
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel bisnaphthalimidopropyl (BNIPs) derivatives as anticancer compounds targeting DNA in human breast cancer cells†
Maria
Kopsida
a,
Gemma A.
Barron
ab,
Giovanna
Bermano
b,
Paul
Kong Thoo Lin
a and
Marie
Goua
 *a
*a
aSchool of Pharmacy and Life Sciences, Robert Gordon University, Garthdee Road, Aberdeen, AB10 7GJ, Scotland, UK. E-mail: m.goua@rgu.ac.uk
bCentre for Obesity Research and Education (CORE), Faculty of Health and Social Care, Robert Gordon University, Garthdee Road, Aberdeen, AB10 7GJ, Scotland, UK
First published on 21st September 2016
Abstract
Bisnaphthalimidopropyl (BNIP) derivatives are a family of compounds that exert anti-cancer activities in vitro and, according to previous studies, variations in the linker sequence have increased their DNA binding and cytotoxic activities. By modifying the linker sequence of bisnaphthalimidopropyl diaminodicyclohexylmethane (BNIPDaCHM), a previously synthesised BNIP derivative with anti-cancer properties, three novel BNIP derivatives were designed. Bisnaphthalimidopropyl-piperidylpropane (BNIPPiProp), a structural isomer of BNIPDaCHM, bisnaphthalimidopropyl ethylenedipiperidine dihydrobromide (BNIPPiEth), an isoform of BNIPDaCHM with a shorter linker chain, and (trans(trans))-bisnaphthalimidopropyl diaminodicyclohexylmethane (trans,trans-BNIPDaCHM), a stereoisomer of BNIPDaCHM, were successfully synthesised (72.3–29.5% yield) and characterised by nuclear magnetic resonance spectroscopy (NMR) and mass spectrometry (MS). Competitive displacement of ethidium bromide (EtBr) and UV binding studies were used to study the interactions of BNIP derivatives with Calf Thymus DNA. The cytotoxicity of these derivatives was assessed against human breast cancer MDA-MB-231 and SKBR-3 cells by MTT assay. Propidium iodide (PI) flow cytometry was conducted in order to evaluate the cellular DNA content in both breast cancer cell lines before and after treatment with BNIPs. The results showed that all novel BNIPs exhibit strong DNA binding properties in vitro, and strong cytotoxicity, with IC50 values in the range of 0.2–3.3 μM after 24 hours drug treatment. Two of the novel BNIP derivatives, BNIPPiEth and trans,trans-BNIPDaCHM, exhibited greater cytotoxicity against the two breast cancer cell lines studied, compared to BNIPDaCHM. By synthesising enantiopures and reducing the length of the linker sequence, the cytotoxicity of the BNIP derivatives was significantly improved compared to BNIPDaCHM, while maintaining DNA binding and bis-intercalating properties. In addition, cell cycle studies indicated that trans,trans-BNIPDaCHM, the most cytotoxic BNIP derivative, induced sub-G1 cell cycle arrest, indicative of apoptotic cell death. Based on these findings, further investigation is under way to assess the potential efficacy of trans,trans-BNIPDaCHM and BNIPPiEth in treating human breast cancer.
Introduction
Breast cancer is the most commonly occurring cancer in women, with incidence rates approaching 1.38 million cases per year worldwide.1 Breast cancer, depending on whether it develops in response to the hormone oestrogen, is divided in two categories: oestrogen dependent (ER+) and oestrogen independent (ER−) cancer.2 ER+ breast cancers respond better to anti-oestrogen (endocrine) therapies, such as tamoxifen and exemestane, by inhibiting the effect of oestrogen and decreasing the uncontrolled proliferation of breast cancer cells.2 On the other hand, ER-breast cancers are more invasive and less responsive to current standard-of-care treatment regimes, such as fluorouracil, epirubicin and doxorubicin, which do not selectively target breast cancer cells hence leading to severe side effects.3 Over the last few decades, there have been numerous attempts to develop novel breast cancer-specific therapies that will act on specific molecular targets, increasing selectivity and potentially reducing treatment resistance and side effects.4 DNA was one of the first biochemical targets identified in anti-cancer therapeutics,5–7 which lead to a new generation of agents with improved efficacy and solubility. In the 1990s naphthalimido compounds, such as mitonafide, were found for the first time to be highly active against cervical cancer and leukaemia cells, however, a Phase I clinical trial revealed that mitonafide treatment at doses above 118 mg m−2 × 5 days lead to central nervous system toxicity.8 Subsequently, bisnaphthalimido compounds were synthesised with improved therapeutic properties. They had also overcome the dose-limiting toxicity issues,9 however, their aqueous insolubility limited their potential use as anti-cancer agents.10 Kong Thoo Lin and Pavlov10 designed and synthesised a number of bisnaphthalimidopropyl derivatives, by incorporating natural polyamines and, diamino or triamino alkyl chains into the bisnaphthalimide structure. This led to improved aqueous solubility, as well as anti-cancer activity.11,12 Additional alterations to the linker were performed to confirm that BNIP moiety was crucial for in vitro anti-cancer activity. One such alteration was the introduction of a bicyclohexylmethane group (in the linker chain). This tended to reduce the flexibility of the linker but it enhanced its DNA binding properties, resulting in the synthesis of BNIPDaCHM (Fig. 1). BNIPDaCHM contained a pseudo-asymmetrical centre with a mixture of three isomers (cis,cis; trans,trans and cis,trans) (indicated by asterisks, Fig. 1).13,14 This linker chain modification resulted in a more cytotoxic BNIP derivative against triple negative breast cancer cells MDA-MB-231 (IC50 value 6.8 μM), compared to chemotherapy drug doxorubicin (IC50 value 14.4 μM), after 24 hours treatment.13 With regards to selectivity, non-tumourigenic breast epithelial MCF-10A cells were found to be less responsive to BNIPDaCHM (IC50 value 6.06 μM) than doxorubicin.13 | ||
| Fig. 1 Chemical structure of BNIP derivatives: BNIPDaCHM with its three stereo isomers, bisnaphthalimidopropyl-piperidylpropane (BNIPPiProp), bisnaphthalimidopropyl-ethylenedipiperidine (BNIPPiEth) and (trans(trans))-4,4′-methylenebis(cyclohexylamine) (trans,trans-BNIPDaCHM). | ||
In the present work, we describe for the first time, the synthesis and characterisation of three novel BNIP derivatives that were designed by considering several modifications to the structure of BNIPDaCHM (Fig. 1). The first BNIP derivative, bisnaphthalimidopropyl-piperidylpropane dihydrobromide (BNIPPiProp) is a structural isomer of BNIPDaCHM that consists of only one species (enantiopure). The aim of synthesising BNIPPiProp was to investigate whether cytotoxicity and DNA binding properties differ among structural isomers and how the position of the ring structure in the linker chain affects cytotoxicity. The second BNIP derivative, bisnaphthalimidopropyl-ethylenedipiperidine (BNIPPiEth), consists of one carbon less between the two piperidine ring structures, compared to BNIPPiProp and was synthesised in order to assess the effect of a shorter linker chain on cytotoxicity and DNA binding properties. In parallel, it is still unknown whether the cytotoxicity of BNIPDaCHM is associated with the existence of the three isomers in its structure, therefore (trans(trans))-4,4′-methylenebis (cyclohexylamine), the only commercially available stereoisomer precursor required to synthesise trans,trans-BNIPDaCHM the latter being, the third BNIP derivative used in this study. The synthesis of trans,trans-BNIPDaCHM lead to the investigation into the importance of this stereoisomer compared to the mixture of three stereoisomers present in BNIPDaCHM in relation to DNA binding affinities, cytotoxicity in MDA-MB-231 and SKBR-3 breast cancer cell lines and the possible mode of cell death via cell cycle analysis were studied for the three novel compounds. By using two cell lines which are unresponsive to currently available anti-cancer regimes, it is possible to extend our knowledge on BNIP derivative cytotoxicity against different breast cancer cells types and to gain more information about their mode of action.
