
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective complexation of divalent cations by a cyclic α,β-peptoid hexamer: a spectroscopic and computational study†
E.
De Santis‡
*a,
A. A.
Edwards
a,
B. D.
Alexander
b,
S. J.
Holder
*c,
A.-S.
Biesse-Martin
de,
B. V.
Nielsen
b,
D.
Mistry
f,
L.
Waters
f,
G.
Siligardi
g,
R.
Hussain
g,
S.
Faure
de and
C.
Taillefumier
*de
aMedway School of Pharmacy, Universities of Kent and Greenwich at Medway, Central Avenue, Chatham Maritime, Kent ME4 4TB, UK. E-mail: emiliana.de.santis@npl.co.uk
bSchool of Science, University of Greenwich, Central Avenue, Chatham Maritime, Kent ME4 4TB, UK
cFunctional Materials Group, School of Physical Sciences, University of Kent, Canterbury, CN2 7NZ, UK. E-mail: S.J.Holder@kent.ac.uk
dUniversité Clermont Auvergne, Université Blaise Pascal, Institut de Chimie de Clermont-Ferrand, BP 10448, F-63000 Clermont-Ferrand, France. E-mail: claude.taillefumier@univ-bpclermont.fr
eCNRS, UMR 6296, ICCF, F-63178 Aubière Cedex, France
fDivision of Pharmacy and Pharmaceutical Sciences, University of Huddersfield, Queensgate, Huddersfield, HD1 3DE, UK
gDiamond Light Source Ltd, Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
First published on 2nd November 2016
Abstract
We describe the qualitative and quantitative analysis of the complexation properties towards cations of a cyclic peptoid hexamer composed of alternating α- and β-peptoid monomers, which bear exclusively chiral (S)-phenylethyl side chains (spe) that have no noticeable chelating properties. The binding of a series of monovalent and divalent cations was assessed by 1H NMR, circular dichroism, fluorescence and molecular modelling. In contrast to previous studies on cations binding by 18-membered α-cyclopeptoid hexamers, the 21-membered cyclopeptoid cP1 did not complex monovalent cations (Na+, K+, Ag+) but showed selectivity for divalent cations (Ca2+, Ba2+, Sr2+ and Mg2+). Hexacoordinated C-3 symmetrical complexes were demonstrated for divalent cations with ionic radii around 1 Å (Ca2+ and Ba2+), while 5-coordination is preferred for divalent cations with larger (Ba2+) or smaller ionic radii (Mg2+).
Introduction
Naturally-occurring macrocycles containing peptidic α-amino acids, including cyclic peptides and depsipeptides, represent a major class of compounds with a broad range of biological activities.1 Their biological properties are often associated to their binding of metal cations. For example, the destruxins are a family of cyclohexadepsipeptides capable of modifying various Ca2+-dependent processes.2 The mycotoxins enniatins are also a class of cyclodepsipeptides ionophores which transport monovalent cations across biological membranes.3In the realm of peptidomimetics, α-peptoids (poly-N-substituted glycines) are particularly attractive architectures that were previously shown to bind metal ions forming metallopeptoids.4 Peptoid synthesis is straightforward and an extremely high diversity of side chains is accessible.5 Their head to tail macrocyclisation is also particularly facile due to the inherent flexibility of the peptoid backbone.6 This also applies to the formation of cyclic β-peptoids7 and analogous cyclic alternated α,β-peptoids.8
Cyclic α-peptoids with 3,9 4, 6, 8 and 10 residues have been shown to bind cations, particularly from the first group alkali metals with selectivity depending on the ring size.10 For example, 18-membered cyclohexamers showed a peak of selectivity for Na+ and metallated structures could be characterised in the solid state.10a,d These include the formation of the first supramolecular 1D metal–organic framework (MOF) based on a cyclic peptoid.10d In this MOF triggered by Na+, N-methoxyethyl coordinating side chains participate to generate the supramolecular assembly. Recently, binding of Gd3+ cations was also demonstrated by cyclic α-hexapeptoids characterized by the presence of six N-carboxyethyl side chains or three N-methoxyethyl and three carboxyethyl side chains in alternation.11 Interestingly, these side chains, which confer aqueous solubility to the complexes, do not appear to have any effective role in Gd3+ coordination.
Herein we present the first reported metal binding ability of a cyclic α,β-peptoid hexamer towards a selection of metal cations. The 21-membered cyclopeptoid cP1 is characterised by six chiral (S)-1-phenylethyl side chains (spe) that lack coordinating sites, so the binding was therefore envisaged via the backbone amides (Fig. 1).
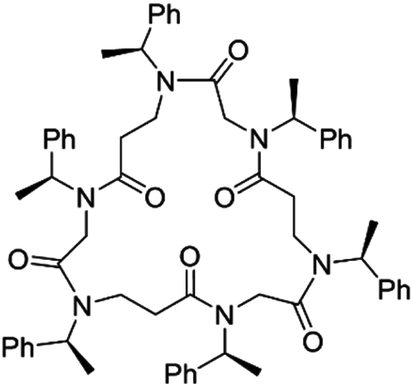 | ||
| Fig. 1 Chemical structure of 21-membered cyclic α,β-peptoid cP1. | ||
Results and discussion
Synthesis
The cyclic α,β-peptoid cP1 was synthesised by head to tail cyclisation of its linear precursor using a previously described methodology.8Briefly, the linear precursor was composed of (S)-N-(1-phenylethyl)glycine and (S)-N-(1-phenylethyl)-β-alanine monomers in alternation and was synthesised by a solution-phase submonomer approach.12 The C-terminal tBu-capping group of the synthesised linear peptoid was cleaved using trifluoroacetic acid and subsequent HATU-mediated cyclisation led to cP1 in 64% yield (see ESI† for details).
Experimental design
Metal binding is a delicate equilibrium between host–guest, host–solvent and guest–solvent interactions. Binding occurs when the interaction between the host and guest overcomes all of the other competing interactions. Thus, the experimental design is essential for the correct evaluation and quantification of the binding event. In this study acetonitrile (CH3CN) was selected due to its low cation-coordinating properties.13 CH3CN has also been extensively employed for conformational studies of α-, β- and α,β-peptoids. Cyclic α-peptoids have already shown the ability to bind monovalent cations in CD3CN by NMR.10a In addition, its transparency in the far and near regions of the UV spectrum makes it an ideal solvent for spectroscopic studies. Picrate salts were used for NMR analysis to facilitate the quantification of the salt concentration, which was calculated from the aromatic region of the spectrum (6–9 ppm). However, due to their limited UV transparency, picrates were not used for circular dichroism (CD) and fluorescence spectroscopies and perchlorates salts were used instead. The choice of the perchlorate counter ions was also supported by their ease of dissociation in CH3CN, which was expected to facilitate the cation-peptoid binding.13,14 A range of metal ions with variable size, polarizability/charge and association constant (Ka) in CH3CN were selected (Table 1) to assess whether cyclic α,β-peptoid cP1 displayed any selective metal binding ability among the cations listed in Table 1.| Cation | Ionic radiusa (Å) | Charge density (charge Å−2) | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kab Kab |
|
|---|---|---|---|---|
|
a Ionic radius for coordination number VI.15a
b logka values for Mn+ + nClO4− → Mn+(ClO4−)n in CH3CN.
