
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A ThDP-dependent enzymatic carboligation reaction involved in Neocarazostatin A tricyclic carbazole formation†
Li
Su‡
a,
Meinan
Lv‡
a,
Kwaku
Kyeremeh
b,
Zixin
Deng
a,
Hai
Deng
c and
Yi
Yu
*a
aKey Laboratory of Combinatorial Biosynthesis and Drug Discovery (Ministry of Education), School of Pharmaceutical Sciences, Wuhan University, 185 East Lake Road, Wuhan 430071, P.R. China. E-mail: yu_yi@whu.edu.cn
bDepartment of Chemistry, University of Ghana, P.O. Box LG56, Legon-Accra, Ghana
cMarine Biodiscovery Centre, Department of Chemistry, University of Aberdeen, Aberdeen AB24 3UE, Scotland, UK
First published on 1st September 2016
Abstract
Although the biosynthetic pathway of Neocarazostatin A (1) has been identified, the detailed enzymatic reactions underlying the assembly of the carbazole ring still remain largely unknown. We demonstrate here that NzsH, a putative thiamine diphosphate dependent enzyme, can catalyze an acyloin coupling reaction between indole-3-pyruvate and pyruvate to generate a β-ketoacid intermediate. Our findings thus shed light on further characterization of the unusual biosynthetic pathway of the bacterial tricyclic carbazole alkaloids.
Introduction
Carbazoles are a group of organic compounds consisting of a tricyclic nucleus with two benzene rings flanking a pyrrole ring.1 The majority of carbazole secondary metabolites have been found in higher plants and fungi.1 However, only a handful of carbazole metabolites of bacterial origin have been identified. Bacterial carbazole natural products generally display strong inhibitory activity against free radical-induced lipid peroxidation.2 Therefore, there has been considerable effort on the part of medicinal chemists to develop this class of compounds as potential radical scavengers. Neocarazostatin A (1) belongs to the bacterial carbazole alkaloids with an isoprene moiety at C-6 of the carbazole ring (Scheme 1).1 It was first isolated from the culture of Streptomyces sp. GP38.2 We have recently confirmed that the Ghanaian soil isolate Streptomyces sp. MA37 produces 1. Genome mining analysis and genetic interrogation in Streptomyces sp. MA37 allowed the identification of the biosynthetic gene cluster of 1, which consists of ten genes (Fig. 1A).3 The genes nzsB, C, D, and E, encoding the putative anthranilate phosphoribosyltransferase, isopentenyl diphosphate delta-isomerase, phenylalanine aminotransferase, and acyl carrier protein, respectively, were predicted to be involved in the precursor supply. Gene knockout experiments demonstrated that the gene products of nzsH, I, and J are essential for the biosynthesis of 1, suggesting that they are responsible for the assembly of the carbazole skeleton of 1.3 Biochemical characterization demonstrated that the phytoene synthase-like prenyltransferase NzsG and the P450 hydroxylase NzsA mediate the coupled reaction of prenylation and hydroxylation in the late stage of the biosynthesis of 1, respectively.3 While the tailoring steps of the biosynthesis of 1 have been characterized, questions concerning the carbon backbone assembly of 1 remain unaddressed, including the chemical logic underlying the construction of the intriguing carbazole “A” ring of 1 (Scheme 1).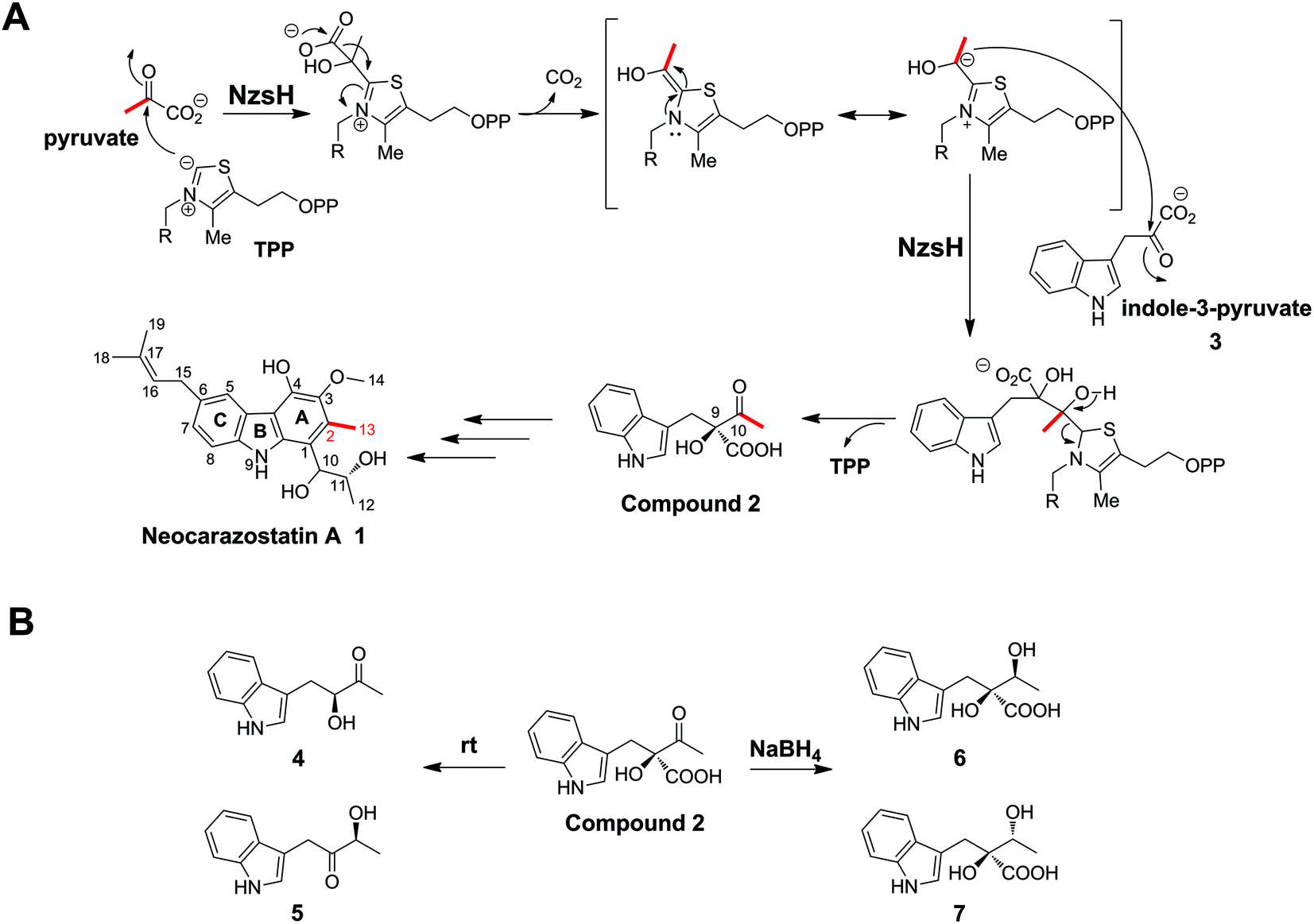 | ||