Results and discussion
Chemical synthesis
The synthesis of bisnaphthalimidopropyl-dipiperidyl-propane and ethane free bases was carried out by reacting 1,3-bis-(4-piperidyl) propane or 1,3-bis-(4-piperidyl)ethane and toluenesulfonyloxypropylnaphthalimide in tetrahydrofuran (THF) under reflux with subsequent addition of cesium carbonate (Scheme 1). The corresponding BNIPPiProp and BNIPPiEth dihydro chloride and bromide salts were prepared by their treatment with HBr/g·CH3CO2H and conc. HCl (72.3% and 29.5% yield), respectively.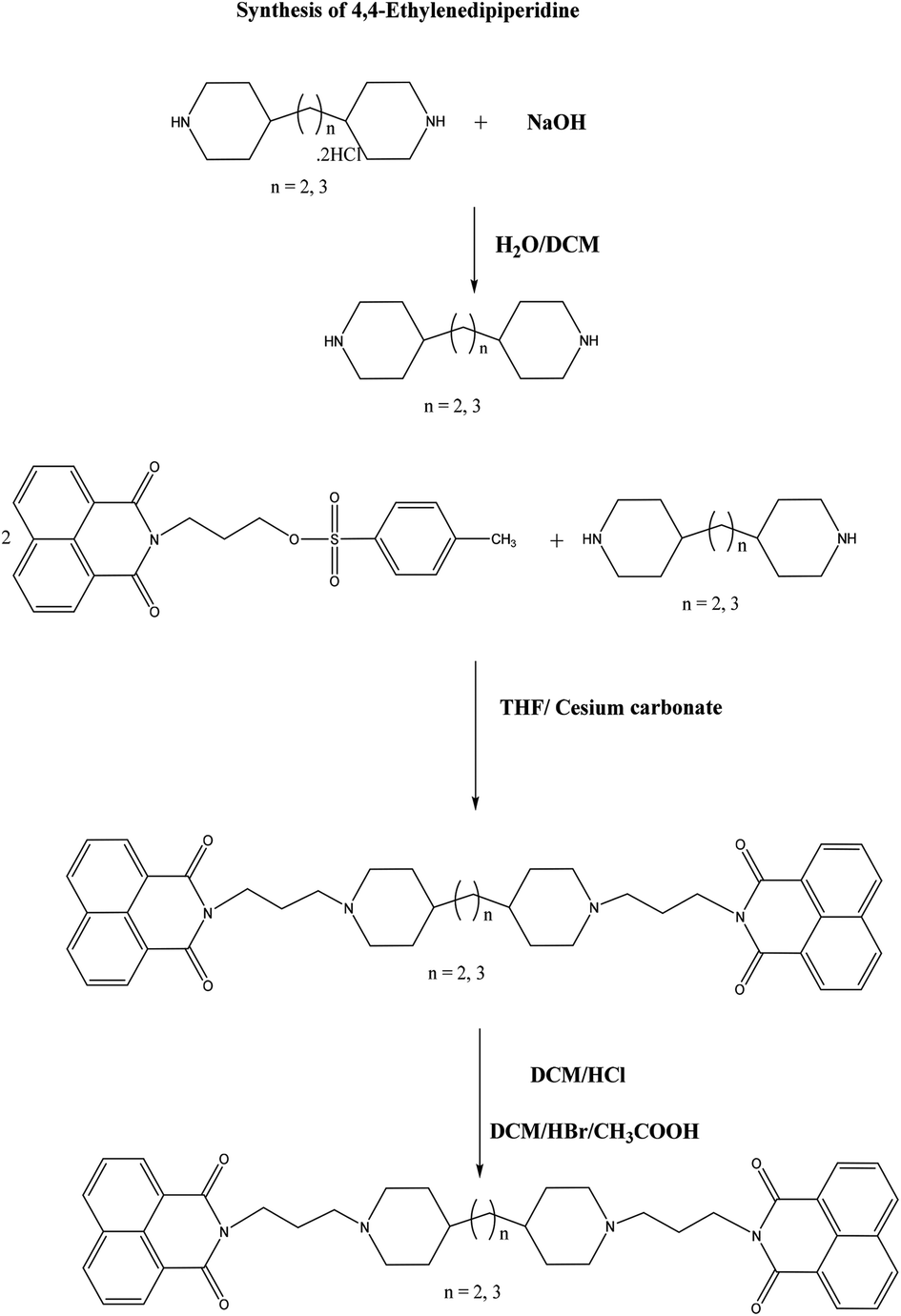 | ||
| Scheme 1 Synthetic pathway of bisnaphthalimidopropyl-piperidylpropane (BNIPPiProp) and bisnaphthalimidopropyl-ethylenedipiperidine dihydrobromide (BNIPPiEth). | ||
The synthetic strategy of trans,trans-BNIPDaCHM (Scheme 2) was based on methods previously developed in our group for the synthesis of BNIPDaCHM.13 Here the single isomer trans,trans-4,4′-methylenebiscyclohexylamine was used as the starting material. The synthesis of trans,trans-N4,N4-dimesityl-dicyclohexylmethane was carried out by reacting trans,trans-4,4′-methylenebis(cyclohexylamine), with 2-mesitylenesulfonyl chloride (Mts-Cl) in anhydrous pyridine (21.1% yield). N-Alkylation was performed by reacting trans,trans-N4,N4-dimesityl-dicyclohexylmethane with toluenesulfonyloxypropylnaphthalimide in DMF (37.0% yield). For the final step, trans,trans-bisnaphthalimido-dimesityl-dicyclohexylmethane was dissolved in DCM, followed by treatment with hydrobromic acid/glacial acetic acid (HBr/g·CH3CO2H). All new compounds were fully characterised by NMR and high resolution mass spectrometry (see Experimental section). The melting point of trans,trans-BNIPDaCHM was found to be 120–125 °C indicating good purity. In contrast, BNIPDaCHM exhibited a higher melting point range (105–130 °C) compared to trans,trans-BNIPDaCHM. BNIPPiProp and BNIPPiEth had melting points in the range of 160–170 °C and 120–125 °C, respectively, indicating their high purity.
 | ||
| Scheme 2 Synthetic pathway of (trans(trans))-4,4′-methylenebis(cyclohexylamine) (trans,trans-BNIPDaCHM). | ||
According to matched molecular pair (MMP) analysis, which has been broadly used in the last few years to investigate the effects of hydrogen bond donors/acceptors and rotatable bonds on the melting point of drug-like compounds,15–17 has shown that an increase in rotatable bonds leads to a decrease of the melting point.18 Therefore, the existence of the three isomers on BNIPDaCHM increases its flexibility and leads to a low melting point, compared to the three novel compounds.
