
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cascade oxime formation, cyclization to a nitrone, and intermolecular dipolar cycloaddition†
Rachel C.
Furnival
,
Rungroj
Saruengkhanphasit
,
Heather E.
Holberry
,
Jonathan R.
Shewring
 ,
Hélène D. S.
Guerrand
,
Harry
Adams
and
Iain
Coldham
*
,
Hélène D. S.
Guerrand
,
Harry
Adams
and
Iain
Coldham
*
Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, S3 7HF, UK. E-mail: i.coldham@sheffield.ac.uk
First published on 1st November 2016
Abstract
Simple haloaldehydes, including enolisable aldehydes, were found to be suitable for the formation of cyclic products by cascade (domino) condensation, cyclisation, dipolar cycloaddition chemistry. This multi-component reaction approach to heterocyclic compounds was explored by using hydroxylamine, a selection of aldehydes, and a selection of activated dipolarophiles. Initial condensation gives intermediate oximes that undergo cyclisation with displacement of halide to give intermediate nitrones; these nitrones undergo in situ intermolecular dipolar cycloaddition reactions to give isoxazolidines. The cycloadducts from using dimethyl fumarate were treated with zinc/acetic acid to give lactam products and this provides an easy way to prepare pyrrolizinones, indolizinones, and pyrrolo[2,1-a]isoquinolinones. The chemistry is illustrated with a very short synthesis of the pyrrolizidine alkaloid macronecine and a formal synthesis of petasinecine.
Introduction
Cycloaddition reactions of nitrones have been known for over 50 years.1 The majority of examples involve the condensation of an aldehyde with an N-alkyl-hydroxylamine or oxidation of an amine to form the nitrone, followed by cycloaddition with an alkene dipolarophile.2 An alternative approach makes use of the condensation of an aldehyde or ketone with hydroxylamine to give an oxime that undergoes subsequent N-alkylation to give the nitrone. Various N-alkylating agents can be used and the most common are unsaturated systems that allow the nitrogen atom of the oxime to undergo conjugate addition.3 We have been exploring the N-alkylation of oximes by intramolecular substitution of an alkyl halide. This cyclization reaction provides the desired nitrone that undergoes intramolecular dipolar cycloaddition with an alkene.4,5So far, we have reported only two examples of intermolecular cycloaddition using this strategy and these make use of a non-enolisable aldehyde as the substrate for reaction with hydroxylamine.6,7 For example, the aldehyde 1 reacts to give the intermediate oxime 2 that undergoes cyclization onto the alkyl chloride to give the nitrone 3 that can be trapped intermolecularly with dimethyl maleate to give the product 4 as a mixture of diastereoisomers (Scheme 1).6 The enolisable aldehyde 5 failed to give the desired tricyclic product and led instead to 3-(pent-4-enyl)pyrrole.4f However, the aldehyde 6 underwent successful condensation with hydroxylamine, followed by cyclization then intramolecular cycloaddition onto the internal unactivated alkene, and this was used in a synthesis of myrioxazine A.4c
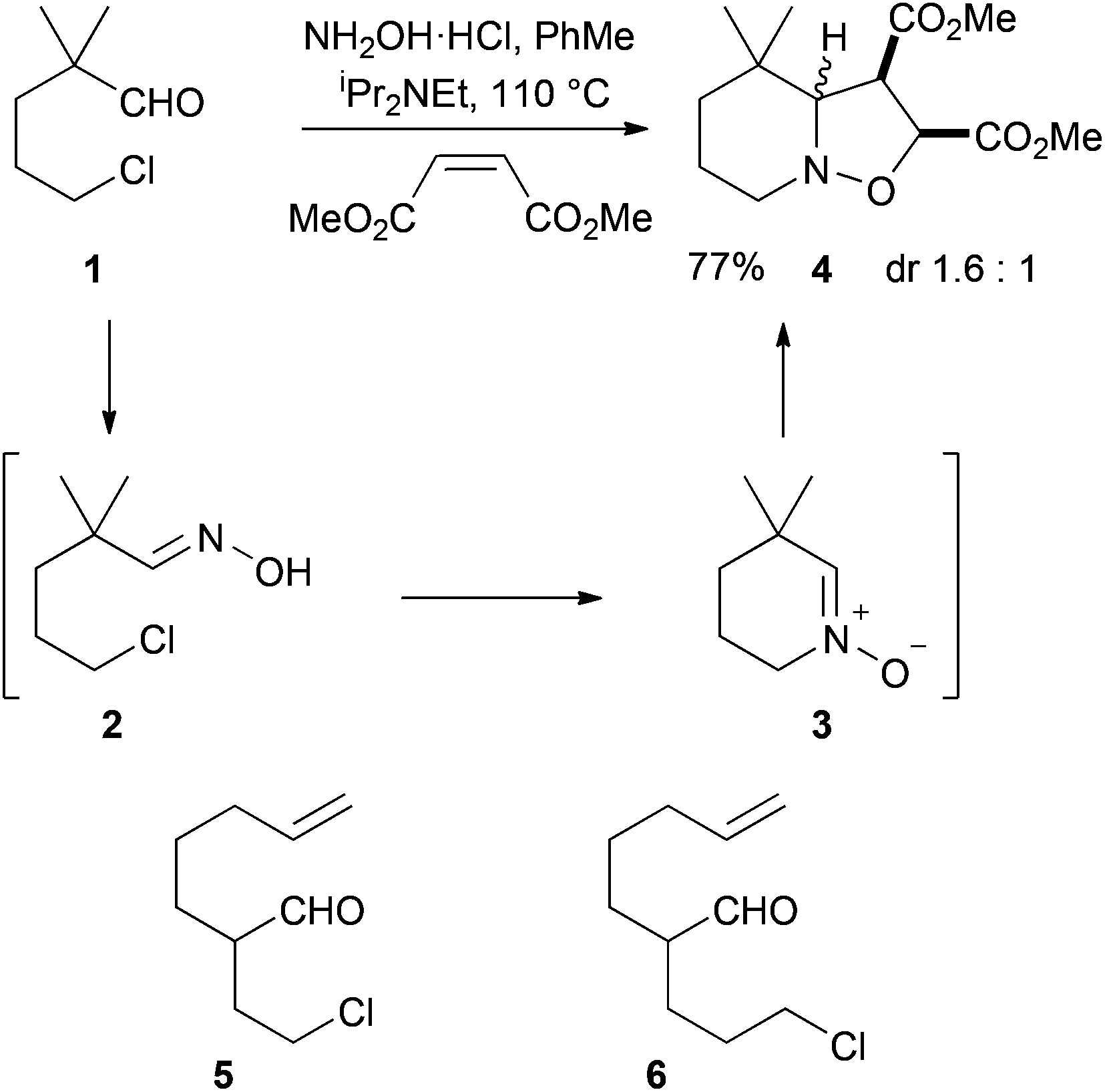 | ||
| Scheme 1 Previous studies with related aldehydes.4,6 | ||
In this paper, we report successful reactions of aldehydes, including enolisable aldehydes, with hydroxylamine, followed by in situ cyclization and intermolecular dipolar cycloaddition. The ability to undergo such chemistry is likely due to the use of reactive, electron-poor alkene dipolarophiles. This chemistry allows the formation of a greater range of substituted products without the need to block enolisation.
