
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDenitrogenative hydrofluorination of aromatic aldehyde hydrazones using (difluoroiodo)toluene†
Kaivalya G.
Kulkarni
,
Boris
Miokovic
,
Matthew
Sauder
and
Graham K.
Murphy
 *
*
Department of Chemistry, University of Waterloo, 200 University Ave. W, Waterloo, ON, Canada N2L3G1. E-mail: graham.murphy@uwaterloo.ca
First published on 26th September 2016
Abstract
An operationally simple conversion of aromatic aldehyde hydrazones to monofluoromethylated arenes is reported. The hypervalent iodine reagent TolIF2 serves as an oxidant, converting the hydrazone to the corresponding diazo compounds. The by-product of the oxidation process, HF, is consumed in situ by a denitrogenative hydrofluorination reaction of the diazo group.
Introduction
There is only a handful of naturally occurring fluorocarbons, so fluorine's use in medicinal chemistry might never have come to pass had chemists limited themselves to naturally-inspired pharmaceuticals.1 As fluorine is small, univalent and makes very strong bonds to carbon, it is now routinely employed as a biostere for hydrogen.2 Over the past few decades, site-specific fluorination has become an essential tool for medicinal chemists due to fluorine's ability to act as a biostere, and because of its desirable effects on the lipophilicity, metabolic stability and bioavailability of pharmaceutical and agrochemical agents.3Given the breadth of biologically active fluorinated compounds,4 the development of new fluorination strategies and reagents remains an important field of research.5 As a complement to nucleophilic fluorination strategies, a large body of research developing electrophilic fluorinating reagents has been completed. While elemental fluorine is the simplest and most direct source of electrophilic fluorine, it is toxic, explosively reactive with organic compounds, and cannot be used without specialized laboratory equipment. To mitigate fluorine's reactivity, chemists have synthesized designer electrophilic fluorinating reagents based on N–F,6 S–F,7 and I–F bonds8,9 (Fig. 1). However, contrary to the N–F reagents (1–3) that serve as sources of electrophilic “F+”, the S–F and I–F compounds (4–8) have umpolung reactivity: attack of the hypervalent sulphur or iodine atom by a Lewis base expels fluoride, and generates an electrophilic adduct to be displaced by the fluoride anion.
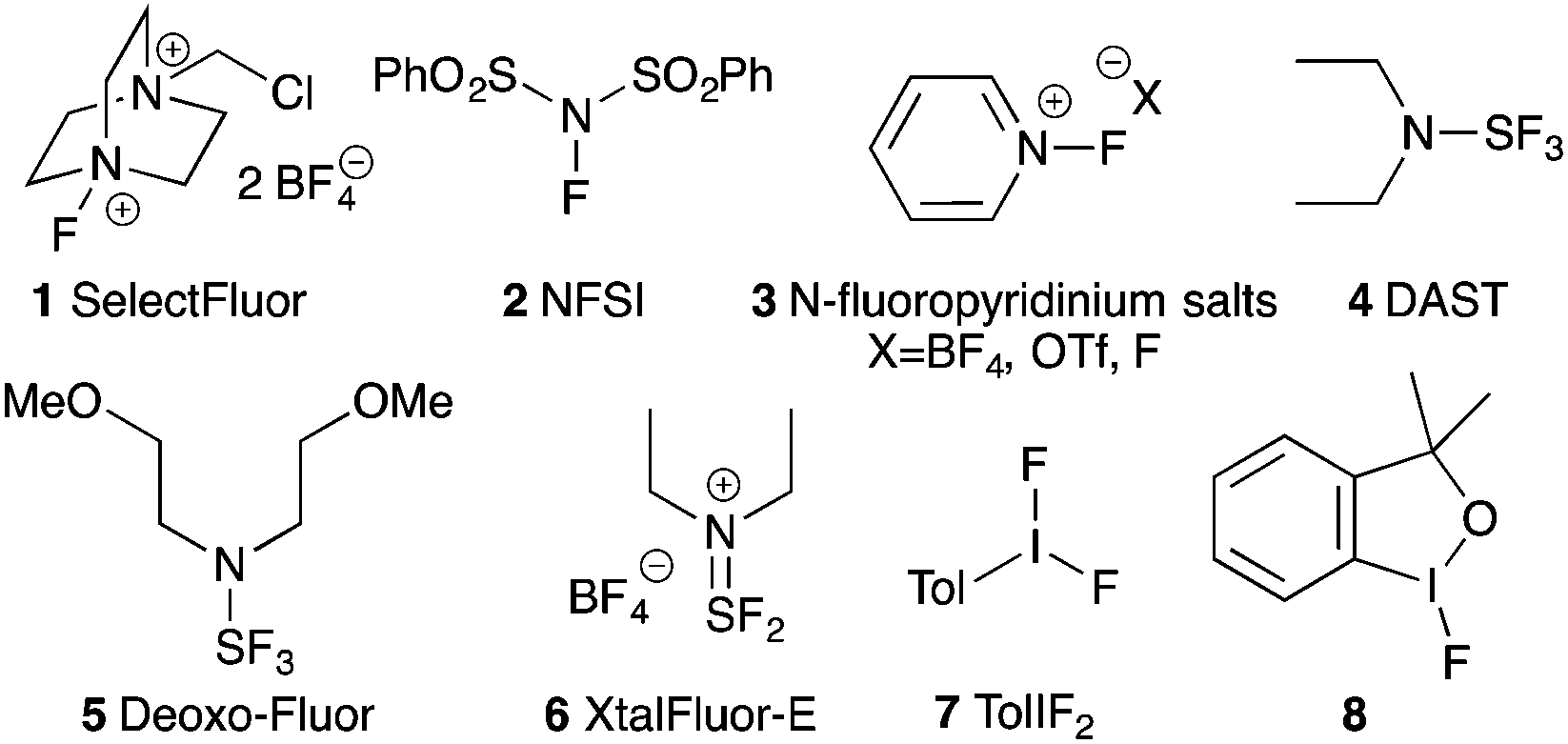 | ||
| Fig. 1 Fluorine transfer reagents based on N–F, S–F or I–F bonds. | ||