| Scheme 1 NzsH catalyzed carboligation reaction. (A) Proposed mechanism underlying the NzsH catalyzed reaction in the biosynthesis of Neocarazostatin A. (B) The transformation of compound 2. Compound 2 can transformed into isomeric acyloins 4 and 5 after incubation at room temperature for 2 h, or transformed into diastereomeric diols 6 and 7 by treating with NaBH4 for 1 h. | ||
 | ||
| Fig. 1 In vivo characterization of NzsH. (A) The biosynthetic gene cluster of Neocarazostatin A. (B) HPLC traces of the extract of (i) the ΔnzsH complementation mutant WDY631; (ii) wild type Streptomyces sp. MA37; and (iii) ΔnzsH mutant WDY630. UV was monitored at 247 nm. | ||
Stable isotopic feeding experiments have shown that the carbon skeleton of ring A in carbazomycin B, a structurally close analogue of 1, originates from tryptophan and pyruvate, and the intact C2 unit of C-2 and C-11 in ring A (corresponding to C-2 and C-13 in 1, see Scheme 1) may result from a condensation reaction of pyruvate and the tryptophan derived indole-3-pyruvate.4 Such an incorporation of pyruvate into natural products led us to speculate that a thiamin diphosphate (ThDP)-dependent enzyme may be involved in the assembly of ring A in 1. ThDP-dependent enzymes are ubiquitously found in the biological system, and have been demonstrated to be involved in diverse biotransformation, including C–C, C–N, C–S, and C–O bond cleavage and formation.5,6 The general mechanism underlying all these reactions is that the enzyme's active site-mediated dissociation of the C2–H proton from the thiazolium ring of ThDP will generate the C2 anion/ylid (ThDP−), which is covalently bound to the donor substrate to give an activated aldehyde intermediate with nucleophilic reactivity (Scheme 1).7,8 The proposed mechanism mediated by these enzymes is the decarboxylation of an α-keto acid, followed by carboligation of the active ThDP-bound enolate intermediate with an acyl acceptor to form the α-hydroxyl ketone (an acyloin). For example, YerE, a ThDP-dependent enzyme involved in the biosynthesis of yersiniose A, is able to catalyze the decarboxylation of pyruvate and then transfer the activated acetaldehyde onto the 3-ketosugar to give the hydroxyethyl branched chain of yesiniose A.9,10 Another example comes from the biosynthetic pathway of scytonemin, a pigment produced by cyanobacteria. The in vitro characterization of the ThDP-dependent enzyme ScyA showed that it is responsible for coupling indole-3-pyruvate and p-hydroxyphenylpyruvate to afford a β-ketoacid product, which is a key intermediate in the biosynthesis of scytonemin.11