|
||||
| +1 | Na | 1.02 | 0.076 | 0.91, 1.0015b |
| Ag | 1.15 | 0.060 | — | |
| K | 1.38 | 0.042 | 1.23, 1.5215b,c | |
| +2 | Mg | 0.72 | 0.307 | 2.26, 2.3715d |
| Zn | 0.74 | 0.291 | 1.6815e | |
| Fe | 0.78 | 0.262 | — | |
| Ca | 1.00 | 0.159 | 2.44, 2.7415d | |
| Sr | 1.18 | 0.114 | 2.5815d | |
| Ba | 1.35 | 0.087 | 2.6915d | |
| +3 | Fe | 0.55 | 0.789 | — |
Metal ion complexation
 | ||
| Fig. 2 Binding of cP1 with perchlorate salts of divalent cations. Qualitative 1H NMR spectra of cP1 alone and in the presence of perchlorate salts at a molar ratio (metal/cP1) of 2. | ||
The 1H NMR spectrum of cyclopeptoid cP1 alone is characterised by a number of benzylic proton resonances between 5 and 6 ppm. This is indicative of the simultaneous presence of cis and trans amides along the backbone. According to literature, for pe side chains, 1H NMR resonances between 5.5 and 6 ppm are associated with benzylic protons on cis amides and those around 5 ppm are from benzylic protons on trans amides.17 The average amide cis/trans ratio was estimated to 75:25 in CD3CN (Fig. 2, 3 and S2†). The addition of a molar excess of metal ion typically resulted in a decrease of the signal around 6 ppm thus indicating an increased population of all-trans conformers. At the saturation point, no further changes of the 1H NMR spectrum and the cis/trans ratio were observed. To confirm this, a step-wise titration of strontium perchlorate into cP1 was performed in CD3CN (Fig. 3 and S3†). A simplification of the 1H NMR spectrum was observed in the region between 5 and 6 ppm. Specifically, increasing the molar ratio (Sr2+/cP1) from 0 to 3 determined a stepwise decrease of the peak around 5.8 ppm with a corresponding increase in the signal around 5 ppm (Fig. 3). This suggested that once the metal complex was formed, the cyclopeptoid cP1 adopted an all-trans conformation. A plot integration of peaks around 5.8 ppm versus the molar ratio clearly indicated that no further changes in the 1H NMR spectrum were observed after a 1:1 stoichiometry was reached. The 1H NMR spectrum of the Sr2+/cP1 complex showed a single set of signals for each α- and β-monomer which therefore was indicative of a discrete conformation with a 3-fold rotational symmetry. NMR temperature studies and 2D experiments enabled full proton assignment of the metal complex (Fig. S4 and Table S1†). Notably, the 1H NMR spectrum for the Ca2+/cP1 complex is very similar to that of the Sr2+/cP1 complex thus also indicative of a 3-fold symmetry (Fig. 2). In contrast the spectra for the Ba2+/cP1 and Mg2+/cP1 complexes, whilst simpler than that of the peptoid cP1 alone, are distinct from those of Sr2+/cP1 and Ca2+/cP1 complexes. For all of the metal complexes the cis to trans conversion was complete upon addition of an excess of metal. A cis to trans isomerisation was previously observed in cyclic α-peptoid binding cations.10a,18
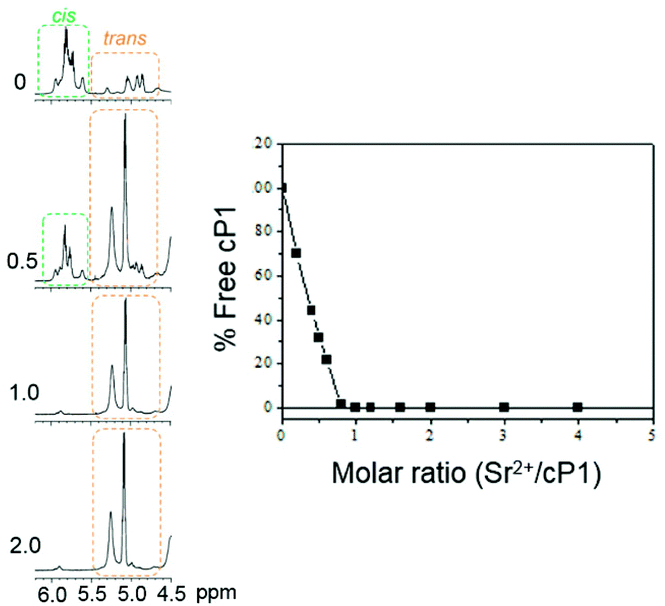 | ||
| Fig. 3 Quantitative titration by 1H NMR of cP1 with Sr(ClO4)2 at the molar ratio indicated (left) and a plot of the area under the peaks at ca. 5.8 ppm against the molar ratio (right). All 1H NMR spectra were recorded in CD3CN at 20 °C and using a peptoid concentration of 8 mM. | ||
The CD spectrum of cP1 is characterised by two negative maxima at 203 and 222 nm (Fig. 4). Similar CD spectra observed for α-peptoids in CH3CN have been assigned to helical conformations.21 However, due to the cyclic nature of the α,β-peptoid cP1, a helical conformation is unlikely and the spectrum may instead arise from a twisted-like conformation. Based on these observations and the complexity of the 1H NMR spectra at room temperature, the CD spectrum of cylopeptoid cP1 is likely to indicate the presence of multiple conformers rather than one dominant species and the [θ]203 nm of 268 × 103 is therefore taken to be indicative of this. In this case, this dynamic behaviour may be due to the intrinsic flexibility of the backbone derived from the presence of β-residues and the cis/trans isomerism of tertiary amides.
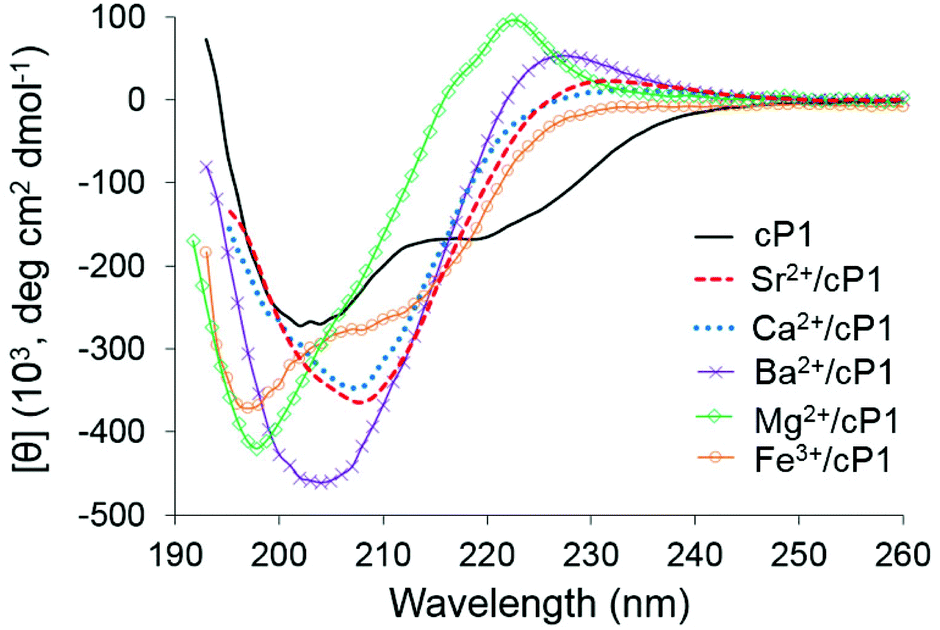 | ||
| Fig. 4 CD spectra of cP1 alone and in the presence of perchlorate salts at a molar ratio of 10. All CD spectra were recorded in CH3CN at 20 °C using a peptoid concentration of 7.5 μM and a 0.4 cm pathlength cell for Ca(ClO4)2, Mg(ClO4)2 and Ba(ClO4)2 and a peptoid concentration of 15 μM using a 1 cm pathlength cell for Sr(ClO4)2 and Fe(ClO4)3. | ||
Consistently with 1H NMR data, no binding was observed with sodium and potassium cations as indicated by the lack of spectral changes in the far UV CD, which was also reported for silver, zinc and iron(II) (Fig. S6†). This was further confirmed for sodium and zinc by quantitative CD titrations in the far UV region (Fig. S7†). CD in the near UV and fluorescence spectroscopy also showed no significant spectral changes upon addition of sodium perchlorate to cP1 (Fig. S8†). In contrast, a significant change to the spectral features in the far UV was observed upon addition of perchlorate salts of Ba2+, Ca2+, Fe3+, Mg2+ and Sr2+ thus indicating metal complex formation (Fig. 4, S8 and S9†). Spectroscopic studies by fluorescence and in the near UV by CD showed significant changes upon addition of magnesium perchlorate to cP1 further supporting the metal binding (Fig. S8†).