Benzylic fluorides, represented by the ArCH2F, ArCF2H and ArCF3 groups,10 are common synthetic targets and strategies towards the ArCH2F and ArCF2H functional groups have received considerable attention in the literature.5a,h,11 General strategies towards their synthesis include deoxygenative fluorination of benzyl alcohol (using 4–6),12 substitution reactions of benzyl halides,13 metal-catalyzed cross-coupling of fluorine-containing units,3a,14 or radical-based fluorination reactions.15 The ArCH2F group might also be synthesized by the hydrofluorination reaction of diazo compounds, simply by treating them with HF sources (Et3N·3HF, Py·HF, HBF4, etc.).16 This reaction is highly effective for stabilized diazo compounds (e.g. flanked by two Ar, carbonyl, CF3 groups); however, the aryldiazomethanes required to synthesize benzylic monofluorides are unstable, rarely isolable, and cannot easily serve as hydrofluorination precursors. Nonetheless, development of this methodology could offer a mild, rapid and metal-free approach to benzylic monofluorides from simple starting materials.
Recently Yadav and co-workers reported a novel reaction where tosylhydrazone derivatives of aromatic aldehydes were converted to diazo intermediates in situ, and then treated with Et3N·3HF to produce hydrofluorination products 10 (Fig. 2a).17 In an earlier study, Myers reported the oxidation of TBS-hydrazones to aryldiazomethanes using (difluoroiodo)benzene, and their subsequent hydroacylation with carboxylic acids (Fig. 2b).18 By combining these two concepts, we have developed the first synthesis of benzyl fluorides where the hypervalent iodine reagent, TolIF2, acts as both an oxidant and as the fluoride source, and gives 10 under mild and operationally simple reaction conditions (Fig. 2c).
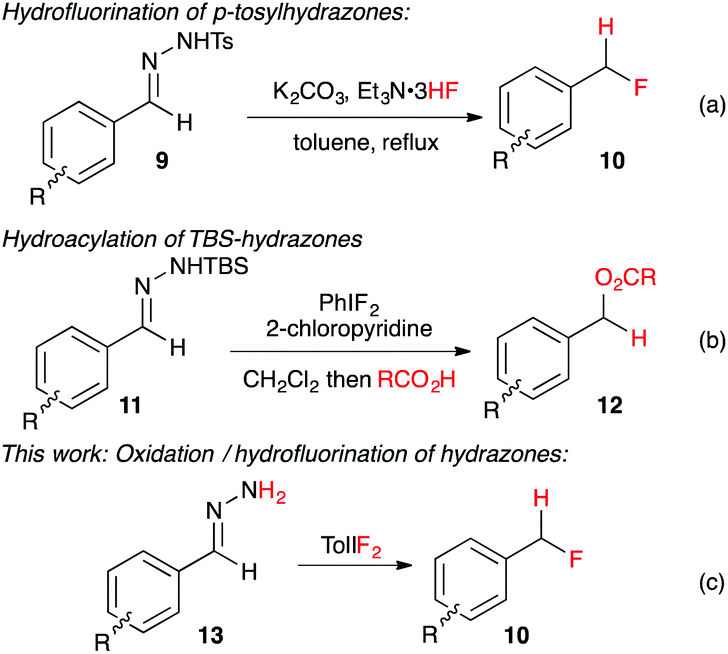 | ||
| Fig. 2 In situ synthesis of aryldiazomethanes with subsequent hydrofunctionalization by acidic nucleophiles. | ||
Results and discussion
Our studies of halogenation reactions using PhICl2 or TolIF2 as halogen sources have led to the gem-dihalogenation of diazocarbonyl compounds19 and the oxidative dichlorination of isatin-3-hydrazones.20 The gem-difluorination reactions of diazoesters proceeded best using Lewis acid activation of TolIF2 with BF3·OEt2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19c or borosilicate glass21 (eqn (1)). Similarly, the oxidative difluorination reactions of benzaldehyde hydrazone 13a proceeded best with TiF3 activation of the iodane (eqn (2)).22 During these latter studies, we observed for the first time traces of hydrofluorination by-product 10a, which we attributed to the HF generated during the oxidation23 competing with TolIF2 as an electrophilic partner for the diazo intermediate. We believed that this intermediate could be induced to react preferentially to give 10a through chemoselectivity-guided modification of the reaction conditions.