In this study, we report a detailed investigation of NzsH, a putative ThDP-dependent enzyme encoded in the biosynthetic gene cluster of 1. Genetic and biochemical characterization of NzsH demonstrates that this enzyme plays a pivotal role in the biosynthesis of 1 by performing an acyloin coupling reaction between indole-3-pyruvate and pyruvate to generate the β-ketoacid intermediate 2 (Scheme 1). We also show that NzsH, while displaying a narrow substrate range, is able to recognize 2-oxobutyrate as the alternative acyl donor to give a novel compound. Evolution analysis of NzsH further suggests that it has diverged from the ancestor of other groups of ThDP-dependent enzymes and represents a new branch of acetolactate synthases.
Results and discussion
We have demonstrated the essential role played by NzsH in the biosynthesis of 1 by showing that the production of 1 was completely abolished in the ΔnzsH mutant (Fig. 1B). To further confirm the in vivo role of NzsH, we constructed the ΔnzsH complement strain (WDY631) in which the gene nzsH was overexpressed under the control of the constitutive promoter ermE*. Surprisingly, high-performance liquid chromatography (HPLC) analysis of the extract from WDY631 revealed that this strain yielded a nearly 5-fold higher titer of 1 than the wild type strain (Fig. 1B). Such a significant improvement of the production of 1 was not observed in other nzs genes’ complement strains, implying that the NzsH mediated reaction is a rate-limiting step during the biosynthesis of 1. To clarify this hypothesis, expression patterns of nzsA-J in both the wild type strain and WDY631 were analysed by reverse transcription polymerase chain reaction (RT-PCR). As shown in Fig. 1C, the transcription level of nzsH was the lowest among all the biosynthetic genes of 1 in the wild type strain, while in WDY631 which produces a much higher titer of 1, the transcription level of nzsH was increased significantly. This result suggests that the expression level of nzsH should directly correlate with the production of the titer of 1 in the host strain. The above finding can also be applied to improve the production of intermediates with trace amounts in the biosynthetic pathway of 1 by transforming the nzsH-overexpression construct into corresponding nzs gene deletion mutants.Sequence alignment indicated that NzsH has moderate similarity (35% identity on average) with the group of acetolactate synthases (ALS), which belongs to the pyruvate decarboxylase (PDC)-like superfamily of ThDP-dependent enzymes.12 ALS generally catalyzes the decarboxylation of pyruvate to yield 2-acetolactate with a second molecule of pyruvate (Fig. S1†).12,13 These pieces of information thus led us to propose that NzsH may be responsible for a carboligation reaction between indole-3-pyruvate (3) and pyruvate to generate the key intermediate 2 during the assembly of ring A (Scheme 1).