Results and discussion
Our initial work in this area focused on the simple aldehyde substrate 7 (Scheme 2). This was prepared by Swern oxidation of commercially available 5-chloro-1-pentanol.8 Treatment of the aldehyde 7 with hydroxylamine hydrochloride salt, the base iPr2NEt, and the dipolarophile dimethyl fumarate gave the desired bicyclic product 8 in high yield as a single diastereoisomer. The stereochemistry could not be determined at this stage, although NMR spectroscopic analysis and later single crystal X-ray analysis of a derivative (see below), revealed the stereochemistry as shown. The 1H NMR spectroscopic data for the product 8 match those reported from oxidation of N-hydroxypiperidine and cycloaddition of the resulting nitrone with dimethyl fumarate.9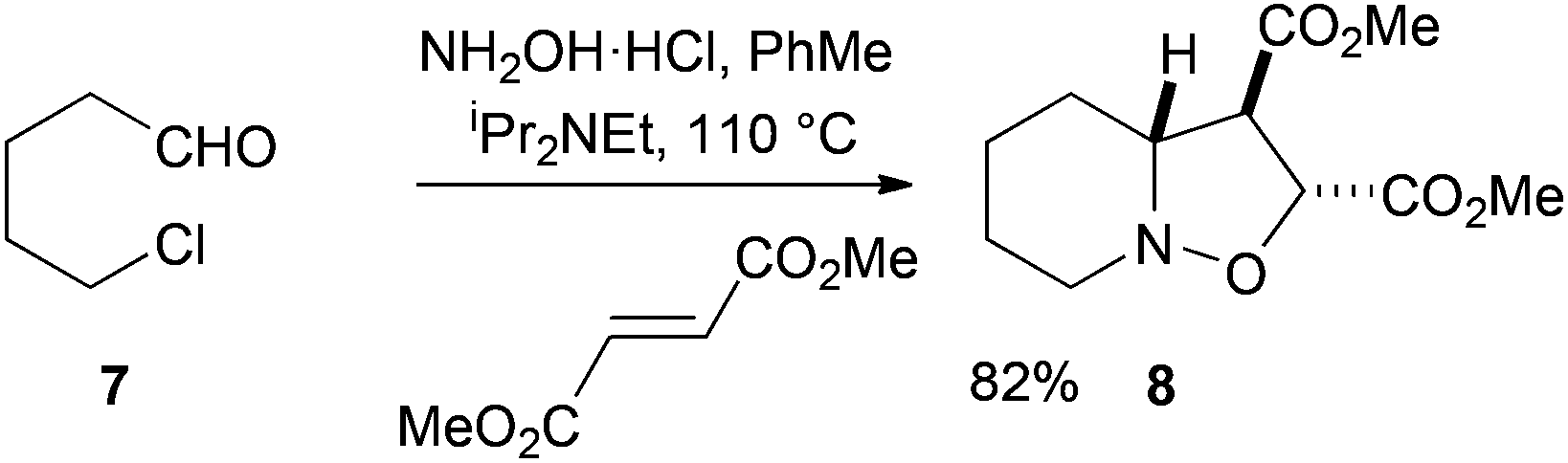 | ||
| Scheme 2 Aldehyde substrate 7 with dimethyl fumarate. | ||
Changing the dipolarophile from dimethyl fumarate to N-phenylmaleimide gave the desired bicyclic product 9 in high yield as the major diastereoisomer (dr 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Scheme 3). The diastereoisomers were separable by column chromatography and the stereochemistry of the major isomer 9 was determined by single crystal X-ray analysis. In a similar way, the use of N-methylmaleimide gave the desired product 10 in high yield as the major diastereoisomer (dr 4:1). The coupling constants for the methine protons in the cycloadduct 10 matched those of the cycloadduct 9, indicating the same relative stereochemistry for the major isomer.
:1) (Scheme 3). The diastereoisomers were separable by column chromatography and the stereochemistry of the major isomer 9 was determined by single crystal X-ray analysis. In a similar way, the use of N-methylmaleimide gave the desired product 10 in high yield as the major diastereoisomer (dr 4:1). The coupling constants for the methine protons in the cycloadduct 10 matched those of the cycloadduct 9, indicating the same relative stereochemistry for the major isomer.
 | ||
| Scheme 3 Aldehyde substrate 7 with N-phenyl- or N-methylmaleimide (major diastereoisomer of product drawn). | ||
Treatment of the cycloadduct 8 with zinc in acetic acid resulted in reductive cleavage of the N–O bond and subsequent cyclization of the amine onto one of the ester groups to give the lactam 11 as a single diastereoisomer (Scheme 4). This product was crystalline but the needles were too fine for X-ray analysis. However, conversion of the alcohol 11 to the ester 12 using p-bromobenzoyl chloride gave crystals suitable for X-ray analysis and allowed the determination of the relative stereochemistry of 12 (and hence of the cycloadduct 8).
 | ||
| Scheme 4 Preparation of lactam 11 and ester 12. | ||
Cyclization of the oxime derived from the aldehyde 7 gives a six-membered ring nitrone. We had previously found that a six-membered ring nitrone could be prepared and reacted intramolecularly (by using aldehyde 6, see Scheme 1).4c However, aldehyde 5 was unsuccessful and resulted in a pyrrole product. To test the feasibility of conducting the intermolecular chemistry with the homologous aldehyde containing one less methylene unit, we prepared the aldehyde 13. We were pleased to find that treatment of aldehyde 13 with hydroxylamine hydrochloride salt, the base iPr2NEt, and the dipolarophile dimethyl fumarate gave the desired bicyclic product 14 (as a mixture of diastereoisomers) in high yield (Scheme 5). The reaction was best conducted by pre-forming the oxime at 60 °C, then heating to 110 °C in the presence of the dipolarophile. Product 14 has been reported to be formed as a mixture of isomers starting from oxidation of N-hydroxypyrrolidine and cycloaddition of the resulting 1-pyrroline-1-oxide.10 The dipolarophiles N-phenylmaleimide and N-methylmaleimide gave the desired cycloaddition products 15 and 16 in good yield. The reaction was cleaner on addition of some MgSO4 and n-Bu4NI. The diastereoisomers were separable by column chromatography, although the minor isomer of the N-phenyl adduct 15 was isolated together with the adduct of hydroxylamine and N-phenylmaleimide. The stereochemistry of the major isomer of 16 was determined by single crystal X-ray analysis. This has the same relative stereochemistry as that for the major isomer of the homolog 10. We assume that the major isomer of 15 also has this relative stereochemistry.
 | ||
| Scheme 5 Aldehyde substrate 13 with dimethyl fumarate, N-phenyl- or N-methylmaleimide. | ||
With the aldehyde substrate 13 we also tested the dipolarophile β-nitrostyrene. This resulted in two products, the minor one of which was amenable to single crystal X-ray analysis, revealing the unexpected isomer 17 (Scheme 6). We have not been able to determine the stereochemistry of the other isomer 18, but on standing in CDCl3, this converts to a mixture of 17 and 18. The isomer 17 could arise either from the isomer 18 (for example by epimerization or via retro-Mannich reaction) or from a stepwise rather than concerted cycloaddition process.11
 | ||
| Scheme 6 Aldehyde substrate 13 with β-nitrostyrene. | ||
From the results above, it is clear that the aldehyde 13 is amenable to the desired cascade chemistry involving oxime formation, cyclization to give a nitrone, followed by intermolecular cycloaddition. This requires an activated alkene to promote cycloaddition, otherwise (as found for compound 5) the five-membered nitrone is prone to conversion to a pyrrole ring.
Treatment of the 2:1 mixture of cycloadducts 14 with Zn/AcOH gave the desired lactams 19 and 20 (ratio 2:1) that were separable by careful column chromatography (Scheme 7). The major product was stereoisomer 19 and the spectroscopic data matched the reported data.12 The major isomer, lactam 19, was reduced with LiAlH4 to give the pyrrolizidine alkaloid (±)-macronecine following a literature procedure (Scheme 7).12 The 1H NMR spectroscopic data matched that reported for the natural product.13 The chemistry also provides a formal synthesis of the alkaloid (±)-petasinecine by reduction.12 Therefore the chemistry allows a rapid access to these natural products in just three steps from 4-chlorobutanal (13).
 | ||
| Scheme 7 Preparation of the alkaloid macronecine. | ||
To complement the examples above, we studied the same cascade chemistry with the benzaldehyde substrate 21. This was prepared in two steps from isochroman.6b,14 Condensation of aldehyde 21 with hydroxylamine at 60 °C followed by addition of dimethyl fumarate, N-phenylmaleimide or N-methylmaleimide gave the cycloadducts 22–24 (Scheme 8). These products were all formed as a single diastereoisomer. The relative stereochemistry for adducts 22 and 24 was confirmed by single crystal X-ray analysis. The preference for the exo adducts matches that reported from reaction of the isolated nitrone.15
 | ||
| Scheme 8 Aldehyde substrate 21 with dimethyl fumarate, N-phenyl- or N-methylmaleimide. | ||
In a similar way, the aldehyde 2514 was treated with hydroxylamine at 60 °C followed by addition of dimethyl fumarate, N-phenylmaleimide or N-methylmaleimide to give the cycloadducts 26–28 (Scheme 9). The relative stereochemistries of the adducts 27 and 28 were confirmed by single crystal X-ray analysis.
