
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and biocatalytic scope of thermophilic flavin-dependent halogenase and flavin reductase enzymes†
Binuraj R. K.
Menon
,
Jonathan
Latham
,
Mark S.
Dunstan
,
Eileen
Brandenburger
,
Ulrike
Klemstein
,
David
Leys
,
Chinnan
Karthikeyan
,
Michael F.
Greaney
,
Sarah A.
Shepherd
and
Jason
Micklefield
*
School of Chemistry and Manchester Institute of Biotechnology, The University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: jason.micklefield@manchester.ac.uk
First published on 6th September 2016
Abstract
Flavin-dependent halogenase (Fl-Hal) enzymes have been shown to halogenate a range of synthetic as well as natural aromatic compounds. The exquisite regioselectively of Fl-Hal enzymes can provide halogenated building blocks which are inaccessible using standard halogenation chemistries. Consequently, Fl-Hal are potentially useful biocatalysts for the chemoenzymatic synthesis of pharmaceuticals and other valuable products, which are derived from haloaromatic precursors. However, the application of Fl-Hal enzymes, in vitro, has been hampered by their poor catalytic activity and lack of stability. To overcome these issues, we identified a thermophilic tryptophan halogenase (Th-Hal), which has significantly improved catalytic activity and stability, compared with other Fl-Hal characterised to date. When used in combination with a thermostable flavin reductase, Th-Hal can efficiently halogenate a number of aromatic substrates. X-ray crystal structures of Th-Hal, and the reductase partner (Th-Fre), provide insights into the factors that contribute to enzyme stability, which could guide the discovery and engineering of more robust and productive halogenase biocatalysts.
Introduction
Halogen substituents are present in a high proportion of the leading pharmaceuticals, as well as many agrochemicals, polymers and other useful synthetic materials.1–3 Moreover, organohalogens are frequently used as intermediates in synthesis, with haloaromatic compounds serving as the key components for a wide range of transition metal catalysed cross-coupling reactions.4–8 Traditional halogenation chemistries often utilise toxic reagents, employing reaction conditions that are becoming increasingly unsustainable, and often lack regioselectivity with aromatic substrates.9–13 Consequently, there has been an interest in developing enzymes that can utilise benign inorganic halides in water to deliver haloaromatic compounds.4–16Nature has evolved halogenase enzymes for the biosynthesis of a diverse array of halogenated natural products including many antibiotics, antitumor agents and other bioactive compounds, with the halogen substituents often contributing to the biological activity of these secondary metabolites.17–20 The Flavin-dependent halogenase (Fl-Hal) enzymes, which typically halogenate aromatic precursors, are particularly common in secondary metabolism. Fl-Hals require an NADPH-dependent flavin reductase enzyme that can reduce flavin-adenine dinucleotide (FAD) to FADH2, which is utilised to oxidise inorganic chloride or bromide to hypohalous acid.21 X-ray crystal structures of the flavin-dependent tryptophan halogenases,22,23 suggest that hypohalous acid is then channelled through an intra-enzymatic tunnel to the substrate binding site, where an active site lysine positions this electrophile in a spatially defined orientation to the aromatic substrate, either through formation of an intermediate chloramine or hydrogen bonding, effectively controlling the regioselectivity.22,24 Fl-Hal have emerged as promising enzymes for use as biocatalysts due to their potential to regioselectively chlorinate or brominate synthetic, as well as natural, aromatic compounds that can also be further derivatised using cross-coupling chemistry.4–8,25–27
The potential of Fl-Hals is best exemplified by the tryptophan 5-, 6- and 7-halogenases that can catalyse the regiodivergent halogenation of the C5, C6 or C7 positions of the substrate indole moiety, which is beyond the scope of current non-enzymatic halogenation methods. The tryptophan halogenases have been shown to exhibit some substrate promiscuity and accept a number of non-native electron rich aromatic substrates. Targeted and random mutagenesis strategies have also been employed to further broaden the substrate scope and switch the regioselectivities of the tryptophan halogenases.28–31 In addition, we recently showed how tryptophan halogenases and other Fl-Hal enzymes can be integrated with palladium-catalyzed cross-coupling chemistry, in one-pot reactions, to affect the direct regioselective arylation or alkenylation of C–H positions in various aromatic scaffolds;32 such transformations are inaccessible using stand-alone chemocatalysis or biocatalysis and further demonstrate the potential of Fl-Hal in synthetic applications (Fig. 1).
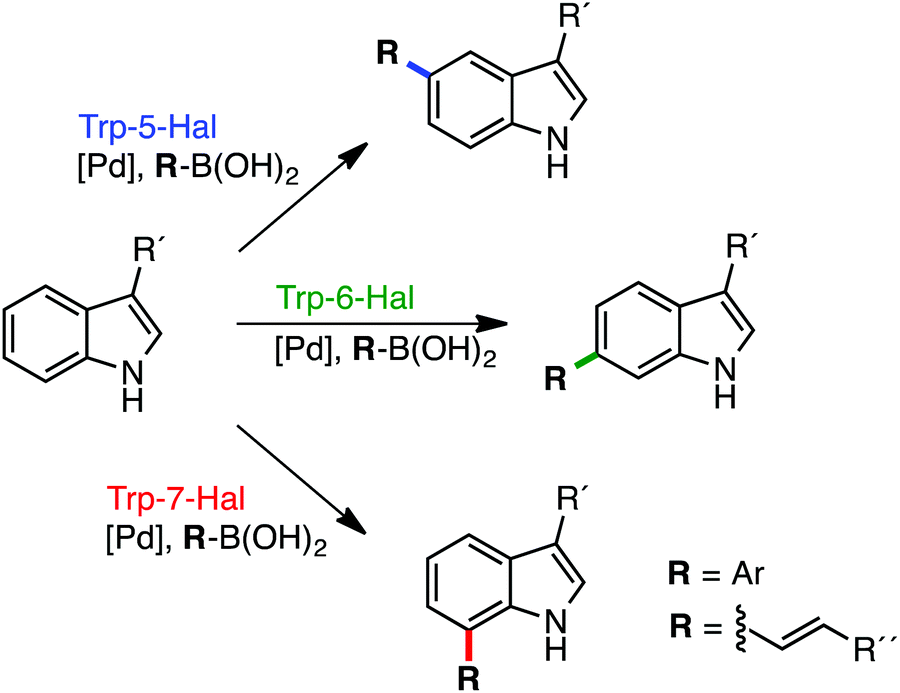 | ||