Nearly identical spectral features were observed for cylopeptoid cP1 in the presence of Ca2+ or Sr2+. The CD spectra of these metal complexes were characterised by one negative maximum around 208 nm which was red shifted in comparison to the free cylopeptoid cP1. The CD spectrum in the presence of Ba2+ has two maxima (negative circa 204 nm, positive circa 227 nm). The CD spectrum of the Ba2+ complex has greater [θ] and the negative maximum is only marginally red shifted relative to cP1 and the corresponding Ca2+/Sr2+ complexes. The Mg2+/cP1 complex was characterised by a negative maximum at 198 nm and a positive maximum centred at 222 nm with a component at 216 nm. The CD spectrum in the presence of Fe3+ has two negative maxima (circa 197 nm and circa 210 nm) and shows similarity to the Mg2+ profile based on the blue shift of the negative maxima relative to cP1.
The similarity in appearance of the Sr2+/cP1 and Ca2+/cP1 CD spectra agrees well with the similarity of their 1H NMR spectra. The differences between the CD spectra of these two metal complexes and those with barium and magnesium were also reflected in their 1H NMR spectra suggesting distinct conformational differences. Notably, all metal complexes were found to have increased [θ] relative to cP1 alone which was also consistent with the 1H NMR observation of a conformational shift to population of all-trans isomers i.e. increased ordering of the conformational preference of cP1.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Os ligating Sr2+) and 6 co-ordinate species (6 COs ligating Sr2+) with the 4 co-ordinate systems possessing consistently higher heats of formation (ΔHf ∼ 275 kJ mol) than the 6 coordinate. At least two distinct co-ordination patterns involving 6 carbonyl co-ordination were observed, one with ΔHf values circa 220 kJ mol−1 involving two COs binding from above and four COs binding from below the Sr2+ cation, relative to the plane of the ring. In contrast, the conformers approaching C3 symmetric arrangement of COs about the Sr2+ (3 above and 3 below the plane of the ring) gave significantly lower ΔHf values, from ∼212 kJ mol−1 down to ∼192 kJ mol−1. Since C3 symmetry was confirmed by 1H NMR system with the lowest ΔHf was chosen to investigate the arrangement of ethylphenyl groups about the ring. Since the ethylphenyl groups displayed full unhindered rotation about the N–C bond this was an extensive conformational space. To facilitate the search a simple system of varying the arrangements of the ethylphenyl groups about the ring was adopted; in essence systematically changing the C–N–C–C dihedral angles to facilitate various conformational starting structures based upon the relative orientation of the phenyl group ‘above’ (up or u) or ‘below’ (down or d) the plane of the ring relative to the other groups. This analysis revealed a relatively flat potential energy surface about these conformers (Fig. S21†) with minimum energy conformers possessing ΔHf values from ∼180 to ∼196 kJ mol−1. However the conformations starting from the alternating ethylphenyl (ududud) starting point all gave lower heats of formation than any of the other arrangements. Two minimum energy conformers possessed C3 symmetry, one with ΔHf = 184.6 kJ mol−1 and one with ΔHf = 180.1 kJ mol−1. The geometries of these two conformers were then optimised using PM6 with the COSMO solvation model inherent in MOPAC. The former conformer did not give a vibrational spectrum without negative frequencies. The latter conformer with further optimisation gave rise to a Sr2+/cP1 complex with three-fold symmetry (C3) about the cyclic axis (Fig. 5 and S10†) with a vibrational spectrum free of negative frequencies. A similar procedure was adopted for the Ba2+ complex giving a Ba2+/cP1 complex with three-fold symmetry (C3) about the cyclic axis. This is in excellent agreement with the NMR spectra (Fig. 2). The average metal–O bond lengths were 2.49 (SD = ±0.05) Å for Sr–O and 2.35 (SD = ±0.04) Å for Ba–O.
Os ligating Sr2+) and 6 co-ordinate species (6 COs ligating Sr2+) with the 4 co-ordinate systems possessing consistently higher heats of formation (ΔHf ∼ 275 kJ mol) than the 6 coordinate. At least two distinct co-ordination patterns involving 6 carbonyl co-ordination were observed, one with ΔHf values circa 220 kJ mol−1 involving two COs binding from above and four COs binding from below the Sr2+ cation, relative to the plane of the ring. In contrast, the conformers approaching C3 symmetric arrangement of COs about the Sr2+ (3 above and 3 below the plane of the ring) gave significantly lower ΔHf values, from ∼212 kJ mol−1 down to ∼192 kJ mol−1. Since C3 symmetry was confirmed by 1H NMR system with the lowest ΔHf was chosen to investigate the arrangement of ethylphenyl groups about the ring. Since the ethylphenyl groups displayed full unhindered rotation about the N–C bond this was an extensive conformational space. To facilitate the search a simple system of varying the arrangements of the ethylphenyl groups about the ring was adopted; in essence systematically changing the C–N–C–C dihedral angles to facilitate various conformational starting structures based upon the relative orientation of the phenyl group ‘above’ (up or u) or ‘below’ (down or d) the plane of the ring relative to the other groups. This analysis revealed a relatively flat potential energy surface about these conformers (Fig. S21†) with minimum energy conformers possessing ΔHf values from ∼180 to ∼196 kJ mol−1. However the conformations starting from the alternating ethylphenyl (ududud) starting point all gave lower heats of formation than any of the other arrangements. Two minimum energy conformers possessed C3 symmetry, one with ΔHf = 184.6 kJ mol−1 and one with ΔHf = 180.1 kJ mol−1. The geometries of these two conformers were then optimised using PM6 with the COSMO solvation model inherent in MOPAC. The former conformer did not give a vibrational spectrum without negative frequencies. The latter conformer with further optimisation gave rise to a Sr2+/cP1 complex with three-fold symmetry (C3) about the cyclic axis (Fig. 5 and S10†) with a vibrational spectrum free of negative frequencies. A similar procedure was adopted for the Ba2+ complex giving a Ba2+/cP1 complex with three-fold symmetry (C3) about the cyclic axis. This is in excellent agreement with the NMR spectra (Fig. 2). The average metal–O bond lengths were 2.49 (SD = ±0.05) Å for Sr–O and 2.35 (SD = ±0.04) Å for Ba–O.