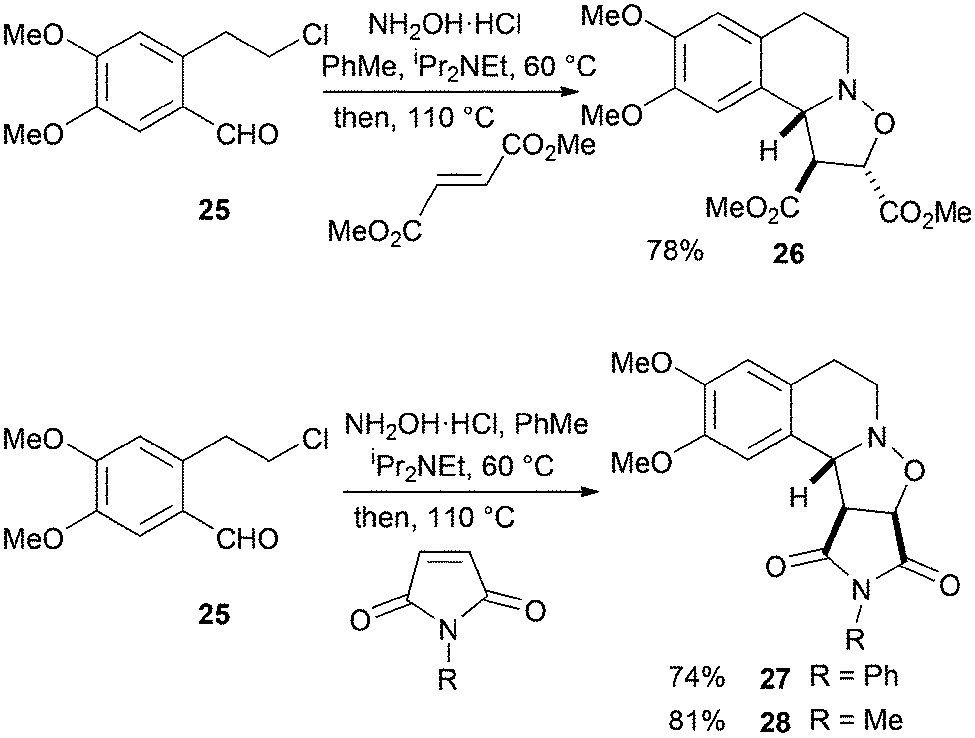 | ||
| Scheme 9 Aldehyde substrate 25 with dimethyl fumarate, N-phenyl- or N-methylmaleimide. | ||
The products 22 and 26 were treated with zinc in acetic acid to give the lactams 29 and 30 respectively as single isomers (Scheme 10).
 | ||
| Scheme 10 Preparation of lactams 29 and 30. | ||
Conclusions
In conclusion, cascade chemistry involving condensation of hydroxylamine, cyclization and intermolecular dipolar cycloaddition has been shown to be successful with a range of aldehydes, including enolisable aldehydes, together with electron-deficient dipolarophiles to give a variety of bicyclic, tricyclic and tetracyclic products. The isoxazolidine cycloadducts from using dimethyl fumarate as the dipolarophile were subjected to N–O bond cleavage to give, after cyclization, α-hydroxylactams. The chemistry provides a rapid entry to nitrogen-containing heterocycles of potential pharmaceutical interest and application to natural product synthesis, as demonstrated by a short synthesis of the alkaloid macronecine and a formal synthesis of petasinecine.Experimental
5-Chloropentanal 7
DMSO (3.4 mL, 48 mmol) in CH2Cl2 (10 mL) was added to freshly distilled oxalyl chloride (2.1 mL, 24 mmol) in CH2Cl2 (60 mL) at −78 °C. After 10 min, 5-chloropentan-1-ol (2.4 mL, 20 mmol) in CH2Cl2 (10 mL) was added slowly. After 30 min, triethylamine (13.9 mL, 100 mmol) was added. The mixture was allowed to warm to room temperature, then CH2Cl2 (20 mL) and water (20 mL) were added. The aqueous layer was washed with CH2Cl2 (3 × 50 mL). The combined organic layers were dried (MgSO4) and evaporated. Purification by column chromatography, eluting with CH2Cl2, gave aldehyde 7 (2.05 g, 85%) as an oil; Rf 0.8 (CH2Cl2); νmax (film)/cm−1 2935, 2865, 2730, 1720; 1H NMR (400 MHz, CDCl3) δ = 9.80 (1H, t, J 1.5 Hz, CHO), 3.60–3.54 (2H, m, CH2), 2.55–2.48 (2H, m, CH2), 1.88–1.77 (4H, m, 2 × CH2); 13C NMR (100 MHz, CDCl3) δ = 201.8, 44.5, 43.0, 31.8, 19.4. Data consistent with the literature.8(2R*,3R*,3aR*)-Dimethyl hexahydro-2H-isoxazolo[2,3-a]pyridine-2,3-dicarboxylate 8
Hydroxylamine hydrochloride (142 mg, 2.04 mmol) and N,N-diisopropylethylamine (0.74 mL, 4.3 mmol) were added to aldehyde 7 (204 mg, 1.70 mmol) in PhMe (17 mL) and the mixture was heated to 110 °C. After 30 min, dimethyl fumarate (368 mg, 2.55 mmol) was added and heating was continued at 110 °C. After 3.5 h, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (98:2), gave the product 8 (340 mg, 82%) as an oil; Rf 0.36 [CH2Cl2–MeOH (98:2)]; νmax (film)/cm−1 2950, 2850, 1730; 1H NMR (400 MHz, CDCl3) δ = 4.84 (1H, d, J 5.5 Hz, CH), 3.81 (3H, s, CH3), 3.79 (3H, s, CH3), 3.60–3.52 (1H, m, CH), 3.42 (1H, dd, J 10, 5.5 Hz, CH), 2.54 (1H, ddd, J 12, 10, 3 Hz, CH), 2.41–2.32 (1H, m, CH), 2.19–2.10 (1H, m, CH), 1.72–1.65 (3H, m, 2 × CH), 1.50 (1H, qd, J 13, 4 Hz, CH), 1.31–1.17 (1H, m, 2 × CH); 13C NMR (100 MHz, CDCl3) δ = 172.0, 171.2, 75.8, 70.5, 56.0, 55.2, 52.7, 52.5, 28.6, 24.3, 23.2; HRMS (ES) Found: MH+, 244.1186. C11H18NO5 requires MH+, 244.1185.
(3aR*,3bS*,8aS*)-2-Phenyl-hexahydro-8-oxa-2,7a-diaza-cyclopenta[a]indene-1,3-dione 9
Hydroxylamine hydrochloride (147 mg, 2.11 mmol) and N,N-diisopropylethylamine (0.77 mL, 4.4 mmol) were added to aldehyde 7 (211 mg, 1.76 mmol) in PhMe (17 mL) and the mixture was heated to 110 °C. After 30 min, N-phenylmaleimide (457 mg, 2.63 mmol) was added and heating was continued at 110 °C. After 2 h, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99:1), gave the product 9 (307 mg, 64%) as a solid and its diastereomer (83 mg, 17%) as a solid. The product 9 was recrystallized from CH2Cl2–hexanes (1:1) as needles; m.p. 157–159 °C; Rf 0.43 [CH2Cl2–MeOH (98:2)]; νmax (film)/cm−1 2960, 2935, 1710; 1H NMR (400 MHz, CDCl3) δ = 7.52–7.47 (2H, m, 2 × CH), 7.45–7.39 (1H, m, CH), 7.36–7.31 (2H, m, 2 × CH), 4.93 (1H, d, J 7.5 Hz, CH), 3.74–3.68 (1H, m, CH), 3.56–3.48 (1H, m, CH), 3.39 (1H, dd, J 7.5, 1.5 Hz, CH), 3.08–2.98 (1H, m, CH), 1.89–1.60 (4H, m, 4 × CH), 1.55–1.31 (2H, m, 2 × CH); 13C NMR (100 MHz, CDCl3) δ = 175.2, 174.5, 131.5, 129.2, 128.8, 126.4, 75.1, 63.4, 54.3, 50.3, 25.7, 22.5, 19.3; HRMS (ES) Found: MH+, 273.1250. C15H17N2O3 requires MH+, 273.1239.
Crystal data deposited at CCDC 1006040.
Unit cell parameters: a 12.9691(19), b 6.6225(11), c 15.583(3), P21/n.
Data for minor stereoisomer: m.p. 135–137 °C; Rf 0.27 [CH2Cl2–MeOH (98:2)]; νmax (film)/cm−1 2960, 2935, 1710; 1H NMR (400 MHz, CDCl3) δ = 7.52–7.45 (2H, m, 2 × CH), 7.45–7.39 (1H, m, CH), 7.34–7.29 (2H, m, 2 × CH), 4.91 (1H, d, J 7.5 Hz, CH), 3.64–3.55 (2H, m, 2 × CH), 2.61–2.50 (2H, m, 2 × CH), 2.28–2.18 (1H, m, CH), 1.85–1.79 (2H, m, 2 × CH), 1.68–1.53 (1H, m, CH), 1.50–1.37 (1H, m, CH), 1.34–1.19 (1H, m, CH); 13C NMR (100 MHz, CDCl3) δ = 176.8, 175.6, 131.5, 129.2, 129.0, 126.4, 75.1, 55.1, 50.6, 26.9, 24.4, 23.5, 19.4; HRMS (ES) Found: MH+, 273.1239. C15H17N2O3 requires MH+, 273.1239.
