the thermodynamically stable [4 + 4]-anti dimer is, in general, produced as a major product, whereas the energetically unfavourable [4 + 4]-syn dimer is regioselectively formed in specific environments, such as within micelles.9 Supramolecular approaches using cyclic host molecules with a rigid concave cavity, such as cyclodextrins5f,10 and cucurbit[n]urils,5d,e,6e,10f,h capable of encapsulating two anthracene molecules in close proximity, and DNA as a template,11 have also been developed to improve the reaction yield and to control the regio- and stereo-selectivities during the photodimerisation of anthracene and its derivatives.
 | ||
| Fig. 1 Photoreaction of (a) 9-substituted anthracene, (b) 9-phenylethynylanthracene (1M) and (c) 9-(4-mercaptoalkylphenylethynyl)anthracenes (1M-SH) and their photoproducts. | ||
On the other hand, when exposed to light in the presence of oxygen, anthracene derivatives readily react with singlet oxygen to produce endoperoxides,12 which eventually revert to the parent anthracenes and oxygen under thermolysis (Fig. 1a). However, anthracene can be effectively protected from photooxidation and dimerised in the cavity of cyclodextrin5f,10 and in the crystal state12e even in the presence of oxygen.
Becker and Andersson reported that the irradiation of 9-phenylethynylanthracene (1M) in solution readily promoted a [4 + 2] Diels–Alder addition reaction between the ethynyl moiety and the central ring of the other anthracene residue of 1M over the [4 + 4] cycloaddition reaction, predominantly producing a [4 + 2]-anti dimer ([4 + 2]-anti-2MM, Fig. 1b).13 In contrast, Weiss and co-workers demonstrated the energetically unfavourable [4 + 4]-syn dimer formation of thiolated 9-phenylethynylanthracenes (1M-SH) (Fig. 1c) upon irradiation that proceeded in a highly regioselective manner when 1M-SH was mixed in self-assembled alkane thiolate monolayers on flat Au surfaces on which the anthracene moieties were favourably arranged in such a way that the intermolecular [4 + 4]-syn cycloaddition ([4 + 4]-syn-2MM) could be possible.14 The regioselective [4 + 4]-syn dimer formation was monitored by scanning tunneling microscopy (STM)14a and surface-enhanced Raman spectroscopy (SERS),14b thus showing a significant decrease in conductivity and disappearance of the peaks due to the anthracene moiety, respectively. Recently, Klajn and co-workers also demonstrated that 1M-SH immobilised on metallic (Au and Pd) nanoparticles dimerised in a regioselective way to yield a [4 + 4]-syn cycloaddition product under photoirradiation (Fig. 1c), based on the time-dependent absorption spectral changes.15
We previously reported a series of m-terphenyl-based complementary double helices that could be rationally designed and synthesised based on a modular strategy we developed using amidinium–carboxylate salt bridges through which the double helices are stabilised by double hydrogen bonds with a well-defined directionality even in polar solvents.16 Therefore, various types of functional linkers, such as Pt(II)-acetylide16b,e,j,l,n and azobenzene linkages,16d,i,m can be introduced between the m-terphenyl units while maintaining the double-helical structures.16 The complementary double-helical framework stabilised by salt bridges has recently been applied to the template-directed synthesis of complementary double helices through imine-bond forming reactions between a carboxylic acid and amidine monomers bearing either a formyl or an amino group at one end.16i,m The complementary dimeric templates linked by an azobenzene residue significantly accelerated the imine-bond forming reactions. However, the reactions along the complementary dimer strands as a template were limited to imine-bond forming reactions.
In this work, we synthesised new carboxylic acid monomers (1C and 3C) bearing an anthracene group at one and both ends (Fig. 2a), respectively, and investigated their template-directed photodimerisation reactions in solution in the absence and presence of the complementary amidine dimer (TAA) connected by a p-diethynylbenzene unit as the template (Fig. 2d). The monomers 1C and 3C possess one or two 9-phenylethynylanthracene moieties identical to 1M, hence we anticipated the regioselective photodimerisation or polymerisation of 1C or 3C, respectively, giving products with a specific regioselectivity among the following four possible regioisomers ([4 + 2]-anti-, [4 + 2]-syn-, [4 + 4]-anti- and [4 + 4]-syn-isomers, Fig. 2b and c) along with acceleration of the photoreactions in the presence of the rigid template TAA through the salt bridges, which permit the 9-phenylethynylanthracene moieties to be arranged in close proximity (Fig. 2d).
 | ||
| Fig. 2 (a) Chemical structures of 9-phenylethynylanthracene-bound carboxylic acid monomers (1C and 3C), monomeric amidine (A) and dimeric amidine template (TAA). (b and c) Possible photoreactions (photooxidation and cycloaddition reactions) of the carboxylic acid monomers ((b) 1C and (c) 3C) and their photoproducts. (d) Schematic illustration for the template-directed photodimerisation of the 9-phenylethynylanthracene-bound monomers (1C and 3C). | ||
For comparison, the photochemical dimerisation of 1M (Fig. 1b),13 a model compound of 1C and 3C, was also thoroughly investigated under various conditions. We found that a [4 + 4]-anti-dimer ([4 + 4]-anti-2MM) was also produced as a minor product along with the major Diels–Alder addition product, [4 + 2]-anti-2MM, (Fig. 1b) and then their structures were unambiguously determined by single-crystal X-ray analysis.