On the other hand, BNIPPiProp, with the longest linker sequence, has the highest melting point (160–170 °C), due to the increase of hydrogen bond donors/acceptors that stabilize the crystal lattice.18
DNA binding studies
| BNIP derivatives | Mean ± SD IC50 value (μM) |
|---|---|
| Calf Thymus DNA alone | – |
| BNIPDaCHM | 2.3 ± 0.1 |
| BNIPPiProp | 3.9 ± 0.3* |
| BNIPPiEth | 1.1 ± 0.2* |
| trans,trans-BNIPDaCHM | 5.6 ± 0.2** |
The order of their binding affinity to Calf Thymus DNA from highest to lowest was BNIPPiEth (1.1 ± 0.2 μM), BNIPDaCHM (2.3 ± 0.1 μM), BNIPPiProp (3.9 ± 0.3 μM) and trans,trans-BNIPDaCHM (5.6 ± 0.2 μM).
BNIPPiEth, which has the shortest linker with two piperidine rings attached to an ethyl group, had the lowest IC50 value. The shorter length of the linker chain, as well as the incorporation of a nitrogen atom within the cyclohexane ring, compared to BNIPDaCHM, resulted in increased DNA binding. On the other hand, the incorporation of the nitrogen atom within the cyclohexane ring was not found to improve the binding properties of BNIPPiProp (3.9 ± 0.3 μM), compared to BNIPDaCHM and BNIPPiEth, confirming that the length of the bridging alkyl linkers is crucial and affects significantly BNIP binding to DNA duplexes.21
The trans,trans-BNIPDaCHM, a stereoisomer of BNIPDaCHM, gave a IC50 value of 5.6 ± 0.2 μM. BNIPDaCHM, which consists of three isomers, gave a IC50 value of 2.3 ± 0.1 μM, which indicated that in absence of cis,cis or/and cis,trans, trans,trans-BNIPDaCHM was not able to achieve as high DNA-binding interactions as BNIPDaCHM. Based on the above results, the planar structure of BNIPDaCHM and its mixture of three isomers was found to improve its interacting properties within the DNA base pairs, compared to trans,trans-BNIPDaCHM. This was confirmed since trans,trans-BNIPDaCHM resulted in a lower IC50 value compared to BNIPDaCHM, revealing that each of the three or more than one (trans,trans/cis,trans or trans,trans/cis,cis) isomers co-existing in BNIPDaCHM, are involved in the intramolecular complexes/interactions with DNA. By isolating one of its isomers (trans,trans-BNIPDaCHM), the DNA binding affinity was decreased (p < 0.01). The planar structure of BNIPDaCHM allows it to fit between the base pairs and in parallel, its rotational freedom within the plane of the aromatic rings, may allow the exposure of more than one intercalating sidechain to DNA.22
UV binding studies
The binding of the novel BNIP derivatives with Calf Thymus DNA was also studied by UV spectroscopy. A continuous decrease in UV absorption was observed at 260 nm, within the range of drug concentrations (0–7 μM) investigated. The apparent binding constants for the compounds under study, were calculated from the intercept and the slope by plotting A0/(A − A0) against BNIP derivative concentrations,23 where A0 and A correspond to the absorbance values in the absence and presence of each BNIP compound (Fig. 2a), respectively. Binding constant values K for the BNIP derivatives range between 3.25 × 104–12.23 × 104 (Fig. 2b), and indicate that all BNIP derivatives interact with the DNA helix. The highest binding constant was observed with BNIPDaCHM (12.23 × 104), followed by trans,trans-BNIPDaCHM (11.38 × 104) and BNIPPiEth (10.85 × 104). The lowest K binding constant was observed for BNIPPiProp (3.25 × 104). This outcome is in agreement with the competitive displacement of EtBr studies (Table 1), highlighting the importance of linker chain length in achieving strong DNA binding interactions. In addition, the UV absorption studies revealed that trans,trans-BNIPDaCHM, the less effective derivative in displacing EtBr from DNA, obtained a high binding constant (11.38 × 104), compared to BNIPDaCHM (12.23 × 104: highest K binding constant). This suggests that trans,trans-BNIPDaCHM exhibits lower intercalation capacity than the other two isomers present in the BNIPDaCHM. However, the trans,trans isomer has similar DNA affinity to the mixture of isomers present in BNIPDaCHM as demonstrated by their binding constant (Fig. 2b). Previous molecular modelling studies have revealed that for the most stable conformation of bis-1,8-naphthalimide in presence of DNA, the naphthalimide rings obtain an antiparallel orientation and are detected in the major groove.24 Furthermore, they have been reported to induce strand cleavage, allowing the electron transfer and formation of hydrogen bonding between the nitrogen atoms and the nucleobases (excluding guanine) of the minor groove,25 suggesting that the high K binding constant of trans,trans-BNIPDaCHM is obtained not only via intercalation, but also via binding on the major and minor groove of DNA. | ||
| Fig. 2 (a) Competitive displacement of EtBr from Calf Thymus DNA. Plot of A0/(A − A0) versus 1/CBNIP of the interaction between BNIP derivatives and Calf Thymus DNA. (b) K constant values of BNIPP derivatives after UV binding studies. | ||
Biological studies
| BNIP derivatives | IC50 values (μM)(Mean ± SEM) | |
|---|---|---|
| MDA-MB-231 | SKBR-3 | |
| BNIPDaCHM | 2.3 ± 0.1 | 0.4 ± 0.1 |
| BNIPPiProp | 3.3 ± 0.1** | 0.7 ± 0.1* |
| BNIPPiEth | 1.8 ± 0.1* | 0.3 ± 0.1* |
| trans,trans-BNIPDaCHM | 1.4 ± 0.1** | 0.2 ± 0.0* |
trans,trans-BNIPDaCHM (1.4 μM) was more active (p < 0.01) than BNIPDaCHM (2.3 μM) against MDA-MB-231 cells, showing that the existence of a single isomer in the linker sequence results in a more cytotoxic compound, compared to a compound which contains a mixture of isomers. BNIPPiEth was more cytotoxic (p < 0.05) than BNIPDaCHM, suggesting that the shorter length of the linker chain, as well as the incorporation of the nitrogen atom within the cyclohexane ring, not only improved the binding properties of BNIPPiEth, but enhanced significantly its in vitro cytotoxicity, too. Regarding BNIPPiProp, the derivative with the longest linker sequence and the highest IC50 value, it was found that the length of the linker chain length plays an important role in the functionality and effectiveness of a BNIP derivative. In addition, all the derivatives followed a similar pattern of cytotoxicity in both cell lines however, they appeared more cytotoxic against SKBR-3 cells. This may be due to the different mutational and tumorigenic statuses.
MDA-MB-231 cells are triple negative breast cancer cells (TNBC) (oestrogen receptor negative (ER−), progesterone receptor negative (PR−), human epidermal growth factor receptor 2 negative HER2−) with four gene mutations being reported (BRAF, CDKN2A, KRAS and TP53) and a fast-growing basal B tumour classification (49.5% proliferation rate),27 which makes them less responsive to anti-cancer treatments, such as docetaxel or carboplatin, compared to cells that are hormone receptor positive.27 On the other hand, SKBR-3 cells are double negative breast cancer cells (DNBC) (ER−, PR−, HER2+) without gene mutations and with a luminal tumour classification (35.2% proliferation rate),27 resulting in a better response to anti-cancer treatments compared to TNBCs, which is in agreement with our findings.
