
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadiation damage to single stranded oligonucleotide trimers labelled with 5-iodopyrimidines†
Kinga
Westphal
a,
Konrad
Skotnicki
b,
Krzysztof
Bobrowski
b and
Janusz
Rak
*a
aFaculty of Chemistry, University of Gdańsk, Wita Stwosza 63, 80-308 Gdańsk, Poland. E-mail: janusz.rak@ug.edu.pl
bCentre of Radiation Research and Technology, Institute of Nuclear Chemistry and Technology, Dorodna 16, 03-195 Warsaw, Poland
First published on 8th September 2016
Abstract
The radiolysis of deoxygenated aqueous solution containing trimeric oligonucleotides labelled with iodinated pyrimidines and Tris-HCl as the hydroxyl radical scavenger leads to electron attachment to the halogenated bases that mainly results in single strand breaks. The iodinated trimers are 2-fold more sensitive to solvated electrons than the brominated oligonucleotides, which is explained by the barrier-free dissociation of the iodinated base anions. The present study fills the literature gap concerning the chemistry triggered by ionizing radiation in the iodinated pyrimidines incorporated into DNA.
Introduction
Solid tumor cells suffer from the lack of oxygen (hypoxia) which makes them significantly more radio-resistant, i.e. they are 2.5–3.0 fold less sensitive to ionizing radiation (IR) than those present in the neighboring oxic tissues.1 Indeed, recently Stewart et al.2 developed a Monte Carlo Damage Simulation (MCDS) model which predicts the decrease of the absolute yield of cluster damage (double strand breaks, Fpg (oxidized purine) and Endo III (oxidized pyrimidine) clusters) with the decrease of oxygen concentration in the cell that agrees pretty well with the experimental findings. Thus, hypoxia protects cellular DNA from damage by IR which leads to an increase of the necessary radiation dose. As a result, radiotherapy is accompanied by a number of side effects among which secondary tumors are the most deleterious.3 Such a situation calls for the employment of substances sensitizing tumors to ionizing radiation which on the one hand enables the tumor hypoxia to be overcome and on the other hand leads to a decrease in the effective IR dose.4The halogenated nucleobases (Hal-NBs) belong to the class of radiosensitizers which owe their reactivity to the substitution for native nucleosides in the genomic DNA.5 Indeed, the radiosensitizing effect is strongly correlated to the recognition of Hal-NBs by cellular kinases and polymerases.6,7
IR interacts with cellular DNA via the radical products of water radiolysis, mainly via hydroxyl radicals (˙OH) and solvated electrons (eaq−).8 In oxic cells, eaq− swiftly react with dissolved oxygen producing relatively inactive O2˙− radicals.8 However, under hypoxic conditions, the amounts of ˙OH and eaq− generated by IR are comparable.8 Unfortunately, the solvated electrons are unable to seriously damage native DNA9,10 and the efficacy of hydroxyl radicals is significantly impaired in the hypoxic environment.1 In a normoxic environment, the initial DNA radicals interact with oxygen or endogenous reductants (e.g. glutathione). The latter process is dubbed chemical repair while interactions between DNA radicals and oxygen, leading to peroxy radicals, are called oxygen fixation and compete with chemical repair. Thus, hypoxia enhances opportunities for chemical repair and as a consequence decreases the extent of DNA damage. This situation changes dramatically for the Hal-NB labelled biopolymers. Indeed, transfer of the solvated electron to a brominated nucleobase results in the formation of a labile radical anion which efficiently dissociates releasing the bromide anion to the environment and leaving a reactive nucleobase radical in DNA.11 The latter species is able to abstract a hydrogen atom from the neighboring 2′-deoxyribose which ultimately results in cytotoxic single strand break formation.12 Hence, the process of the formation of a highly reactive nucleobase radical as a result of dissociative electron attachment (DEA) is a key factor responsible for the radiosensitizing properties of halogenated nucleosides.10 It is also worth noticing that DEA is strongly enhanced to Hal-NBs in the presence of water,13 which emphasizes the importance of cellular milieu for the radiosensitizing effects.