Results and discussion
Synthesis
9-Phenylethynylanthracene (1M) was prepared according to the reported method.17 The achiral carboxylic acid monomers (1C and 3C) bearing an anthracene group at one and both ends were newly synthesised according to Schemes S3 and S5,† respectively, (see the ESI†). The p-phenylene-linked optically active amidine dimer (TAA)16g and its monomeric amidine (A)16a were also prepared according to the reported methods.Photoreactions of model monomer (1M)
The photoreaction of 1M (0.50 mM) was first investigated in undegassed CDCl3 at 25 °C (run 1 in Table 1) upon light irradiation over 400 nm. Reaction progress was monitored by 1H NMR spectroscopy. The peak intensity of the anthracene proton (Ha) at the 10-position in 1M decreased with time, whereas a new singlet signal appeared in a relatively low magnetic field of 6.08 ppm, which can be assigned to the bridge-head proton (Hb) of endoperoxide (1M-O2) and its peak intensity gradually increased as the reaction progressed, reaching 89% after irradiation for 3 min (Fig. 3a and S6a†). This assignment was supported by a molecular ionic peak at m/z = 309.19 ([1M-O2–H]−) in its negative-mode electron-spray ionisation (ESI) mass spectrum of the reaction mixture and also by the absorption spectrum of 1M after irradiation for 3 min, in which the peaks due to the anthracene moiety almost disappeared (Fig. S6†). It was noted that the obtained 1M-O2 was not stable in solution and was thermally converted back to 1M without decomposition at 25 °C.12b,d,f Thus, the rate constant for the retro-photooxidation reaction was estimated to be 4.7 × 10−6 s−1 on the basis of the time-dependent 1H NMR spectral changes (Fig. S7†). | ||
| Fig. 3 Partial 1H NMR (500 MHz, 25 °C) spectra of 1M (0.50 mM) in (a) (run 1 in Table 1) undegassed and (b) (run 5 in Table 1) degassed CDCl3 before (top) and after (bottom) irradiation of light (>400 nm). Full-scale spectra are shown in Fig. S6a and S11a,† respectively. | ||
| Run | 1M | Product yield (%) (Reaction rate 10−3k (s−1)) | Sup. Fig. no. | |||||
|---|---|---|---|---|---|---|---|---|
| Solvent (conc. (mM)) | Irradiation time (min) | Conv. (%) (consumption rate 10−3k (s−1)) | 1M-O2 | [4 + 2]-anti-2MM | [4 + 4]-anti-2MM | [4 + 2]/[4 + 4] | ||
| a The maximum yield of 1M-O2 was 18% after 8 min irradiation of light (>400 nm) (see Fig. S8a). b The maximum yield of 1M-O2 was 38% after 8 min irradiation of light (>400 nm) (see Fig. S9a). c The maximum yield of 1M-O2 was 15% after 8 min irradiation of light (>400 nm) (see Fig. S10a). d Not available in ref. 13. | ||||||||
| 1 | Undegassed CDCl3 (0.50) | 3 | 89 (16) | 89 (16) | — | — | — | Fig. S6 |
| 2 | Undegassed CDCl3 (8.0) | 30 | 88 (2.0) | 12a (0.71) | 60 (0.93) | 14 (0.14) | 4.3 | Fig. S8 |
| 3 | Undegassed benzene-d6 (0.50) | 16 | 88 (3.2) | 32b (1.0) | 42 (0.96) | 8 (0.14) | 5.3 | Fig. S9 |
| 4 | Undegassed benzene-d6 (8.0) | 30 | 96 (1.9) | 9c (0.37) | 70 (1.0) | 12 (0.13) | 5.8 | Fig. S10 |
| 5 | Degassed CDCl3 (0.50) | 30 | 88 (1.7) | — | 64 (1.1) | 23 (0.21) | 2.8 | Fig. S11 |
| 6 | Degassed CDCl3 (8.0) | 45 | 95 (1.5) | — | 77 (1.0) | 18 (0.15) | 4.3 | Fig. S12 |
| 7 | Degassed benzene-d6 (0.50) | 30 | 90 (1.6) | — | 74 (1.1) | 15 (0.16) | 4.9 | Fig. S13 |
| 8 | Degassed benzene-d6 (8.0) | 45 | 97 (1.3) | — | 80 (1.0) | 15 (0.13) | 5.3 | Fig. S14 |
| 9 | Degassed benzene (8.0) | 90 | —d | —d | 75 | 8 | 9.4 | Ref. 13 |
In contrast, after light irradiation of 1M (0.50 mM) for 30 min in degassed CDCl3 at 25 °C (run 5 in Table 1), the 1H NMR spectrum showed two new sets of singlet peaks due to the bridge-head protons (H′b and H′′b) along with a new anthracene proton (Ha′), indicating the formation of two photodimers (Fig. 3b and S11a†), which were unambiguously identified as [4 + 2]-anti-2MM and [4 + 4]-anti-2MM, respectively, by X-ray crystallographic analyses (Fig. 4) together with 2D NMR spectroscopy (Fig. S41 and S44†). Becker and Andersson reported that the photoreaction of 1M (8.0 mM) in degassed benzene gave [4 + 2]-anti-2MM in ca. 75% yield together with a by-product (ca. 8%) which was assumed to be an intermolecular [4 + 4] cycloadduct in spite of no structural information (run 9 in Table 1).13 Thus, we examined the photoreaction of 1M (8.0 mM) under almost the same conditions in degassed benzene-d6, affording [4 + 2]-anti-2MM (80%) and [4 + 4]-anti-2MM (15%) after 45 min irradiation (run 8 in Table 1 and Fig. S14†). The regioisomer ratio (5.3) was different from that reported by Becker and Andersson (9.4), but the difference may not be important.
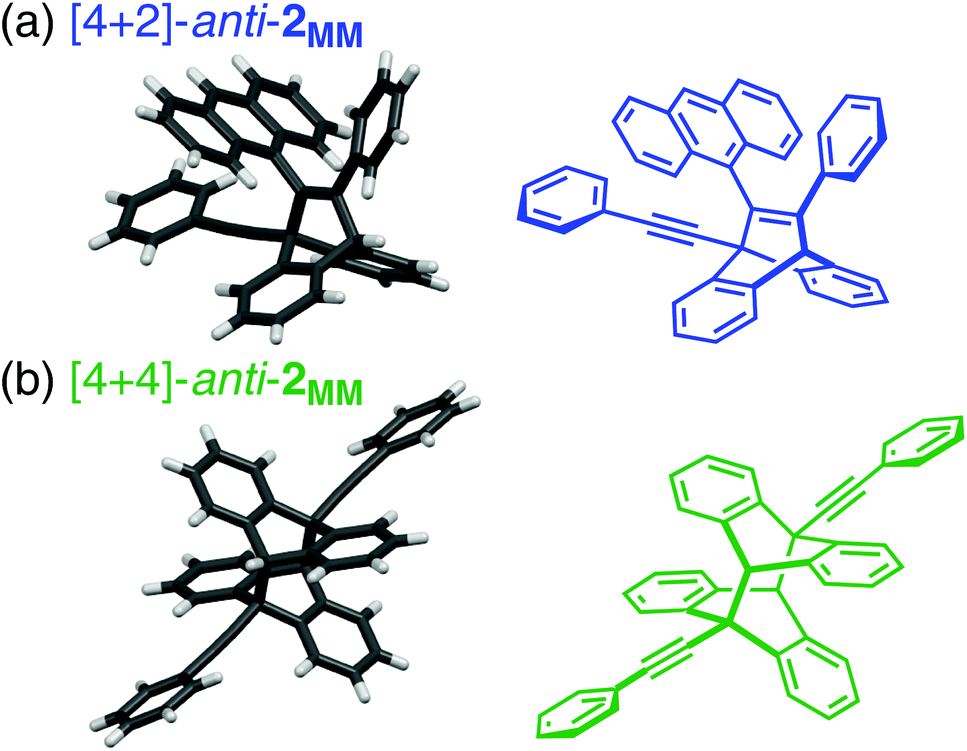 | ||
| Fig. 4 The crystal structures of (a) [4 + 2]-anti-2MM (CCDC 1503775) and (b) [4 + 4]-anti-2MM (CCDC 1503774). | ||
The effects of oxygen, solvent (CDCl3 and benzene-d6) and concentration (0.50 and 8.0 mM) on the photooxidation and regioselectivity ([4 + 2]/[4 + 4]) during the photoreaction of 1M were further investigated at 25 °C and the results are summarised in Table 1. In dilute undegassed solution, the photooxidation of 1M preferentially took place more than that in the concentrated solution. In particular, irradiation of a dilute solution of 1M (0.50 mM) in undegassed CDCl3 (run 1 in Table 1) quantitatively produced 1M-O2 due to the heavy-atom effect of the solvent, which inhibits the photodimerisation by promoting intersystem crossing.12c Although the concentration and solvent effects of the photodimerisation on regioselectivity were not significant, the yield of the major product, [4 + 2]-anti-2MM, tended to increase with the increasing concentration of 1M from 0.50 to 8.0 mM on changing the solvent from CDCl3 to benzene-d6.