| Fig. 1 Regiodivergent functionalisation of aromatic scaffolds using Fl-Hal C–H activation. Tryptophan 5-, 6- & 7-halogenases and other Fl-Hal enzymes have been integrated with Pd-catalysis to deliver functionally diverse arylated and alkenylated products, in one-pot reactions, under mild, aqueous conditions, via aryl bromide intermediate formed in situ.32 | ||
Despite their promise, the synthetic utility of Fl-Hal enzymes is limited by their relatively low activity and poor stability.30,33 In common with other enzymes from secondary metabolism, Fl-Hal exhibit lower catalytic efficiencies than many of the enzymes from primary metabolism, which have presumably been under greater evolutionary pressure.34 Thus, while Fl-Hal have been used in vivo to produce large quantities of halogenated natural products including antibiotics, such as vancomycin and griseovulfin,35–38 their low activity and instability currently precludes their deployment in vitro for larger scale synthetic applications. To address the low productivity of Fl-Hal, cross-linked enzyme aggregates (CLEAS) containing Fl-Hal have been developed,39 which have enhanced stability and can be recycled to increase the scale of biohalogenation reactions.31,32,39 It has also been demonstrated that increasing the thermal stability of Fl-Hal by directed evolution could be a way to improve the limited product yield and catalytic lifetime of these enzymes.33 However, this approach is usually time consuming and requires multiple rounds of mutagenesis to achieve the desired levels of improvement. In this paper, we take the alternative strategy of exploring natural biosynthetic pathways in thermophilic bacteria, to identify more robust and productive variants of Fl-Hal. Thermostable enzyme variants have been shown to possess many advantages, including prolonged catalyst life, as well as increased tolerance to proteolysis and organic solvents;33,40–43 all of which would make these enzymes better candidates for biocatalysis and further engineering. In addition, a thermostable Fl-Hal would be more compatible with chemocatalysts that often require elevated temperatures, which could enable development of more integrated one-pot chemobio-transformations.32
Results and discussion
Substrate scope and regioselectivity of a thermophilic halogenase
BLAST analysis of published genome sequences revealed the presence of putative Fl-Hal encoding gene from a thermophilic halotolerant Streptomyces violaceusniger strain SPC6 (Table S1†).44,45 The putative halogenase (Th-Hal) shows similarity to tryptophan halogenases (Fig. S1†), and is present within an uncharacterised biosynthetic gene cluster that includes gene(s) encoding a hybrid nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) assembly line. To explore the properties of Th-Hal, the protein was overproduced in E. coli. In addition, a thermophilic flavin reductase (Th-Fre) enzyme from Bacillus subtilis WU-S2B, which had been identified previously,46,47 was also overproduced in E. coli to provide a potentially thermostable halogenase system.The substrate specificity of Th-Hal was tested with tryptophan (1) and a series of related compounds including 1-methyltryptophan (2), 5-hydroxytryptophan (3), kynurenine (4), anthranilic acid (5) and anthranilamide (6) (Fig. 2). With tryptophan, halogenation occurs solely at the C6 position indicating that Th-Hal is a tryptophan 6-halogenase enzyme, similar to SttH from Streptomyces toxytricini and KtzR from Kutzneride biosynthetic pathways.48,49 Halogenation of 1-methyltryptophan (2) also occurred at the C6 position, whilst 5-hydroxytryptophan (3) was halogenated at C4, suggesting a different mode of substrate binding.50 The non-indolic substrates (4–6) are all halogenated at the more chemically activated C5 position. Overall, this reveals that Th-Hal shows similar substrate specificity and regioselectivity to the SttH halogenase which we explored previously.31
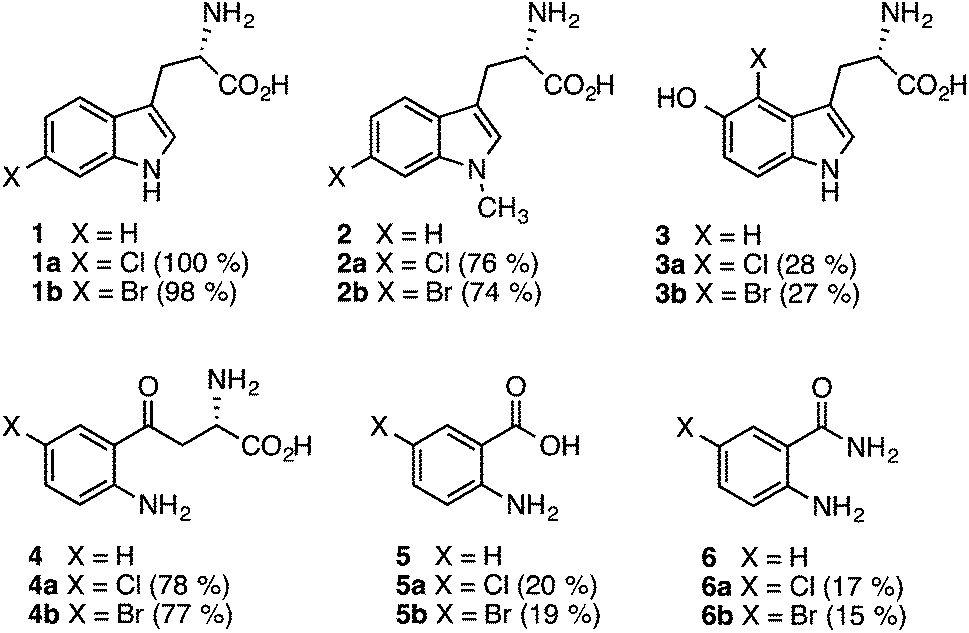 | ||
| Fig. 2 Substrates accepted by Th-Hal and products obtained. HPLC conversations are shown in brackets. Reaction conditions: Th-Hal (2.5 μM), E. coli flavin reductase (Fre) (1 μM), FAD (1 μM), NADH (2.5 mM), MgCl2 (10 mM) and substrate (0.5 mM) in 10 mM potassium phosphate buffer, pH 7.4, Reactions (2–6) were measured in HPLC after 3 hours incubation at 30 °C and 200 rpm. Reactions with tryptophan (1) were complete after incubation for 1 hour. | ||
As anticipated, tryptophan is the best substrate for Th-Hal affording quantitative chlorination and bromination (Fig. 2). Activity of Th-Hal was reduced upon introduction of a methyl group on to the substrate indole nitrogen (2), presumably through the loss of a hydrogen bonding contact to indole moiety. Activity is further decreased when an hydroxyl group is introduced into the substrate adjacent to the halogenation position (3). Of the non-indolic substrates tested kynurenine (4) is preferred over anthranilic acid (5) and anthranilamide (6), which gave only modest conversions over the same time period. In all cases, conversions for bromination reactions were comparable with those for chlorination (Fig. 2).