5-Bromo-2′-deoxyuridine (5-BrdU) and 5-iodo-2′-deoxyuridine (5-IdU) are the most widely studied radiosensitizers of the Hal-NB type. Their radiosensitizing action in vitro (at the cellular level) was discovered as early as in 1960 by Djordjevic and Szybalski.14 Investigators demonstrated that the mechanisms of radiosensitization by 5-bromo and 5-iodopyrimidines are similar and mainly result in radiation-induced irreparable double strand breaks in DNA.15,16In vitro results concerning halogenated pyrimidines were so promising that several in vivo studies followed the cellular experiments. For instance, it was demonstrated that both 5-BrdU and 5-IdU are efficiently incorporated into cellular DNA in animal models.17,18 Finally, the compounds were tested in patients, e.g. for the radiotherapy of head-and-neck cancer19 and soft tissue sarcomas.20,21
In the following, we will demonstrate, using the HPLC and LC-MS techniques, that the main reaction channel opened by the attachment of electrons to the iodinated pyrimidines incorporated into single stranded DNA is associated with the formation of single strand breaks (SSBs). Moreover, the iodinated trimers are overall 2-fold more sensitive to the solvated electrons than the brominated ones. Our present studies fill the literature gap. Indeed, since the Djordjevic and Szybalski work,14 many other in vitro studies22 on animal models17,18 and even clinical studies19,20,23 have demonstrated the radiosensitizing properties of bromo- and iodopyrimidines. Similarly, data devoted to the radiation chemistry of both bromo- and iodouridine are also accessible.24 However, radiation chemistry investigations on the aqueous solutions of model oligonucleotides comprising modified pyrimidines are limited to DNA labeled with bromonucleobases.10,25,26 Thus, our work represents the very first report concerning the radiation chemistry of model oligonucleotides labeled with iodopyrimidines. One should emphasize that it is difficult to overestimate the role of such studies in comprehending the chemistry triggered by radiolysis.
Materials and methods
General
All the trinucleotides of HPLC purity were synthesized using the phosphoramidite method and were purchased from Future Synthesis, Poland. The irradiated aqueous solutions (ultrapure water obtained using a Milli-Q system from Merck, Poland HLP) containing ca. 3 × 10−5 M of a trinucleotide were freshly prepared before radiolysis. HPLC reagents (triethylammonium acetate (TEAA) and HPLC grade acetonitrile (ACN)) were purchased from Sigma-Aldrich (Poland) and POCh (Gliwice, Poland), respectively.γ-Irradiation
γ-Irradiation was carried out in a solution that contained 30 mM Tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) as a scavenger of the OH radicals. Each sample was deoxygenated by purging with argon for ca. 3 min and irradiated for one hour. In analogy to our recent project,10 the irradiation was carried out with the use of the cobalt source (Issliedowatiel) 60Co (1.17 and 1.33 MeV) with a dose of 140 Gy. In our previous radiation chemical studies on brominated TZT trimers,10 we demonstrated that a Tris-HCL concentration of 30 mM is sufficient to scavenge the hydroxyl radicals quantitatively.Chromatography
The Dionex UltiMate 3000 System with a diode array detector, which was set at 260 nm for monitoring the effluents was used for all the HPLC separations. The analytes were separated at a flow rate of 0.5 ml min−1 on a Waters® XBridge™ OST C18 column (4.6 mm × 50 mm; 2.5 μm in particle size). The linear gradient (the flow rate was set at 0.5 ml min−1) of 0–20% B over 20 min was used (phase A: 50 mM TEAA + 1% ACN, phase B: 80% ACN).Preparation of chromatographic standards