We then calculated the structures of four possible photodimers (Fig. 1b) using the density functional theory (DFT) and found that their stabilities decrease in the following order: [4 + 2]-anti-2MM > [4 + 2]-syn-2MM > [4 + 4]-anti-2MM > [4 + 4]-syn-2MM (Fig. S5†), thus supporting the experimental results that the major product, [4 + 2]-anti-2MM, is much more stable than the minor one, [4 + 4]-anti-2MM, by 170.6 kJ mol−1, whereas the corresponding syn-photodimers ([4 + 2]-syn-2MM and [4 + 4]-syn-2MM) could not be observed at all in the 1H NMR spectra under the present conditions even though the syn-Diels–Alder adduct, [4 + 2]-syn-2MM, is the second most stable photodimer. The reason is not clear, but it was suggested that it was due to the more favourable centrosymmetric-oriented complex formation ((1M)2), which could generate [4 + 2]-syn-2MM,13 or these may be thermally unstable and immediately revert to the parent 1M in solution.
On the other hand, an energetically unfavourable [4 + 4]-syn-2MM derivative was reported to form in a highly regioselective fashion on self-assembled flat metal surfaces upon photoirradiation (Fig. 1c) as revealed by STM,14a Raman14b and the time-dependent absorption spectral changes.15
Fig. S15† shows the experimental (top) and DFT simulated (bottom) Raman spectra of 1M, 1M-O2, [4 + 2]-anti-2MM and [4 + 4]-anti-2MM, which are in good agreement, although 1M-O2 contains 1M generated from 1M-O2via thermolysis (Fig. S15b†).
The peaks due to the anthracene residue of 1M completely disappeared in the Raman spectrum of [4 + 4]-anti-2MM (Fig. S15d†), while these remained in the Raman spectrum of [4 + 2]-anti-2MM (Fig. S15c†). Similar distinct spectral changes were also observed in the absorption spectra of 1M, [4 + 2]-anti-2MM and [4 + 4]-anti-2MM (Fig. S2a†). We note, however, that the Raman spectra of 1M-O2 and [4 + 4]-anti-2MM, in which an anthracene moiety no longer exists, were quite similar to each other except for a weak peak at 921 cm−1 observed for 1M-O2, which can be assigned to the –O–O– bond vibration (Fig. S15b†).18 These results combined with the absorption spectra of 1M-O2 and [4 + 4]-anti-2MM (Fig. S2a†) suggested that it might be difficult to assign the photochemical reaction products of 1M and its derivatives, 1M-O2 or [4 + 4]-anti-2MM, based on the Raman14b and absorption spectroscopies.15 However, it is apparent that the photooxidation is protected in the absence of oxygen.
Template effects on the photoreactions of mono-9-phenylethynylanthracene-bound carboxylic acid monomer
The mono-9-phenylethynylanthracene-bound carboxylic acid monomer (1C) used in this study contains a 9-phenylethynylanthracene moiety identical to 1M, probably showing a similar photoreactivity to 1M in the absence of the template. Based on the photoreaction results of the model monomer 1M (Table 1), we employed a dilute CDCl3 (0.50 mM) solution throughout the following photoreactions. As anticipated, the irradiation of 1C in undegassed CDCl3 at 25 °C (run 1 in Table 2) resulted in the formation of endoperoxide (1C-O2)19 in 89% yield after 3 min as evidenced by the bridge-head proton (Hb) that newly appeared as a singlet at 6.08 ppm (Fig. 5a and S16a†); the chemical shift was very similar to that of 1M-O2 (Fig. 3a and S6a†). The structure of 1C-O2 was characterised and identified by comparing the 1H NMR spectrum of 1C-O2 with that of 1M-O2 (Fig. S1†) and ESI-mass measurements (Fig. S16c†). | ||
| Fig. 5 Partial 1H NMR (500 MHz, undegassed CDCl3, 25 °C) spectra of 1C (0.50 mM) in the absence (run 1 in Table 2) (a) and presence of A (0.50 mM) (run 2 in Table 2) (b) and TAA (0.25 mM) (run 3 in Table 2) (c) before (top) and after (bottom) irradiation of light (>400 nm). Full-scale spectra are shown in Fig. S16a, S18a and S19a.† | ||
| Run | 1C | Product yield (%) (reaction rate 10−3k (s−1)) | Sup. Fig. no. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Additive (conc. (mM)) | Solvent (conc. (mM)) | Irradiation time (min) | Conv. (%) (consumption rate 10−3k (s−1)) | 1C-O2 | [4 + 2]-anti-2CC | [4 + 4]-anti-2CC | [4 + 2]/[4 + 4] | ||
| a 1C-O2 formed during the initial stage was gradually converted into an unknown compound probably due to photolysis products of 1C-O2 upon further photoirradiation. The reaction rate was therefore estimated based on the time–conversion relationship during the initial stage. The maximum yield of 1C-O2 was 10% after 30 s irradiation of light (>400 nm) (see Fig. S19a). b The peaks for (A)2·[4 + 2]-anti-2CC were too broad to estimate its reaction rate (see Fig. S22a). | |||||||||
| 1 | — | Undegassed CDCl3 (0.50) | 3 | 89 (11) | 89 (11) | — | — | — | Fig. S16 |
| 2 | A (0.50) | Undegassed CDCl3 (0.50) | 3 | 89 (10) | 89 (10) | — | — | — | Fig. S18 |
| 3 | TAA (0.25) | Undegassed CDCl3 (0.50) | 2 | 92 (25) | 10a (4.4) | 63 (11) | 18 (2.5) | 3.5 | Fig. S19–20 |
| 4 | — | Degassed CDCl3 (0.50) | 60 | 85 (0.66) | — | 60 (0.40) | 25 (0.11) | 2.4 | Fig. S21 |
| 5 | A (0.50) | Degassed CDCl3 (0.50) | 90 | 68 (0.18) | — | 19 (—)b | 17 (0.036) | 1.1 | Fig. S22–24 |
| 6 | TAA (0.25) | Degassed CDCl3 (0.50) | 2 | 89 (20) | — | 68 (12) | 20 (2.9) | 3.4 | Fig. S25–26 |
The photoreactions of 1C (0.50 mM) in the presence of the template TAA (0.25 mM) and its monomeric amidine A (0.50 mM)20 were then investigated in undegassed CDCl3 at 25 °C. The monomeric amidine A was used for the control experiment to evaluate the template effect of TAA on the photoreaction. The 1H NMR spectra of 1C (0.50 mM) in the presence of A (0.50 mM) or TAA (0.25 mM) showed the characteristic peaks for the NH protons in the low magnetic field at ca. 13.3 ppm, indicating the salt bridge formation (Fig. S18a and S19a†).