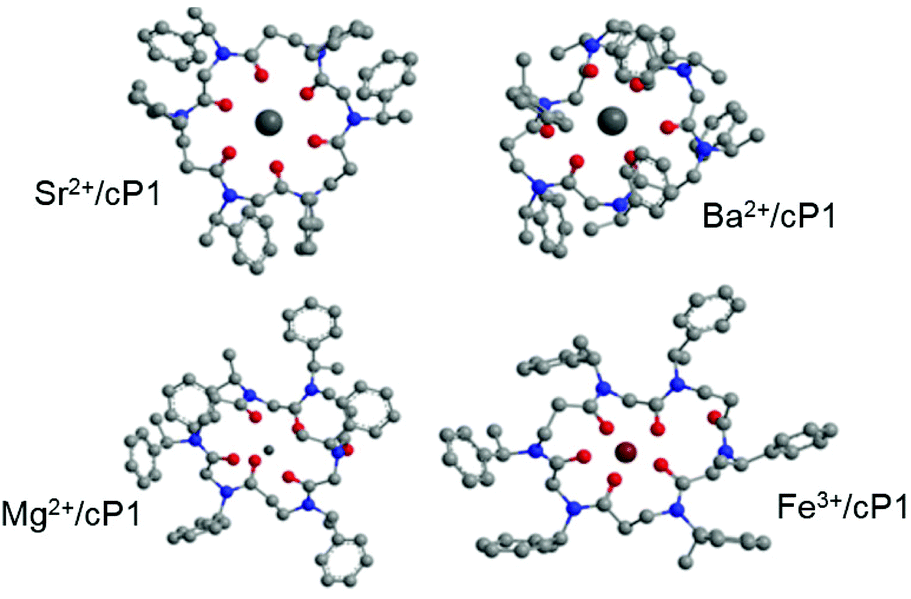 | ||
| Fig. 5 Minimum energy conformations for metal complexes predicted by PM6 (COSMO) calculations. Hydrogens are omitted for clarity. The Ca2+/cP1 complex is nearly identical to the Sr2+/cP1 complex (see ESI†). | ||
In contrast, the lowest energy conformers for both Mg2+/cP1 and Fe3+/cP1 complexes involved only 5 of the 6 carbonyls coordinating to the metal centre. For these complexes the average metal–O bond lengths for the 5 bonding carbonyls were 1.98 (SD = ±0.03) Å for Mg2+/cP1 and 1.91 (SD = ±0.06) Å for Fe3+/cP1 (Fig. 5 and S11a, c†). Consequently, these complexes possessed no symmetry beyond their identity; again, this is in accordance with the results from the 1H NMR for the Mg2+/cP1 complex (Fig. 2). Replacing the Mg2+ and Fe3+ ions in the sites of the Ca2+ or Sr2+ ions of the C3 symmetrical complexes and optimising the geometry led to stable symmetrical C3 complexes of these metals (Fig. S11b and d†) but their energies were significantly higher (Table S2†) than those illustrated in Fig. 5. Similarly transposing the Ca2+/cP1 and Sr2+/cP1 complexes into those with Mg2+ or Fe3+ led to rearrangement of the cyclic peptoid cP1 to facilitate 6 coordinate carbonyl binding once again. It is uncertain as to whether the metal complexes with Mg2+ and Fe3+ shown in Fig. 5 are absolutely the lowest energy conformers due to the range of conformational possibilities available to these complexes however they are significantly lower in energy than any 6 coordinate species found.
The Ba2+/cP1 complex also showed 5 coordinate carbonyl binding but unusually the cation itself was displaced from the plane of the peptoid resulting in close contacts with three of the phenyl groups (Fig. 5 and S12†). Thus average metal–O bond lengths for 3 of the bonding carbonyls were 2.76 (SD = ±0.03) Å and for the other two = 2.56 (SD = ±0.01), with the average C–Ba distance for the three coordinating phenyls being 2.74 (SD = ±0.12) Å. Again, this lack of symmetry was mirrored in the 1H NMR and support for the phenyl coordination comes from the appearance of an upfield shifted aromatic resonance at 6.51 ppm.
Overall, the semi-empirical modelling largely agrees with the CD spectra in that the spectra for Ca2+/cP1 and Sr2+/cP1 complexes are virtually identical in line with identical predicted conformations. The maxima for the complexes of Ba2+, Mg2+ and Fe3+ are distinct from those of Ca2+ and Sr2+. However, whereas the negative maxima occur at similar positions for Mg2+/cP1 and Fe3+/cP1 (circa 197 nm) that of the Ba2+/cP1 complex is unique (circa 204 nm) which is in line with the distinct conformation predicted by modelling. Interestingly the CD spectrum of the Fe3+/cP1 complex appears to consist of at least one more distinct peak at circa 210 nm; one possibility for this is that this peak arises from a certain proportion of the complexes in solution adopting the higher energy 6-coordinate C3 symmetry in equilibrium with the predominately 5-coordinate species.
Quantification of binding
Given the conformational changes observed for cylopeptoid cP1 upon binding and the sensitivity of CD to subtle conformational changes, a stepwise titration of Sr(ClO4)2 into cylopeptoid cP1 was performed to give further insight into the binding event (Fig. 6A). The titration showed a gradual increase of the intensity of the CD spectrum for cP1 together with a red shift by 5 nm of the negative maxima around 205 nm and the inversion of the sign for the maximum around 230 nm. An isodichroic point was also observed at 218 nm suggesting a conformational change between two (or more) different conformational states. The differential CD (ΔCD) signal was obtained by subtracting the CD spectrum of cP1 and that of Sr2+ from that of the Sr2+/cP1 metal complex for the different molar ratios (Fig. S13†). Using the CD data, a plot of the ΔAbsorbance (ΔA, where ΔA = ΔCD/32980) at 230 nm as a function of the Sr2+ concentration yielded a dissociation constant (Kd) of 11.9 ± 0.5 μM when fitted to the Hill equation (R2 = 0.99) (Fig. 6B).22 It is of note that the value of the Kd was independent of the wavelength selected and the maximum at 231 nm was chosen due to the absence of any significant wavelength shift. The Kd was of the same order of magnitude as that previously reported for an α-peptoid hexamer with benzyloxyethyl (be) side chains which was shown to bind monovalent cations by quantitative 1H NMR.10a This similarity indicated that the α,β-peptoid backbone retained the ability to bind metal ions in the presence of the more flexible β-residues and different side chains, albeit with a different cation selectivity. No binding to the monovalent cations investigated was observed for cP1.
 | ||
| Fig. 6 Quantitative titration of cP1 with Sr(ClO4)2 and Mg(ClO4)2. (A) SRCD spectra of cP1 and Sr2+/cP1 complexes at the molar ratio indicated. (B) Plot of ΔAbsorbance as a function of the Sr2+ concentration and Sr2+/cP1 molar ratio fitted to the Hill equation (R2 = 0.99). All spectra were recorded in CH3CN at 20 °C with a cP1 concentration of 15 μM using a 1 cm pathlength cell. (C) ITC binding isotherm of Mg(ClO4)2 titrated into cP1 (107 μM) in CH3CN at 20 °C. Differential power signal measured for each injection during the experiment (top) and integrated peak areas corresponding to the measured heat absorbed per injection and the theoretical fit to a one binding site model (bottom). | ||
A Yoe–Jones plot23 where the differential molar ellipticity at the positive and negative maxima was plotted versus the molar ratio (Sr2+/cP1) gave a stoichiometric point of 1.6 as indicated by the projection of the inflection point on the x-axis (Fig. S13†). This was not consistent with the stoichiometry obtained by 1H NMR and may be rationalised on the basis of the higher sensitivity of CD to subtle conformational changes. Specifically, 1H NMR indicated changes to the cis/trans isomerisation up to a Sr2+/cP1 molar ratio of 1 indicating a 1:1 stoichiometry. Any further structural rigidification, which does not alter the cis/trans ratio or does not modify significantly the ring conformation would not be detected by 1H NMR. By contrast, these would be detected by CD (as an increased signal intensity) and may give rise to the higher stoichiometry observed in comparison to 1H NMR.