19c or borosilicate glass21 (eqn (1)). Similarly, the oxidative difluorination reactions of benzaldehyde hydrazone 13a proceeded best with TiF3 activation of the iodane (eqn (2)).22 During these latter studies, we observed for the first time traces of hydrofluorination by-product 10a, which we attributed to the HF generated during the oxidation23 competing with TolIF2 as an electrophilic partner for the diazo intermediate. We believed that this intermediate could be induced to react preferentially to give 10a through chemoselectivity-guided modification of the reaction conditions.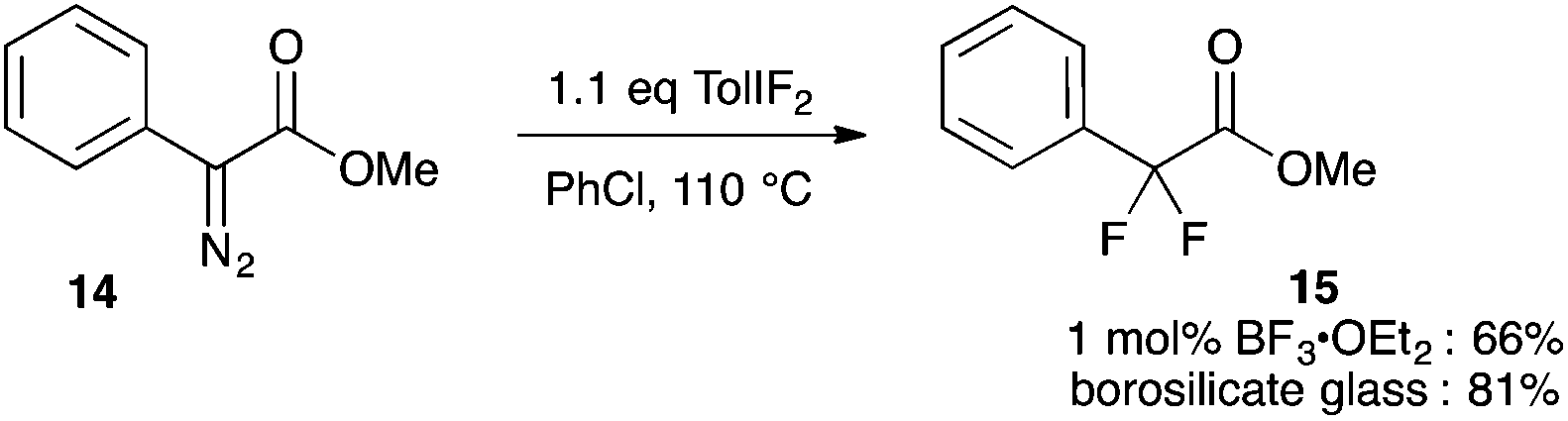 | (1) |
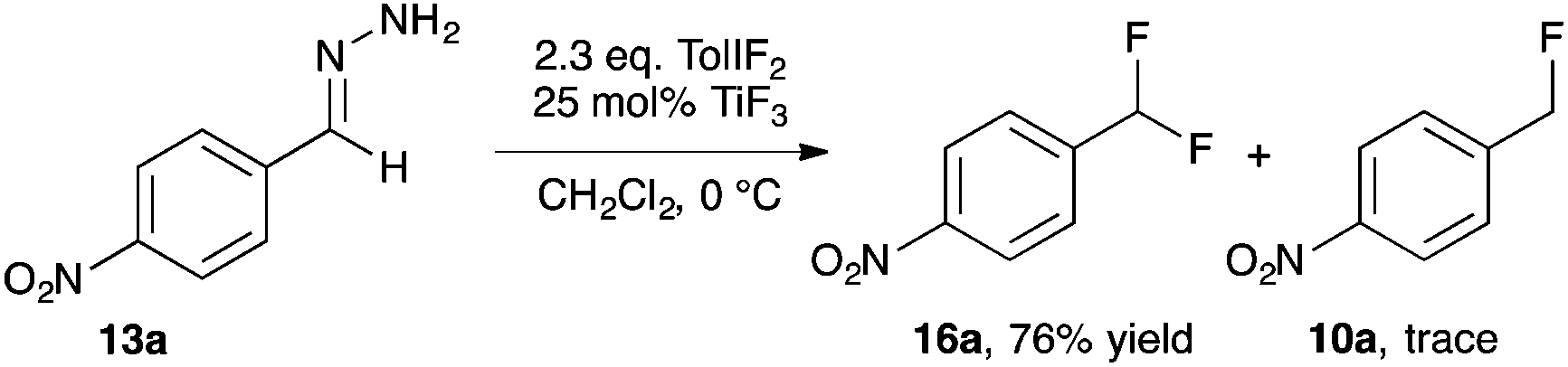 | (2) |
Due to the increased stability of the putative diazo intermediate, hydrazone 13a was used as our model substrate during the reaction optimization. We first studied the effects of Lewis acid activators on the reaction, in both laboratory glassware and PFA vials, using the optimal conditions previously determined in our gem-difluorination reactions.19c,21,22 Treating 13a with TolIF2 and 1 mol% BF3·OEt2 generally failed in both types of reaction vessel, and gave aldehyde 17a as the major product (Table 1, entries 1 and 2). The reactions were repeated using TiF3 as the activator, and while the reaction in glassware was very low yielding, the reaction in PFA gave 10a in 48% yield, along with 25% of aldehyde 17a (Table 1, entries 3 and 4). The experiment conducted in glassware without a Lewis acidic activator was also very low yielding,21 but the analogous reaction in a PFA vial proceeded in 47% yield (Table 1, entries 5 and 6).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Vessel | TolIF2 (equiv.) | Additive (mol%) | Solvent | Temp. (°C) | Yielda10a | Yielda16a | Yielda17a |
| a Isolated yields. b Reaction carried out in a borosilicate round bottom flask. c Reaction carried out in a 4 mL PFA vial. d Reverse addition of hydrazone to TolIF2. | ||||||||