The irradiation of A·1C (run 2 in Table 2) also produced only one set of 1H NMR signals, such as Hb (6.09 ppm), resulting from the preferential formation of 1C-O2 complexed with A (Fig. 5b and S18a†) as observed in the photooxidation of 1C in the absence of A (run 1 in Table 2). In sharp contrast, upon the irradiation of TAA·(1C)2 (run 3 in Table 2), the peak intensity of Ha decreased and almost disappeared within 2 min, whereas two new sets of singlet peaks due to the bridge-head protons (H′b and H′′b) appeared at 6.21 and 5.10 ppm, respectively (Fig. 5c and S19a†). These peaks could be clearly identified as the complexes of TAA·[4 + 2]-anti-2CC (63%) and TAA·[4 + 4]-anti-2CC (18%) based on the 1H NMR spectra of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of the isolated [4 + 2]-anti-2CC and [4 + 4]-anti-2CC after purification with TAA, respectively (Fig. S28†). Interestingly, the photooxidation reaction of 1C was significantly protected in the presence of the template TAA, affording 1C-O2 in less than 10% yield (Fig. S19a†) and the intermolecular photodimerisation selectively proceeded due to the close proximity of the two anthracene moieties arranged along the template, as supported by the decrease and red-shift of the absorption peaks of 1C upon the addition of TAA, indicative of the π-stacked arrangements of the anthracene moieties (Fig. S20†).
:1 mixtures of the isolated [4 + 2]-anti-2CC and [4 + 4]-anti-2CC after purification with TAA, respectively (Fig. S28†). Interestingly, the photooxidation reaction of 1C was significantly protected in the presence of the template TAA, affording 1C-O2 in less than 10% yield (Fig. S19a†) and the intermolecular photodimerisation selectively proceeded due to the close proximity of the two anthracene moieties arranged along the template, as supported by the decrease and red-shift of the absorption peaks of 1C upon the addition of TAA, indicative of the π-stacked arrangements of the anthracene moieties (Fig. S20†).
In the same way, the photoreactions of 1C (0.50 mM) in the absence and presence of A (0.50 mM) or TAA (0.25 mM) were performed in degassed CDCl3 at 25 °C (runs 4–6 in Table 2), in which the photooxidation was completely prohibited as observed in the model reaction of 1M under the same conditions. In the absence of A and TAA, 1C was gradually converted to the photodimers, producing [4 + 2]-anti-2CC and [4 + 4]-anti-2CC in 60 and 25% yields, respectively, after irradiation for 60 min (run 4 in Table 2 and Fig. 6a and S21a†).
 | ||
| Fig. 6 Partial 1H NMR (500 MHz, degassed CDCl3, 25 °C) spectra of 1C (0.50 mM) in the absence (run 4 in Table 2) (a) and presence of A (0.50 mM) (run 5 in Table 2) (b) and TAA (0.25 mM) (run 6 in Table 2) (c) before (top) and after (bottom) irradiation of light (>400 nm). * denotes the peak due to 1,1,2,2-tetrachloroethane used as an internal standard. Full-scale spectra are shown in Fig. S21a, S22a and S25a.† The peaks for (A)2·[4 + 2]-anti-2CC were too broad to estimate its yield. The yield of (A)2·[4 + 2]-anti-2CC was estimated to be 19% from the integral ratios of the peaks for isolated [4 + 4]-anti-2CC and [4 + 2]-anti-2CC (see Fig. S22b†). | ||
Quite interestingly, the photodimerisation of 1C in the presence of the template TAA (run 6 in Table 2) took place much faster than that in the absence of TAA, affording the TAA·[4 + 2]-anti-2CC (68%) and TAA·[4 + 4]-anti-2CC (20%) complexes after 2 min of light irradiation (Fig. 6c and S25a†).21 Contrary to our expectation, however, we could not observe a specific regioselectivity during the photodimerisation, producing photodimers with an almost similar regioselectivity ([4 + 2]/[4 + 4]) to that in the absence of the template TAA.
To estimate the reaction rate constant (k) of 1C, the conversions of 1C were estimated from the integral ratios of the peaks for Ha (TAA·(1C)2), H′′b ([4 + 2]-anti-2CC), H′′b ([4 + 4]-anti-2CC) and the internal standard (1,1,2,2-tetrachloroethane) based on the 1H NMR spectral changes and were plotted versus the reaction time (Fig. 7, S21, S23 and S25†). The experimental data were then fitted to a first-order kinetic model using eqn (1),
| ln(C/C0) = −kt | (1) |
 | ||
| Fig. 7 Kinetic plots of the photodimerisation of 1C (0.50 mM) in degassed CDCl3 at 25 °C in the absence (blue line) and presence of A (black line) and TAA (red line). The reaction rates were estimated from the integral ratios of the peaks for Ha (1C) and the internal standard (1,1,2,2-tetrachloroethane) based on the 1H NMR spectral changes shown in Fig. S21a, S22a and S25a.† | ||
In the presence of A, however, the photodimerisation (run 5 in Table 2) of 1C took place very slowly with the k value of 0.18 × 10−3 s−1 (Fig. 7 and S23†). The 1H NMR spectrum of A·1C after irradiation for 90 min was rather complicated except for the signals of (A)2·[4 + 4]-anti-2CC (17%), and those of (A)2·[4 + 2]-anti-2CC were not clearly observed because the signals were too broad (Fig. 6b and S22a†). The resulting photodimers with carboxylic acid residues were then isolated from the reaction mixture by silica gel column chromatography and the yield of (A)2·[4 + 2]-anti-2CC was estimated to be 19% based on the integral ratio ([4 + 2]-anti-2CC/[4 + 4]-anti-2CC) in the 1H NMR spectrum (Fig. S22b†). Molecular mechanics (MM) calculations for the A·1C salt-bridged complex suggested that one of the terminal TMS units of A is located close to the anthracene moiety of 1C and a reactive acetylene residue of 1C is likely sandwiched between the TMS and the phenyl group of the amidine residue of A (Fig. S24†). Thus, the photodimerisation of 1C, in particular, the formation of [4 + 2]-anti-2CC may be considerably retarded once complexed with A due to such steric effects, leading to the decrease in the reaction rate.