Cell cycle analysis
Cell cycle distribution of MDA-MB-231 and SKBR-3 cells after cell synchronisation was studied using flow cytometry following PI staining28 with the most active compound trans,trans-BNIPDaCHM (Table 2), together with BNIPDaCHM (mixture of isomers) that has been reported to induce cell cycle instability.29An increase in the proportion of MDA-MB-231 cells in sub-G1 phase (122.3%, 139.3% and 142.2% increase, respectively) was exhibited relative to the control, whilst in G1 phase, the cell population was decreased after treatment with BNIPDaCHM (1 μM) and trans,trans-BNIPDaCHM (1 μM) (31.3% and 29.4% decrease, both p < 0.05) (Fig. 3a). The above results indicate that both BNIPDaCHM and trans,trans-BNIPDaCHM induced sub-G1 cell cycle arrest to a greater extent than camptothecin. Camptothecin is a well known positive control for sub-G1 arrest30,31 and for inducing apoptosis.32,33
 | ||
Fig. 3 Cell cycle distribution of MDA-MB-231 and SKBR-3 cells after BNIP treatment. (a) Quantification of MDA-MB-231 and (b) SKBR-3 cell cycle profiles by flow cytometry following PI staining after 24 hours treatment with BNIPDaCHM, trans,trans-BNIPDaCHM and camptothecin. DMSO/dH2O (50% v/v) was used as the solvent control. The percentage of the cell population in sub-G1, G1, S and G2/M were calculated from histograms of linear FL-2 plots in the ungated regions (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events). Data are mean ± SEM of three independent experiments (n = 3), conducted in duplicates. *P < 0.05, compared to solvent control. 000 events). Data are mean ± SEM of three independent experiments (n = 3), conducted in duplicates. *P < 0.05, compared to solvent control. | ||
Therefore, this suggests that BNIPDaCHM and trans,trans-BNIPDaCHM could use similar mechanisms of action compared to camptothecin and may trigger apoptotic cell death in MDA-MB-231 and SKBR-3 human breast cancer cells.
Similar experiments were carried out with synchronised SKBR-3 cells (Fig. 3b), where the cell population was significantly increased in sub-G1 phase only after treatment with BNIPDaCHM (136.4%) and camptothecin (210.2%), but not for trans,trans-BNIPDaCHM, demonstrating that the effect of the two BNIP derivatives was different in each cell line and that the existence of one isomer has a different effect on cell cycle distribution. Therefore, it is suggested that the mechanisms of cell death among trans,trans-BNIPDaCHM and BNIPDaCHM may differ, even though they belong to the same family of compounds.
Conclusions
In this study, we have shown that three BNIP derivatives, BNIPPiProp, BNIPPiEth and trans,trans-BNIPDaCHM could have potential as anti-cancer agents. DNA binding affinity was confirmed as each BNIP derivative had the ability to competitively displace EtBr from DNA and quenched UV absorption in presence of DNA in a dose dependent manner. Linker sequence modifications showed stronger cytotoxic effects against MDA-MB-231 and SKBR-3 cells, compared to the parental compound BNIPDaCHM. In addition, cell cycle analysis showed that trans,trans-BNIPDaCHM induced sub-G1 arrest in MDA-MB-231 cells, but not in SKBR-3 cells, suggesting that its mode of action could be cell line dependent, in contrast to BNIPDaCHM that shows a similar trend in cell cycle arrest in both cell lines. This study has confirmed that BNIPPiProp, BNIPPiEth and trans,trans-BNIPDaCHM have the ability to interact with DNA, intercalate and stabilise the double helix, and exhibit better cytotoxic activities, compared to previously synthesised BNIP derivatives. Further research is ongoing into the mode of DNA damage, cell death or pathway deregulations in human breast cancer cell lines.Experimental
Chemical synthesis
BNIPPiProp, BNIPPiEth and trans,trans-BNIPDaCHM were synthesised according to the methods (with some modifications) previously described by Kong Thoo Lin and Pavlov.10 The chemical structure, purity and stability of the derivatives were confirmed by TLC, NMR, MS and melting point determination. All reagents were purchased from Fisher Scientific or Sigma-Aldrich, unless otherwise stated. TLC was performed on silica gel 60 F254 aluminium plates (EMD/Merck) in chloroform/methanol (95:5). NMR was recorded on a Bruker 400 Ultrashield spectrometer operating at 400.1 MHz for 1H and 100.6 MHz for 13C. Accurate mass spectra were obtained by were obtained on Thermo Scientific LTQ Orbitrap XL or Waters Xevo G2-S analytical instruments (EPSRC National Mass Spectrometry Service Centre at Swansea University, Swansea).
Synthesis of bisnaphthalimidopropyl-dipiperidyl-propane base
1,3-Bis-(4-piperidyl)propane (1.19 × 10−3 mol, 0.25 g) and toluenesulfonyloxypropylnaphtalamide (2.39 × 10−3 mol, 0.98 g) were dissolved in tetrahydrofuran (THF) (6 mL). Using a reflux condenser, the reaction was stirred at 50 °C for 15 minutes and after the addition of caesium carbonate (3.069 × 10−3 mol, 1 g), the reaction was left to stir overnight at 50 °C. The reaction was monitored with TLC and once complete, the solution was poured into icy water (100 mL). A precipitate was formed and after vacuum filtration, the product was dried in a vacuum oven at 45 °C overnight. The crude product (base of BNIPPiProp) was recrystallized from ethanol and the pure product was characterised by 1H-NMR (64.8% yield).1H-NMR (CDCl3): δH 8.53–8.51 (2H, CH aromatic protons), 8.14–8.12 (2H, CH aromatic protons), 7.70–7.66 (2H, CH aromatic protons), 4.17–4.14 (2, CH2 protons), 2.84–2.82 (2H, CH2 protons), 2.41–2.37 (2H, CH2 protons), 1.91–1.83 (2H, CH2 protons), 1.79–1.74 (2H, CH2 protons), 1.51–1.48 (2H, CH2 protons), 1.71–1.09 (H, CH proton) ppm (parts per million).
Synthesis of BNIPPiProp salt
The base of BNIPPiProp (1.459 × 10−3 mol, 1 g) was dissolved in DCM (20 mL) and HBr/CH3CO2H (2 mL) was added slowly. The reaction was stirred for 2 hours at room temperature and reaction completion was monitored by TLC yielding a precipitate. The precipitate was filtered by vacuum filtration and washed with DCM (30 mL) and ether (10 mL). The BNIPPiProp salt was dried under negative pressure in a vacuum oven set at 45 °C for 2 hours (72.3% yield).1H-NMR (CDCl3): δH 8.53–8.51 (2H, CH aromatic protons), 8.14–8.12 (2H, CH aromatic protons), 7.70–7.66 (2H, CH aromatic protons), 4.69 (2H, CH2 protons), 4.17–4.14 (2H, CH2 protons), 2.84–2.82 (2H, CH2 protons), 2.41–2.37 (2H, CH2 protons), 1.91–1.83 (2H, CH2 protons), 1.79–1.74 (2H, CH2 protons), 1.51–1.48 (2H, CH2 protons), 1.71–1.09 (H, CH proton) ppm.