Interestingly, when looking at the intensity and wavelength position of the negative maximum at 207, whilst the intensity increases up to a molar ratio of 2, a significant wavelength shift only occurs up to a molar ratio of 1 (Fig. 6A and S14†). Based on these observations and supported by the PM6 calculation showing the 1:1 complex as the most favourable, it is reasonable to assume that the complex is characterised by a 1:1 stoichiometry and any further change in intensity of the CD signal is likely to result from further structural rigidification and an increased population of metal complexes. A similar trend was also observed for the other metal complexes (Fig. S14†) for which similar binding affinity was observed between each metal and cP1 (Table 2 and Fig. S15–S18†).
| Complex | K d (μM) | Stoichiometry (Mtn+/cP1) | |
|---|---|---|---|
| >210 nm | <210 nm | ||
| Sr2+/cP1 | 11.9 ± 0.5 | 1.6 | 1.6 |
| Mg2+/cP1 | 12 ± 0.2 | 1.5 | 1.5 |
| Ca2+/cP1 | 8.2 ± 0.3 | 1.1 | 1.04 |
| Ba2+/cP1 | 12.7 ± 8.6 | 1.1 | 1.2 |
| Fe3+/cP1 | 1.2 ± 0.9 | — | 1.5 |
The Mg2+/cP1 complex gave a Kd of 12 ± 0.2 μM by CD and this was largely comparable with that calculated by isothermal titration calorimetry (ITC),24 which yielded a Kd of the same order of magnitude (2.3 μM) (Fig. 6C). ITC also showed that the binding event was endothermic, as indicated by the positive values for the heat exchange and the enthalpy value (ΔHcal) of 6902 cal mol−1 with an entropic contribution (ΔScal) of 49.2 cal mol−1. Using ITC, the projection of the mid-point of the titration curve on the x axis provided the stoichiometry of the binding which was found to be close to 1:1 (metal/peptoid) suggesting an approximately equimolecular interaction. This stoichiometry was consistent with that deduced by CD and with the PM6 calculations for the Mg2+/cP1 complex. Unfortunately, quantitative studies by 1H NMR were hampered by the significant signal overlap (Fig. S19†).
Binding selectivity
Based on size, there appears to be no correlation in the selectivity of cP1 for divalent over monovalent cations, for example the ionic radius of Na+ (not complexed by cP1), is very close to that of Ca2+ (1.02 and 1.00 Å, respectively). However, selectivity observed for divalent cations, seems to be at least in part correlated with their greater charge density (Table 1). Amide groups, especially the non-hydrogen-bonded carbonyl groups of peptoid amides, represent polar coordination sites with enhanced divalent ion sensitivity by comparison to peptides and proteins.25 Our results agree well with the observations of Tinès et al. on the selectivity of tetralactam systems for alkaline-earth over alkaline cations.26 Wipff et al., by working on calix[4]arene amide systems have also observed that complexation of cations is strongly dependent on the nature of the amide groups (primary, secondary and tertiary), with the complexes involving tertiary amides being the most stable.27 Consistent with our results, the higher Sr2+/Na+ and Ca2+/Na+ selectivities (ca. 500 and 125, respectively) were measured from calix[4]arene with tertiary amide-based pendant groups. A comparison can also be made between the symmetrical binding mode of Sr2+/cP1 and Ca2+/cP1 and the S6-symmetry Sr2+ complex identified in the solid state for a cyclic α-hexapeptoid bearing be side chains.10a It is of note that in this case the coordinating be side chains assume a pseudo equatorial orientation and do not intervene in the binding. Affinity of cyclic α-hexapeptoids (18-membered rings) for alkali metals has also been described, with a peak of selectivity for Na+. However, a direct comparison with our results is not fully relevant since the host cyclopeptoids studied carry chelating side chains that were shown to participate in the binding.10dWith regard to the divalent cations, between each other the charge density is not the only criterion to be considered, as their difference in size also plays a significant role in their binding selectivity. The divalent cations with the smallest ionic radii (Mg2+, Zn2+ and Fe2+) bind to cP1 only if the associated charge density is very high. Thus, the hard Mg2+ cation binds to cP1 whereas the Zn2+ and Fe2+ cations having slightly lower charge density do not bind (Table 1). For the other divalent cations tested (Ca2+, Sr2+ and Ba2+), even though their charge density is lower, they have ionic radii ≥1 Å, thus are more adapted to the cavity size, which facilitate their complexation to cP1. Size of the cations also appears to influence the final conformation of the complexes i.e. when the cation radius is too large or too small the binding is 5 coordinate and 6 coordinate only obtainable for a specific range of ionic radii. Despite these observations, the case for the role of the ionic radius and charge density in predicting binding is not clear cut. It would appear that a number of factors involving size, charge density, enthalpy and entropy play a part for all of the species involved, namely the cation, anion, acetonitrile and peptoid. Surprisingly a clear correlation between the occurrence of binding is observed with the association constants of the metal perchlorates in acetonitrile (Table 1, Fig. 7). The metal perchlorates with low association constants in acetonitrile (logKa < circa 2) are not bound whereas all of the salts possesing Ka values roughly an order of magnitude larger were observed to bind.
 | ||
| Fig. 7 The average Ka for Mn+ + nClO4− → Mn+(ClO4−)n in CH3CN for the metal cations illustrating binding and non-binding regimes. | ||
The Ka values of the salts in acetonitrile can be taken as an indication of the free energy change in solvating the ions. A high dissociation (low association) is a reflection of the affinity of the salts for acetonitrile. The low dissociation constants (high association) indicate a lower free energy change upon solvation i.e. the solvation energy for the non-binding salts is higher than that of the binding salts. Thus, there is a larger thermodynamic penalty being paid by taking the non-binding salts out of acetonitrile into the macrocyclic complex and consequently the overall free energy change is positive. Since the binding salts are highly associated in acetonitrile it can be taken that the free energy penalty in removing them from the coordination/solvation sphere of acetonitrile is lower and a negative free energy results from this process. This is by no means a conclusive correlation or interpretation of binding selectivity but the correlation between Ka and binding is striking.