Activity and stability of the thermophilic halogenase and reductase
The kinetic parameters of the thermophilic halogenase enzyme, with the natural tryptophan substrate, were compared with related Fl-Hal including the tryptophan 5-halogenase PyrH, tryptophan 6-halogenases SttH and KtzR along with tryptophan 7-halogenases PrnA and RebH which are all derived from mesophilic bacteria (Table 1). By selecting halogenases with different regioselectivities, a better comparison of kinetic parameters for Fl-Hal in this class could be obtained. The kcat values of Th-Hal with tryptophan was found to be 4.3 min−1 indicating that this enzyme is more active towards tryptophan than any of the other halogenases tested at 30 °C. Th-Hal was found to be ca. 2 times faster than PyrH, which is the most efficient of the other Fl-Hal tested here, with similar substrate binding (Km) values. Th-Hal exhibits both improved catalytic turnover and binding parameters, compared with the remaining Fl-Hal tested, resulting in between 5- to 35-fold higher catalytic efficiency (kcat/Km). Though Th_hal showed a higher catalytic turnover value at 45 °C, it was found that catalytic efficiency decreases with increase in temperature mainly due to poor binding of tryptophan at higher temperature (Table 1).| Enzyme | k cat (min−1) | K M (μM) | k cat/KM |
|---|---|---|---|
| Th-Hal (30 °C) | 4.3 ± 0.5 | 12.2 ± 1.8 | 0.35 ± 0.07 |
| Th-Hal (45 °C) | 5.1 ± 0.4 | 20.4 ± 1.3 | 0.25 ± 0.03 |
| PyrH | 2.4 ± 0.4 | 15.2 ± 4.2 | 0.16 ± 0.05 |
| SttH | 1.7 ± 0.1 | 25.3 ± 3.2 | 0.07 ± 0.01 |
| PrnA | 1.1 ± 0.1 | 20.7 ± 0.1 | 0.05 ± 0.005 |
| KtzR | 0.4 ± 0.1 | 34.1 ± 2.1 | 0.01 ± 0.003 |
| RebH | 0.6 ± 0.1 | 28.7 ± 1.3 | 0.02 ± 0.004 |
In order to explore the feasibility of establishing a thermophilic biocatalytic halogenation system, melting temperatures of individual halogenase enzymes and flavin reductase enzymes were determined using Circular Dichroism (CD) spectroscopy (Fig. 3, S2 & S3†). Comparison of the Th-Hal melting temperature with that of PyrH, SttH, PrnA, KtzR and RebH revealed that the Th-Hal has a higher thermal stability, with a melting temperature (47.8 °C) that is approximately 10 °C above that of the other halogenase enzymes. The melting temperature of the thermophilic flavin reductase (Th-Fre) from Bacillus subtilis WU-S2 was also compared with two reductase partners from E. coli which are commonly used with Fl-Hal in vitro, the NAD(P)H-flavin reductase Fre and the NAD(P)H-dependent FMN reductase SsuE.51,52 This showed that the Th-Fre has a melting temperature of 49.0 °C which was 9 °C above that of the E. coli Fre and 15.4 °C above that for SsuE, indicating that Th-Fre has a higher thermostability, as previously reported,46,47 and should be more compatible with Th-Hal in high temperature biocatalytic halogenation reactions.
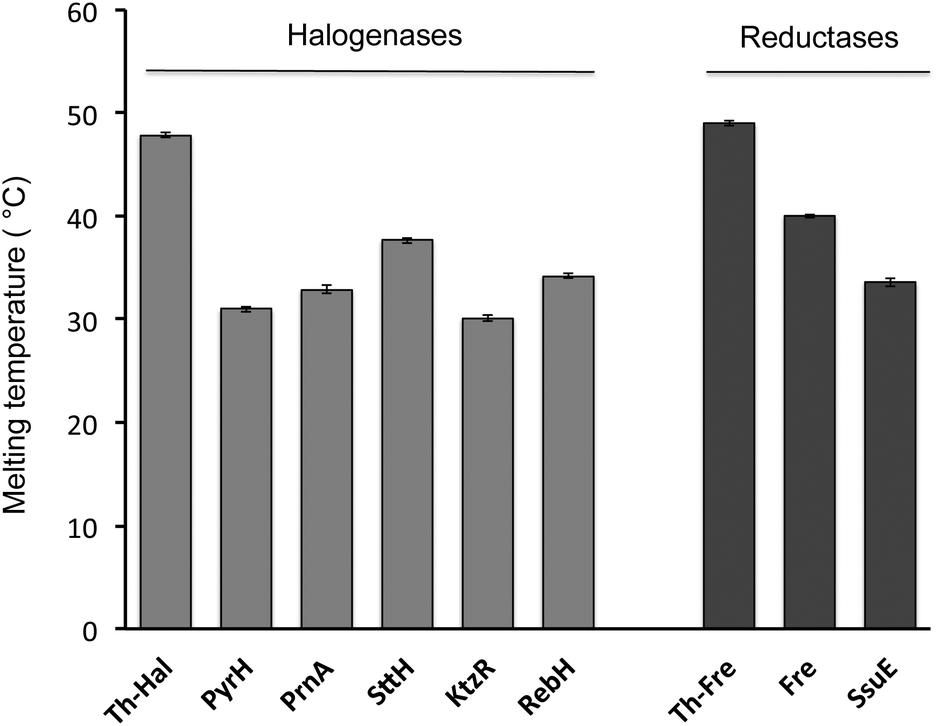 | ||
| Fig. 3 Thermal stability of halogenase and flavin reductase enzymes determined by CD. The CD spectrum was measured every 2 °C, from 20 °C up to 80 °C with 10 μM enzyme in a 0.5 mm path length cuvette at fixed temperature for 2 minute with the temperature ramp of 0.2 °C per minute. The absorbance changes at 222 nm was plotted against temperature, and fitted with a nonlinear curve fitting function to determine protein melting temperatures. | ||
The kinetic parameters and co-factor specificities of the flavin reductase enzymes were determined, showing that Th-Fre exhibited a higher flavin reduction rate and reactivity towards both FMN and FAD in the presence of NADH compared to Fre and SsuE (Tables 2, S2 & S3†). The KM values indicated that Th-Fre has a higher preference and binding affinity towards FMN (KM = 2.5 μM) over FAD (16.3 μM). The kinetic parameters for flavin reductase enzymes with FMN and NADH were also obtained at 30 °C and 45 °C (Table 2), showing that Th-Fre has a higher activity and stability over the E. coli flavin reductase enzymes and confirmed Th-Fre to be suitable partner for Th-Hal or other Fl-Hal in higher temperature reactions.