Template effects on the photoreactions of di-9-phenylethynylanthracene-bound carboxylic acid monomer
The photoreactions of the di-9-phenylethynylanthracene-bound carboxylic acid monomer (3C) (0.50 mM) in the absence and presence of the template TAA (0.25 mM) were also investigated in undegassed and degassed CDCl3 at 25 °C (Table 3). The irradiation of 3C in undegassed CDCl3 (run 1 in Table 3 and Fig. S29†) readily promoted the photooxidation to produce a mixture of mono- (3C-O2) and di-endoperoxides (3C-2O2) in 50% yield as identified by the ESI-mass (Fig. S29c†) and NMR analysis (Fig. S1†), which eventually reverted to the parent 3C and further converted in part to unknown compounds by thermolysis (Fig. S30†). In the presence of TAA, the photooxidation of 3C was significantly suppressed as anticipated (12% yield, run 2 in Table 3)22 and the [4 + 2]- and [4 + 4]-anti-4CC photodimers were mainly produced in 60 and 17% yields, respectively, on the basis of the 1H NMR spectra of the products by comparing them with those of the photodimers of 2CC formed in the presence of TAA (Fig. S37a–d†). We found that one of the terminal anthracene units of 4CC complexed with TAA was further oxidised (Fig. S31b†), resulting in the formation of the mono- and/or di-oxidised dimers, [4 + 2]-anti-4CC-O2 and [4 + 4]-anti-4CC-O2.| Run | 3C | Products yield (%) (reaction rate 10−3k (s−1)) | Sup. Fig. no. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Additive (conc. (mM)) | Solvent (conc. (mM)) | Irradiation time (min) | Conv. (%) (consumption rate 10−3k (s−1)) | poly-3C | 3C-O2 and 3C-2O2 | [4 + 2]-anti-4CC | [4 + 4]-anti-4CC | [4 + 2]/[4 + 4] | |||
| [4 + 2]-anti | [4 + 4]-anti | ||||||||||
| a Total yield of mono- (3C-O2) and di-endoperoxides (3C-2O2) of 3C, which were gradually converted into unknown compounds probably due to photolysis upon further photoirradiation (see Fig. S30). b One of the terminal anthracene units of [4 + 2]- and [4 + 4]-anti-4CC was oxidised (see Fig. S31). c The conversion of 3C and its consumption rate were estimated based on the formation of poly-3C using 1H NMR (see Fig. S35a). d The number-average molecular weight (Mn) was determined by SEC using polystyrene standards in THF containing 0.1 wt% TBAB as the eluent (see Fig. 8B(a)). e It was difficult to estimate the yields of [4 + 2]- and [4 + 4]-anti-4CC because their proton NMR signals were overlapped with those of poly-3C (see Fig. S35a). | |||||||||||
| 1 | — | Undegassed CDCl3 (0.50) | 3 | 82 (9.9) | — | 50a (4.2) | — | — | — | Fig. S29 | |
| 2 | TAA (0.25) | Undegassed CDCl3 (0.50) | 2 | 97 (38) | — | 12 (1.4) | 60b (19) | 17b (3.6) | 3.5 | Fig. S31–34 | |
| 3 | — | Degassed CDCl3 (0.50) | 60 | 22c (0.61)c | 72dMn = 3.2 × 103 | — | —e | —e | 3.0 | Fig. S35 | |
| 54 (0.39) | 18 (0.010) | ||||||||||
| 4 | TAA (0.25) | Degassed CDCl3 (0.50) | 2 | 95 (37) | — | — | 74 (23) | 20 (4) | 3.7 | Fig. S36–38 | |
In strong contrast, the 1H NMR spectrum of 3C (0.50 mM) after irradiation for 60 min in degassed CDCl3 (run 3 in Table 3) became significantly broadened, giving signals due to the [4 + 2]- and [4 + 4]-anti dimerisations (H′b and H′′b), indicating the formation of a random copolymer (poly-3C) composed of [4 + 2]- and [4 + 4]-anti units (Fig. 8A(a) and S35a†). The size-exclusion chromatography (SEC) analysis further supported the polymerisation of 3C (Fig. 8B(a)).23 Interestingly, the photoreaction of TAA·(3C)2 in degassed CDCl3 (run 4 in Table 3) showed two sharp bridge-head peaks (H′b and H′′b), suggesting the formation of the photodimers, TAA·[4 + 2]-anti-4CC and TAA·[4 + 4]-anti-4CC, while the other anthracene moieties remained unreacted as evidenced by the 1H NMR and absorption spectra (Fig. 8A(b), S36a and S38†). The SEC analysis of the products further supported this conclusion (Fig. 8B(b–d)).
 | ||
| Fig. 8 (A) Partial 1H NMR (500 MHz, degassed CDCl3, 25 °C) spectra of 3C (0.50 mM) in the absence (run 3 in Table 3) (a) and presence of TAA (run 4 in Table 3) (b) before (top) and after (bottom) irradiation of light (>400 nm). * denotes the peak due to 1,1,2,2-tetrachloroethane used as an internal standard. Full-scale spectra are shown in Fig. S35a and S36a.† (B) SEC chromatograms of (a) poly-3C produced in a degassed CDCl3 solution of 3C (0.50 mM) upon irradiation with light (>400 nm) for 60 min (see Fig. S35†), (b) isolated carboxylic acid dimers obtained from TAA·(3C)2 (0.25 mM) after irradiation of light (>400 nm) for 120 s in degassed CDCl3 (see Fig. S37g†), (c) dimer [4 + 2]-anti-2CC, and (d) monomer 3C. These carboxylic acids were converted into the corresponding methyl esters by treatment with (trimethylsilyl)diazomethane before SEC analysis. | ||
These results indicated that the dimeric template TAA selectively promoted the photodimerisation of the anthracene units of the bimolecular 3C located in close proximity at the center (Fig. S38†), whereas the further polymerisation of the resultant TAA·[4 + 2]-anti-4CC and TAA·[4 + 4]-anti-4CC dimers was exclusively protected due to the steric effects between the remaining anthracene units at the ends and the TMS and phenyl groups of the amidine residues of TAA as observed in the photodimerisation of A·1C (run 5 in Table 2).