Formation of strand breaks due to the exposure of the labeled oligonucleotides to ionizing radiation results in the formation of the respective monomeric (pT, Tp) and dimeric (pXT, TXp) fragments originating from the irradiated TYT trimers (X, Y and p stand for native, and iodinated nucleobase, and the phosphate group, respectively). Dimeric and monomeric standards were obtained by the enzymatic digestion of 2 μg of a TXT trimer. The fragments with the 3′ terminal phosphate group were obtained by the digestion of the native trimers with micrococcal nuclease (Sigma-Aldrich, Poland) (0.02 U μl−1) while the fragments containing the 5′ terminal phosphate were obtained by the digestion with P1 nuclease (Sigma-Aldrich, Poland) (0.2 U μl−1) – see Scheme S1 (ESI†). The retention times for the particular fragments are given in Table S1 (ESI†).Mass spectrometry
All analyses were conducted on an Agilent 1200 Technologies HPLC System coupled directly to an HCTultra ion-trap mass spectrometer (Bruker Daltonics). The same column and separation conditions were used as those described in the Chromatography section. MS operation parameters were as follows: the spray voltage was ±4.0 kV, the drying gas (N2) pressure: 50 psi, the flow rate: 11 l min−1 and the temperature was 365 °C. The spectra were registered in negative-ion mode. Each spectrum was obtained by averaging 3 scans, the time of each scan being 0.1 s.Quantum-chemical calculations
All quantum-chemical calculations were performed with the use of Gaussian 09 code. For all systems studied, (1) hydrogen of a nucleobase (C/U) from the C5 position was substituted with a Br or I atom and (2) the N1 position of each compound was substituted with the methyl group to mimic the N-glyosidic bond between the nucleobase and deoxyribose ring. The process of halogen anion release from its nucleobase anion radical was studied at the DFT/B3LYP level27–29 with the DGDZVP basis set30,31 augmented with one extra set of sp symmetry functions (DGDZVP++). The exponent of the diffuse set basis function was equal to 1/3 of the smallest exponent in the original basis for every symmetry. To mimic aqueous solution effects, we applied the polarizable continuum model (PCM,32 water). No geometrical constraints were used for geometry optimization and the stationary points’ characters (minima and first-order saddle point) were evaluated with harmonic frequency analysis. The differences between the electronic energies of the transition state and the stable radical anion, including zero-point vibration terms, thermal corrections to energy, pV and entropy terms were calculated as changes in the Gibbs free energy (ΔG*) for particular reactions.Results and discussion
Very recently we demonstrated, using the LC-MS technique, the radiosensitivity of four oligonucleotide TZT trimers labelled with bromonucleosides (Z stands for the brominated nucleoside).10 In order to compare those results with these concerning radiation-induced damage to oligonucleotides labelled with iodonucleosides, in the current work the deoxygenated aqueous solutions of trinucleotides labelled with 5-iodocytidine (5-IdC) or 5-iodouridine (5IdU) (TIUT or TICT, respectively; for structures see Fig. 1) were irradiated with IR. The present study is limited to 5-IdU and 5-IdC since oligonucleotides labelled with the remaining iodinated nucleosides, 