| Enzyme | Temp. | k cat (s−1) | K M (μM) | k cat/KM |
|---|---|---|---|---|
| Th-Fre | 30 °C | 16.0 ± 1.0 | 2.5 ± 0.3 | 6.4 ± 0.9 |
| 45 °C | 26.0 ± 2.0 | 2.8 ± 0.2 | 9.3 ± 1.0 | |
| SsuE | 30 °C | 8.1 ± 0.20 | 8.3 ± 0.1 | 0.98 ± 0.03 |
| 45 °C | 1.9 ± 0.02 | 10.2 ± 0.4 | 0.02 ± 0.002 | |
| Fre | 30 °C | 4.3 ± 0.4 | 14.7 ± 0.3 | 0.29 ± 0.03 |
| 45 °C | 0.3 ± 0.03 | 16.2 ± 0.5 | 0.02 ± 0.002 |
The activity of the Th-Hal, was next compared the other mesophilic tryptophan halogenases (PyrH, SttH, PrnA, KtzR and RebH), in combination with the thermostable reductase, Th-Fre, at two different temperatures. This showed (Fig. 4) that Th-Hal/Th-Fre affords slightly higher conversion of tryptophan to 6-chlorotryptophan at 45 °C (74%) than at 30 °C (69%) over 30 min time period of the assay (Fig. 4). In contrast the mesophilic halogenases all gave significantly lower conversions to the corresponding chlorinated tryptophan derivatives, particularly at the higher temperature. In addition, the other substrates (2–6) were tested with Th-Hal/Th-Fre at different temperatures, revealing that improved conversions to chlorinated products (2a–6a) could be obtained at 45 °C compared with the normal operating temperature (Fig. S4†). The halogenation efficiency of Th_hal was then tested with the addition of different solvents to understand the solvent tolerance (Fig. S5†). Th_hal retained significant activity in different polar solvents and the activity pattern showed that Th_hal is more tolerant and stable towards the solvents tested than its mesophilic counterpart SttH. Both of these enzymes however showed a reduced activity in non-polar solvents such as dichloromethane and acetonitrile. Taken together these results indicate that Th-Hal in combination with Th-Fre, provides a more efficient, stable and robust halogenase system, than the previously characterised tryptophan halogenase, for use in vitro with both the natural and non-native substrates.
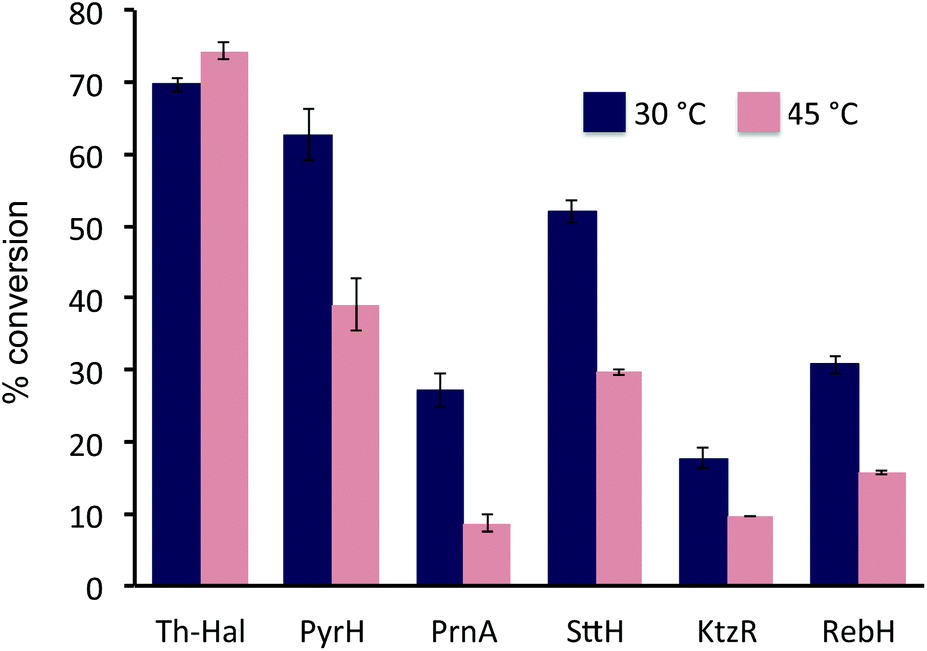 | ||
| Fig. 4 Effects of temperature on % chlorination of tryptophan with halogenase enzymes and Th-Fre. Reaction conditions: halogenase enzyme (2.5 μM), Th-Fre (1 μM), FAD (1 μM), NADH (2.5 mM), MgCl2 (10 mM) and tryptophan (0.5 mM) in 10 mM potassium phosphate buffer, pH 7.4, Reactions were measured by HPLC after 30 minutes incubation at 30 °C (or 45 °C) and 200 rpm. | ||
Crystal structure and amino acid variability of Th-Hal
An X-ray crystal structure of apo Th-Hal was obtained at 2.33 Å using tryptophan 7-halogenase (PrnA, PDB 2AQJ) as the molecular replacement model (Fig. 5, Table S4†). Th-Hal, shares 76.2% sequence identity with SttH the structure of which we had determined previously.31 The crystal structure indicates that Th-Hal forms a dimer, similar to other Fl-Hal, which was also observed in gel filtration chromatography.48,49 The overall structure of Th-Hal is similar to other tryptophan halogenases – a single-domain protein in which several helices and two large β-sheets form a “box” and a triangular pyramid dominated by helixes. The secondary structural arrangements of other tryptophan halogenase enzymes are conserved in Th-Hal, despite the fact that Th-Hal sequences has a divergence from PyrH and PrnA sequences reflecting the individual regioselectivity (Fig. 5B). The catalytic lysine (K75) and glutamate (E359) residues of Th-Hal align well with those in SttH (K79, E363), PyrH (K75, E354) and