13C-NMR (CDCl3): δH 164.24 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 131.87 (CH aromatic), 131.60 (C aromatic), 131.24 (CH aromatic), 127.75 (C aromatic), 126.94 (CH aromatic), 122.80 (C aromatic), 56.58 (CH2), 54.03 (CH2), 39.10 (CH2 aromatic), 36.77–35.69 (CH), 32.34 (CH2), 25.33 (CH2) and 23.76 (CH2) ppm.
O), 131.87 (CH aromatic), 131.60 (C aromatic), 131.24 (CH aromatic), 127.75 (C aromatic), 126.94 (CH aromatic), 122.80 (C aromatic), 56.58 (CH2), 54.03 (CH2), 39.10 (CH2 aromatic), 36.77–35.69 (CH), 32.34 (CH2), 25.33 (CH2) and 23.76 (CH2) ppm.
Mass spectrum (HRMS), m/z = 685.3739 (M + H)+ C43H48N4O4 requires 685.3748 (M + H)+.
Synthesis of 4,4-ethylenedipiperidine
4,4-Ethylenedipiperidine dihydrochloride (7.428 × 10−4 mol, 0.2 g) was dissolved in distilled water (2 mL), sodium hydroxide (2 M, 1 mL) was added, which resulted in the formation of a precipitate. The pH was 14. The resulting was transferred into a separating funnel (100 mL) followed by extraction with DCM (300 mL). The organic layer was collected, dried with sodium sulfate and filtered. The solvent was evaporated by a rotary film evaporator. The final white solid product was left to dry under negative pressure in a vacuum oven at 45 °C for 30 minutes (90.6% yield). The synthesis of the free base was confirmed by 1H-NMR.1H-NMR (CDCl3): δ 2.98–2.95 (2H, CH2 protons), 2.52–2.45 (2H, CH2 protons), 1.8 (NH), 1.60–1.57 (2H, CH2 protons), 1.25–1.21 (H, CH protons) and 1.19–1.16 (2H, CH2 protons) ppm.
Synthesis of BNIPPiEth base
4,4-Ethylenedipiperidine (5.044 × 10−4 mol, 0.1 g) was reacted with toluenesulfonyloxypropyl-naphthalamide (1.001 × 10−3 mol, 0.41 g, 2.01 excess). Caesium carbonate (3.069 × 10−3 mol, 1 g) was added in the reaction. All the reagents were dissolved in THF (6 mL) and the solution refluxed overnight at 60 °C. The reaction was monitored using TLC. Once the reaction was complete, the solution was poured into icy water (100 mL), which resulting in the formation of a precipitate. The precipitate was filtered using a Buchner funnel and the product was left to dry under negative pressure in a vacuum oven at 45 °C for 60 minutes (72.3% yield). The crude product was recrystallised from ethanol and the pure product was characterised by 1H-NMR.1H-NMR (CDCl3): δH 8.52–8.50 (2H, CH aromatic protons), 8.13–8.11 (2H, CH aromatic protons), 7.69–7.65 (2H, CH aromatic protons), 4.18–4.14 (2H, CH2 protons), 2.83–2.80 (2H, CH2 protons), 2.40–2.36 (2H, CH2 protons), 1.90–1.83 (2H, CH2 protons), 1.79–1.71 (2H, CH2 protons), 1.48 (2H, CH2 protons), 1.2 (H, CH protons) and 1.00–0.80 (2H, CH2 protons) ppm.
Synthesis of BNIPPiEth
BNIPPiEth (0.1 g) was dissolved in DCM (20 mL). Then, conc. HCl (1.5 mL) was added dropwise and the solution stirred at room temperature for 60 minutes, which resulted in the formation of a pale, blue precipitate. The latter was filtered and washed with ether (50 mL) and afterwards with ethanol (50 mL). The product was left under negative pressure in a vacuum oven at 45 °C for 60 minutes (29.5% yield).1H-NMR (CDCl3): δH 8.42–8.38 (2H, CH aromatic protons), 7.82–7.78 (2H, CH aromatic protons), 4.05–4.01 (2H, CH2 protons), 2.7 (2H, CH2 protons), 2.44–2.43 (2H, CH2 protons), 2.05 (2H, CH2 protons), 1.70–1.67 (2H, CH2 protons), 1.35–1.32 (H, CH protons) and 1.090 (2H, CH2 protons) ppm.
13C-NMR (CDCl3): δH 164.10 (CO), 134.81 (CH aromatic), 131.76 (C aromatic), 131.18 (CH aromatic), 127.92 (C aromatic), 127.68 (CH aromatic), 122.59 (C aromatic), 55.40 (CH2), 52.27 (CH2), 37.79 (CH), 29.43 (CH2) 23.04 (CH2) and 22.54 (CH2) ppm.
Mass spectrum (HRMS), m/z = 671.5372 (M + H)+ C42H46N4O4 requires 671.3592 (M + H)+.
Synthesis of N-(3-hydroxypropyl)naphthalimide
1,8-Naphthalic anhydride (0.050 mol, 10 g) was dissolved in dimethylformamide (DMF) (140 mL). Once the 1,8-naphthalic anhydride was totally dissolved, 3-amino-1-propanol (0.050 mol, 3.75 g) and 1,8-diazabicylo[5.4.0]undec-7-ene (DBU) (13 mL) were added. The reaction was left to stir for 5 hours at 85 °C. Reaction completion was monitored with TLC and once completed; the solution was poured into icy water (200 mL) while stirring with a glass rod to form a precipitate. The precipitate was filtered and washed with water. The pure product was characterised using 1H-NMR. Yield was calculated as: 53.9%.1H-NMR (CDCl3): δH 8.66–8.63 (2H, aromatic H, doublet, J = 7.2 Hz), 8.29–8.25 (2H, aromatic H, doublet, J = 8.4 Hz), 7.83–7.78 (2H, aromatic H, doublet, J = 8.0 Hz), 4.41–4.38 (3H, triplet, J = 5.6 Hz), 3.64–3.59 (2H, CH2, multiplet) 3.23 (OH, singlet) and 2.06–2.00 (2H, CH2 multiplet) ppm.
Synthesis of toluenesulfonyloxypropylnaphthalimide
N-(3-Hydropropyl)naphthalimide (0.0196 mol, 5.0 g) was dissolved in anhydrous pyridine (70 mL), whilst stirring on ice. Once the N-(3-hydroxypropyl)naphthalimide was completely dissolved, p-toluenesulfonyl chloride (Ts-Cl) (0.0394 mol, 7.51 g, 2.01 excess) was slowly added to the reaction. The reaction was left at 4 °C overnight. The reaction was monitored using TLC and once it was complete, the solution was poured into icy water (200 mL) to form a precipitate which was filtered and washed thoroughly with water. The crude product was recrystallised from ethanol (68.2% yield).1H-NMR (CDCl3): δH 8.59–8.57 (2H, aromatic H, doublet, 8.2 Hz), 8.25–8.23 (2H, aromatic H, triplet, 7.2 Hz), 7.81–7.76 (2H, multiplet), 7.30 (2H, aromatic H, doublet, 1.2 Hz), 4.27–4.20 (2H, CH2, multiplet), 2.44 (3H, CH3, singlet) and 2.19–2.12 (2H, CH2, multiplet) ppm.