To explore the validity of this hypothesis, NzsH was overexpressed in E. coli as an N-terminal hexahistidine-tagged fusion protein and purified to near homogeneity by Ni2+-affinity chromatography (Fig. S2†). The molecular weight of the resultant protein was measured to be ∼64 kDa on SDS-PAGE. Further native-PAGE analysis of NzsH indicated an approximate molecular mass of 120 kDa (data not shown), suggesting that the quaternary structure of NzsH appears to be homodimeric. The appearance of the purified NzsH was bright yellow, and the UV-vis spectroscopic analysis of the protein solution showed an absorbance maximum at ∼450 nm, indicating the presence of a flavin cofactor. We then performed a biochemical reaction by incubating NzsH with both indole-3-pyruvate and pyruvate in the presence of MgCl2 and ThDP (see the ESI†). HPLC analysis of the reaction revealed the appearance of a new peak with the retention time of 12.5 min (Fig. 2A). The chemical formula of the new product was determined by High Resolution-Electrospray Ionization Mass Spectrometry (HR-ESIMS) to be C13H14NO4+ ([M + H]+ calc. 248.0914, −1.2 ppm error), and its MS/MS fragmentation pattern is in good agreement with the structure of the predicted intermediate 2 (Scheme 1, Fig. 2B). To elucidate the real structure of the new product, we set out to isolate and purify the compound from a large-scale enzymatic preparation. However, HPLC analysis of the purified sample showed a peak with a different retention time than the original product (Fig. 2A). HR-ESIMS analysis indicated that the calculated molecular formula of this peak is C12H14NO2+ (compounds 4 and 5, [M + H]+ obsd: 204.1020, calc. 204.1020, 0 ppm error, Fig. S3A†), the molecular mass of which is 44 Da different from the proposed compound 2. It was speculated that the new compound may arise from a spontaneous decarboxylation of 2 during the chemical isolation; this reaction was also observed in a previous report.11 To prevent the decarboxylation event, we then treated the NzsH reaction mixture with NaBH4. HPLC analysis of the resulting sample revealed two new compounds (Fig. S3A†), 6 and 7, which have identical molecular formula (C14H18NO4+) and molecular masses ([M + H]+ obsd: 250.1075, calc. 250.1074, 0.4 ppm error) as observed in HR-ESI/MS and tandem MS/MS. The structure of 6 was further characterized through 1-D and 2-D NMR analyses (1H, 13C, HSQC, HMBC, and 1H–1H COSY, Fig. S4–S8†). By a comparison of the 1H and 13C NMR spectra of 6 and 7 (Fig. S4, S5 and S9†), we confirmed that 7 is the enantiomer of 6. Both compounds should have derived from the reduction of a single β-ketoacid regioisomer 2 (Scheme 1). Therefore, NzsH should belong to acetohydroxy acid synthase (AHAS), a subgroup of the ThDP-dependent ALS superfamily. AHAS has been characterized to have a strong preference for catalyzing the formation of (S)-acetolactate (Fig. S1†).14 For example, incubation of pyruvate with 2-oxobutyrate in the presence of YerE led to the formation of both (S)-acetolactate and (S)-acetohydroxybutyrate.15 ScyA has also been shown to catalyze a regioselective acyloin coupling reaction to afford a β-ketoacid product in (S) configuration.11 Hence, there is a strong preference for generating products with (S) configuration in ALS-type enzyme catalyzed reactions. A comparison of the 1H NMR data of 6 and 7 with the two diastereomeric diols generated from the reduction of a single β-ketoacid regioisomeric product from the reaction mixture of ScyA indicated similar chemical shifts,11 suggesting that the relative stereochemistry at C9 should be the (S) configuration. Taken together, we proposed that the absolute stereochemistry at C9 of 2 should be the (S) configuration. These results demonstrate that NzsH catalyzes an acyloin coupling reaction between indole-3-pyruvate and pyruvate to generate the intermediate 2 during the biosynthesis of 1. Significantly, this carboligation reaction between indole-3-pyruvate and pyruvate has not been known for other reported ThDP-dependent enzymes.