8-iodo-2′-deoxyadenine (8-IdA) and 8-iodo-2′-deoxyguanine (8-IdG), are unstable and not offered by commercial oligonucleotide providers. The obtained radiolytes were analysed by the HPLC and LC-MS (LC/MS/MS) methods. Our results demonstrate unequivocally that iodopyrimidines are better sensitizers than bromopyrimidines. Although similar products are formed in the irradiated solutions containing the brominated trimers,10 the extent of damage is significantly larger in the iodinated systems. There are also several qualitative differences concerning the identity of products formed in the brominated and iodinated oligonucleotides.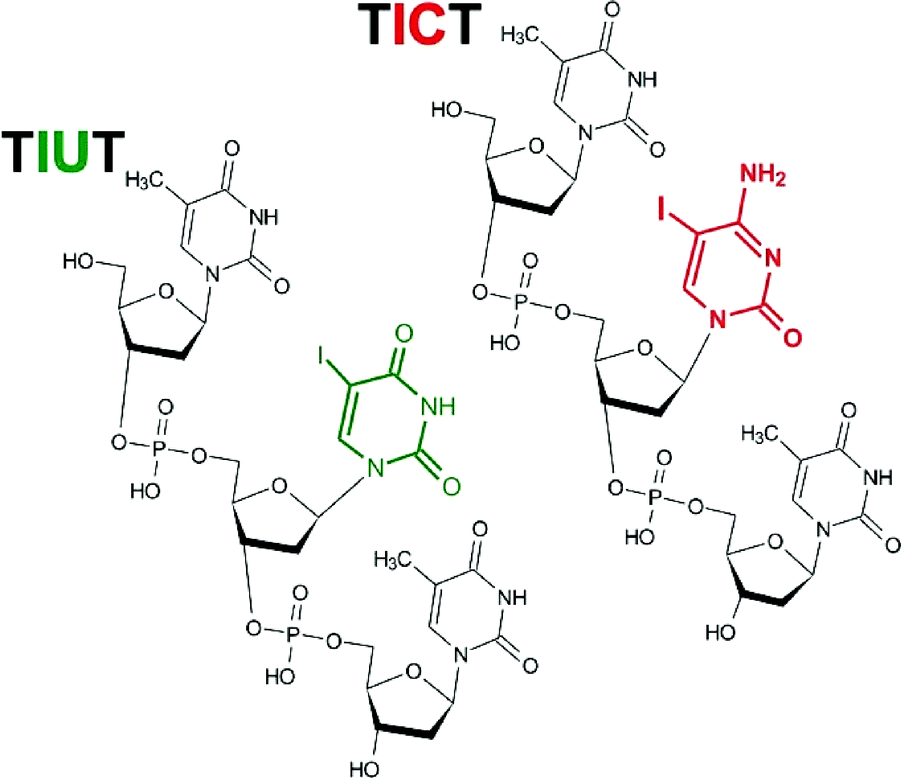 | ||
| Fig. 1 Chemical structures of the studied oligonucleotides. | ||
In Fig. 2 (upper panel) the exemplar chromatograms of radiolytes are shown. As indicated by the chromatograms, the applied dose of γ-radiation leads to the substantial and rather complex degradation of the studied trimers (cf.Fig. 2 with Fig. S1 and S2†). Since hydroxyl radicals were efficiently scavenged by Tris-HCl under our experimental conditions, the observed damage was exclusively induced by solvated electrons. In the lower panel of Fig. 2, the chemical structures corresponding to the particular chromatographic signals are displayed. The structures were deciphered using the chromatographic standards and/or LC-MS/MS analysis.
 | ||
| Fig. 2 HPLC analysis of γ-irradiated aqueous solution containing TIUT and TICT. | ||
The identity of HPLC signals denoted with numbers 3–5 and 7–11 (see Fig. 2 and Table 1) was determined by using chromatographic standards. Standards 3, 4, 5 and 7 were obtained via the enzymatic digestion of the TUT and TCT oligonucleotides, while the compounds 8 to 11 were purchased from Future Synthesis, Poland. Our assignments were additionally confirmed by the MS/MS analysis of the particular 3–5 and 7–11 HPLC signals (see Fig. S3 and S4 in the ESI†). On the other hand, the compounds corresponding to the remaining HPLC peaks, i.e. 1, 2 and 6 (see Fig. 2), were assigned to the use of LC-MS and MS/MS analysis alone (see Fig. S3 and S4 in the ESI†). In the later approach, we employed the fragmentation pattern of oligonucleotides described in the literature.33 Namely, the major fragmentation of electrospray-generated oligodeoxynucleotide anions involves the release of a base coupled to the cleavage of the 3′ C–O bond in the sugar moiety to which this base belongs. Then, a subsequent base is detached along with the phosphate group followed again by the release of the sugar moiety.27 Such a mechanism explains well the MS/MS spectra shown in Fig. S3 and S4.† For instance, in the course of fragmentation of pXTOH oligonucleotide, (m/z equal to 611 or 610 for pUTOH or pCTOH, respectively) (see Scheme 1 and Fig. S3 and S4†) 5′-base is released which leads to m/z = 499 for both oligonucleotides. Next, the phosphate group and sugar moiety are detached giving rise to m/z = 401 and 321 signals, respectively (see Fig. S3 and S4†). This mechanism is slightly modified for the iodinated trimers. Due to low energy of the C–I bond, the fragmentation process begins with the release of the iodine atom (see TIUT and TICT MS/MS spectra in Fig. S3 and S4,† respectively). Then thymine along with uracil for TIUT or along with cytosine for TICT is detached. Finally, the fragmentation pathway comprises the loss of the phosphate group followed by the release of the sugar residue which gives rise to the MS/MS signals corresponding to m/z = 401 and 321 (see Fig. S3 and S4†).