In the same way, the k values of 3C in degassed CDCl3 in the presence and absence of TAA were estimated to be 37 × 10−3 s−1 and 0.61 × 10−3 s−1, respectively, (Fig. S35d and S36c†). Thus, the photodimerisation of 3C was accelerated 61-fold (k(TAA·(3C)2)/k(3C) = 61) in the presence of TAA. Moreover, the dimerisation of 3C was found to take place faster than that of 1C in the presence of TAA by a factor of 1.9 (k(TAA·(3C)2)/k(TAA·(1C)2) = 1.9). This indicated that 3C can more effectively dimerise along the template than 1C because 3C possesses two anthracene moieties at both ends (Fig. S37e†).
Conclusions
In conclusion, we have performed a close inspection of the photochemical reactions of 9-phenylethynylanthracene under various reaction conditions and proved the [4 + 4]-anti and [4 + 2]-anti dimers structures produced during the photodimerisation13 by single-crystal X-ray analysis. We also found a remarkable template effect of the amidine dimer on the photoreactions of the mono- and di-9-phenylethynylanthracene-bound carboxylic acid monomers. The detailed kinetic and structural studies revealed that the [4 + 4]- and [4 + 2]-anti photodimers (2CC and 4CC) were selectively produced along the template and the reaction rate was accelerated 30 or 61-fold without side reactions, oxidation and polymerisation. The observed noticeable enhancement of the dimerisation rate constants could be ascribed to the two anthracene moieties of the monomers located in close proximity at the reaction site along the rigid template assisted by the amidinium–carboxylate salt bridges. Although the template-directed, specific regioselective-photodimerisation was not achieved, the present template-directed photodimerisation system based on salt bridges will be further applied to the template-directed enantioselective photodimerisation of prochiral anthracene derivatives10,24 because the amidine dimer used in the present study is optically active.25 Studies along this line are now underway in our laboratory.Acknowledgements
This work was supported in part by JSPS KAKENHI (Grant-in-Aid for Scientific Research (S), No. 25220804 (E. Y.) and Grant-in-Aid for Young Scientists (B), No. 26810048 (D. T.)). J. T. expresses his thanks for the JSPS Research Fellowship for Young Scientists (No. 8886). The authors thank Professor Hiroki Iida for his help with the single crystal X-ray diffraction analysis.Notes and references
- (a) N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed; (b) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (c) N. Hoffmann, J. Photochem. Photobiol., C, 2014, 19, 1–19 CrossRef CAS; (d) N. Hoffmann, Synthesis, 2016, 1782–1802 CrossRef CAS.
- (a) N. D. McClenaghan and D. M. Bassani, Int. J. Photoenergy, 2004, 6, 185–192 CrossRef CAS; (b) J. Svoboda and B. König, Chem. Rev., 2006, 106, 5413–5430 CrossRef CAS PubMed; (c) C. Yang, Chin. Chem. Lett., 2013, 24, 437–441 CrossRef; (d) B. Bibal, C. Mongin and D. M. Bassani, Chem. Soc. Rev., 2014, 43, 4179–4198 RSC; (e) N. Vallavoju and J. Sivaguru, Chem. Soc. Rev., 2014, 43, 4084–4101 RSC; (f) C. Yang and Y. Inoue, Chem. Soc. Rev., 2014, 43, 4123–4143 RSC; (g) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS PubMed; (h) V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS PubMed.
- (a) H.-D. Becker, Chem. Rev., 1993, 93, 145–172 CrossRef CAS; (b) H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC; (c) H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2001, 30, 248–263 RSC.
- (a) P. Kissel, R. Erni, W. B. Schweizer, M. D. Rossell, B. T. King, T. Bauer, S. Götzinger, A. D. Schlüter and J. Sakamoto, Nat. Chem., 2012, 4, 287–291 CrossRef CAS PubMed; (b) R. Bhola, P. Payamyar, D. J. Murray, B. Kumar, A. J. Teator, M. U. Schmidt, S. M. Hammer, A. Saha, J. Sakamoto, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2013, 135, 14134–14141 CrossRef CAS PubMed; (c) P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed; (d) M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779–784 CrossRef CAS PubMed; (e) P. Payamyar, K. Kaja, C. Ruiz-Vargas, A. Stemmer, D. J. Murray, C. J. Johnson, B. T. King, F. Schiffmann, J. VandeVondele, A. Renn, S. Götzinger, P. Ceroni, A. Schütz, L.-T. Lee, Z. Zheng, J. Sakamoto and A. D. Schlüter, Adv. Mater., 2014, 26, 2052–2058 CrossRef CAS PubMed; (f) D. J. Murray, D. D. Patterson, P. Payamyar, R. Bhola, W. Song, M. Lackinger, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2015, 137, 3450–3453 CrossRef CAS PubMed; (g) P. Payamyar, M. Servalli, T. Hungerland, A. P. Schütz, Z. Zheng, A. Borgschulte and A. D. Schlüter, Macromol. Rapid Commun., 2015, 36, 151–158 CrossRef CAS PubMed.
- (a) J.-F. Xu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Org. Lett., 2013, 15, 6148–6151 CrossRef CAS PubMed; (b) P. Wang, J. Hu, S. Yang, B. Song and Q. Wang, Chem. – Asian J., 2014, 9, 2880–2884 CrossRef CAS PubMed; (c) P. Wei, X. Yan and F. Huang, Chem. Commun., 2014, 50, 14105–14108 RSC; (d) Z. Ji, Y. Li, Y. Ding, G. Chen and M. Jiang, Polym. Chem., 2015, 6, 6880–6884 RSC; (e) Z. Yu, J. Zhang, R. J. Coulston, R. M. Parker, F. Biedermann, X. Liu, O. A. Scherman and C. Abell, Chem. Sci., 2015, 6, 4929–4933 RSC; (f) X. Zhang, Y. Gao, Y. Lin, J. Hu and Y. Ju, Polym. Chem., 2015, 6, 4162–4166 RSC.