Synthesis of N4,N4-dimesityl-dicyclohexylmethane
4,4′-Methylenebis(cyclohexylamine) (4.75 × 10−3 mol, 1.0 g) was dissolved in anhydrous pyridine (10 mL) and left to stir for 15 minutes. 2-Mesitylenesulfonyl chloride (Mts-Cl) (9.56 × 10–3 mol; 2.1 g) was added. The reaction was left to stir overnight at room temperature and TLC confirmed completion of reaction. The solution was poured into icy water (150 mL) with the formation of a precipitate. The latter was filtered by vacuum filtration, left to dry in a vacuum oven set at 45 °C for 24 hours (43.3% yield).1H-NMR (CDCl3): δH 7.30 (NH), 6.97 (4H, CH aromatic protons), 2.99 (4H, CH protons), 2.67–2.66 (6H, CH3-Mts protons) and 1.73–1.49 (CH2 and cyclohexane protons) ppm.
Synthesis of protected bisnaphthalimido-dimesityl-dicyclohexylmethane
N 4,N4-Dimesityl-dicyclohexylmethane (6.968 × 10−4 mol, 0.4 g) and toluenesulfonyloxypropylnaphthalimide (1.400 × 10−3 mol, 2.01 excess) were dissolved in DMF (8 mL). Afterwards, excess of caesium carbonate (1.13 g, 3.5 × 10−3 mol, 2.5 g excess) was added slowly. The reaction was left to stir for 48 hours at 60 °C. After TLC confirmed the reaction was complete, the solution was poured into icy water (150 mL) to form a precipitate. After vacuum filtration and several washes with water, the product was left to dry under negative filtration in a vacuum oven at 45 °C for 3 hours (92.2% yield). The product was recrystallised from ethanol and characterised by NMR.1H-NMR (CDCl3): δH 8.50–8.47 (2H, aromatic H), 8.18–8.15 (2H, aromatic H), 7.72–7.67 (2H, aromatic H), 6.87 (2H, CH-Mts H), 6.55–6.53 (2H, CH-Mts H), 3.94–3.92 (2H, CH2), 3.12–3.10 (2H, CH2), 2.87–2.80 (2H, CH2), 2.35–2.32 (3H, CH3), 2.05–2.04 (3H, CH3), 1.61–1.58 (H, CH), 1.61 (2H, CH2) 1.49–1.37 (2H, CH) and 1.04 (2H, CH2) ppm.
13C-NMR (CDCl3): δH 163.87–162.76 (CO), 134.36 (C aromatic), 131.40–130.74 (C aromatic), 127.75 (C aromatic), 35.75 (CH2), 30.41 (CH2), 21.97 (CH3) and 20.32 (CH3) ppm.
Synthesis of bisnaphthalimidopropyl-diamino-dicyclohexylmethane dihydro-bromide salt
Bisnaphthalimido-dimesityl-dicyclohexylmethane (3.813 × 10−4 mol, 0.4 g) was dissolved in dichloromethane (DCM) (8 mL). Afterwards, hydrobromic acid/glacial acetic acid (HBr/CH3CO2H) (1 mL) was added drop wise. The reaction was stirred overnight at room temperature. TLC was used to confirm that the reaction was complete. The precipitate formed, was filtered by vacuum filtration and washed with DCM (15 mL) and ether (5 mL). The final product was dried under negative pressure in a vacuum oven set at 45 °C for 3 hours (37.5% yield).1H-NMR (CDCl3): δH 8.32–8.25 (2H, aromatic Hs), 7.73–7.65 (2H, aromatic Hs), 3.70–3.64 (2H, CH2 protons), 3.13 (2H, CH2 protons), 2.90–2.30 (3H, CH3 protons), 2.09–2.02 (3H, CH3 protons), 1.83–1.80 (H, CH proton), 1.43–1.33 (2H, CH2 protons) ppm.
13C-NMR(CDCl3): δ 163.87–162.76 (CO), 134.36 (C aromatic), 131.40–130.74 (C aromatic), 127.75 (C aromatic), 35.75 (CH2) and 30.41 (CH2) ppm.
Mass spectrum (HRMS), m/z = 685.3764 (M + H)+ C43H48N4O4 requires 685.3748 (M + H)+.
Synthesis of trans,trans-N4,N4-dimesityl-dicyclohexylmethane
trans,trans-4,4′-Methylenebis(cyclohexylamine) (2.38 × 10−4 mol, 0.05 g) was added in anhydrous pyridine (1.5 mL) and left to stir for 15 minutes with warming. After dissolution, 2-mesitylenesulfonyl chloride (Mts) (4.76 × 10−4 mol; 0.10 g) was added. The reaction was left to stir overnight at room temperature and TLC confirmed reaction completion. The solution was poured into icy water (10 mL) and stirred with a glass rod until the formation of a precipitate. The suspension was centrifuged and was washed 3 times with distilled water and the precipitate was left to dry under negative pressure in a vacuum oven set at 45 °C for overnight (21% yield). The product was afterwards characterised by 1H-NMR.1H-NMR (CDCl3): δH 7.31 (NH), 6.98 (4H, CH aromatic protons), 3.87 (4H, CH protons), 2.67 (6H, CH3-Mts protons), 2.33 (3H, CH3-Mts protons), 1.84–1.82 (CH2-cyclohexane ring), 1.65–1.62 (CH2-cyclohexane ring), 1.16–1.10 (CH-cyclohexane ring) and 0.99–0.95 (CH2 cyclohexane protons) ppm.
Synthesis of protected trans,trans-bisnaphthalimidopropyl-dimesityl-dicyclohexylmethane
trans,trans-N4,N4-Dimesityl-dicyclohexylmethane (6.44 × 10−5 mol, 0.037 g) and toluenesulfonyloxypropylnaphthalimide (1.29 × 10−4 mol, 0.053 g, 2.01 excess) were dissolved in DMF (1 mL). Afterwards, excess of caesium carbonate (n = 3.07 × 10−4 mol, 0.1 g, 5.0 excess) was added slowly. The reaction was left to stir for 48 hours at 60 °C. After TLC confirmed the reaction was complete, the solution was poured into icy water (10 mL) to form a precipitate. The suspension was centrifuged and was washed twice with distilled water. The product was left to dry under negative pressure in a vacuum oven at 45 °C for 24 hours. The crude product (59 mg, 88% yield) was purified using column chromatography and the final product (25 mg, 37% yield) was characterised by 1H-NMR.1H-NMR (CDCl3): δH 8.65–8.59 (2H, CH aromatic protons), 8.30–8.25 (2H, CH aromatic protons), 7.84–7.78 (2H, CH aromatic protons), 6.65 (4H, CH-Mts protons), 4.41–4.38 (2H, CH2 protons), 4.06–4.04 (2H, CH2 protons), 3.71–3.68 (2H, CH2 protons), 2.68 (3H, CH3 protons), 2.46–2.44 (3H, CH3 protons), 2.34 (H, CH protons), 2.16–2.15 (2H, CH2 protons) 1.84 (2H, CH protons) and 1.73–1.71 (2H, CH2 protons) ppm.