 | ||
| Scheme 1 Fragmentation pattern of PXTOH. | ||
| No. | Fragment | TIUT | TICT |
|---|---|---|---|
| 1 | dTI | 7.4 | 6.5 |
| 2 | dT![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
16.9 | 16.7 |
| 3 | PTOH | 1.5 | 3.0 |
| 4 | HOTP | 2.2 | 1.8 |
| 5 | PXTOH | 17.1 | 17.4 |
| 6 | OXTOH |
3.0 | 2.8 |
| 7 | HOXTOH | 6.9 | 4.5 |
| 8/9 | THOXT | 2.8 | 2.3 |
| 10/11 | TXT | 11.5 | 8.1 |
The γ-irradiation of deoxygenated aqueous solutions containing the iodinated trimer and a hydroxyl radical scavenger results in a complex mixture of degradation products (see the HPLC chromatograms shown in Fig. 2). The relative molar contributions of individual products gathered in Table 1 were calculated using the absorption coefficient presented in Table S1† and ref. 10. Adding up these contributions one obtains 58 and 55% of degradation for TIUT and TICT, respectively, which demonstrates that the iodinated pyrimidines are about twice as much better hypoxic radiosensitizers than the brominated ones. Indeed, the radiolysis of an aqueous solution of TBrUT and TBrCT under similar experimental conditions and with the same dose of IR leads to only 29 and 27 percent of degradation, respectively.10 It is worthy of note that such distinctive sensitizing effects are observed only in the single stranded form of DNA. Indeed, as was demonstrated by Cecchini et al.25 for BrdU labelled oligonucleotides, the double stranded form of the biopolymer strongly inhibits formation of single strand breaks induced by solvated electrons. Therefore, one can expect a similar effect for oligonucleotides labeled with 5-iodouridine or 5-iodocytosine. It should be emphasized that although double stranded DNA is a dominant form of the biopolymer in the cell, its single-stranded regions, such as those occurring in transcription bubbles, replication forks, DNA bulges, and the loop region of telomeres do exist under physiological conditions.13
Data gathered in Table 1 show that deiodination, leading to the TXT trimers, is only the third most abundant process induced by electron attachment while in the irradiated solutions of brominated trimers debromination is by far the most significant degradation channel.10 Thus, electron attachment to the iodinated trimers mainly triggers single strand breaks (SSBs). Indeed, the pXTOH dimer, the most copious product, as well as OXTOH and HOXTOH dimers, all occur due to the dissociation of the diphosphate bond at the 5′ side of the middle nucleoside. Their monomeric counterparts constitute fragments 1, 2 and 4 (see Fig. 2 and Table 1). The results gathered in Table 1 indicate a strong preference (similarly to the brominated TYT trimers10) toward cleavage of the 5′ side diphosphate bond (only a small amount of monomer 3 reveals the breakage of the phosphate bond at the 3′ side of the iodinated nucleoside (see Table 1)).
The analogue of dTI (see Fig. 2 and Table 1), dTBr, was not observed in the studies on the brominated trimers.10 Its formation observed in the current study may be explained by the attack of the iodide anion on the C3′ of the cation radical which is formed due to the beta elimination of the phosphate anion from the 2′ sugar radical in the trioligonucleotide.34,35 The latter radical is formed via hydrogen atom transfer from the 2′ site of 5′-neighbouring sugar to the uracil/cytosine radical (this uracil/cytosine radical is a product of the primary dissociative electron attachment (DEA) process).36 The formation of dTI in the current studies and no evidence for dTBr formation in the previous one10 may be explained by the difference in masses between the iodide and bromide anions. Probably, the dT radical cation forms before the heavier iodide leaves the reaction centre while the lighter bromide anion moves too far, in the time needed for the formation of the dT cation radical, to interact with it effectively (forming dTBr).