(3aR*,3bS*,8aS*)-2-Methyl-hexahydro-8-oxa-2,7a-diaza-cyclopenta[a]indene-1,3-dione 10
Hydroxylamine hydrochloride (142 mg, 2.04 mmol) and N,N-diisopropylethylamine (0.74 mL, 4.3 mmol) were added to aldehyde 7 (204 mg, 1.70 mmol) in PhMe (17 mL) and the mixture was heated to 110 °C. After 30 min, N-methylmaleimide (283 mg, 2.55 mmol) was added and heating was continued at 110 °C. After 3.5 h, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (98:2), gave the product 10 (218 mg, 61%) as a solid and its diastereomer (54 mg, 15%) as a solid. Data for the major stereoisomer 10: m.p. 80–83 °C; Rf 0.44 [CH2Cl2–MeOH (95:5)]; νmax (film)/cm−1 2925, 2860, 1695; 1H NMR (400 MHz, CDCl3) δ = 4.78 (1H, d, J 7 Hz, CH), 3.60–3.54 (1H, m, CH), 3.51–3.47 (1H, m, CH), 3.22 (1H, dd, J 7, 1.5 Hz, CH), 3.05 (3H, s, CH3), 3.01–2.90 (1H, m, CH), 1.84–1.74 (2H, m, 2 × CH), 1.67–1.54 (2H, m, 2 × CH), 1.51–1.38 (1H, m, CH), 1.37–1.20 (1H, m, CH); 13C NMR (100 MHz, CDCl3) δ = 176.0, 175.4, 75.1, 62.8, 54.4, 50.2, 25.7, 25.0, 22.5, 19.2; HRMS (ES) Found: MH+, 211.1084. C10H15N2O3 requires MH+, 211.1083.
(1R*,2R*,8aR*)-Methyl 2-hydroxy-3-oxo-octahydroindolizine-1-carboxylate 11
Zinc powder (186 mg, 2.85 mmol) was added to cycloadduct 8 (165 mg, 0.678 mmol) in AcOH/H2O (14 mL, 5:9). The mixture was heated to 70 °C for 2 h before being cooled to room temperature. The zinc salts were filtered, washed with CH2Cl2 and the solvent was evaporated. The solute was partitioned between aqueous ammonia (5 mL) and CH2Cl2 (10 mL), and the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). The organic fractions were dried (MgSO4), filtered and evaporated to give the lactam 11 (102 mg, 70%) as a solid, which was recrystallized from CH2Cl2–hexanes (1:1) as needles; m.p. 124–127 °C; Rf 0.10 [CH2Cl2–MeOH (99:1)]; νmax (film)/cm−1 3220, 2950, 2920, 2895, 2865, 1740, 1680; 1H NMR (400 MHz, CDCl3) δ = 4.50 (1H, d, J 7.5 Hz, CH), 4.13 (1H, dt, J 13, 5 Hz, CH), 3.95–3.87 (1H, m, CH), 3.80 (3H, s, CH3), 2.93 (1H, t, J 7.5 Hz, CH), 2.78 (2H, dt, J 13, 3.5 Hz, 2 × CH), 2.14–2.06 (1H, m, CH), 1.96–1.87 (1H, m, CH), 1.81–1.72 (1H, m, CH), 1.50 (1H, qt, J 13, 3.5 Hz, CH), 1.40–1.24 (1H, m, CH), 1.11 (1H, qd, J 13, 3.5 Hz, CH); 13C NMR (100 MHz, CDCl3) δ = 170.4, 169.8, 70.2, 56.8, 52.2, 50.4, 40.5, 32.4, 24.1, 23.4; HRMS (ES) Found: MH+, 214.1070. C10H16NO4 requires MH+, 214.1079.
(1R*,2R*,8aR*)-Methyl 2-(4-bromobenzoyloxy)-3-oxo-octahydroindolizine-1-carboxylate 12
p-Bromobenzoyl chloride (875 mg, 5.0 mmol) and DMAP (60 mg, 0.5 mmol) were added to the lactam 11 (425 mg, 2.0 mmol) in pyridine (2 mL) at room temperature. After 16 h, the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99:1), gave ester 12 (687 mg, 87%) as fine needles; m.p. 90–92 °C; Rf 0.2 [CH2Cl2–MeOH (99:1)]; νmax (film)/cm−1 2940, 2855, 1735, 1695; 1H NMR (400 MHz, CDCl3) δ = 7.92 (2H, d, J 8.5 Hz, 2 × CH), 7.60 (2H, d, J 8.5 Hz, 2 × CH), 5.76 (1H, d, J 6.5 Hz, CH), 4.23 (1H, d, J 14 Hz, CH), 3.75–3.66 (2H, m, 2 × CH), 2.75 (1H, t, J 14 Hz, CH), 2.04–1.97 (1H, m, CH), 1.92–1.85 (1H, m, CH), 1.83–1.76 (1H, m, CH), 1.55–1.38 (3H, m, 3 × CH); 13C NMR (100 MHz, CDCl3) δ = 169.0, 166.1, 164.5, 131.8, 131.4, 129.7, 128.6, 70.2, 56.9, 52.4, 48.7, 40.8, 32.4, 24.0, 23.4; HRMS (ES) Found: MH+, 396.0444. C17H19NO579Br requires MH+ 396.0447.
Crystal data deposited at CCDC 1022997.
Unit cell parameters: a 11.0353(7), b 21.5973(13), c 15.0726(9), P21/c.
4-Chlorobutanal 13
DMSO (2.9 mL, 40.6 mmol) in CH2Cl2 (10 mL) was added dropwise to oxalyl chloride (1.8 mL, 20.3 mmol) in CH2Cl2 (60 mL) at −78 °C. After 10 min, 4-chlorobutanol (1.8 mL, 16.9 mmol) in CH2Cl2 (10 mL) was added dropwise. After 30 min, Et3N (11.8 mL, 84.5 mmol) was added. After 30 min, the mixture was allowed to warm to room temperature then water (20 mL) and CH2Cl2 (20 mL) were added. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography, eluting with CH2Cl2, gave aldehyde 13 (1.09 g, 61%) as an oil; Rf 0.9 (CH2Cl2); 1H NMR (400 MHz, CDCl3) 9.84 (1H, s, CHO), 3.62 (2H, t, J 6.5 Hz, CH2), 2.70 (2H, t, J 6.5 Hz, CH2), 2.13 (2H, pent., J 6.5 Hz, CH2). Data consistent with the literature.16(2S*,3S*,3aS*)-2,3-Dimethyl hexahydropyrrolo[1,2-b][1,2]oxazole-2,3-dicarboxylate 14a and (2S*,3S*,3aR*)-2,3-dimethyl hexahydropyrrolo[1,2-b][1,2]oxazole-2,3-dicarboxylate 14b
To aldehyde 13 (106 mg, 1 mmol) in PhMe (10 mL) was added hydroxylamine hydrochloride (85 mg, 1.2 mmol) and iPr2NEt (0.41 mL, 2.4 mmol) and the mixture was warmed to 60 °C. After 30 min, dimethylfumarate (217 mg, 1.5 mmol) was added and the mixture was heated under reflux. After 3.5 h, the mixture was allowed to cool to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99:1) gave cycloadducts 14a and 14b (200 mg, 0.87 mmol, 87%) as an oil, as an inseparable mixture of diastereoisomers (dr 2:1); alternatively, aldehyde 13 (500 mg, 4.7 mmol) in PhMe (50 mL), hydroxylamine hydrochloride (390 mg, 5.6 mmol), iPr2NEt (2.0 mL, 11 mmol) and dimethylfumarate (1.0 g, 7.0 mmol) gave, after heating for 17 h then purification as above, the cycloadducts 14a and 14b (770 mg, 3.4 mmol, 72%) as an oil (dr 2:1); Rf 0.5 [CH2Cl2–MeOH (98:2)]; νmax (film)/cm−1 2955, 1735; 1H NMR (500 MHz, CDCl3) δ = 4.98 (0.35H, d, J 5.5 Hz, CH), 4.85 (0.65H, d, J 7.5 Hz, CH), 4.13–4.08 (0.35H, dd, J 7.5, 5.0 Hz, CH), 3.93–3.85 (1H, m, CH), 3.81 (3H, s, 2 × Me), 3.79 (3H, s, 2 × Me), 3.57–3.49 (0.35H, ddd, J 14.0, 8.0, 3.5 Hz, CH), 3.39 (0.65H, dd, J 7.5, 5.0 Hz CH), 3.39–3.43 (0.35H, m, CH), 3.13–3.02 (1.3H, m, 2 × CH), 2.13–1.99 (1.65H, m, 3 × CH), 1.86–1.72 (2H, m, 2 × CH), 1.63–1.53 (0.35H, m, CH); 13C (125.7 MHz, CDCl3) δ = 171.7, 170.9, 170.5, 169.9, 78.9, 76.0, 69.7, 68.3, 57.4, 56.3, 56.2, 55.1, 52.7, 52.65, 52.6, 52.4, 29.8, 26.6, 24.3, 23.2; HRMS (ES) Found: MH+, 230.1022. C10H16NO5 requires MH+ 230.1028. 1H NMR data consistent with the literature.9
(1S*,2S*,6R*)-4-Phenyl-7-oxa-4,8-diazatricyclo[6.3.0.01]undecane-3,5-dione 15
To aldehyde 13 (106 mg, 1 mmol) in PhMe (10 mL) was added hydroxylamine hydrochloride (76 mg, 1.1 mmol), iPr2NEt (0.41 mL, 2.4 mL), dry MgSO4 (∼200 mg) and Bu4NI (37 mg, 0.1 mmol) and the mixture was warmed to 60 °C. After 30 min, N-phenylmaleimide (260 mg, 1.5 mmol) was added and the mixture was heated under reflux. After 3.5 h, the mixture was allowed to cool to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99:1), gave cycloadduct 15 (168 mg, 65%) as an oil; Rf 0.4 [CH2Cl2–MeOH (99:1)]; νmax (film)/cm−1 2960, 1710, 1498; 1H NMR (400 MHz, CDCl3) δ = 7.55–7.33 (5H, m, Ph), 4.97 (1H, d, J 7.5 Hz, CH), 3.90 (1H, t, J 8.0 Hz, CH), 3.73 (1H, d, J 7.5 Hz, CH), 3.66–3.58 (2H, m, 2 × CH), 3.07 (1H, dt, J 14.5, 8.5 Hz, CH), 2.07–1.98 (1H, m, CH), 1.93–1.73 (2H, m, 2 × CH); 13C NMR (101 MHz, CDCl3) δ = 174.8, 174.4, 131.4, 129.2, 128.9, 126.4, 75.9, 70.8, 55.9, 54.2, 30.0, 24.3; HRMS (ES) Found: MH+, 259.1074. C14H15N2O3 requires MH+, 259.1083.