| 1 | Glassb | 1.1 | 1% BF3·OEt2 | PhCl | 110 | 5% | 18% | 71% |
| 2 | PFAc | 1.1 | 1% BF3·OEt2 | DCM | 40 | 22% | — | 55% |
| 3 | Glass | 1.1 | 25% TiF3 | DCM | 40 | 10% | 19% | 34% |
| 4 | PFA | 1.1 | 25% TiF3 | DCM | 40 | 48% | — | 25% |
| 5 | Glass | 1.1 | — | PhCl | 110 | 22% | 10% | 44% |
| 6 | PFA | 1.1 | — | DCM | 40 | 47% | — | 6% |
| 7d | PFA | 1.1 | 25% TiF3 | DCM | 40 | 48% | — | 19% |
| 8d | PFA | 1.1 | — | DCM | 40 | 60% | 5% | 12% |
| 9d | PFA | 1.5 | — | DCM | 40 | 61% | 10% | 15% |
| 10d | PFA | 1.7 | — | DCM | 40 | 74% | 11% | 12% |
| 11d | PFA | 2.1 | — | DCM | 40 | 71% | 15% | 11% |
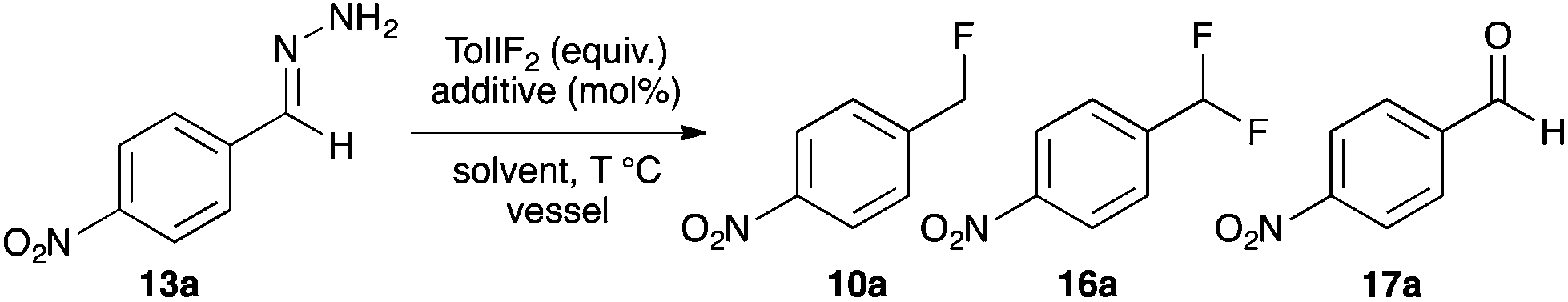
Mass spectrometry analysis of the crude reaction mixtures of these preliminary trials revealed the occasional occurrence of a hydrofluorinated dimerization product. To preclude dimer formation by minimizing the concentration of substrate relative to electrophile, the two most promising trial reactions (entries 4 and 6) were repeated using reverse addition of the hydrazone. The reaction using TiF3 was unchanged (entry 7), but the reaction without a Lewis acid activator gave 10a in 60% yield (entry 8). These two trials (entries 6 and 8) were our first examples of fluorination reactions occurring in a PFA vessel in the absence of a Lewis-acidic activator. Presumably this is due to TolIF2 acting solely as an oxidant, instead of as an electrophilic fluorine source, for which Lewis acid activation is unnecessary.18 We completed our reaction optimization by varying the loading of the TolIF2 (entries 9–11). While the production of the aldehyde by-product 17a was unchanged with increasing TolIF2 loading, the amount of benzylic difluoride (16a) increased with increasing TolIF2 loading. This is consistent with our expectation that the gem-difluorination reaction should compete with hydrofluorination in the presence of excess TolIF2. The highest yield (74%) of the desired benzylic fluoride 10a was achieved when 1.7 equivalents of iodane was used.