- (a) Y. Zheng, M. Micic, S. V. Mello, M. Mabrouki, F. M. Andreopoulos, V. Konka, S. M. Pham and R. M. Leblanc, Macromolecules, 2002, 35, 5228–5234 CrossRef CAS; (b) L. A. Connal, R. Vestberg, C. J. Hawker and G. G. Qiao, Adv. Funct. Mater., 2008, 18, 3315–3322 CrossRef CAS; (c) P. Froimowicz, H. Frey and K. Landfester, Macromol. Rapid Commun., 2011, 32, 468–473 CrossRef CAS PubMed; (d) L. A. Wells, M. A. Brook and H. Sheardown, Macromol. Biosci., 2011, 11, 988–998 CrossRef CAS PubMed; (e) F. Biedermann, I. Ross and O. A. Scherman, Polym. Chem., 2014, 5, 5375–5382 RSC; (f) L. López-Vilanova, I. Martinez, T. Corrales and F. Catalina, Eur. Polym. J., 2014, 56, 69–76 CrossRef; (g) H. Xie, C.-Y. Cheng, L. Du, C.-J. Fan, X.-Y. Deng, K.-K. Yang and Y.-Z. Wang, Macromolecules, 2016, 49, 3845–3855 CrossRef CAS; (h) H. Xie, M.-J. He, X.-Y. Deng, L. Du, C.-J. Fan, K.-K. Yang and Y.-Z. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 9431–9439 CrossRef CAS PubMed.
- (a) F. Moriwaki, A. Ueno, T. Osa, F. Hamada and K. Murai, Chem. Lett., 1986, 1865–1868 CrossRef CAS; (b) Y. Molard, D. M. Bassani, J.-P. Desvergne, P. N. Horton, M. B. Hursthouse and J. H. R. Tucker, Angew. Chem., Int. Ed., 2005, 44, 1072–1075 CrossRef CAS PubMed; (c) C. Schäfer, R. Eckel, R. Ros, J. Mattay and D. Anselmetti, J. Am. Chem. Soc., 2007, 129, 1488–1489 CrossRef PubMed; (d) C.-K. Liang, G. V. Dubacheva, T. Buffeteau, D. Cavagnat, P. Hapiot, B. Fabre, J. H. R. Tucker and D. M. Bassani, Chem. – Eur. J., 2013, 19, 12748–12758 CrossRef CAS PubMed; (e) H. Wang, F. Liu, R. C. Helgeson and K. N. Houk, Angew. Chem., Int. Ed., 2013, 52, 655–659 CrossRef CAS PubMed.
- (a) R. O. Al-Kaysi, A. M. Müller and C. J. Bardeen, J. Am. Chem. Soc., 2006, 128, 15938–15939 CrossRef CAS PubMed; (b) R. O. Al-Kaysi and C. J. Bardeen, Adv. Mater., 2007, 19, 1276–1280 CrossRef CAS; (c) J. T. Good, J. J. Burdett and C. J. Bardeen, Small, 2009, 5, 2902–2909 CrossRef CAS PubMed; (d) L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, J. Am. Chem. Soc., 2011, 133, 12569–12575 CrossRef CAS PubMed; (e) T. Kim, L. Zhu, L. J. Mueller and C. J. Bardeen, J. Am. Chem. Soc., 2014, 136, 6617–6625 CrossRef CAS PubMed.
- (a) T. Wolff, J. Photochem., 1981, 16, 343–346 CrossRef CAS; (b) T. Wolff and N. Müller, J. Photochem., 1983, 23, 131–140 CrossRef CAS; (c) T. Wolff, N. Müller and G. von Bünau, J. Photochem., 1983, 22, 61–70 CrossRef CAS; (d) A. Schütz and T. Wolff, J. Photochem. Photobiol., A, 1997, 109, 251–258 CrossRef.
- (a) T. Tamaki, Chem. Lett., 1984, 13, 53–56 CrossRef; (b) T. Tamaki and T. Kokubu, J. Inclusion Phenom., 1984, 2, 815–822 CrossRef CAS; (c) A. Ueno, F. Moriwaki, Y. Iwama, I. Suzuki, T. Osa, T. Ohta and S. Nozoe, J. Am. Chem. Soc., 1991, 113, 7034–7036 CrossRef CAS; (d) A. Nakamura and Y. Inoue, J. Am. Chem. Soc., 2003, 125, 966–972 CrossRef CAS PubMed; (e) C. Yang, A. Nakamura, G. Fukuhara, Y. Origane, T. Mori, T. Wada and Y. Inoue, J. Org. Chem., 2006, 71, 3126–3136 CrossRef CAS PubMed; (f) C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2008, 130, 8574–8575 CrossRef CAS PubMed; (g) C. Ke, C. Yang, T. Mori, T. Wada, Y. Liu and Y. Inoue, Angew. Chem., Int. Ed., 2009, 48, 6675–6677 CrossRef CAS PubMed; (h) C. Yang, C. Ke, W. Liang, G. Fukuhara, T. Mori, Y. Liu and Y. Inoue, J. Am. Chem. Soc., 2011, 133, 13786–13789 CrossRef CAS PubMed; (i) J. Yao, Z. Yan, J. Ji, W. Wu, C. Yang, M. Nishijima, G. Fukuhara, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2014, 136, 6916–6919 CrossRef CAS PubMed.
- T. Ihara, T. Fujii, M. Mukae, Y. Kitamura and A. Jyo, J. Am. Chem. Soc., 2004, 126, 8880–8881 CrossRef CAS PubMed.
- (a) N. Sugiyama, M. Iwata, M. Yoshioka, K. Yamada and H. Aoyama, Bull. Chem. Soc. Jpn., 1969, 42, 1377–1379 CrossRef CAS; (b) N. J. Turro, M.-F. Chow and J. Rigaudy, J. Am. Chem. Soc., 1981, 103, 7218–7224 CrossRef CAS; (c) N. Toshima, T. Sugano and H. Hirai, Can. J. Chem., 1984, 62, 2047–2053 CrossRef CAS; (d) J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS PubMed; (e) E. Berni, C. Dolain, B. Kauffmann, J.-M. Léger, C. Zhan and I. Huc, J. Org. Chem., 2008, 73, 2687–2694 CrossRef CAS PubMed; (f) W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS PubMed.
- H.-D. Becker and K. Andersson, J. Photochem., 1984, 26, 75–77 CrossRef CAS.
- (a) M. Kim, J. N. Hohman, Y. Cao, K. N. Houk, H. Ma, A. K.-Y. Jen and P. S. Weiss, Science, 2011, 331, 1312–1315 CrossRef CAS PubMed; (b) Y. B. Zheng, J. L. Payton, T.-B. Song, B. K. Pathem, Y. Zhao, H. Ma, Y. Yang, L. Jensen, A. K.-Y. Jen and P. S. Weiss, Nano Lett., 2012, 12, 5362–5368 CrossRef CAS PubMed.
- T. Zdobinsky, P. S. Maiti and R. Klajn, J. Am. Chem. Soc., 2014, 136, 2711–2714 CrossRef PubMed.