The minor diastereomer was isolated as a mixture with the adduct formed from conjugate addition of hydroxylamine with the maleimide.
(1S*,2S*,6R*)-4-Methyl-7-oxa-4,8-diazatricyclo[6.3.0.01]undecane-3,5-dione 16a and (1R*,2S*,6R*)-4-methyl-7-oxa-4,8-diazatricyclo[6.3.0.01]undecane-3,5-dione 16b
To aldehyde 13 (106 mg, 1 mmol) in PhMe (10 mL) was added hydroxylamine hydrochloride (76 mg, 1.1 mmol), iPr2NEt (0.41 mL, 2.4 mL), dry MgSO4 (∼200 mg) and Bu4NI (37 mg, 0.1 mmol) and the mixture was warmed to 60 °C. After 30 min, N-methylmaleimide (260 mg, 1.5 mmol) was added and the mixture was heated under reflux. After 3.5 h, the mixture was allowed to cool to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99:1), gave cycloadduct 16a (96 mg, 49%) as an oil and the cycloadduct 16b (43 mg, 22%) as an oil, both of which recrystallised from EtOAc as cubes.
Data for adduct 16a: m.p. 91–93 °C; Rf 0.4 [CH2Cl2–MeOH (95:5)]; νmax (film)/cm−1 2955, 1705; 1H NMR (400 MHz, CDCl3) δ = 4.86 (1H, d, J 7.0 Hz, CH), 4.00–3.91 (2H, m, 2 × CH), 3.38–3.30 (1H, m, CH), 3.18–3.06 (1H, m, CH), 3.04 (3H, s, CH3), 2.07–1.94 (2H, m, 2 × CH), 1.90–1.80 (1H, m, CH), 1.75–1.64 (1H, m, CH); 13C NMR (101 MHz, CDCl3) δ = 174.8, 172.0, 77.6, 67.9, 55.8, 53.6, 29.7, 25.0, 24.1; HRMS (ES) Found: MH+, 197.0922. C9H13N2O3 requires MH+, 197.0926.
Crystal data deposited at CCDC 1051097.
Unit cell parameters: a 7.9208(3), b 16.5681(9), c 6.7713(3), Pna21.
Data for adduct 16b: m.p. 88–91 °C; Rf 0.3 [CH2Cl2–MeOH (95:5)]; νmax (film)/cm−1 2955, 1700; 1H NMR (400 MHz, CDCl3) δ = 4.83 (1H, d, J 7.0 Hz, CH), 3.77 (1H, t, J 8.5 Hz, CH), 3.61–3.52 (2H, m, 2 × CH), 3.06 (3H, s, CH3), 3.04–2.96 (1H, m, CH), 2.23–2.12 (2H, m, 2 × CH), 1.88–1.80 (1H, m, CH), 1.75–1.68 (1H, m, CH); 13C NMR (101 MHz, CDCl3) δ = 174.8, 172.1, 77.6, 67.9, 55.8, 53.6, 25.7, 25.0, 24.1; HRMS (ES) Found: MH+, 197.0917. C9H13N2O3 requires MH+, 197.0926.
(2R*,3R*,3aR*)-3-Nitro-2-phenyl-hexahydropyrrolo[1,2-b][1,2]oxazole 17 and another isomer of 3-nitro-2-phenyl-hexahydropyrrolo[1,2-b][1,2]oxazole 18
To aldehyde 1 (106 mg, 1 mmol) in PhMe (10 mL) was added hydroxylamine hydrochloride (76 mg, 1.1 mmol), iPr2NEt (0.41 mL, 2.4 mL), MgSO4 (∼200 mg) and Bu4NI (37 mg, 0.1 mmol) and the mixture was warmed to 60 °C. After 30 min, trans-β-nitrostyrene (224 mg, 1.5 mmol) was added and the mixture was heated under reflux. After 3.5 h, the mixture was allowed to cool to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with petrol–EtOAc (8:2 to 1:1), gave cycloadducts 17 (56 mg, 24%) as a solid and 18 (152 mg, 65%) as an oil.
Data for adduct 17: m.p. 89–92 °C; Rf 0.4 [petrol–EtOAc (1:1)]; νmax (film)/cm−1 2980, 1550, 1455; 1H NMR (400 MHz, CDCl3) δ = 7.41–7.34 (5H, m, Ph), 5.50 (1H, d, J 5.5 Hz, CH), 5.24 (1H, dd, J 5.5, 1.5 Hz, CH), 4.51 (1H, broad t, J 7.5 Hz, CH), 3.50 (1H, dt, J 11.5, 5.5 Hz, CH), 3.21 (1H, dt, J 11.5, 7.5 Hz, CH), 2.35–2.25 (1H, m, CH), 2.08–1.99 (1H, m, CH), 1.94–1.77 (2H, m, 2 × CH); 13C NMR (101 MHz CDCl3) δ = 132.0, 129.3, 128.6, 126.5, 98.4, 80.8, 68.7, 56.4, 29.9, 24.0; HRMS (ES) Found: MH+, 235.1073. C12H15N2O3 requires MH+, 235.1083; Anal. Calcd for C12H14N2O3: C, 61.5; H, 6.0; N, 12.0. Found: C, 61.9; H, 6.0; N, 11.7.
Crystal data deposited at CCDC 1006037.
Unit cell parameters: a 9.0398(3), b 7.2748(3), c 17.0449(6), P21/c.
Data for adduct 18: Rf 0.6 [petrol–EtOAc (4:1)]; νmax (film)/cm−1 2980, 1545, 1500; 1H NMR (400 MHz, CDCl3) δ = 7.49–7.35 (5H, m, Ph), 5.75 (1H, d, J 6.0 Hz, CH), 5.43 (1H, dd, J 8.5, 6.0 Hz, CH), 4.29 (1H, br q, J 7.5 Hz, CH), 3.50 (1H, dt, J 12.5, 6.5 Hz, CH), 3.32 (1H, dt, J 12.5, 7.0 Hz, CH), 2.08–1.97 (4H, m, 4 × CH); 13C NMR (101 MHz CDCl3) δ = 132.3, 128.9, 128.8, 126.3, 96.4, 79.4, 68.1, 56.7, 26.4, 23.9; HRMS (ES) Found: MH+, 235.1088. C12H15N2O3 requires MH+, 235.1083.
(1S*,2S*,6aR*)-Methyl 2-hydroxy-3-oxo-hexahydro-1H-pyrrolizine-1-carboxylate 19 and (1S*,2S*,6aS*)-methyl 2-hydroxy-3-oxo-hexahydro-1H-pyrrolizine-1-carboxylate 20
To a 2:1 mixture of cycloadducts 14 (215 mg, 0.9 mmol) in H2O (3.5 mL) and AcOH (2.5 mL) was added zinc (280 mg, 4.3 mmol) and the mixture was heated under reflux. After 22 h, the mixture was cooled to room temperature. The mixture was diluted with CH2Cl2 (50 mL) and aqueous NH3 (5 mL, 35%), the layers separated and the aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure. Purification by column chromatography, eluting with CH2Cl2–iPrOH (97:3), gave the lactams 19 and 20 (combined yield of 142 mg, 79%). The pure lactams were isolated after careful chromatography: 19 (72 mg, 40%), 20 (32 mg, 18%) and both were recrystallized from hot EtOAc as needles.
Alternatively, a 2:1 mixture of cycloadducts 14 (500 mg, 2.2 mmol), H2O (11 mL), AcOH (5.5 mL) and zinc (600 mg, 9.2 mmol) gave, after heating for 22 h then purification as above, the lactams 19 (129 mg, 30%) and 20 (70 mg, 16%).