PrnA (K79, E346), indicating that the alterations in tryptophan-binding mode leads to different regioselectivity of these enzymes (Fig. 5B).22,31,48,49 The crystal structures of other tryptophan halogenase enzymes shows that substrate binding in the active site is such that the chlorination site (C6 of tryptophan in Th-Hal) is always directed towards the reactive chlorinating species controlled by the catalytic lysine.22,31,48,49,52 A possible tryptophan bound model for the resolved Th-Hal structure could therefore be predicted (Fig. 5B) using this fact.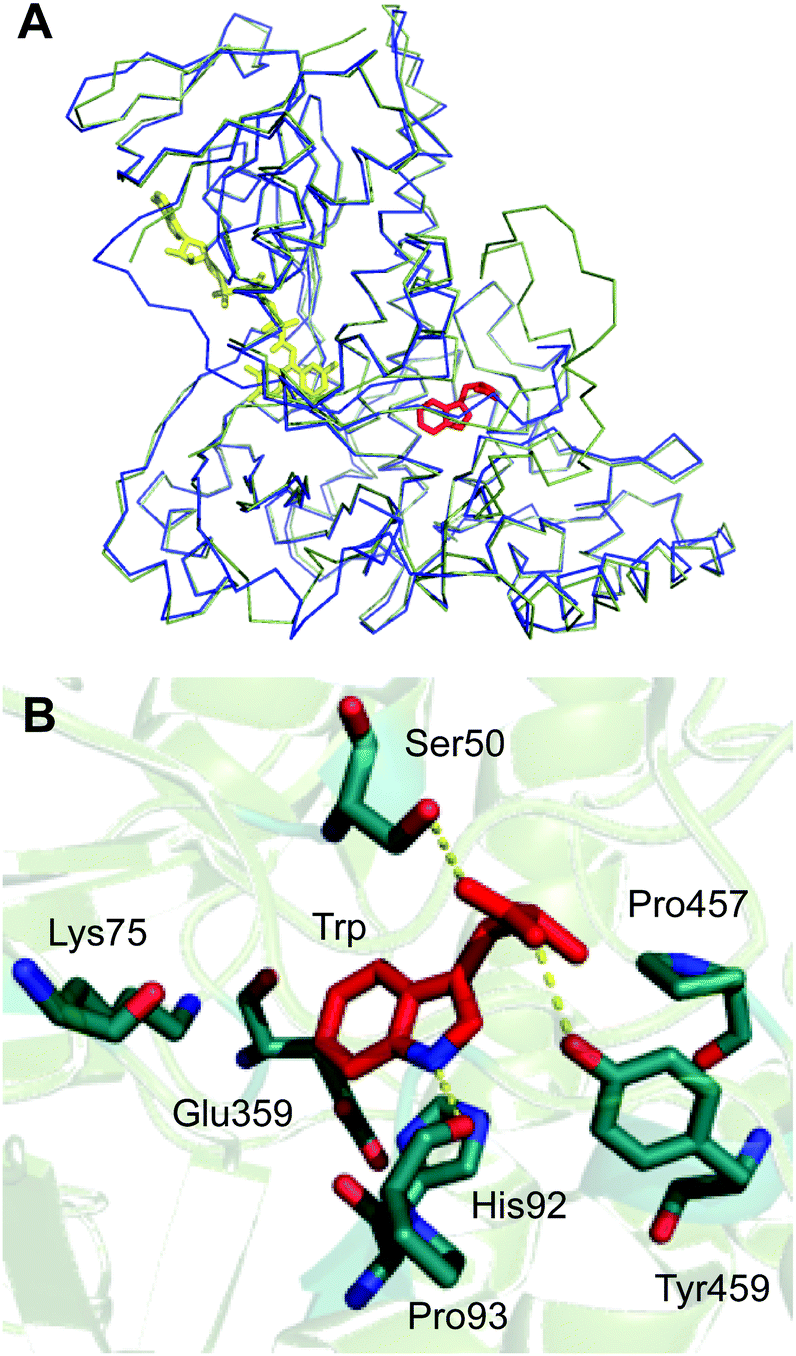 | ||
| Fig. 5 Crystal structure of thermophilic tryptophan halogenase (Th-Hal) enzyme. (A) Structural alignment of Th-Hal (green ribbon) with SttH (blue ribbon). (B) Active site residues and the predicted tryptophan binding mode in the active site of Th-Hal. | ||
Similar to the PyrH, PrnA and SttH structures, the benzene portion of the tryptophan indole ring is sandwiched between the π systems of H92 and F94 residues. The indole nitrogen is hydrogen bonded to the carbonyl of P93, as in PyrH, whereas the hydroxyl of Y454 and S50 could form hydrogen bonding interactions with the tryptophan carboxyl group. Residues L455, P456 and P457 in Th-Hal, which are comparable to residues L460, P461 and P462 in SttH, are also in a close proximity to the tryptophan-binding site and are oriented similar to that in SttH. The loop region involving these residues has been previously shown to be important in contributing towards the regioselectivity of SttH and PrnA.31,48,53,54 The residues near to and part of the FAD binding strap region (A41 to E46 in Th-Hal, T45 to E50 in SttH) is completely disordered in apo Th-Hal structure as the monomer structures are without FAD bound.31 The inherent structural flexibility of this region was reported previously as the FAD binding region is solvent exposed in tryptophan halogenases.53,54 Other than this, the secondary structural arrangements of FAD binding modules of Th-Hal are very similar to that of SttH. The signature motifs of the flavin dependent halogenases GxGxxG (residues 9–14, FAD consensus sequence) and WxWxIP (residues 284–289, to prevent monoxygenase activity) that are located in the FAD module are also structurally conserved in Th-Hal and in SttH.
The lower thermal stability of SttH means that the structural and sequence comparison between Th-Hal with SttH could shed more light on the structural features that are responsible for the increased thermal stability and reactivity in Th-Hal. Conversely, this information could also provide insight into the poor catalytic efficiency and stability of the tryptophan halogenase family. The amino acid variability in Th-Hal was analysed against SttH. Sequence alignment with SttH showed that the sequence variations are present in the entire length of protein (Fig. S8†). However, when these changes were marked in the crystal structure of Th-Hal; the identified pattern showed that most of the variations are at surface exposed residues (Fig. 6). Supporting this observation, there were no mutations at the dimeric interface region. About 40% of these changes were to introduce polar residues on the Th-Hal surface regions. It was noted before that introducing surface polar residues could influence surface hydrogen bonds and thus increase the stability of these proteins by inhibiting aggregation.55–58 As more diverse chemical substitutions are easily incorporated in surface residues rather than the catalytic inner core residues of proteins during evolution, it is possible that nature has adopted this strategy to produce a more active and thermally stable halogenase enzyme.55–58