13C-NMR (CDCl3): δH 141.93 (CO), 138.76 (C aromatic), 135.17 (C aromatic), 131.95 (C aromatic), 43.72 (CH2), 33.93–31.99 (CH2), 23.02 (CH3) and 20.97 (CH3) ppm.
Synthesis of trans,trans-bisnaphthalimidopropyl-diamino-dicyclohexylmethane dihydro-bromide salt
trans,trans-Bisnaphthalimido-dimesityl-dicyclohexylmethane (2.38 × 10−5 mol, 25 mg) was dissolved in dichloromethane (DCM) (1.0 mL). Afterwards, hydrobromic acid/glacial acetic acid (HBr/CH3CO2H) (0.2 mL) was added slowly. The reaction was stirred overnight at room temperature. TLC was used to confirm that the reaction was complete. The suspension formed was transferred to Eppendorf tubes and centrifuged, was with DCM (1.0 mL) and ether (1.0 mL). The final product was dried under negative pressure in a vacuum oven set at 45 °C for 3 hours to give the product as a white solid (8.3 mg, 41% yield).1H-NMR (CDCl3): δ 8.53–8.49 (2H, CH aromatic protons), 4.16–4.12 (2H, CH aromatic protons), 3.42 (2H, CH aromatic protons) 3.04 (2H, CH2 protons), 2.94 (2H, CH2 protons), 2.51–2.49 (3H, CH3 protons), 2.09–1.99 (3H, CH3 protons), 1.75–1.72 (H, CH proton), 1.31–1.28 (2H, CH2 protons) ppm.
13C-NMR(CDCl3): δ 164.26 (CO), 131.82–131.23 (C aromatic), 127.99–127.75 (C aromatic), 122.66 (C aromatic), 42.45 (CH2) and 40.61–37.61 (CH2) ppm.
Mass spectrum (HRMS), m/z = 685.3732 (M + H)+ C43H48N4O4 requires 685.3748 (M + H)+.
Ethidium bromide (EtBr) fluorescence displacement studies
BNIPDaCHM, BNIPPiProp, BNIPPiEth and trans,trans-BNIPDaCHM working solutions (100 μM) were prepared from stock solutions (10 mM in 50% DMSO/H20) and were further diluted to a final concentration of 50 μM in 0.01 M saline sodium citrate (SSC) buffer. Calf Thymus DNA (0.5 g) was dissolved in 0.01 M SSC buffer (100 mL). EtBr solution (200 μM) was prepared by dissolving 3.94 mg of EtBr in distilled water (50 mL) and was further diluted in 0.01 SSC buffer to give the final concentration of 20 μM. Test solutions were prepared by adding varying volumes of SSC buffer, Calf Thymus DNA solution, EtBr solution and BNIP derivative solution. The final solutions were thoroughly mixed and analysed at 510 nm (excitation) and 520 nm (emission) using a Shimadzu RF-5301 spectrophotometer. The IC50 values were determined as the concentration (μM) required to decrease the fluorescence of DNA bound EtBr by 50%.UV binding studies
BNIPDaCHM, BNIPPiProp, BNIPPiEth and BNIPDaCHM working solutions (100 μM) were prepared from their stock solutions (10 mM) as before, and were further diluted to 20 μM final concentration in 0.01 M SSC buffer. Calf Thymus DNA and 0.01 SSC buffer were prepared as described before (EtBr Fluorescence Displacement studies). Test solutions were prepared by adding Calf Thymus DNA solution (1 mL) in a quartz cuvette and BNIP derivative solution (100 μL) was added. The final solutions were thoroughly mixed and analysed at 260 nm using an Agilent 8453 UV-visible spectrophotometer. The values of apparent binding constants (K) were calculated from the intercept and slope by plotting A0/(A − A0) against the BNIP derivative concentrations, where A0 and A correspond to the absorbance values in absence and presence of a compound.Cell culture maintenance
MDA-MB-231 (ECACC, Public Health England, UK, 92020424) and SKBR-3 (ATCC, HTB-30) cells were maintained in Roswell Park Memorial Institute 1640 medium (RPMI-1640) (containing GlutaMAX-1 with 25 mM HEPES), supplemented with 10% (v/v) Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin (10000 μg mL−1). Cells were grown at 37 °C (5% CO2).
Cytotoxicity
Colourimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazodium bromide (MTT) assay was performed to access the growth/inhibitory effects of each BNIP compound. MDA-MB-231 and SKBR-3 cells (7.5 × 103 cells per 100 μL) were treated with different concentrations (0–10 μM) of BNIP derivatives. After 24 hours treatment, sterile-filtered MTT solution (1 mg mL−1) was added to each well. After 4 hours incubation at 37 °C, the MTT solution was removed and formazan crystals solubilised in DMSO. The plates were shaken for 20 minutes at room temperature and absorbance measured at 560 nm (Synergy/HT, BIOTEK, UK). For each compound, three independent experiments were carried out and each treatment consisted of six replicates per plate. Curves were used to represent the percentage growth inhibition of MDA-MB-231 and SKBR-3 cells treated with BNIP derivatives, compared to DMSO/H2O control that represented 100% cell viability. IC50 values were defined as the drug concentration that reduces absorbance compared to control values by 50%.Cell cycle analysis
MDA-MB-231 and SKBR-3 cells (1 × 106 cells per T75 flask) cells were washed with PBS and serum free medium added in the flasks in order to achieve cell synchrony. The cells were incubated in serum free medium for 24 hours. BNIP derivatives (IC25 concentrations) were added and incubated for 24 hours, at 37 °C. After 24 hours treatment, the medium was removed and collected. The cells were washed twice with PBS. Both washes were collected and the cells trypsinised, mixed with the collected washes and centrifuged at 2500 rpm for 5 minutes at 4 °C. The supernatant of each sample was discarded, the pellet was resuspended in PBS (1 mL) and centrifuged at 2500 rpm for 5 minutes at 4 °C, the supernatant was discarded and the pellet resuspended in PBS (100 μL). Then, 70% (v/v) ice-cold ethanol (900 μL) was added and samples incubated for 2 hours at −20 °C. The cells were centrifuged at 5000 rpm for 5 minutes at 4 °C, the supernatant was discarded and the pellet resuspended in PBS (1 mL), followed by re-centrifugation at 5000 rpm for 5 minutes at 4 °C. The supernatant was discarded once more and the pellet resuspended in PBS (500 μL) and DNA extraction buffer (0.2 M Na2HPO4, 4 mM citric acid, pH 7.8, 500 μL) and incubated for 5 minutes at room temperature. The extracts were centrifuged at 5000 rpm for 5 minutes at 4 °C, the supernatant was removed and the pellets were resuspended in DNA staining solution (0.2 mg mL−1 Ribonuclease A (DNAse-free) and 20 μg mL−1 propidium iodide (PI) in PBS). The samples were incubated for 30 minutes at 4 °C in the dark and PI nuclei were examined by flow cytometry (EXPO32 ADC XL 4 color, Beckman Coulter, UK). For cell cycle analysis, EXPO32 ADC analysis software (Beckman Coulter, UK) was used to record and analyse 10000 single events. The percentage of cells with DNA content in sub-G1, G1, S and G2/M phases was calculated from histograms of linear FL-2 plots (575 nm) in the gated region.