Data for lactam 19: m.p. 138–141 °C, lit.12 m.p. for (+)-19: 191–192 °C; Rf 0.6 [CH2Cl2–iPrOH–NH3 (90:9:1)]; 1H NMR (400 MHz, CDCl3) δ = 4.63 (1H, dd, J 6.0, 4.0 Hz, CH), 4.42 (1H, ddd, J 9.0, 8.0, 5.5 Hz, NCH), 3.85 (1H, d, J 4.0 Hz, OH), 3.80 (3H, s, CH3), 3.52 (1H, dt, J 12.0, 8.0 Hz, NCH), 3.22 (1H, ddd, J 12.0, 8.5, 3.5 Hz, NCH), 2.95 (1H, dd, J 8.0, 6.0 Hz, CH), 2.28–2.13 (3H, m, 3 × CH), 1.44–1.32 (1H, m, CH). Data correspond to the literature.12
Data for lactam 20: m.p. 182–185 °C, lit.12 m.p. for 20 was not reported; Rf 0.5 [CH2Cl2–iPrOH–NH3 (90:9:1)]; 1H NMR (400 MHz, CDCl3) δ = 4.76 (1H, br s, CH), 3.98 (1H, dt, J 8.0, 6.5 Hz, NCH), 3.74 (3H, s, CH3), 3.67 (1H, t, J 6.5 Hz, CH), 3.63 (1H, dt, J 11.5, 7.0 Hz, NCH), 3.10–3.22 (2H, m, NCH and OH), 2.13–1.97 (3H, m, 3 × CH), 1.59–1.47 (1H, m, CH). Data correspond to the literature.12
Macronecine
The lactam 19 (126 mg, 0.63 mmol) in THF (9 mL) was added to a solution of LiAlH4 (148 mg, 3.91 mmol) in THF (4 mL). The grey suspension was then heated under reflux. After 17 h, the mixture was cooled to room temperature and aqueous NaOH (0.3 mL, 2 M) was added. After 1 h, the suspension was filtered and was washed with MeOH (60 mL). The filtrate was concentrated to give a white solid which was purified by column chromatography on silica with a plug of Celite, eluting with CH2Cl2–MeOH–NH3 (10:5:1), to give macronecine (70 mg, 70%) as an amorphous solid; m.p. 106–108 °C, lit.13g m.p. 107–108 °C; Rf 0.1 [CH2Cl2–MeOH–NH3 (10:5:1)]; 1H NMR (400 MHz, CDCl3) δ = 4.50 (1H, t, J 4.0 Hz, CH), 4.34 (2H, br s, 2 × OH), 3.89–3.82 (2H, m, 2 × CH), 3.56–3.50 (1H, m, CH), 3.17 (1H, d, J 11.0 Hz, CH), 2.97 (1H, td, J 11.0, 6.5 Hz, CH), 2.67 (1H, dd, J 11.0, 3.5 Hz, CH), 2.59–2.54 (1H, m, CH), 2.00–1.92 (1H, m, CH), 1.87–1.76 (3H, m, 3 × CH), 1.57–1.50 (1H, m, CH). Data corresponds to the literature.12,13
(1S*,2S*,10bR*)-1,2-Dimethyl 2,5,6,10b-tetrahydro-1H-isoxazolo[3,2-a]isoquinoline-1,2-dicarboxylate 22
Hydroxylamine hydrochloride (56 mg, 0.81 mmol) and N,N-diisopropylethylamine (0.28 mL, 1.62 mmol) were added to aldehyde 2114 (154 mg, 0.67 mmol) in PhMe (7 mL) and the mixture was heated to 60 °C. Dimethyl fumarate (117 mg, 0.81 mmol) was added and the mixture was heated under reflux. After 1 h, the mixture was cooled to room temperature and the solvent evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99.7:0.3), gave the product 22 (150 mg, 76%), which was recrystallised from CH2Cl2–hexanes as needles; m.p. 83–85 °C, lit.13 m.p. 89–90 °C; Rf 0.5 [CH2Cl2–MeOH (99.5:0.5)]; νmax (film)/cm−1 2955, 1735; 1H NMR (400 MHz, CDCl3) δ = 7.26–7.19 (2H, m, 2 × CH), 7.16–7.13 (1H, m, CH), 7.07 (1H, d, J 7.0 Hz, CH), 4.95 (1H, d, J 7.5 Hz, CH), 4.89 (1H, d, J 8.5 Hz, CH), 3.88 (3H, s, CH3), 3.80–3.76 (1H, m, CH), 3.77 (3H, s, CH3), 3.42–3.38 (1H, m, CH), 3.23–3.15 (1H, m, CH), 3.08–3.00 (1H, m, CH), 2.96–2.92 (1H, m, CH); 13C NMR (100 MHz, CDCl3) δ = 171.5, 171.1, 133.2, 132.5, 128.9, 127.6, 127.1, 126.5, 80.9, 67.4, 57.7, 52.8, 52.7, 49.7, 27.9; HRMS (ES) Found MH+, 292.1172, C15H18NO5 requires MH+, 292.1185.
Crystal data deposited at CCDC 1006038.
Unit cell parameters: a 9.1248(8), b 32.242(3), c 9.4579(9), P21/c.
(8aR*,11aS*,11bR*)-10-Phenyl-5,8a,11a,11b-tetrahydropyrrolo[3′,4′:4,5][1,2]oxazolo[3,2-a]isoquinoline-9,11(6H,10H)-dione 23
Hydroxylamine hydrochloride (124 mg, 1.78 mmol) and N,N-diisopropylethylamine (0.62 mL, 3.55 mmol) were added to aldehyde 2114 (339 mg, 1.48 mmol) in PhMe (15 mL) and the mixture was heated to 60 °C. After 2 h, N-phenylmaleimide (308 mg, 1.78 mmol) was added and the mixture was heated under reflux. After 1 h, the mixture was allowed to cool to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99.8:0.2), gave cycloadduct 23 (291 mg, 61%) as an amorphous solid; m.p. 172–174 °C, lit.15 m.p. 178–179 °C; Rf 0.8 [CH2Cl2–MeOH (99.5:0.5)]; νmax (film)/cm−1 2935, 1715; 1H NMR (400 MHz, CDCl3) δ = 7.55–7.51 (2H, m, 2 × CH), 7.47–7.44 (2H, m, 2 × CH), 7.41–7.39 (1H, m, CH), 7.35–7.31 (1H, m, CH), 7.28–7.25 (1H, m, CH), 7.16 (1H, d, J 7.5, CH), 4.98–4.95 (2H, m, 2 × CH), 3.93 (1H, dd, 7.5, 2.0 Hz, CH), 3.74–3.66 (1H, m, CH), 3.33–3.21 (2H, m, CH), 2.69–2.62 (1H, m, CH); 13C NMR (101 MHz, CDCl3) δ = 174.7, 173.8, 133.9, 132.9, 131.4, 129.2, 128.9, 128.8, 127.3 (2 × CH), 127.2, 126.4, 75.7, 65.8, 56.8, 47.8, 22.9; HRMS (ES) Found MH+, 321.1232. C19H17N2O3 requires MH+, 321.1239.
(8aR*,11aS*,11bR*)-10-Methyl-5,8a,11a,11b-tetrahydropyrrolo[3′,4′:4,5][1,2]oxazolo[3,2-a]isoquinoline-9,11(6H,10H)-dione 24
Hydroxylamine hydrochloride (120 mg, 1.73 mmol) and N,N-diisopropylethylamine (0.60 mL, 3.47 mmol) were added to aldehyde 2114 (327 mg, 1.43 mmol) in PhMe (15 mL) and the mixture was heated to 60 °C. N-Methylmaleimide (192 mg, 1.73 mmol) was added and the mixture was heated under reflux. After 1 h, the mixture was allowed to cool to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99.7:0.3), gave the product 24 (280 mg, 76%), which was recrystallised from CH2Cl2–hexanes as needles; m.p. 162–164 °C, lit.11 m.p. 164–166 °C; Rf 0.5 [CH2Cl2–MeOH (99.5:0.5)]; νmax (film)/cm−1 2915, 2825, 1700; 1H NMR (400 MHz, CDCl3) δ = 7.40 (1H, d, J 7.5, CH), 7.31 (1H, t, J 7.5, CH), 7.24 (1H, t, J 7.5, CH), 7.14 (1H, d, J 7.5, CH), 4.82–4.80 (2H, m, 2 × CH), 3.76 (1H, dd, J 7.5, 2.0 Hz, CH), 3.76–3.66 (1H, m, CH), 3.26–3.20 (2H, m, 2 × CH), 3.12 (3H, s, CH3), 2.64–2.56 (1H, m, CH); 13C NMR (100 MHz, CDCl3) δ = 175.6, 174.9, 133.8, 132.9, 128.8, 127.3, 127.2, 127.1, 75.7, 65.2, 56.7, 47.7, 25.2, 22.9; HRMS (ES) Found MH+, 259.1084. C14H15N2O3 requires MH+, 259.1083.