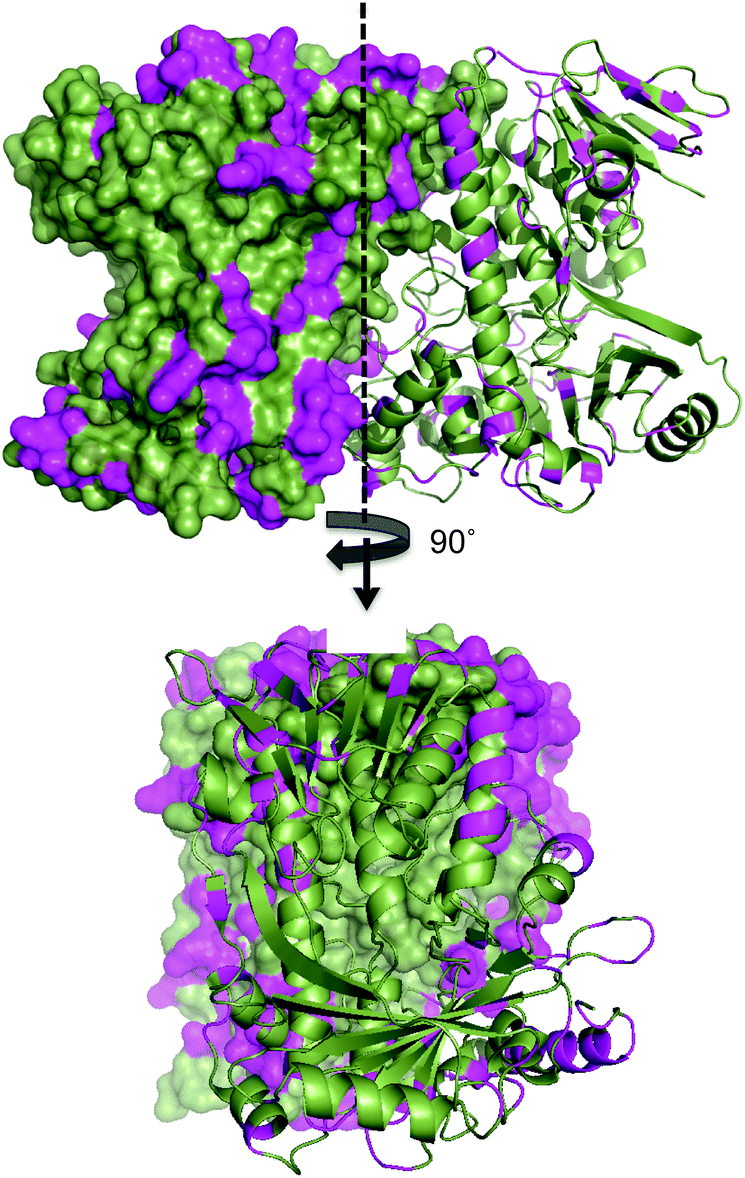 | ||
| Fig. 6 Surface map structure of Th-Hal (PDB 5LV9) showing its amino acid variation from SttH structure. One monomeric unit is shown as surface map format while the other unit as a cartoon. The sequence variation is shown in magenta colour. The Th-Hal dimeric interface (after 90° rotation) is also shown for clarity. | ||
Crystal structure of Th-Fre
The crystal structure of Th-Fre was resolved at 2.53 Å using the general stress protein 14 (PDB 3F2 V) as the molecular replacement model (Fig. 7, Table S4†). Th-Fre is a homodimer in the asymmetric unit and both monomeric units contained FMN that was incorporated into the protein from E. coli cells during recombinant expression. Each monomeric unit comprises of a classical flavodoxin_2 domain formed from five β-sheets that connected with loops and helixes. FMN is bound at the apex of this flavodoxin_2 domain and in the asymmetric unit monomers are arranged in antiparallel directions. The planar stacking interactions with the FMN isoalloxazine ring are mainly from the phenyl ring of Tyr67. Along with this two additional aromatic residues Trp68 and Tyr69 are thought to be involved in substrate stacking and associated dynamics. Tyr114 forms a hydrogen bonding interaction with isoalloxazine ketonic group at position 2. His9, Val16, Val16 and Asn18 residues are in direct contact with phosphate tail of FMN (Fig. 7).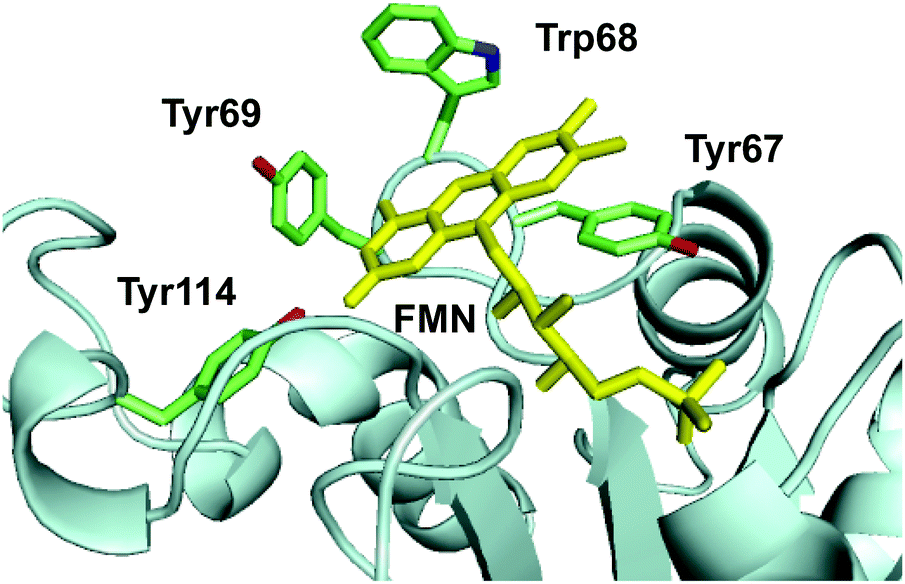 | ||
| Fig. 7 The active site structure of thermophilic flavin reductase (Th-Fre) enzyme (PDB 5LVA). The aromatic core formed by residues Tyr67, Trp68, Tyr69, Tyr114 in the active site around FMN cofactor of Th-Fre is shown. FMN is shown as yellow sticks. | ||
The resolved structure showed that Th-Fre is structurally similar to quinone reductase 2 enzymes (Fig. 8). In this category the closest structurally identical protein is NADPH-quinone reductase from P. pentosaceus (PDB 3HA2). Quinone reductase 2 enzymes are one of known human targets for antimalarial drugs, primaquine and chloroquine.59,60 The most well-characterised quinone reductase 2 enzyme is human quinone reductase 2 (Nqo2) (PDB 2FGL, 2FGI), which is unique in that it uses dihydronicotinamide riboside (NRH) as a reducing coenzyme rather than NADH or NADPH.59,60 The structural and sequence alignment of Th-Fre with human quinone reductase 2 shows that Th-Fre is a shorter protein and there is a 30 amino acid insertion at the N terminal of human quinone reductase 2 that forms an extended helix above the flavin domain of opposite monomer (Fig. 8, S9 and S10†). Whether this extended region in human quinone reductase 2 provide specific interactions for NRH binding and π stacking over flavin isoalloxazine ring is not clear. The mesophilic flavin reductase enzymes, Fre and SsuE from E. coli, are not homologous to Th-Fre (25.2% and 22.4% sequence identitity with Fre and SsuE respectively). Crystal structure of Fre (PDB 1QFJ) also showed that Th-fre is structurally different to Th-Fre making it difficult to plot the sequence variability of other known flavin reductase enzymes on to Th-Fre for comparison.