- (a) Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2005, 44, 3867–3870 CrossRef CAS PubMed; (b) Y. Furusho, Y. Tanaka and E. Yashima, Org. Lett., 2006, 8, 2583–2586 CrossRef CAS PubMed; (c) M. Ikeda, Y. Tanaka, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 6806–6807 CrossRef CAS PubMed; (d) Y. Furusho, Y. Tanaka, T. Maeda, M. Ikeda and E. Yashima, Chem. Commun., 2007, 3174–3176 RSC; (e) T. Hasegawa, Y. Furusho, H. Katagiri and E. Yashima, Angew. Chem., Int. Ed., 2007, 46, 5885–5888 CrossRef CAS PubMed; (f) H. Ito, Y. Furusho, T. Hasegawa and E. Yashima, J. Am. Chem. Soc., 2008, 130, 14008–14015 CrossRef CAS PubMed; (g) T. Maeda, Y. Furusho, S.-i. Sakurai, J. Kumaki, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2008, 130, 7938–7945 CrossRef CAS PubMed; (h) H. Iida, M. Shimoyama, Y. Furusho and E. Yashima, J. Org. Chem., 2010, 75, 417–423 CrossRef CAS PubMed; (i) H. Yamada, Y. Furusho, H. Ito and E. Yashima, Chem. Commun., 2010, 46, 3487–3489 RSC; (j) H. Ito, M. Ikeda, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2011, 133, 3419–3432 CrossRef CAS PubMed; (k) H. Yamada, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 7250–7253 CrossRef CAS PubMed; (l) H. Yamada, Z.-Q. Wu, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 9506–9520 CrossRef CAS PubMed; (m) J. Tanabe, D. Taura, H. Yamada, Y. Furusho and E. Yashima, Chem. Sci., 2013, 4, 2960–2966 RSC; (n) M. Horie, N. Ousaka, D. Taura and E. Yashima, Chem. Sci., 2015, 6, 714–723 RSC.
- C.-W. Wan, A. Burghart, J. Chen, F. Bergstrom, L. B.-Å. Johansson, M. F. Wolford, T. G. Kim, M. R. Topp, R. M. Hochstrasser and K. Burgess, Chem. – Eur. J., 2003, 9, 4430–4441 CrossRef CAS PubMed.
- (a) Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS; (b) H. Zhang, L.-H. Guo, L. Zhao, B. Wan and Y. Yang, J. Phys. Chem. Lett., 2015, 6, 958–963 CrossRef CAS PubMed.
- The photooxidised 1C-O2 gradually decomposed under shielded light, producing the monomer 1C together with a small amount of an unknown compound (Fig. S17†).
- The association constant (Ka) between 1C and A could be roughly estimated to be ca. 3.5 × 106 M−1 in CDCl3 by comparison of the Ka value between analogous amidine and carboxylic acids.16.
- The template TAA possesses four alkyne units that are not bonded to the anthracene residue. However, these alkyne units did not participate in the photoreactions because the conversion (89%) of 1C after photoirradiation for 2 min in the presence of TAA is almost consistent with the total yield (68 and 20%) of the resulting carboxylic acid photodimers ([4 + 2]-anti-2CC and [4 + 4]-anti-2CC) (run 6 in Table 2). In addition, the structures of the dimer products isolated were fully characterised, indicating that the photodimers were not composed of TAA at all. Becker and Andersson also reported that the intermolecular photodimerisation between anthracene and diphenylacetylene did not take place, giving no [4 + 2]-cycloadduct upon light irradiation over 400 nm.13 This also suggests that the alkyne units of TAA will not react with the anthracene unit(s) of 1C and 3C upon photoirradiation (>400 nm).
- The peak assignments were performed by comparing the 1H NMR spectrum of a mixture of 3C-O2 and 3C-2O2 in the presence of TAA (Fig. S33†).
- Poly-3C was converted to the corresponding methyl esters by treatment with (trimethylsilyl)diazomethane. The number average molecular weight (Mn) and its distribution (Mw/Mn) were estimated to be 3.2 × 103 and 1.5, respectively, by SEC using polystyrene standards and THF containing 0.1 wt% tetrabutylammonium bromide as the eluent. The Mn value corresponds to ca. 4 repeating units in a single polymer chain.
- (a) Y. Ishida, Y. Kai, S.-y. Kato, A. Misawa, S. Amano, Y. Matsuoka and K. Saigo, Angew. Chem., Int. Ed., 2008, 47, 8241–8245 CrossRef CAS PubMed; (b) A. Dawn, T. Shiraki, S. Haraguchi, H. Sato, K. Sada and S. Shinkai, Chem. – Eur. J., 2010, 16, 3676–3689 CrossRef CAS PubMed; (c) Y. Ishida, A. S. Achalkumar, S.-y. Kato, Y. Kai, A. Misawa, Y. Hayashi, K. Yamada, Y. Matsuoka, M. Shiro and K. Saigo, J. Am. Chem. Soc., 2010, 132, 17435–17446 CrossRef CAS PubMed; (d) D. Fuentealba, H. Kato, M. Nishijima, G. Fukuhara, T. Mori, Y. Inoue and C. Bohne, J. Am. Chem. Soc., 2013, 135, 203–209 CrossRef CAS PubMed; (e) Y. Ishida, Y. Matsuoka, Y. Kai, K. Yamada, K. Nakagawa, T. Asahi and K. Saigo, J. Am. Chem. Soc., 2013, 135, 6407–6410 CrossRef CAS PubMed; (f) Y. Kawanami, H. Umehara, J.-i. Mizoguchi, M. Nishijima, G. Fukuhara, C. Yang, T. Mori and Y. Inoue, J. Org. Chem., 2013, 78, 3073–3085 CrossRef CAS PubMed; (g) G. Fukuhara, H. Umehara, S. Higashino, M. Nishijima, C. Yang, T. Mori, T. Wada and Y. Inoue, Photochem. Photobiol. Sci., 2014, 13, 162–171 RSC; (h) G. Fukuhara, K. Iida, Y. Kawanami, H. Tanaka, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2015, 137, 15007–15014 CrossRef CAS PubMed; (i) G. Fukuhara, K. Iida, T. Mori and Y. Inoue, J. Photochem. Photobiol., A, 2016, 331, 76–83 CrossRef; (j) M. M. Maturi, G. Fukuhara, K. Tanaka, Y. Kawanami, T. Mori, Y. Inoue and T. Bach, Chem. Commun., 2016, 52, 1032–1035 RSC.
- The complexes of the achiral 1C and 3C with the optically active amidine template TAA exhibited apparent Cotton effects in the absorption region of the anthracene residues (ca., 350–440 nm) accompanied by a decrease and red-shift in their absorption spectra (Fig. S20, S26, S34 and S38†), indicating that the two anthracene units of the monomers appear to be arranged in a chiral fashion along the chiral template. These results seem to be promising for realising such a template-directed enantioselective photodimerisation.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional spectroscopic data. CCDC 1503774 and 1503775. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02087a |
| This journal is © The Royal Society of Chemistry 2016 |