Crystal data deposited at CCDC 1006039.
Unit cell parameters: a 6.8159(2), b 26.7835(7), c 7.0628(2), P21/c.
(1S*,2S*,10bR*)-1,2-Dimethyl 8,9-dimethoxy-2,5,6,10b-tetrahydro-1H-isoxazolo[3,2-a]isoquinoline-1,2-dicarboxylate 26
Hydroxylamine hydrochloride (56 mg, 0.81 mmol) and N,N-diisopropylethylamine (0.28 mL, 1.62 mmol) were added to aldehyde 2514 (154 mg, 0.67 mmol) in PhMe (7 mL) and the mixture was heated to 60 °C. After 2.5 h, dimethyl fumarate (126 mg, 0.88 mmol) was added and the mixture was heated under reflux. After 1 h, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99.5:0.5), gave the cycloadduct 26 (182 mg, 78%) as an amorphous solid; m.p. 96–99 °C; Rf 0.5 [CH2Cl2–MeOH (98:2)]; νmax (film)/cm−1 2940, 1730, 1610; 1H NMR (400 MHz, CDCl3) δ = 6.61 (1H, s, CH), 6.58 (1H, s, CH), 4.96 (1H, d, J 7.5 Hz, CH), 4.80 (1H, d, J 8.5 Hz, CH), 3.87 (6H, s, 2 × CH3), 3.84 (3H, s, CH3), 3.78 (3H, s, CH3), 3.78–3.74 (1H, m, CH), 3.40–3.37 (1H, m, CH), 3.17–3.13 (1H, m, CH), 3.01–2.94 (1H, m, CH), 2.85–2.80 (1H, m, CH); 13C NMR (101 MHz, CDCl3) δ = 171.7, 171.2, 148.5, 147.7, 125.5, 124.0, 110.8, 109.7, 80.9, 67.2, 57.6, 55.9 (2 × CH3), 52.8 (2 × CH3), 49.9, 27.7; HRMS (ES) Found: MH+, 352.1381. C17H22NO7 requires MH+, 352.1396.
(8aR*,11aS*,11bR*)-2,3-Dimethoxy-10-phenyl-5,8a,11a,11b-tetrahydropyrrolo[3′,4′:4,5][1,2]oxazolo[3,2-a]isoquinoline-9,11(6H,10H)-dione 27
Hydroxylamine hydrochloride (150 mg, 2.1 mmol) and N,N-diisopropylethylamine (0.75 mL, 4.3 mmol) were added to aldehyde 2514 (410 mg, 1.8 mmol) in PhMe (18 mL) and the mixture was heated to 60 °C. After 2.5 h, N-phenylmaleimide (370 mg, 2.1 mmol) was added and the mixture was heated under reflux. After 1 h, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99.5:0.5), gave the cycloadduct 27 (500 mg, 74%) as an amorphous solid; m.p. 185–187 °C (decomposed); Rf 0.7 [CH2Cl2–MeOH (98:2)]; νmax (film)/cm−1 2985, 1710, 1615; 1H NMR (400 MHz, CDCl3) δ = 7.55–7.51 (2H, m, 2 × CH), 7.47–7.44 (1H, m, CH), 7.40–7.38 (2H, m, 2 × CH), 6.90 (1H, s, CH), 6.63 (1H, s, CH), 5.03 (1H, d, J 7.5 Hz, CH), 4.90 (1H, br, CH), 3.93 (3H, s, CH3), 3.90 (3H, s, CH3), 3.91–3.89 (1H, m, CH), 3.63–3.57 (1H, m, CH), 3.31–3.25 (1H, m, CH), 3.15–3.08 (1H, m, CH), 2.67–2.60 (1H, m, CH); 13C NMR (101 MHz, CDCl3) δ = 174.9, 173.5, 148.4, 148.3, 131.4, 129.3, 129.0, 126.4, 125.8, 124.3, 111.0, 109.6, 76.0, 65.9, 56.5, 56.2, 56.0, 48.0, 23.4; HRMS (ES) Found: MH+, 381.1447. C21H21N2O5 requires MH+, 381.1450; Anal. Calcd for C21H20N2O5: C, 66.3; H, 5.3; N, 7.4. Found: C, 66.6; H, 5.1; N, 7.4.
Crystal data deposited at CCDC 1006041.
Unit cell parameters: a 10.6709(10), b 7.6760(7), c 22.1886(18), P21/c.
(8aR*,11aS*,11bR*)-2,3-Dimethoxy-10-methyl-5,8a,11a,11b-tetrahydropyrrolo[3′,4′:4,5][1,2]oxazolo[3,2-a]isoquinoline-9,11(6H,10H)-dione 28
Hydroxylamine hydrochloride (91 mg, 1.3 mmol) and N,N-diisopropylethylamine (0.45 mL, 2.6 mmol) were added to aldehyde 2514 (250 mg, 1.1 mmol) in PhMe (11 mL) and the mixture was heated to 60 °C. After 2.5 h, N-methylmaleimide (150 mg, 1.3 mmol) was added and the mixture was heated under reflux. After 1 h, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography, eluting with CH2Cl2–MeOH (99.5:0.5), gave the cycloadduct 28 (280 mg, 81%) as an amorphous solid; m.p. 154–156 °C (decomposed); Rf 0.7 [CH2Cl2–MeOH (98:2)]; νmax (film)/cm−1 2940, 1700, 1610; 1H NMR (400 MHz, CDCl3) δ = 6.84 (1H, s, CH), 6.60 (1H, s, CH), 4.86 (1H, d, J 7.5 Hz, CH), 4.75 (1H, br, CH), 3.93 (3H, s, CH3), 3.89 (3H, s, CH3), 3.73 (1H, dd, J 7.5, 2.5 Hz, CH), 3.60–3.57 (1H, m, CH), 3.24–3.17 (1H, m, CH), 3.12 (3H, s, CH3), 3.13–3.06 (1H, m, CH), 2.58–2.52 (1H, m, CH); 13C NMR (101 MHz, CDCl3) δ = 175.7, 174.7, 148.3, 148.2, 125.8, 124.3, 111.0, 109.5, 75.8, 65.2, 56.2, 55.9, 55.5, 47.9, 25.2, 23.0; HRMS (ES) Found: MH+, 319.1308. C16H19N2O5 requires MH+, 319.1294.
Crystal data deposited at CCDC 1006042.
Unit cell parameters: a 9.8243(10), b 8.6128(8), c 17.8328(18), P21/c.
(1S*,2S*,10bR*)-Methyl 2-hydroxy-3-oxo-1,2,3,4,5,10b-hexahydropyrrolo[2,1-a]isoquinoline-1-carboxylate 29
Zinc powder (56 mg, 0.85 mmol) was added to cycloadduct 22 (59 mg, 0.20 mmol) in AcOH/H2O (1.5 mL, 1:2). The mixture was heated to 70 °C for 1.5 h before being cooled to room temperature. The zinc salts were filtered, washed with CH2Cl2 (8 mL) and the solvent was evaporated. The solute was partitioned between aqueous ammonia (4 mL, 18 M) and CH2Cl2 (8 mL), and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The organic fractions were dried (MgSO4), filtered and evaporated to give the lactam 29 (39 mg, 75%) as needles; m.p. 181–184 °C, lit.15 m.p. 190–192 °C; Rf 0.4 [CH2Cl2–MeOH (95:5)]; νmax (film)/cm−1 3250, 2950, 1730, 1680; 1H NMR (400 MHz, CDCl3) δ = 7.27–7.15 (4H, m, 4 × CH), 5.66 (1H, br, OH), 5.48 (1H, d, J 8.0 Hz, CH), 4.61 (1H, d, J 7.5, CH), 4.26 (1H, ddd, J 13.0, 6.0, 3.0, CH), 3.90 (3H, s, CH3), 3.25–3.18 (2H, m, 2 × CH), 3.00–2.92 (1H, m, CH), 2.87–2.81 (1H, m, CH); 13C NMR (100 MHz, CDCl3) δ = 170.8, 170.1, 135.6, 133.4, 129.2, 127.4, 127.2, 125.2, 71.5, 56.5, 52.6, 52.1, 37.4, 28.5; HRMS (ES) Found MH+, 262.1075. C14H16NO4 requires MH+, 262.1079.