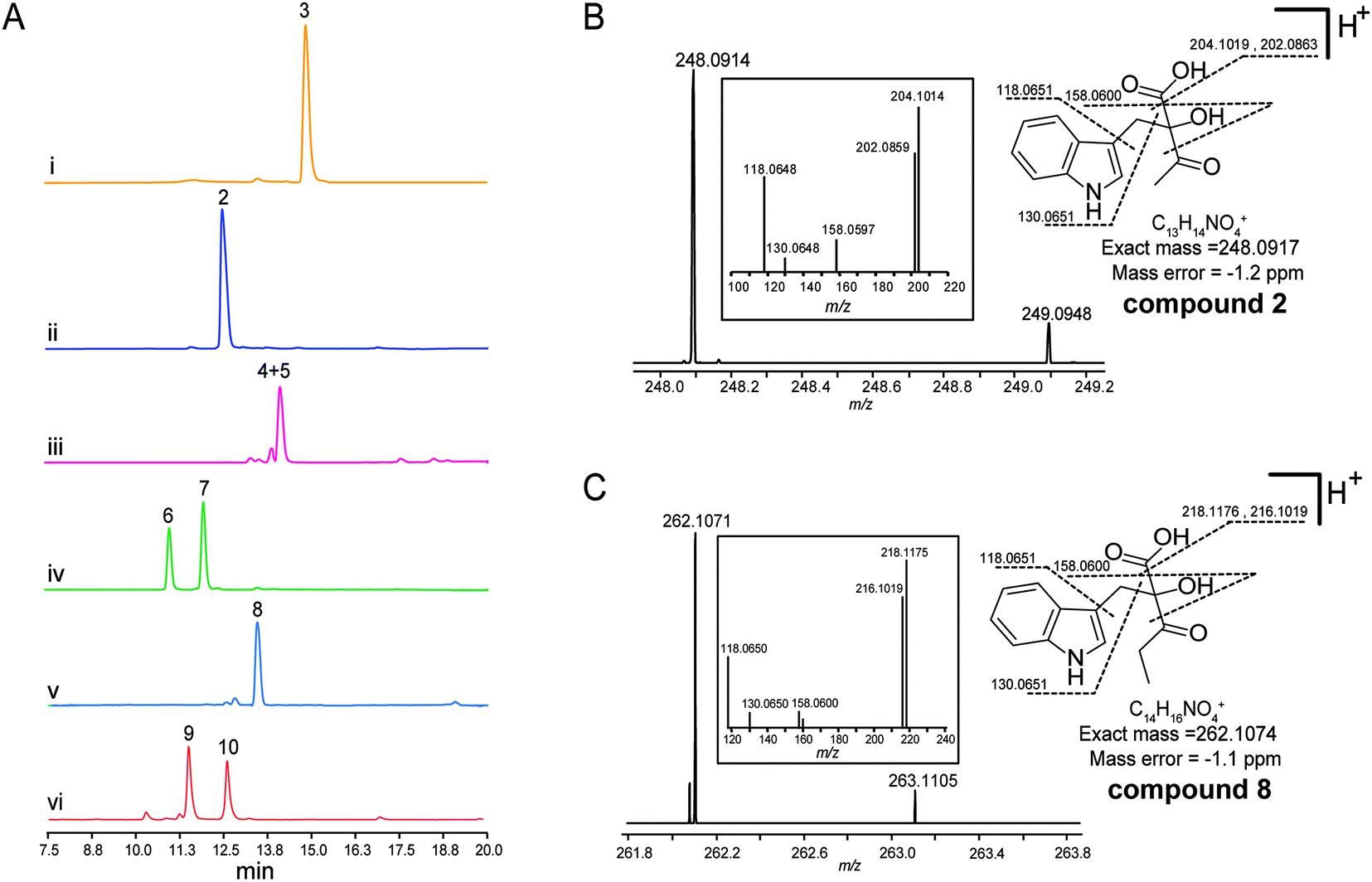 | ||
| Fig. 2 In vitro characterization of NzsH catalyzed reactions. (A) HPLC profiles of the reaction samples. (i) Control reaction using pyruvate and indole-3-pyruvate (3) and boiled NzsH; (ii) NzsH catalyzed reaction with pyruvate and indole-3-pyruvate, detected immediately after quenching; (iii) NzsH catalyzed reaction with pyruvate and indole-3-pyruvate, detected after incubation of the quenched reaction mixtures at room temperature; (iv) NaBH4 catalyzed reductive transformation of compound 2; (v) NzsH catalyzed reaction with 2-oxobutyrate and indole-3-pyruvate, detected immediately after quenching; (vi) NaBH4 catalyzed reductive transformation of compound 8. UV was monitored at 280 nm. (B) and (C) HR-ESI-MS analysis of compounds 2 and 8. | ||
Several ThDP-dependent enzymes have been shown to be able to accept a broad range of substrates; this expands their applicability in asymmetric chemoenzymatic synthesis.5 Müller and colleagues have found that besides the physiological substrate CDP-3,6-di(deoxy)-4-keto-D-glucose, YerE can also accept cyclic and open-chain ketones as well as diketones and α- and β-ketoesters as the acetaldehyde acceptor in the ThDP mediated carboligation reaction.10 The same research group also found that another ThDP-dependent enzyme, cyclohexane-1,2-dione hydrolase (CDH), can catalyze the asymmetric cross-benzoin reaction utilizing