
A comparative computational study of N-heterocyclic olefin and N-heterocyclic carbene mediated carboxylative cyclization of propargyl alcohols with CO2†
Abstract
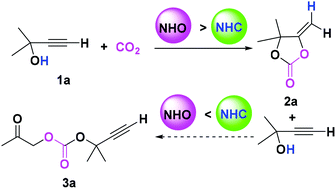
The carboxylative cyclization of a propargyl alcohol with CO2 mediated by a N-heterocyclic olefin (NHO) or N-heterocyclic carbene (NHC) has been comparatively studied using density functional theory (DFT) calculations. The calculations show that the advantageous catalytic performance of the NHO in the title reaction can be attributed to two aspects: (i) the active site of the NHO extends outside the imidazolium ring, which enhances the reactivity and stability of the [NHOH]+[carbonate]− ionic pair intermediate. Thus, the turnover frequency (TOF)-determining intramolecular cyclization step is kinetically more favorable in the NHO system. (ii) As the basicity of the NHO is weaker than the NHC, deprotonation of the propargyl alcohol by the NHO is relatively more difficult. Consequently, the side reaction of ring-opening transesterification of the α-alkylidene cyclic carbonate with the nucleophilic [NHOH]+[alkoxide]− ionic pair intermediate can be inhibited using the NHO system. The present mechanistic study provides a basis for further application of these promising organocatalysts in more organic transformations.
 Please wait while we load your content...
Please wait while we load your content...