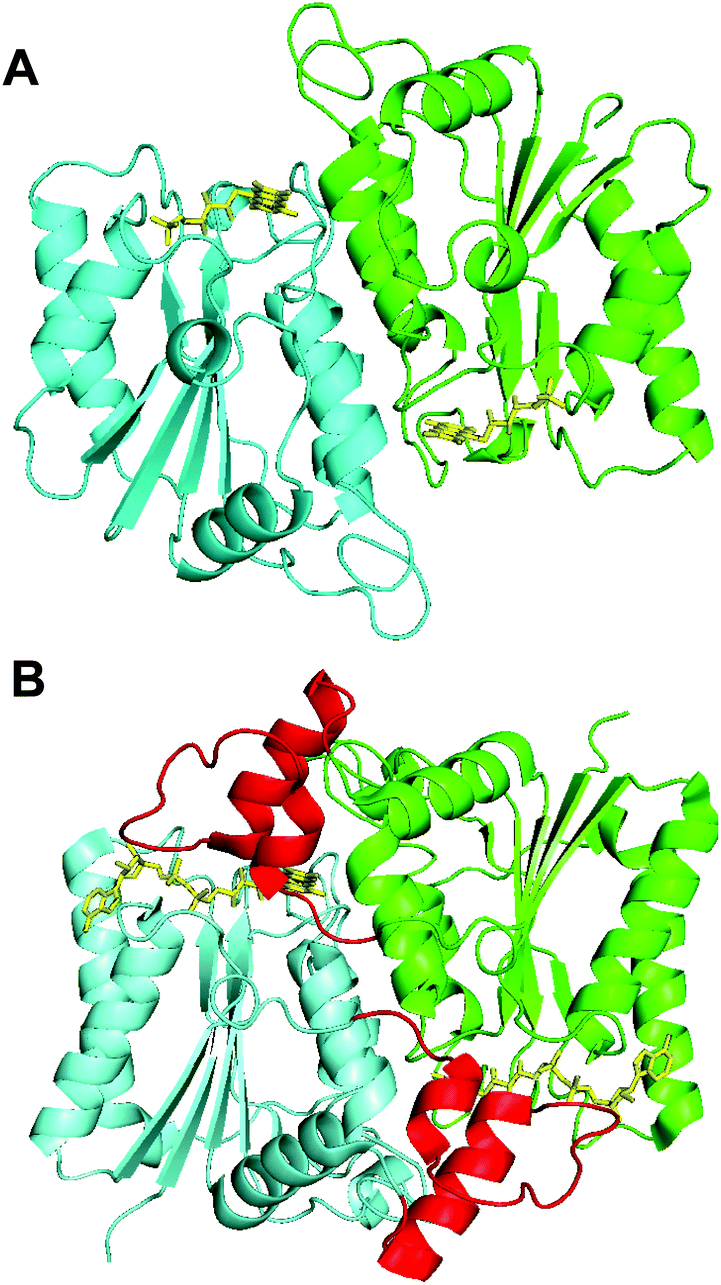 | ||
| Fig. 8 Crystal structures of Th-Fre and a quinone reductase 2 enzyme. The structural comparison between Th-Fre (A) and human quinone reductase 2 enzyme Nqo2 (B) are also shown. The monomeric units in each protein are shown in different colours (Cyan and Green) while flavin cofactor as yellow sticks. The extra amino acid region at the N terminal of human quinone reductase 2 that forms an extended helix above the flavin domain of opposite monomer is shown in red colour (PDB 2FGL, 2FGI). | ||
Conclusions
In nature, there are many flavin dependent halogenases (Fl-Hal) encoded within various microbial biosynthetic gene clusters, which are involved in the biosynthesis of medicinally important halogenated secondary metabolites. Whilst biosynthetic pathways incorporating these Fl-Hal have been used to produce chlorinated antibiotics on an industrial scale, through fermentation,35–38 the deployment of these enzymes in vitro for chemoenzymatic synthesis remains a major challenge. The exploitation of Fl-Hal for large-scale chemo-enzymatic synthesis of pharmaceuticals and other valuable chemicals is encumbered by the low activity, stability and conversion rates that these enzymes exhibit.In order to generate more robust and efficient Fl-Hal biocatalysts, we set out to find and characterise thermally stable variants of Fl-Hal. Our search led to the identification of a thermostable tryptophan halogenase (Th-Hal) from a thermophilic and halotolerant strain of Streptomyces. In order to develop this enzyme for direct biocatalytic applications, we have combined it with a thermophilic flavin reductase (Th-Fre) enzyme from Bacillus. Kinetic characterisation and thermal stability assays showed that these enzymes have a higher stability and reaction rate compared to the other halogenases and reductases that have previously been utilised for in vitro biohalogenation. The crystal structures of both of these enzymes are also reported here. Th-Hal showed a similar overall structure to SttH, a mesophilic Fl-Hal with similar substrate scope and regioselectivity. Notably, however, the structural comparisons revealed that there are significant variations of amino acids residues on the surface of the proteins, with Th-Hal possessing many more polar surface residues than SttH, which could influence surface hydrogen bonding, reduce aggregation, and contribute to the higher stability of Th-Hal. These insights could be used in genome mining for more stable, naturally evolved, Fl-Hal and could also provide guidance for the rational engineering of more effective halogenase enzymes. Establishing structure–activity relationships for Fl-Hal from thermophilic vs. mesophilic environments, could enable targeted mutagenesis approaches to improve the properties of the many other less productive Fl-Hal, from mesophilic microorganisms. Finally, the availability of more stable enzymes could allow for the more effective integration of halogenase biocatalysis with chemocatalytic cross-coupling reactions for scaffold diversification and synthesis of high value targets.
Acknowledgements
We acknowledge BBSRC (grant BB/K00199X/1), GlaxoSmithKline and CoEBio3 for financial support. We acknowledge Diamond Light Source for time on beamlines.Notes and references
- M. C. Ford and P. S. Ho, J. Med. Chem., 2016, 59, 1655 CrossRef CAS PubMed.
- P. Jeschke, Pest Manage. Sci., 2010, 66, 10 CrossRef CAS PubMed.
- M. Hernandes, S. Cavalcanti, D. Moreira, W. Azevedo and C. Leite, Curr. Drug Targets, 2010, 11, 303 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed.
- C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS PubMed.
- F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- Z. Zou, D. T. Ahneman, L. Chi, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef PubMed.
- M. C. White, Synlett, 2012, 2746 CAS.
- G. K. Dewkar, S. V. Narina and A. Sudalai, Org. Lett., 2003, 5, 4501 CrossRef CAS PubMed.
- D. C. Braddock, G. Cansell and S. A. Hermitage, Synlett, 2004, 461 CrossRef CAS.
- G. K. S. Prakash, T. Matthew, D. Hoole, P. M. Esteeves, Q. Wang, G. Rasul and G. A. Olah, J. Am. Chem. Soc., 2004, 126, 15770 CrossRef CAS PubMed.
- Y. Zhang, K. Shibatomi and H. Yamamoto, Synlett, 2005, 2837 CAS.
- S. Brown and S. E. O'Connor, ChemBioChem, 2015, 16, 2129 CrossRef CAS PubMed.
- V. Weichold, D. Milbredt and K.-H. Van Pée, Angew. Chem., Int. Ed., 2016, 55, 6374 CrossRef CAS PubMed.
- J. T. Payne and J. C. Lewis, Synpacts, 2014, 25, 1345 Search PubMed.
- F. H. E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364 CrossRef PubMed.
- C. S. Neumann, D. G. Fujimori and C. T. Walsh, Chem. Biol., 2008, 15, 99 CrossRef CAS PubMed.
- A. Butler and M. Sandy, Nature, 2009, 460, 848 CrossRef CAS PubMed.
- K.-H. Van Pée, C. Dong, S. Flecks, J. Naismith, E. P. Patallo and T. Wage, Adv. Appl. Microbiol., 2006, 59, 127 Search PubMed.
- E. Yeh, L. J. Cole, E. W. Barr, J. M. Bollinger, D. P. Ballou and C. T. Walsh, Biochemistry, 2006, 45, 7904 CrossRef CAS PubMed.