Conclusion
We have shown for the first time clear evidence of the selective complexation of divalent over monovalent cations by a 21-membered cyclopeptoid hexamer composed of α- and β-monomers in alternation. This selectivity is different to that generally observed with α-cyclopeptoid hexamers (18-membered rings) which have mainly shown selectivity for monovalent cations. Besides NMR analyses, a detailed analysis of cation binding was undertaken by CD, an invaluable technique to assess subtle conformational changes. From detailed discussion of the combined spectroscopic data, in conjunction with modelling, it is clear that a holistic approach is needed to deduce the reasons for the unique binding behaviour of this cyclic peptoid. Application of this work in a hydrophobic environment could be envisaged e.g. to support selective ion transport in membranes. Alternatively modifications of the side chains could be explored to ascertain if aqueous solubility can be achieved whilst maintaining complexation ability which would allow a wider range of biological applications to be considered.Experimental
Synthesis and characterisation
CH2Cl2 was distilled under N2 from CaH2 and stored over 4 Å molecular sieves. MeOH was distilled under N2 from CaH2 and stored over 4 Å molecular sieves. EtOAc, CH2Cl2, cyclohexane, and MeOH for column chromatography were distilled before use. DMF and iPr2NEt were dried over 4 Å molecular sieves. All other solvents and chemicals obtained from commercial sources were used as received. Melting points were determined on a Reichert microscope apparatus and are uncorrected. Specific rotations were measured on a Jasco DIP-370 polarimeter using a 10 cm cell. IR spectra were recorded on a Shimadzu FTIR-8400S spectrometer equipped with a Pike Technologies MIRacle™ ATR and ν are expressed in cm−1. NMR spectra were recorded on a Bruker AC-400 spectrometer or a Bruker AC-500 spectrometer. Chemical shifts are referenced to the residual solvent peak and J values are given in Hz. The following multiplicity abbreviations are used: (s) singlet, (d) doublet, (t) triplet, (q) quartet, (m) multiplet, and (br) broad. Where applicable, assignments were based on COSY, HMBC, HSQC and J-mod-experiments. Flash chromatography was performed with Merck silica gel 60, 40–63 μm. HRMS were recorded on a Micromass Q-Tof Micro (3000 V) apparatus. HPLC analysis was performed on a Waters 590 instrument equipped with an Acclaim® 120 column (C18, 5 μm, 120 Å, 4.6 × 250 mm) and a Waters 484 UV detector.Synthesis of cyclic α,β-peptoid cP1
To a solution of the linear α,β-hexapeptoid12 (427 mg, 0.40 mmol, 1.0 equiv.) in CH2Cl2 (0.10 M) at rt under Ar was added TFA (1 mL per 1 mL CH2Cl2) and the resulting mixture was stirred for 2 h at rt. The solvents were evaporated under reduced pressure and the residue was dried in vacuo, yielding the crude termini deprotected peptoid. To a solution of the crude peptoid (5.0 mM) in CH2Cl2/DMF (4:1) at 0 °C under Ar, enough iPr2NEt (approx. 5.0 equiv.) was added to turn the mixture slightly basic. HATU (0.48 mmol, 1.2 equiv.) was added and the resulting mixture was stirred for 3 d at rt. The solvents were evaporated under reduced pressure and the residue was taken up in EtOAc (approx. 1/4 of the volume used for cyclization). The organic layer was washed with an equal amount of water and the aqueous layer was extracted with EtOAc (×1). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Flash column chromatography in EtOAc until the impurities had passed followed by change to EtOAc/MeOH 97:3 yielded cyclic peptoid cP1 (258 mg, 64%) as a colorless solid: Rf (EtOAc/MeOH 97:3) = 0.85; mp = 131–133 °C; [α]21D = –156.2 (c 0.74, CHCl3); 1H NMR (400 MHz, acetone-d6): δ 7.84–6.70 (30H, m, PhH), 6.08–5.60 (4.85H, m, NCHCH3), 5.50–5.42 (0.10H, m, NCHCH3), 5.24–4.93 (1.05H, m, NCHCH3), 4.56–3.56 (6H, m, 3 × NCH2CO), 3.52–2.14 (12H, m, 3 × NCH2CH2CO and 3 × NCH2CH2CO), 1.70–1.34 (18H, m, 6 × NCHCH3); 13C NMR (100 MHz, acetone-d6): δ 173.4, 173.2, 173.2, 173.0, 172.9, 172.7, 172.7, 172.4, 172.1, 171.3, 171.2, 170.3, 169.8, 169.7, 169.5, 169.2 (6C, 6 × CO), 144.0, 143.4, 143.2, 143.0, 142.9, 142.1, 141.8, 141.4, 141.1 (6C, Ph), 130.6, 130.5, 130.3, 130.2, 130.1, 130.0, 129.9, 129.8, 129.2, 129.0, 128.9, 128.8, 128.4, 128.1 (30CH, Ph), 56.8, 55.9, 55.8, 54.7, 54.3, 53.9, 53.5, 53.0, 52.8, 52.3, 51.9 (6CH, 6 × NCHCH3), 47.3, 46.9, 46.6, 46.5, 46.2 (3CH2, 3 × NCH2CO), 42.5, 42.3, 42.0, 41.8, 40.8, 39.3, 39.1, 37.4, 36.9, 36.0, 35.1, 33.6 (6CH2, 3 × NCH2CH2CO and 3 × NCH2CH2CO), 20.1, 19.2, 18.7, 18.3, 18.2, 17.9, 17.8, 17.6, 17.5, 17.4, 17.0 (6CH3, 6 × NCHCH3); IR (ATR): 1647 (CO, amide), 1495, 1451, 1414, 1377, 1341, 1283, 1234, 1206, 1186, 1074, 1030, 986, 914, 843, 783, 746; HRMS (TOF MS ES+) calcd for C63H72N6O6Na [M + Na]+m/z 1031.5411, found 1031.5417. HPLC (Water (0.1% TFA)/MeCN 20:80, flow = 0.80): tr = 15.79 min, purity = 99.5%.
NMR study
1H NMR experiments were undertaken using a peptoid concentration of 4 or 8 mM for all the spectra of the cyclopeptoid cP1 and the corresponding metal complexes. 1H NMR spectra were measured in deuterated solvents (CDCl3, acetone-d6 and CD3CN) on a 400 MHz Bruker AC-400 spectrometer or a Bruker AC-500 spectrometer. NMR temperature studies were conducted on a Bruker AC-500 spectrometer equipped with a Bruker BVT3000 Variable Temperature Unit (using a Eurotherm 902S temperature controller).Sr2+/cP1 complex: 1H NMR (500 MHz, CD3CN, 293 K): δ 7.42–7.15 (30H, m, PhH), 5.25 (3H, m, NCHCH3), 5.07 (3H, q, J = 6.5 Hz, NCHCH3), 4.47 (3H, m, 3 × NCH2aCO), 3.86 (6H, m, 3 × NCH2bCO, 3 × NCH2aCH2CO), 3.25 (3H, m, 3 × NCH2bCH2CO), 2.71 (3H, m, 3 × NCH2CH2aCO), 2.32 (3H, m, 3 × NCH2CH2bCO), 1.62, 1.57 and 1.49 (18H, 3xd, J = 6.8 Hz, 6 × NCHCH3); 13C NMR (100 MHz, CD3CN, 293 K): δ 177.1 (3C, 3 × CO), 171.9 (3C, 3 × CO), 141.0, 140.1 (6C, Ph), 129.8, 129.6, 128.8, 128.6, 127.9, 127.4 (30CH, Ph), 57.0 (3CH, 3 × NCHCH3), 56.4 (3CH, 3 × NCHCH3), 45.6 (3CH2, 3 × NCH2CO), 40.3 (3CH2, 3 × NCH2CH2CO), 35.0 (3CH2, 3 × NCH2CH2CO), 19.0 (3CH3, 3 × NCHCH3), 18.2 (3CH3, 3 × NCHCH3).