Having demonstrated that benzaldehyde hydrazones could be induced to react preferentially via the denitrogenative hydrofluorination reaction, we next investigated the reaction efficacy on a variety of substituted benzaldehyde hydrazone derivatives (Scheme 1). The para-, meta- and ortho-nitro substituted substrates all performed well, as did the di-substituted derivatives having a meta-NO2 substituent, giving the products 10a–e in 51–74% yield. The para-carbomethoxy and para- or meta-cyano substituted benzylic fluorides (10f–h) were isolated in good yield, as were the benzylic fluorides of the para-tosylate, para-phenyl and 3,4-dichloro substrates (10i–k). The para-methoxy and para-dimethylamino benzaldehyde hydrazones substrates were consumed by the action of TolIF2; however, no traces of the benzylic fluorides (10l,m) were observed. Interestingly, when the electron-releasing –OMe functional group was in the meta-position, the hydrofluorination reaction gave 10n in 45% NMR yield, and 33% isolated yield. Presuming the volatility of 10n to be problematic, we investigated the bulkier substrates 13o and 13p, which also possessed electron releasing –OBn and –OMe groups in their respective meta-positions, and in both cases, the products 10o,p were recovered in moderate yield.24
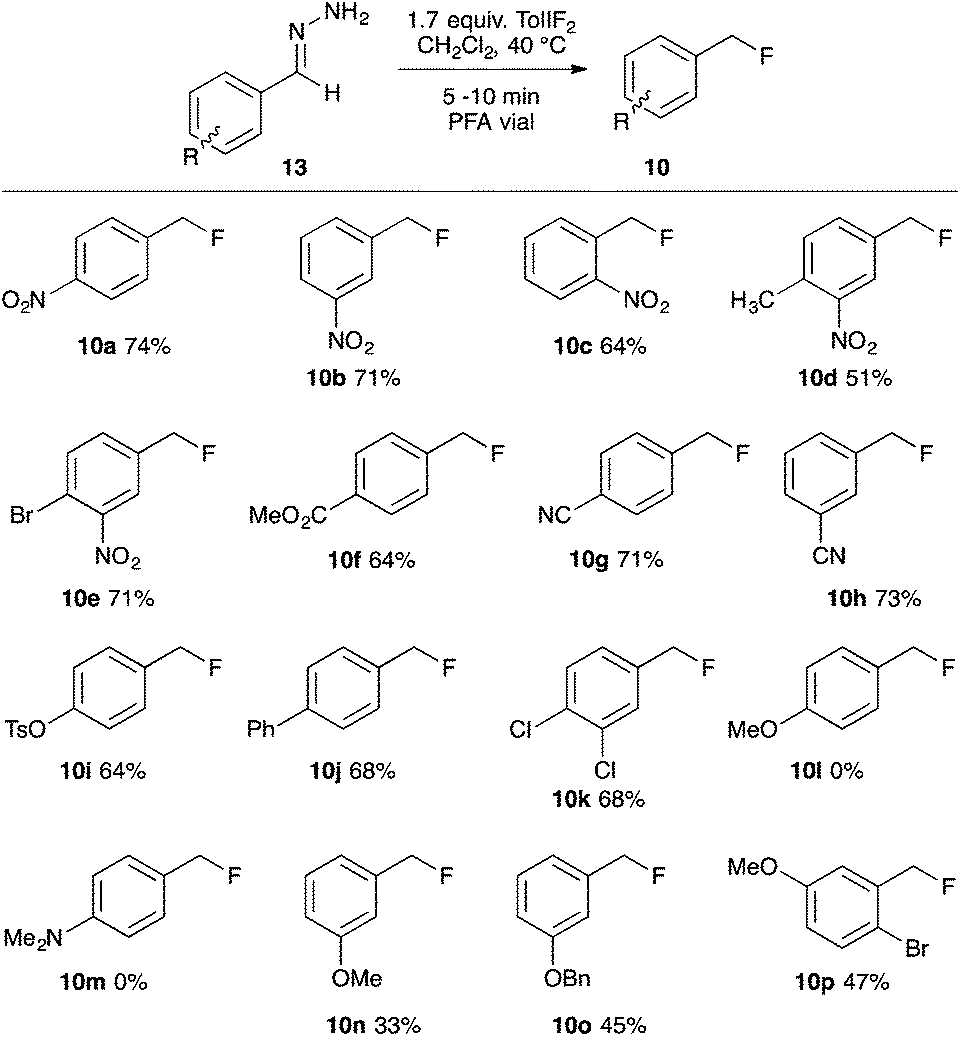 | ||
| Scheme 1 Denitrogenative hydrofluorination of benzaldehyde hydrazone derivatives. | ||
These experiments indicate that substrates bearing electron-neutral or electron-withdrawing substituents are well-tolerated in the hydrofluorination reaction. And while substrates with electron-releasing substituents at the para-position were not compatible, substrates with the same electron-donating groups in the meta-position were moderately effective in the reaction. We attribute this trend to the destabilizing effect of electron-donating groups on the aryldiazomethane intermediates, and the decreased stability of the benzyl fluoride products that bear an electron-donating group at the para-position. Our experimental observations, coupled with the unlikelihood of free carbene formation at this low reaction temperature, led us to propose an ionic mechanism for the oxidative hydrofluorination reaction (Fig. 3). Upon oxidation of hydrazone 13 to aryldiazomethane intermediate A, iodotoluene and HF (excess) would be generated as by-products. Protonation of the diazo group would give diazonium ion B, from which N2 gas could be expelled upon fluoride attack, leading to benzylic fluoride 10.
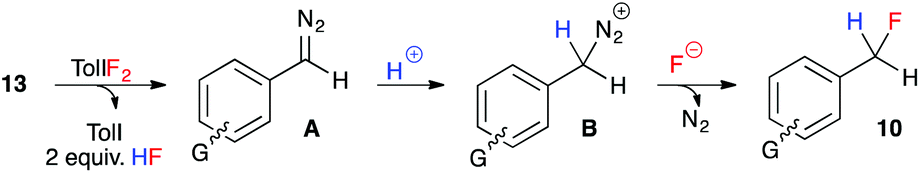 | ||