- C. J. Dong, S. Flecks, S. Unversucht, C. Haupt, K.-H. Van Pée and J. H. Naismith, Science, 2005, 309, 2216 CrossRef CAS PubMed.
- X. Zhu, W. De Laurentis, K. Leang, J. Herrmann, K. Ihlefeld, K.-H. Van Pée and J. H. Naismith, J. Mol. Biol., 2009, 391, 74 CrossRef CAS PubMed.
- E. Yeh, L. C. Blasiak, A. Koglin, C. L. Drennan and C. T. Walsh, Biochemistry, 2007, 46, 1284 CrossRef CAS PubMed.
- A. D. Roy, S. Grüschow, N. Cairns and R. J. M. Goss, J. Am. Chem. Soc., 2010, 132, 12243 CrossRef CAS PubMed.
- L. J. Durak, J. T. Payne and J. C. Lewis, ACS Catal., 2016, 6, 1451 CrossRef CAS PubMed.
- M. Frese, C. Schepel, H. Minges, H. Voβ, R. Feiner and N. Sewald, ChemCatChem, 2016, 8, 1799 CrossRef CAS.
- A. Lang, S. Polnick, T. Nicke, P. William, E. P. Patallo, J. H. Naismith and K.-H. Van Pée, Angew. Chem., Int. Ed., 2011, 50, 2951 CrossRef CAS PubMed.
- S. A. Shepherd, C. Karthikeyan, J. Latham, A.-W. Struck, M. L. Thompson, B. R. K. Menon, M. Q. Styles, C. Levy, D. Leys and J. Micklefield, Chem. Sci., 2015, 6, 3454 RSC.
- J. T. Payne, C. B. Poor and J. C. Lewis, Angew. Chem., Int. Ed., 2015, 54, 4226 CrossRef CAS PubMed.
- S. A. Shepherd, B. R. K. Menon, H. Fisk, A.-W. Struck, C. Levy, D. Leys and J. Micklefield, ChemBioChem, 2016, 17, 821 CrossRef CAS PubMed.
- J. Latham, J.-M. Henry, H. H. Sharif, B. R. K. Menon, S. A. Shepherd, M. F. Greaney and J. Micklefield, Nat. Commun., 2016, 7, 11873 CrossRef PubMed.
- C. B. Poor, M. C. Andorfer and J. C. Lewis, ChemBioChem, 2014, 15, 1286 CrossRef CAS PubMed.
- A. Bar-Even, E. Noor, Y. Savir, W. Liebermeister, D. Davidi, D. S. Tawfix and R. Milo, Biochemistry, 2011, 50, 4402 CrossRef CAS PubMed.
- J. J. McIntyre, A. T. Bull and A. W. Bunch, Biotechnol. Bioeng., 1996, 49, 412 CrossRef CAS PubMed.
- V. V. Dasu and T. Panda, Bioproc. Bioeng., 1999, 21, 489 CrossRef.
- H.-M. Jung, S.-Y. Kim, H.-J. Moon, D.-K. Oh and J.-K. Lee, Appl. Microbiol. Biotechnol., 2007, 77, 789 CrossRef CAS PubMed.
- L. B. Pickens, Y. Tang and Y.-H. Chooi, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 211 CrossRef CAS PubMed.
- M. Frese and N. Sewald, Angew. Chem., Int. Ed., 2014, 54, 298 CrossRef PubMed.
- H. H. Liao, Enzyme Microb. Technol., 1993, 15, 286 CrossRef CAS PubMed.
- H. Zhao and F. H. Arnold, Prot. Eng., 1999, 12, 47 CrossRef CAS.
- G. D. Haki and S. K. Rakshit, Bioresour. Technol., 2003, 89, 17 CrossRef CAS PubMed.
- I. Wu and F. H. Arnold, Biotechnol. Bioeng., 2013, 110, 1874 CrossRef CAS PubMed.
- X. Chen, B. Zhang, W. Zhang, X. Wu, M. Zhang, T. Chen, G. Liu and P. Dyson, Genome Announcements, 2013, 1, 4 Search PubMed.
- J. Harrison and D. J. Studholme, Microb. Biotechnol., 2014, 7, 373 CrossRef PubMed.
- T. Ohshiro, Y. Ishii, T. Matsubara, K. Ueda, Y. Izumi, K. Kino and K. Kirimura, J. Biosci. Bioeng., 2005, 100, 266 CrossRef CAS PubMed.
- S. Takahashi, T. Furuya, Y. Ishii, K. Kino and K. Kirimura, J. Biosci. Bioeng., 2009, 107, 38 CrossRef CAS PubMed.
- J. Zeng and J. Zhan, Biotechnol. Lett., 2011, 33, 1607 CrossRef CAS PubMed.
- J. R. Heemstra and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 14024 CrossRef CAS PubMed.
- M. Frese, P. H. Guzowska, H. Voβ and N. Sewald, ChemCatChem, 2014, 6, 1270 CAS.
- Z. T. Campbell and T. O. Baldwin, J. Biol. Chem., 2009, 284, 8322 CrossRef CAS PubMed.
- T. Matsubara, T. Oshiro, Y. Nishina and Y. Izumi, Appl. Environ. Microbiol., 2001, 67, 1179 CrossRef CAS PubMed.
- X. Zhu, W. D. Laurentis, K. Leang, J. Herrmann, K. Ihlefeld, K.-H. Van Pée and J. H. Naismith, J. Mol. Biol., 2010, 391, 74 CrossRef PubMed.
- E. Bitto, Y. Huang, C. A. Bingman, S. Singh, J. S. Thorson and G. N. Phillis, Proteins, 2007, 70, 289 CrossRef PubMed.
- P. R. Pokkuluri, R. Raffen, L. Dieckman, C. Boogaard, F. J. Stevens and M. Schiffer, Biophys. J., 2002, 82, 391 CrossRef CAS PubMed.
- S. S. Strickler, A. V. Gribenko, A. V. Gribenko, T. R. Keiffer, J. Tomlinson, T. Reihle, V. V. Loladze and G. I. Makhatadze, Biochemistry, 2006, 45, 2761 CrossRef CAS PubMed.
- M. D. Smith, M. A. Rosenow, M. Wang, J. P. Allen, J. W. Szostak and J. C. Chaput, PLoS One, 2007, 2, 467 Search PubMed.
- C.-H. Chan, C. C. Wilbanks, G. I. Makhatadze and K.-B. Wong, PLoS One, 2012, 7, 30296 Search PubMed.
- K. K. K. Leung and B. H. Shilton, J. Biol. Chem., 2013, 288, 11242 CrossRef CAS PubMed.
- C. F. Megarity, J. R. E. Gill, M. C. Caraher, I. J. Stratford, K. A. Nolan and D. J. Timson, FEBS Lett., 2014, 588, 1666 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental methods, supplementary figures, tables and NMR spectra. See DOI: 10.1039/c6ob01861k |
| This journal is © The Royal Society of Chemistry 2016 |