It is worth emphasizing that under identical experimental conditions (the same oligonucleotide concentration, OH˙ scavenger, IR dose etc.) the native trimers, TUT and TCT, do not undergo fragmentation.10 The only product observed in almost negligible amounts (1–3% at 140 Gy)10 is attributed to the hydrogenation of thymine that results in trimers containing 5,6-dihydrothymine.37 This fact demonstrates that some type of DNA radiosensitization is necessary, especially under hypoxia characteristics for solid tumor cells, in order to make radiotherapy work. Actually, the genotoxic properties of hydroxyl radicals are significantly impaired in hypoxic cells,1 while solvated electrons are not able to induce strand breaks (SBs) in native DNA (see the above discussion and ref. 10). Although the formation of SBs in DNA due to electron attachment under ultra-high vacuum was demonstrated many times in the past,38–40 the situation is very different in an aqueous solution, i.e. under physiological conditions, where the polar solvent forms activation barriers to SB formation and enables the nucleobase anion formed due to electron attachment to be protonated.41,42 Despite the fact that solvated electrons are easily transferred to nucleobases,36 a swift protonation of the anions leads to a significant increase of the activation barriers as well as the driving forces for strand breaks.43 Only modification of natural bases in a way which makes the respective anions unstable (note that the radical anions of brominated and iodinated uracil are, as opposed to the uracil radical anion itself, very unstable and dissociate forming a reactive uracil-5-yl radical and the halide anion with half-times of 7.0 and 1.7 ns, respectively)24 so that the protonation is kinetically hindered and electron attachment to a modified nucleobase becomes an irreversible process, which enables the solvated electrons to damage labelled DNA.
As was mentioned above the life-time of the BrU radical anion is ca. 4-fold longer than that of the IU anion.24 This experimental picture is corroborated by our quantum-chemical calculations. Namely, the activation energy for the release of the bromide anion from the 5-bromouracil (BrU) and 5-bromocytosine (BrC) anions in the free energy scale amounts to 0.6 and 0.4 kcal mol−1, respectively. The presence of a small kinetic barrier was confirmed at slightly different levels of theory both in the gas phase44,45 and aqueous solution44,46 as well as by our ab initio molecular dynamics calculations using a cubic box comprising the brominated bases and 67 explicit water molecules.47 On the other hand, the B3LYP/DGDZVP++ calculations for the IU/IC anions revealed that the iodide anion release occurs in a barrier-free fashion. These findings (a barrier for the brominated bases and the lack of barrier for the iodinated ones) correlate qualitatively with the difference of life-times of the BrU and IU anions in water as well as with the larger reactivity of the iodinated trimers toward solvated electrons demonstrated in the current work.
Conclusions
The propensity of iodopyrimidines incorporated into DNA in relation to damage induced by hydrated electrons has been studied for the first time. Using the LC-MS approach we analyzed the product mixture formed within the radiolysis of the aqueous solutions of iodinated trimers under hypoxic conditions and in the presence of a hydroxyl radical scavenger. Our results indicate unequivocally that substitution of the native pyrimidine nucleobase with the iodinated one makes the substituted oligonucleotide sensitive to solvated electrons. Attachment of an electron to the iodinated pyrimidine base triggers a barrier-free dissociation of the C5–I bond that ultimately leads mainly to strand breaks, i.e. to the dissociation of the diphosphate bond preferentially at the 5′ side of the iodinated base. The comparison between the yield of damage for the previously studied brominated trimers and the iodinated oligonucleotides investigated in the present work demonstrates that iodination leads to almost 2-fold higher radiosensitization as compared to bromination. The results of the present studies together with the previous radiation chemistry investigations on the brominated oligonucleotides as well as numerous clinical studies carried out so far with bromo- and iodouridine suggest that an appropriately modified nucleoside(s) may play a role of universal radiosensitizer(s). We, therefore, believe that our efforts in searching for a radiosensitizing nucleoside will eventually result in a molecular system that will be commonly used in future radiotherapy.Acknowledgements
J. R. would like to thank Miss Samanta Makurat for quantum chemical calculations concerning the DEA process in the iodinated uracil and cytosine. The work was supported by the Polish National Science Centre (NCN) under the Grant No. 2012/05/B/ST5/00368 (J. R.). The support by the CMST COST Action CM1201 “Biomimetic Radical Chemistry” is kindly acknowledged (K. B. & J. R.).References
- B. T. Oronsky, T. Bryan, S. J. Knox and J. Scicinski, Transl. Oncol., 2011, 4, 189–198 CrossRef PubMed.