| Fig. 3 Plausible mechanistic pathway for the conversion of hydrazones to benzylic fluorides. | ||
Conclusions
In conclusion, we have developed the first use of a hypervalent iodine reagent (TolIF2) as both an oxidant and source of fluorine in the hydrofluorination of aromatic aldehyde hydrazones. The one-pot reaction made use of the oxidative potential of the iodane, converting the hydrazone to a diazo group, and generated an excess of the HF by-product which was consumed by a denitrogenative hydrofluorination reaction. The reaction proceeded best with electron-neutral or -withdrawing substituents, and failed with electron-donating groups capable of destabilizing the diazo intermediate. This reaction is a mild, rapid, metal-free and operationally simple alternative to other deoxygenative fluorinating strategies in the synthesis of benzyl fluorides. Further exploration of this reaction, including expanding the substrate and functional group scope, is underway and will be disclosed in due course.Experimental
A 4 mL PFA vial containing (difluoroiodo)toluene (1.7 equiv.) was placed under nitrogen and immersed in a pre-heated 40 °C bath. To this was added a pre-made solution of hydrazone 13a (50 mg, 1.0 equiv.) in CH2Cl2 (1.5 mL) via a syringe pump over ∼10 minutes. The reaction was monitored by TLC analysis, and upon consumption of the hydrazone (5–10 min), the reaction mixture was cooled to RT and concentrated by rotary evaporation. The resulting crude reaction mixture was purified by flash silica gel chromatography (10% EtOAc in hexanes) to provide benzyl fluoride 10a (35 mg) in 74% yield.Acknowledgements
The Natural Sciences and Engineering Research Council (NSERC) of Canada and the University of Waterloo provided financial support for this work. We would like to thank the mass spec facility of the University of Waterloo for sample analysis.References
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- (a) J. Hu, B. Gao, L. Li, C. Ni and J. Hu, Org. Lett., 2015, 17, 3086 CrossRef CAS PubMed; (b) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16 CrossRef CAS; (c) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) Y. Zhou, J. Wang, Z. N. Gu, S. N. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (b) K. V. Turcheniuk, V. P. Kukhar, G. V. Roschenthaler, J. L. Acena, V. A. Soloshonok and A. E. Sorochinsky, RSC Adv., 2013, 3, 6693 RSC; (c) B. Manteau, S. Pazenok, J. P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140 CrossRef CAS; (d) S. Fustero, J. F. Sanz-Cervera, J. L. Acena and M. Sanchez-Rosello, Synlett, 2009, 525 CrossRef CAS.