Circular dichroism (CD)
CD samples were prepared by weighing accurately small amounts of the appropriate compound using a calibrated balance with accuracy of 0.002 mg (Sartorius CPA26P) or 0.001 mg (Mettler Toledo XS3DU). The solvent was added volumetrically to obtain a sample solution of appropriate concentration. Circular dichroism analyses were performed either by SRCD or ECD as indicated. SRCD spectra were recorded at beamline B23 of the Diamond Light Source (UK) using an Olis CD spectropolarimeter (Module B) equipped with a Peltier temperature control system and fitted with a bespoke thermostated cell holder.28 The following parameters were used for SRCD: 4 acquisitions, 10 nm min−1 scan speed, 0.1 nm data interval, 1.0 nm spectral band width, 4 s time constant, 260 nm–190 nm scan range. ECD spectra were recorded using a Chirascan Plus instrument (Applied Photophysics Ltd) equipped with a Peltier temperature control system. The following parameters were used for ECD: 4 aquisitions, 1 nm step, 1 s time per point, 1.0 nm spectral band width, 260 nm–190 nm range. The qualitative and quantitative CD titrations were carried out in a stepwise manner by adding small aliquots (3–80 μl) of stock solutions of perchlorate salt of varied concentrations (1, 2 or 3 mM) into a solution of the peptoid of a fixed concentrations (7.5 or 15 μM in the far UV region using 0.4 or 1 cm rectangular pathlength quartz cuvettes from Hellma and 600 μM in the far UV region using a 1 cm cylindrical quartz cuvette). The titration was designed to reach the saturation of the peptoid and a final dilution of the peptoid inferior to 15%. An accurate and precise addition of the small aliquots of the guest, was achieved using positive displacement pipettes. Peptoid spectra (4 accumulations) were recorded at 20 °C in CH3CN. Solvent baseline spectra (4 accumulations) were recorded in the same cell at proximal time and averaged. The peptoid spectra were averaged to give a spectrum from which the averaged baseline was subtracted. The resulting baseline corrected spectrum was normalised for pathlength and concentration to give the molar ellipticity ([θ], deg cm2 dmol−1). CD data from quantitative metal titrations were processed and analysed using a proprietary software developed at the beamline B23 at Diamond Light Source (UK). The software processed the data by taking into account the baseline subtraction, the dilution of the host (peptoid) upon titration and any contributions to the CD deriving from the guest (none for the perchlorate). The binding constant was obtained by fitting of experimental data expressed as ΔAbsorbance (ΔA, where ΔA = ΔCD/32980) into the Hill equation.22
Fluorescence spectroscopy
Fluorescence data were obtained at 20 °C using a Varian Eclipse instrument equipped with a Peltier temperature control system and a cell with 1 cm path length using a peptoid concentration of 600 μM. Emission spectra were recorded using the following parameters: 250 nm excitation wavelength, 5 nm excitation and emission slit width. Excitation spectra were recorded using the following parameters: 284 nm excitation wavelength, 5 nm excitation and emission slit width. Solvent and metal blanks were also recorded at a proximal time and showed no contribution to the fluorescence in the spectral region investigated.Isothermal titration calorimetry (ITC)
ITC experiments were undertaken using a GE Healthcare microcalorimeter. The reference cell was filled with CH3CN, the sample cell (1.8 mL) was filled with a solution of the peptoid at a concentration of 107 μM and titrated with aliquotes (2 μL) of a solution of Mg(ClO4)2 placed in the syringe (2.5 mM). ITC experiments were undertaken at 20 °C using the following experimental parameters: 145 injections, 200 seconds spacing between injections, 307 rpm stirring speed and 5 μcal s−1 reference power. Titrations of the Mg(ClO4)2 into the solvent and of solvent into the peptoids were carried out separately to account for the heats of dissolution and solution respectively and subtracted from the metal – peptoid titration. Each titration was repeated in triplicate to ensure accuracy of the data obtained but data presented herein were not averaged.Molecular modelling (PM6)
Initial models of the complexes were made using ChemDraw (Version 15.0.0.106, Perkin Elmer Informatics) and Chem3D (Version 15.0.0.106, Perkin Elmer Informatics). Initial geometry and conformer searches were conducted using the integral MM2 force field facility in Chem3D.29 Fuller geometry and conformer searches were conducted using the semi-empirical method PM6 in MOPAC2009 via the Chem3D MOPAC interface. Final geometries were first minimised in the gas phase and then minimised and assessed using PM6 with the COSMO model for acetonitrile (dielectric constant set at 37.5).30Acknowledgements
We acknowledge the Medway School of Pharmacy for funding, and Diamond Light Source for access to B23 beamline (experiment numbers: SM7104 and SM6657). We acknowledge T. Hjelmgaard for his contribution to the synthetic work.References
- R. Maharani, B. E. Sleebs and A. B. Hughes, Macrocyclic N-methylated cyclic peptides and depsipeptides, in Studies in Natural Product Chemistry, ed. Atta-ur-Rahman, Elsevier, 2015, vol. 44, pp. 113–249 Search PubMed.
- M. Hinaje, M. Ford, L. Banting, S. Arkle and B. Khambay, Arch. Biochem. Biophys., 2002, 405, 73–77 CrossRef CAS PubMed.
- Y. A. Ovchinnikov, V. T. Ivanov, A. V. Evstratov, I. I. Mikhaleva, V. F. Bystrov, S. L. Portnova, T. A. Balashova, E. N. Meshcheryakova and V. M. Tulchinsky, Int. J. Pept. Protein Res., 1974, 6, 465–498 CrossRef CAS PubMed.
- (a) G. Maayan, Eur. J. Org. Chem., 2009, 5699–5710 CrossRef CAS; (b) Metallofoldamers. Supramolecular Architectures from Helicates to biomimetics, ed. G. Maayan and M. Albrecht, John Wiley& Sons, Ltd, 2013 Search PubMed; (c) B. C. Lee, T. K. Chu, K. A. Dill and R. N. Zuckermann, J. Am. Chem. Soc., 2008, 130, 8847–8855 CrossRef CAS PubMed; (d) G. Maayan, M. D. Ward and K. Kirshenbaum, Chem. Commun., 2009, 56–58 RSC; (e) T. Zabrodski, M. Baskin, J. K. Prathap and G. Maayan, Synlett, 2014, A–F Search PubMed; (f) M. Baskin and G. Maayan, Biopolymers, 2015, 104, 577–584 CrossRef CAS PubMed; (g) A. S. Knight, E. Y. Zhou, J. G. Pelton and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17488–17493 CrossRef CAS PubMed; (h) A. S. Knight, E. Y. Zhou and M. B. Francis, Chem. Sci., 2015, 6, 4042–4048 RSC; (i) M. Baskin and G. Maayan, Chem. Sci., 2016, 7, 2809–2820 RSC.
- A. S. Culf and R. J. Ouellette, Molecules, 2010, 15, 5282–5335 CrossRef CAS PubMed.
- (a) S. B. Shin, B. Yoo, L. J. Todaro and K. Kirshenbaum, J. Am. Chem. Soc., 2007, 129, 3218–3225 CrossRef CAS PubMed; (b) B. Yoo, S. B. Y. Shin, M. L. Huang and K. Kirshenbaum, Chem. – Eur. J., 2010, 16, 5528–5537 CrossRef CAS PubMed; (c) A. S. Culf, M. Cuperlovic–Culf, D. A. Léger and A. Decken, Org. Lett., 2014, 16, 2780–2783 CrossRef CAS PubMed.
- O. Roy, S. Faure, V. Thery, C. Didierjean and C. Taillefumier, Org. Lett., 2008, 10, 921–924 CrossRef CAS PubMed.