(1S*,2S*,10bR*)-Methyl 2-hydroxy-8,9-dimethoxy-3-oxo-1,2,3,4,5,10b-hexahydropyrrolo[2,1-a]isoquinoline-1-carboxylate 30
Zinc powder (145 mg, 2.2 mmol) was added to cycloadduct 26 (185 mg, 0.53 mmol) in AcOH/H2O (5 mL, 1:2) and the mixture was heated to 70 °C. After 1.5 h, the zinc salts were filtered, washing the filtrate with CH2Cl2 (15 mL) and the solvent was evaporated. The solute was partitioned between aqueous ammonia (5 mL) and CH2Cl2 (10 mL), and the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). The organic fractions were dried (MgSO4), filtered and evaporated. The residue was recrystallized with hot MeOH to give the lactam 30 (81 mg, 47%) as an amorphous solid; m.p. 190–192 °C; Rf 0.29 [CH2Cl2–MeOH (97:3)]; νmax (film)/cm−1 3300, 2990, 1730, 1665; 1H NMR (400 MHz, DMSO-d6) δ = 6.76 (1H, s, CH), 6.66 (1H, s, CH), 6.30 (1H, d, J 6.0 Hz, OH), 5.12 (1H, d, J 8.0 Hz, CH), 4.26 (1H, dd, J 7.0, 6.0 Hz, CH), 4.06–4.00 (1H, m, CH), 3.75 (3H, s, CH3), 3.72 (3H, s, CH3), 3.66 (3H, s, CH3), 3.17 (1H, dd, J 8.0, 7.0 Hz, CH), 3.07–2.98 (1H, m, CH), 2.70–2.68 (2H, m, 2 × CH); 13C NMR (101 MHz, DMSO-d6) δ = 171.0, 169.9, 148.2, 148.1, 128.2, 126.4, 112.8, 108.7, 71.5, 55.95, 55.9, 55.5, 53.0, 52.4, 36.9, 28.1; HRMS (ES) Found: MH+, 322.1301. C16H20NO6 requires MH+, 322.1291; Anal. Calcd for C16H19NO6: C, 59.8; H, 6.0; N, 4.4. Found: C, 60.1; H, 5.8; N, 4.3.
Acknowledgements
We acknowledge support from the University of Sheffield, the EPSRC, and the Thai Government. We are grateful to Alexandra Kirchner for related studies.Notes and references
- (a) R. Grashey, R. Huisgen and H. Leitermann, Tetrahedron Lett., 1960, 1, 9 CrossRef; (b) N. A. LeBel, M. E. Post and J. J. Whang, J. Am. Chem. Soc., 1964, 86, 3759 CrossRef CAS.
- For some reviews on nitrone cycloadditions, see: (a) J. Revuelta, S. Cicchi, A. Goti and A. Brandi, Synthesis, 2007, 485 CAS; (b) A. J. M. Burrell and I. Coldham, Curr. Org. Synth., 2010, 7, 312 CrossRef CAS.
- (a) R. Grigg, J. Markandu, S. Surendrakumar, M. Thornton-Pett and W. J. Warnock, Tetrahedron, 1992, 48, 10399 CrossRef CAS; (b) E. C. Davison, M. E. Fow, A. B. Holmes, S. D. Roughley, C. J. Smith, G. M. Williams, J. E. Davies, P. R. Raithby, J. P. Adams, I. T. Forbes, N. J. Press and M. J. Thompson, J. Chem. Soc., Perkin Trans. 1, 2002, 1494 RSC; (c) R. A. Stockman, A. Sinclair, L. G. Arini, P. Szeto and D. L. Hughes, J. Org. Chem., 2004, 69, 1598 CrossRef CAS PubMed; (d) M. S. Wilson and A. Padwa, J. Org. Chem., 2008, 73, 9601 CrossRef CAS PubMed; (e) A. C. Flick, M. J. A. Caballero and A. Padwa, Tetrahedron, 2010, 66, 3643 CrossRef CAS.
- (a) I. Coldham, A. J. M. Burrell, L. E. White, H. Adams and N. Oram, Angew. Chem., Int. Ed., 2007, 46, 6159 CrossRef CAS PubMed; (b) A. J. M. Burrell, I. Coldham, L. Watson, N. Oram, C. D. Pilgram and N. G. Martin, J. Org. Chem., 2009, 74, 2290 CrossRef CAS PubMed; (c) A. J. M. Burrell, I. Coldham and N. Oram, Org. Lett., 2009, 11, 1515 CrossRef CAS PubMed; (d) A. J. M. Burrell, L. Watson, N. G. Martin, N. Oram and I. Coldham, Org. Biomol. Chem., 2010, 8, 4530 RSC; (e) I. Coldham, A. J. M. Burrell, H. D. S. Guerrand and N. Oram, Org. Lett., 2011, 13, 1267 CrossRef CAS PubMed; (f) I. Coldham, L. Watson, H. Adams and N. G. Martin, J. Org. Chem., 2011, 76, 2360 CrossRef CAS PubMed; (g) I. Coldham, A. J. M. Burrell, L. Watson, N. Oram and N. G. Martin, Heterocycles, 2012, 84, 597 CrossRef CAS.
- For recent examples of cascade nitrone formation by cyclization then intramolecular cycloaddition, see: (a) X. Peng, B. M. K. Tong, H. Hirao and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 1959 CrossRef CAS PubMed; (b) C. V. Suneel Kumar and C. V. Ramana, Org. Lett., 2014, 16, 4766 CrossRef CAS PubMed.
- (a) I. Coldham, S. Jana, L. Watson and C. D. Pilgram, Tetrahedron Lett., 2008, 49, 5408 CrossRef CAS; (b) I. Coldham, S. Jana, L. Watson and N. G. Martin, Org. Biomol. Chem., 2009, 7, 1674 RSC.
- For a related cascade nitrone formation by cyclization then intermolecular cycloaddition, see: A. Siriwardena, D. P. Sonawane, O. P. Bande, P. R. Markad, S. Yonekawa, M. B. Tropak, S. Ghosh, B. A. Chopade, D. J. Mahuran and D. D. Dhavale, J. Org. Chem., 2014, 79, 4398 CrossRef CAS PubMed.
- For an alternative method, see: N. C. Deno, K. A. Eisenhardt, D. G. Pohl, H. J. Spinnelli and R. C. White, J. Org. Chem., 1974, 39, 520 CrossRef CAS.
- (a) T. Shono, Y. Matsumura and K. Inoue, J. Org. Chem., 1986, 51, 549 CrossRef CAS; (b) S. A. Ali and M. I. M. Wazeer, J. Chem. Soc., Perkin Trans. 1, 1988, 597 RSC.
- (a) J. J. Tufariello and J. P. Tette, J. Org. Chem., 1975, 40, 3866 CrossRef CAS PubMed; (b) G. Pandey, G. Kumaraswamy and A. Krishna, Tetrahedron Lett., 1987, 28, 2649 CrossRef CAS; (c) S. A. Ali, J. H. Khan, M. I. M. Wazeer and H. P. Perzanowski, Tetrahedron, 1989, 45, 5979 CrossRef CAS.
- M. Burdisso, A. Gamba, R. Gandolfi and P. Pevarello, Tetrahedron, 1987, 43, 1835 CrossRef CAS.
- S. E. Denmark and A. R. Hurd, J. Org. Chem., 1998, 63, 3045 CrossRef CAS.
- For other syntheses of macronecine and petasinecine, see: (a) A. J. Aasen and C. C. J. Culvenor, J. Org. Chem., 1969, 34, 4143 CrossRef CAS; (b) H. Rüeger and M. Benn, Heterocycles, 1983, 20, 235 CrossRef; (c) J. Mulzer and M. Shanyoor, Tetrahedron Lett., 1993, 35, 6545 CrossRef; (d) H. Ito, Y. Ikeuchi, T. Taguchi and Y. Hanzawa, J. Am. Chem. Soc., 1994, 116, 5469 CrossRef CAS; (e) D.-C. Ha, J.-B. Ahn and Y.-E. Kwon, Bull. Korean Chem. Soc., 1998, 19, 514 CAS; (f) D. Ma and J. Zhang, J. Chem. Soc., Perkin Trans. 1, 1999, 1703 RSC; (g) T. K. Sarkar, A. Hazra, P. Gangopadhyay, N. Panda, Z. Slanina, C.-C. Lin and H.-T. Chen, Tetrahedron, 2005, 61, 1155 CrossRef CAS; (h) T. Sengoku, T. Suzuki, T. Kakimoto, M. Takahashi and H. Yoda, Tetrahedron, 2009, 65, 2415 CrossRef CAS.
- (a) M. Yamato, K. Hashigaki, N. Qais and S. Ishikawa, Tetrahedron, 1990, 46, 5909 CrossRef CAS; (b) P. C. B. Page, G. A. Rassias, D. Barros, A. Ardakani, B. Buckley, D. Bethell, T. A. D. Smith and A. M. Z. Slawin, J. Org. Chem., 2001, 66, 6926 CrossRef CAS PubMed.
- (a) M. Burdisso, A. Gamba and R. Gandolfi, Tetrahedron, 1988, 44, 3735 CrossRef CAS. See also: (b) R. Huisgen, H. Hauck, R. Grashey and H. Seidl, Chem. Ber., 1969, 102, 736 CrossRef CAS.
- K. M. Laemmerhold and B. Breit, Angew. Chem., Int. Ed., 2010, 49, 2367 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1006037–1006042, 1022997 and 1051097. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01871h |
| This journal is © The Royal Society of Chemistry 2016 |