- For recent reviews of various fluorination strategies see: (a) P. A. Champagne, J. Desroches, J. D. Hamel, M. Vandamme and J. F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed; (b) C. Chatalova-Sazepin, R. Hemelaere, J. F. Paquin and G. M. Sammis, Synthesis, 2015, 2554 CAS; (c) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS PubMed; (d) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed; (e) S. Barata-Vallejo, S. M. Bonesi and A. Postigo, Org. Biomol. Chem., 2015, 13, 11153 RSC; (f) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415 CrossRef CAS; (g) Y. B. Dudkina, M. N. Khrizanforov, T. V. Gryaznova and Y. H. Budnikova, J. Organomet. Chem., 2014, 751, 301 CrossRef CAS; (h) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed.
- NFSI: (a) E. Differding and H. Ofner, Synlett, 1991, 187 CrossRef CAS; Selectfluor: (b) R. E. Banks, S. N. Mohialdinkhaffaf, G. S. Lal, I. Sharif and R. G. Syvret, J. Chem. Soc., Chem. Commun., 1992, 595 RSC; (c) R. E. Banks, J. Fluorine Chem., 1998, 87, 1 CrossRef CAS N-Fluoropyridinium salts: (d) T. Umemoto and K. Tomita, Tetrahedron Lett., 1986, 27, 3271 CrossRef CAS; (e) T. Umemoto, K. Kawada and K. Tomita, Tetrahedron Lett., 1986, 27, 4465 CrossRef CAS. For reviews of N–F reagents see: (f) X.-S. Xue, Y. Wang, M. Li and J.-P. Cheng, J. Org. Chem., 2016, 81, 4280 CrossRef CAS PubMed; (g) G. S. Lal, G. P. Pez and R. G. Syvret, Chem. Rev., 1996, 96, 1737 CrossRef CAS PubMed.
- C. F. Ni, M. Y. Hu and J. B. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed.
- For TolIF2 see: (a) R. F. Weinland and W. Stille, Liebigs Ann. Chem., 1903, 328, 132 CrossRef CAS; (b) M. A. Arrica and T. Wirth, Eur. J. Org. Chem., 2005, 395 CrossRef CAS; (c) P. Conte, B. Panunzi and M. Tingoli, Tetrahedron Lett., 2006, 47, 273 CrossRef CAS; (d) T. Inagaki, Y. Nakamura, M. Sawaguchi, N. Yoneda, S. Ayuba and S. Hara, Tetrahedron Lett., 2003, 44, 4117 CrossRef CAS; (e) J. Yu, J. Tian and C. Zhang, Adv. Synth. Catal., 2010, 352, 531 CrossRef CAS; (f) W. B. Motherwell, M. F. Greaney and D. A. Tocher, J. Chem. Soc., Perkin Trans. 1, 2002, 2809 RSC.
- For fluoro iodoxole 8 see: (a) C. Y. Legault and J. Prevost, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, 1238 Search PubMed; (b) G. C. Geary, E. G. Hope, K. Singh and A. M. Stuart, Chem. Commun., 2013, 49, 9263 RSC; (c) V. Matoušek, E. Pietrasiak, R. Schwenk and A. Togni, J. Org. Chem., 2013, 78, 6763 CrossRef PubMed; (d) G. C. Geary, E. G. Hope and A. M. Stuart, Angew. Chem., Int. Ed., 2015, 54, 14911 CrossRef CAS PubMed; (e) N. O. Ilchenko, B. O. A. Tasch and K. J. Szabo, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS PubMed; (f) A. Ulmer, C. Brunner, A. M. Arnold, A. Pothig and T. Gulder, Chem. – Eur. J., 2016, 22, 3660 CrossRef CAS PubMed.
- (a) X. Y. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (b) G. Landelle, A. Panossian, S. Pazenok, J. P. Vors and F. R. Leroux, Beilstein J. Org. Chem., 2013, 9, 2476 CrossRef PubMed; (c) X. F. Wu, H. Neumann and M. Beller, Chem. – Asian J., 2012, 7, 1744 CrossRef CAS PubMed; (d) F. L. Qing, Chin. J. Org. Chem., 2012, 32, 815 CrossRef CAS; (e) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed.
- (a) A. Koperniku, H. Q. Liu and P. B. Hurley, Eur. J. Org. Chem., 2016, 871 CrossRef CAS; (b) A. J. Lin, C. B. Huehls and J. Yang, Org. Chem. Front., 2014, 1, 434 RSC; (c) T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed; (d) M. C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2015, 21, 12836 CrossRef CAS PubMed; (e) J. B. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC.
- (a) G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048 CrossRef CAS; (b) M. J. Koen, F. Le Guyader and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1995, 1241 RSC; (c) H. Koroniak, J. Walkowiak, K. Grys, A. Rajchel, A. Alty and R. Du Boisson, J. Fluorine Chem., 2006, 127, 1245 CrossRef CAS; (d) D. Obermayer, M. Damm and C. O. Kappe, Chem. – Eur. J., 2013, 19, 15827 CrossRef CAS PubMed; (e) H. Zhao and F. P. Gabbaï, Org. Lett., 2011, 13, 1444 CrossRef CAS PubMed; (f) G. Ung and G. Bertrand, Chem. – Eur. J., 2012, 18, 12955 CrossRef CAS PubMed.