Statistics
Three independent experiments were conducted and each experiment was comprised of at least two internal replicates. Data are presented as mean ± SD or mean ± SEM. Statistical analysis was performed by using an unpaired Student's t-test. Statistically significant data were detailed when P < 0.05).Acknowledgements
The authors acknowledge the Robert Gordon University for financial support, and the EPSRC National Mass Spectrometry Service Centre at Swansea University, Swansea for mass spectral analysis.References
- R. Siegel, D. Naishadham and A. Jemal, CA-Cancer J. Clin., 2013, 63, 11–30 CrossRef PubMed.
- A. Redmond, C. Byrne, F. Bane, G. Brown, P. Tibbitts, K. O'Brien, A. Hill, J. Carroll and L. Young, Oncogene, 2015, 34, 3871–3880 CrossRef CAS PubMed.
- A. Becorpi, G. Sisti, F. Sorbi, E. Malosso and S. Guaschino, Clin. Cases Miner. Bone Metab., 2014, 11, 110–113 Search PubMed.
- B. Lehmann and J. Pietenpol, J. Pathol., 2013, 232, 142–150 CrossRef PubMed.
- S. F. Chen, D. L. Behrens, C. H. Behrens, P. M. Czerniak, D. L. Dexter, B. L. Dusak, J. R. Fredericks, K. C. Gale, J. L. Gross, J. B. Jiang, M. Kirshenbau, R. J. McRipley, L. M. Papp, A. D. Patten, F. W. Perrella, S. P. Seitz, M. P. Stafford, J. H. Sun, T. Sun, M. A. Wuonola and D. D. Von Hoff, Anti-Cancer Drugs, 1993, 4, 4 CrossRef.
- M. F. Braña, M. Cacho, A. Gradillas, B. de Pascual-Teresa and A. Ramos, Curr. Pharm. Des., 2001, 7, 1745–1780 CrossRef.
- V. Pavlov, P. Kong Thoo Lin and V. Rodilla, Chem.-Biol. Interact., 2001, 137, 15–24 CrossRef CAS PubMed.
- M. Llombart, A. Poveda, E. Forner, C. Fernández Martos, C. Gaspar, M. Muñoz, T. Olmos, A. Ruiz, V. Soriano, A. Benavides, M. Martin, E. Schlick and V. Guillem, Invest. New Drugs, 1992, 10, 177–181 CrossRef CAS PubMed.
- M. F. Braña and A. Ramos, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 237–255 CrossRef.
- P. Kong Thoo Lin and V. Pavlov, Bioorg. Med. Chem. Lett., 2000, 10, 1609–1612 CrossRef.
- V. Pavlov, P. Kong Thoo Lin and V. Rodilla, Chem.-Biol. Interact., 2001, 137, 15–24 CrossRef CAS PubMed.
- J. Oliveira, L. Ralton, J. Tavares, A. Codeiro-Da-Silva, C. S. Bestwick, A. MacPherson and P. Kong Thoo Lin, Bioorg. Med. Chem., 2007, 15, 541–545 CrossRef CAS PubMed.
- G. A. Barron, G. Bermano, A. Gordon and P. Kong Thoo Lin, Eur. J. Med. Chem., 2010, 45, 1430–1437 CrossRef CAS PubMed.
- R. Lima, G. A. Barron, J. Grabowska, G. Bermano, S. Kaur, N. Roy, M. Vasconcelos and P. Kong Thoo Lin, Anti-Cancer Agents Med. Chem., 2013, 3, 414–421 Search PubMed.
- A. Birch, P. Kenny, I. Simpson and P. Whittamore, Bioorg. Med. Chem. Lett., 2009, 19, 850–853 CrossRef CAS PubMed.
- P. Gleeson, G. Bravi, S. Modi and D. Lowe, Bioorg. Med. Chem., 2009, 17, 5906–5919 CrossRef CAS PubMed.
- P. Hajduk and D. Sauer, J. Med. Chem., 2008, 51, 553–564 CrossRef CAS PubMed.
- S. Scultes, C. de Graaf, H. Berger, M. Mayer, A. Steffen, E. Haaksma, I. de Esch, R. Leurs and O. Kramer, MedChemComm, 2012, 3, 584–591 RSC.
- P. Scaria and R. J. Shafer, J. Biol. Chem., 1991, 266, 5417–5423 CAS.
- W. Chen, J. Turro and D. Tomalia, Langmuir, 2000, 16, 15–19 CrossRef CAS.
- P. Liu, B. Wu, J. Liu, Y. Dai, Y. Wang and K. Wang, Inorg. Chem., 2016, 55, 1412–1422 CrossRef CAS PubMed.
- M. F. Braña, M. Cacho, A. Ramos, M. Dominguez, J. Pozuelo, C. Abradelo, M. Rey-Stolle, M. Yuste, C. Carrasco and C. Bailly, Org. Biomol. Chem., 2003, 1, 648–654 Search PubMed.
- T. Zhi-Yong, L. Jing-Hua, L. Qian, Z. Feng-Lei, Z. Zhong-Hua and W. Chao-Jie, Molecules, 2014, 19, 7646–7668 CrossRef PubMed.
- M. F. Braña, M. Cacho, M. Garcia, B. de Pascual-Teresa, A. Ramos, M. Dominguez, J. Pozuelo, C. Abradelo, M. Rey-Stolle, M. Yuste, M. Banez-Coronel and J. Lacal, J. Med. Chem., 2004, 47, 1391–1399 CrossRef PubMed.
- C. Bailly, C. Carrasco, A. Joubert, C. Bal, N. Wattez, M. Hildebrand, A. Lansiaux, P. Colson, C. Houssier, M. Cacho, A. Ramos and M. F. Braña, Biochemistry, 2003, 42, 4136–4150 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 16, 55–63 CrossRef.
- P. Kenny, G. Lee, C. Myers, R. Neve, J. Semeiks, P. Spellman, K. Lorenz, E. Lee, M. Barcellos-Hoff, O. Petersen, J. Greay and M. Bissell, Mol. Oncol., 2007, 1, 84–96 CrossRef CAS PubMed.
- C. M. Henry, E. Hollville and S. J. Martin, Methods, 2013, 61(2), 90–97 CrossRef CAS PubMed.
- G. A. Barron, M. Goua, I. Kuraoka, G. Bermano, S. Iwai and P. Kong Thoo Lin, Chem.-Biol. Interact., 2015, 242, 307–315 CrossRef CAS PubMed.
- C. Chu, J. Xu, D. Cheng, X. Li, S. Tong, J. Yan and Q. Li, Mol., 2014, 19(4), 4941–4955 CrossRef PubMed.
- R. Doddapaneni, K. Patel and M. Singh, Cancer Res., 2015, 75(15), 2619 Search PubMed.
- M. Kang, J. N. Ho, H. R. Kook, S. Lee, J. J. Oh, S. K. Hong, S. E. Lee and S. S. Byun, Oncol. Rep., 2016, 35(3), 1463–1472 Search PubMed.
- J. U. Hong, H. J. Chung, S. Y. Bae, T. N. Trung, K. Bae and S. K. Lee, J. Cancer Prev., 2004, 19(4), 273–278 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob01850e |
| This journal is © The Royal Society of Chemistry 2016 |