- R. D. Stewart, K. Y. Victor, A. G. Georgakilas, C. Koumenis, J. H. Park and D. J. Carlson, Radiat. Res., 2011, 176, 587–602 CrossRef CAS PubMed.
- M. Joiner and A. van der Kogel, Basic Clinical Radiobiology, Hodder Arnold, London, 2009 Search PubMed.
- J. Rak, L. Chomicz, J. Wiczk, K. Westphal, M. Zdrowowicz, P. Wityk, M. Żyndul, S. Makurat and Ł. Golon, J. Chem. Phys. B, 2015, 119, 8227–8238 CrossRef CAS PubMed.
- P. Wardman, Clin. Oncol., 2007, 19, 397–417 CrossRef CAS PubMed.
- L. S. Lee and Y. Cheng, Biochemistry, 1976, 15, 3686–3690 CrossRef CAS PubMed.
- B. Goz, Pharmacol. Rev., 1977, 29, 249–272 CAS.
- C. von Sonntag, Free-Radical-Induced DNA Damage and Its Repair, A Chemical Perspective, Springer-Verlag, Heidelberg, Germany, 2006 Search PubMed.
- F. J. Nabben, J. P. Karman and H. Loman, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1982, 42, 23–30 CrossRef CAS.
- K. Westphal, J. Wiczk, J. Miloch, G. Kciuk, K. Bobrowski and J. Rak, Org. Biomol. Chem., 2015, 13, 10362–10369 CAS.
- J. Zimbrick, J. Ward and L. Myers, Int. J. Radiat. Biol., 1969, 16, 505–523 CAS.
- W. Knapp-Pogozelski and T. D. Tullius, Chem. Rev., 1998, 98, 1089–1108 CrossRef.
- L. Sanche, Radiat. Phys. Chem., 2016 DOI:10.1016/j.radphyschem , 2016.05.008.
- B. Djordjevic and W. Szybalski, J. Exp. Med., 1960, 112, 509–531 CrossRef CAS PubMed.
- R. Watanabe and H. Nikjoo, Int. J. Radiat. Biol., 2002, 78, 953–966 CrossRef CAS PubMed.
- D. S. Shewach and T. S. Lawrence, in Deoxynucleoside Analogs in Cancer Therapy, ed. G. J. Peters, Humana Press Inc., Totowa, New Jersey USA, 2006, pp. 289–331 Search PubMed.
- J. J. Jaffe and W. H. Prusoff, Cancer Res., 1960, 20, 1383–1388 CAS.
- J. Sohaller, M. Gordon and S. S. Stenberg, Proc. Soc. Exp. Biol. Med, 1965, 93, 124–128 CrossRef.
- A. H. Epstein, R. S. Lebovics, T. Goffman, D. Teague, E. S. Fuetsch, E. Glatstein, P. Okunieff and J. A. Cook, J. Natl. Cancer Inst., 1994, 86, 1775–1780 CrossRef CAS PubMed.
- T. Goffman, Z. Tochner and E. Glatstein, Cancer, 1991, 67, 572–576 CrossRef CAS PubMed.
- J. M. Robertson, V. K. Sondak, S. A. Weiss, J. J. Sussman, A. E. Chang and T. S. Lawrence, Int. J. Radiat. Oncol., Biol., Phys., 1995, 31, 87–92 CrossRef CAS.
- R. L. Erikson and W. Szybalski, Radiat. Res., 1963, 20, 252–262 CrossRef CAS PubMed.