- (a) S. Bouvet, B. Pégot, J. Marrot and E. Magnier, Tetrahedron Lett., 2014, 55, 826 CrossRef CAS; (b) M. E. Hirschberg, N. V. Ignat'ev, A. Wenda and H. Willner, J. Fluorine Chem., 2012, 137, 50 CrossRef CAS; (c) R. Mirabdolbaghi, T. Dudding and T. Stamatatos, Org. Lett., 2014, 16, 2790 CrossRef CAS PubMed; (d) T. Sawamura, S. Kuribayashi, S. Inagi and T. Fuchigami, Adv. Synth. Catal., 2010, 352, 2757 CrossRef CAS; (e) T. Kitazume and T. Ebata, J. Fluorine Chem., 2004, 125, 1509 CrossRef CAS; (f) M. Mąkosza and R. Bujok, Tetrahedron Lett., 2004, 45, 1385 CrossRef; (g) H. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050 CrossRef CAS PubMed.
- (a) H. Doi, I. Ban, A. Nonoyama, K. Sumi, C. Kuang, T. Hosoya, H. Tsukada and M. Suzuki, Chem. – Eur. J., 2009, 15, 4165 CrossRef CAS PubMed. For an example of coupling between benzyl carbanion and carbonyl compounds, see: (b) Y. Arroyo, M. A. Sanz-Tejedor, A. Parra and J. L. García Ruano, Chem. – Eur. J., 2012, 18, 5314 CrossRef CAS PubMed.
- (a) J.-J. Ma, W.-B. Yi, G.-P. Lu and C. Cai, Org. Biomol. Chem., 2015, 13, 2890 RSC; (b) J. B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed; (c) P. Xu, S. Guo, L. Wang and P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955 CrossRef CAS PubMed; (d) S. Bloom, M. McCann and T. Lectka, Org. Lett., 2014, 16, 6338 CrossRef CAS PubMed; (e) J. C. T. Leung, C. Chatalova-Sazepin, J. G. West, M. Rueda-Becerril, J.-F. Paquin and G. M. Sammis, Angew. Chem., Int. Ed., 2012, 51, 10804 CrossRef CAS PubMed.
- (a) E. Emer, J. Twilton, M. Tredwell, S. Calderwood, T. L. Collier, B. Liegault, M. Taillefer and V. Gouverneur, Org. Lett., 2014, 16, 6004 CrossRef CAS PubMed; (b) R. Pasceri, H. E. Bartrum, C. J. Hayes and C. J. Moody, Chem. Commun., 2012, 48, 12077 RSC; (c) C. Qin and H. M. L. Davies, Org. Lett., 2013, 15, 6152 CrossRef CAS PubMed.
- A. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Chem. Commun., 2013, 49, 2154 RSC.
- M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 12222 CrossRef CAS PubMed.
- (a) K. E. Coffey and G. K. Murphy, Synlett, 2015, 1003 CAS; (b) G. K. Murphy, F. Z. Abbas and A. V. Poulton, Adv. Synth. Catal., 2014, 356, 2919 CrossRef CAS; (c) J. Tao, R. Tran and G. K. Murphy, J. Am. Chem. Soc., 2013, 135, 16312 CrossRef CAS PubMed.
- (a) K. E. Coffey, R. Moreira, F. Z. Abbas and G. K. Murphy, Org. Biomol. Chem., 2015, 13, 682 RSC; (b) C. Hepples and G. K. Murphy, Tetrahedron Lett., 2015, 56, 4971 CrossRef CAS.
- G. S. Sinclair, R. Tran, J. Tao, W. S. Hopkins and G. K. Murphy, Eur. J. Org. Chem., 2016, 4603 CrossRef CAS.
- Murphy lab, unpublished results.
- (a) M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 5436 CrossRef CAS PubMed; (b) D. H. R. Barton, J. C. Jaszberenyi, W. S. Liu and T. Shinada, Tetrahedron, 1996, 52, 14673 CrossRef CAS; (c) P. A. S. Smith and E. M. Bruckmann, J. Org. Chem., 1974, 39, 1047 CrossRef CAS; (d) A. Cnossen, J. C. M. Kistemaker, T. Kojima and B. L. Feringa, J. Org. Chem., 2014, 79, 927 CrossRef CAS PubMed; (e) L. Lapatsanis, G. Milias and S. Paraskewas, Synthesis, 1985, 513 CrossRef CAS.
- Hydrazones of 1-naphthaldehyde, pentafluorobenzaldehyde and 3-bromo-4-methoxybenzaldehyde were also effective in the reaction, proceeding in 40–50% NMR yield. The products were not sufficiently purified to determine isolated yields.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR spectra. See DOI: 10.1039/c6ob02074g |
| This journal is © The Royal Society of Chemistry 2016 |