- (a) T. Hjelmgaard, S. Faure, C. Caumes, E. De Santis, A. A. Edwards and C. Taillefumier, Org. Lett., 2009, 11, 4100–4103 CrossRef CAS PubMed; (b) E. De Santis, T. Hjelmgaard, S. Faure, O. Roy, C. Didierjean, B. D. Alexander, G. Siligardi, R. Hussain, T. Javorfi, A. A. Edwards and C. Taillefumier, Amino Acids, 2011, 41, 663–672 CrossRef PubMed; (c) C. Caumes, C. Fernandes, O. Roy, T. Hjelmgaard, E. Wenger, C. Didierjean, C. Taillefumier and S. Faure, Org. Lett., 2013, 15, 3626–3629 CrossRef CAS PubMed.
- H. Hioki, H. Kinami, A. Yoshida, A. Kojima, M. Kodama, S. Takaoka, K. Ueda and T. Katsu, Tetrahedron Lett., 2004, 45, 1091–1094 CrossRef CAS.
- (a) N. Maulucci, I. Izzo, G. Bifulco, A. Aliberti, C. De Cola, D. Comegna, C. Gaeta, A. Napolitano, C. Pizza, C. Tedesco, D. Flot and F. De Riccardis, Chem. Commun., 2008, 3927–3929 RSC; (b) C. De Cola, S. Licen, D. Comegna, E. Cafaro, G. Bifulco, I. Izzo, P. Tecilla and F. De Riccardis, Org. Biomol. Chem., 2009, 7, 2851–2854 RSC; (c) G. Della Sala, B. Nardone, F. De Riccardis and I. Izzo, Org. Biomol. Chem., 2013, 11, 726–731 RSC; (d) I. Izzo, G. Ianniello, C. De Cola, B. Nardone, L. Erra, G. Vaughan, C. Tedesco and F. De Riccardis, Org. Lett., 2013, 15, 598–601 CrossRef CAS PubMed; (e) R. Schettini, F. De Riccardis, G. Della Sala and I. Izzo, J. Org. Chem., 2016, 81, 2494–2505 CrossRef CAS PubMed.
- C. De Cola, G. Fiorillo, A. Meli, S. Aime, E. Gianolio, I. Izzo and F. De Riccardis, Org. Biomol. Chem., 2014, 12, 424–431 CAS.
- E. De Santis, T. Hjelmgaard, C. Caumes, S. Faure, B. D. Alexander, S. J. Holder, G. Siligardi, C. Taillefumier and A. A. Edwards, Org. Biomol. Chem., 2012, 10, 1108–1122 CAS.
- P. Eberspaecher, E. Wismeth, R. Buchner and J. Barthel, J. Mol. Liq., 2006, 129, 3–12 CrossRef CAS.
- S. Devarajan and K. R. K. Easwaran, Biopolymers, 1981, 20, 891–899 CrossRef CAS.
- (a) R. D. Shannon, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1976, 32, 751–767 CrossRef; (b) F. Conti and G. Pistoia, J. Phys. Chem., 1968, 72, 2245–2248 CrossRef CAS; (c) A. Mishustin, Russ. J. Inorg. Chem., 2013, 58, 684–690 CrossRef CAS; (d) O. N. Kalugin, V. N. Agieienko and N. A. Otroshko, J. Mol. Liq., 2012, 165, 78–86 CrossRef CAS; (e) A. Diamond, A. Fanelli and S. Petrucci, Inorg. Chem., 1973, 12, 611–619 CrossRef CAS.
- Ba picrate was not used due its limited solubility in CD3CN.
- (a) Q. Sui, D. Borchardt and D. L. Rabenstein, J. Am. Chem. Soc., 2007, 129, 12042–12048 CrossRef CAS PubMed; (b) P. Armand, K. Kirshenbaum, R. A. Goldsmith, S. Farr–Jones, A. E. Barron, K. T. V. Truong, K. A. Dill, D. F. Mierke, F. E. Cohen, R. N. Zuckermann and E. K. Bradley, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4309–4314 CrossRef CAS PubMed; (c) B. C. Gorske, B. L. Bastian, G. D. Geske and H. E. Blackwell, J. Am. Chem. Soc., 2007, 129, 8928–8929 CrossRef CAS PubMed.
- M. Baskin, L. Panz and G. Maayan, Chem. Commun., 2016, 52, 10350–10353 RSC.
- (a) F. Zsila, Z. Bikadi, I. Fitos and M. Simonyi, Curr. Drug Discovery Technol., 2004, 1, 133–153 CrossRef CAS; (b) G. Siligardi, B. Panaretou, P. Meyer, S. Singh, D. N. Woolfson, P. W. Piper, L. H. Pearl and C. Prodromou, J. Biol. Chem., 2002, 277, 20151–20159 CAS; (c) G. Siligardi and R. Hussain, Applications of circular dichroism, in Encyclopedia of spectroscopy and spectrometry, Elsevier, Oxford, 2nd edn, 2010, vol. 1, pp. 9–14 Search PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751, 119–139 CrossRef CAS PubMed.
- (a) K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS PubMed; (b) C. W. Wu, T. J. Sanborn, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 2958–2963 CrossRef CAS PubMed; (c) C. W. Wu, T. J. Sanborn, K. Huang, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 6778–6784 CrossRef CAS PubMed.
- S. Goutelle, M. Maurin, F. Rougier, X. Barbaut, L. Bourguignon, M. Ducher and P. Maire, Fundam. Clin. Pharmacol., 2008, 22, 633–648 CrossRef CAS PubMed.
- (a) J. Yoe and A. Jones, Ind. Eng. Chem., Anal. Ed., 1944, 16, 111–115 CrossRef CAS; (b) J. M. Bosque-Sendra, E. Almansa-López, A. M. García-Campaña and L. Cuadros-Rodríguez, Anal. Sci., 2003, 19, 1431–1439 CrossRef CAS PubMed; (c) I. Durán Merás, A. Muñoz de la Peña, F. Salinas López and M. I. Rodríguez Cáceres, Analyst, 2000, 125, 1471–1476 RSC.
- (a) Y. B. Zeng, N. Yang and H. Sun, Chem. – Eur. J., 2011, 17, 5852–5860 CrossRef CAS PubMed; (b) D. Han, S. Huh and H. Myung, J. Pept. Sci., 2011, 17, 565–568 CrossRef CAS PubMed.
- M. D. Daily, M. D. Baer and C. J. Mundy, J. Phys. Chem. B, 2016, 120, 2198–2208 CrossRef CAS PubMed.
- T. Pigot, M.-C. Duriez, C. Picard, L. Cazaux and P. Tinès, Tetrahedron, 1992, 48, 4359–4368 CrossRef CAS.
- F. Arnaud-Neu, S. Barboso, F. Berny, A. Casnati, N. Muzet, A. Pinalli, R. Ungaro, M.-J. Schwing-Weill and G. Wipff, J. Chem. Soc., Perkin Trans. 2, 1999, 1727–1738 RSC.
- T. Javorfi, R. Hussain, D. Myatt and G. Siligardi, Chirality, 2010, 22(1), E149–E153 CrossRef CAS PubMed.
- M. J. Dudek and J. W. Ponder, J. Comput. Chem., 1995, 16, 791–816 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, CD and fluorescence spectra and additional modelling data. See DOI: 10.1039/c6ob01954d |
| ‡ Currently affiliation is National Physical Laboratory, Hampton road, Teddington, Middlesex, TW11 0LW, UK. |
| This journal is © The Royal Society of Chemistry 2016 |