- T. J. Kinsella, A. Russo, J. B. Mitchell, J. M. Collins, J. Rowland, D. Wright and E. Glastein, Int. J. Radiat. Oncol., Biol., Phys., 1985, 11, 194–196 Search PubMed.
- E. Rivera and R. H. Schuler, J. Phys. Chem., 1983, 87, 3966–3971 CrossRef CAS.
- S. Cecchini, S. Girouard, M. A. Huels, L. Sanche and D. J. Hunting, Radiat. Res., 2004, 162, 604–615 CrossRef CAS PubMed.
- S. Cecchini, S. Girouard, M. A. Huels, L. Sanche and D. J. Hunting, Biochemistry, 2005, 44, 1932–1940 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- C. Sosa, J. Andzelm, B. C. Elkin, E. Wimmer, K. D. Dobbs and D. A. Dixon, J. Phys. Chem., 1992, 96, 6630–6636 CrossRef CAS.
- A. Yurieva, O. K. Poleshchuk and V. Filimonov, J. Struct. Chem., 2008, 49, 548–552 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- K. X. Wan, J. Gross, F. Hillenkamp and M. L. Gross, J. Am. Soc. Mass Spectrom., 2001, 12, 193–205 CrossRef CAS PubMed.
- M. Dizdaroglu, C. von Sonntag and D. J. Schulte-Frohlinde, J. Am. Chem. Soc., 1975, 97, 2277–2278 CrossRef CAS PubMed.
- R. Glatthar, M. Spichty, A. Gugger, R. Batra, W. Damm, M. Mohr, H. Zipse and B. Giese, Tetrahedron, 2000, 56, 4117–4128 CrossRef CAS.
- P. Schyman, R. B. Zhang, L. A. Eriksson and A. Laaksonen, Phys. Chem. Chem. Phys., 2007, 9, 5975–5979 RSC.
- J. M. Falconea, D. Becker, M. D. Sevilla and S. G. Swarts, Radiat. Phys. Chem., 2005, 72, 257–264 CrossRef.
- B. Boudaiffa, P. Cloutier, D. Hunting, M. A. Huels and L. Sanche, Science, 2000, 287, 1658–1660 CrossRef CAS PubMed.
- L. Sanche, in Radical and Radical Ion Reactivity in Nucleic Acid Chemistry, ed. M. M. Greenberg, Johns Wiley and Sons Inc., Hoboken, New Jersey, USA, 2009, pp. 239–295 Search PubMed.
- E. Alizadeh, T. M. Orlando and L. Sanche, Annu. Rev. Phys. Chem., 2015, 66, 379–398 CrossRef CAS PubMed.
- C. von Sonntag and H. P. Schuchmann, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1986, 49, 1–34 CrossRef CAS.
- S. Steenken, Chem. Rev., 1989, 89, 503–520 CrossRef CAS.
- M. McAllister, M. Smyth, B. Gu, G. A. Tribello and J. Kohanoff, J. Phys. Chem. Lett., 2015, 6, 3091–3097 CrossRef CAS PubMed.
- S. D. Wetmore, R. J. Boyd and L. A. Eriksson, Chem. Phys. Lett., 2001, 343, 151–158 CrossRef CAS.
- X. Li, L. Sanche and M. D. Sevilla, J. Phys. Chem. A, 2002, 106, 11248–11253 CrossRef CAS.
- L. Chomicz, J. Rak and P. Storoniak, J. Phys. Chem. B, 2012, 116, 5612–5619 CrossRef CAS PubMed.
- M. Wieczór, P. Wityk, J. Czub, L. Chomicz and J. Rak, Chem. Phys. Lett., 2014, 595, 133–137 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Digestion of a TXT trimer by Micrococcal (MC) or P1 nuclease (P1), HPLC chromatograms of aqueous solutions of TICT and TIUT, MS/MS spectra of gamma irradiated aqueous solution of TIUT and TICT, molar absorption coefficients of iodinated compounds. See DOI: 10.1039/c6ob01713d |
| This journal is © The Royal Society of Chemistry 2016 |