hydroxybenzaldehydes and nitrobenzaldehydes as the acceptor substrates.16 With the recombinant protein on hand, we then set out to probe the substrate tolerance of NzsH using different derivatives of indole-3-pyruvate and pyruvate, respectively (Table S1†). Unlike other biochemically characterized ThDP-dependent enzymes with broad substrate promiscuity, NzsH showed a rather narrow substrate range, and was unable to utilize most of the derivatives we tested as the substrates. However, NzsH is able to mediate the biotransformation of indole-3-pyruvate and 2-oxobutyrate to generate a new product as observed in the HPLC analysis (Fig. 2A). HR-ESIMS analysis and MS/MS fragmentation suggest that the molecular formula of this compound is C14H16NO4+ ([M + H]+ calc. 262.1074, −1.1 ppm error), which is consistent with the protonated form of compound 8 (Fig. 2C). Direct isolation from the enzymatic mixture failed to generate the expected compound due to spontaneous decarboxylation. Thus we used the aforementioned NaBH4-based derivatization method and generated its reductive derivatives (9 and 10). These two diol derivatives were then isolated and their structures were confirmed through 1H-NMR analysis and by a comparison with that of compound 6 (Fig. S3B and S10†). This finding indicates that NzsH can also catalyze the coupling of indole-3-pyruvate and 2-oxobutyrate, which prompted us to examine whether 2-oxobutyrate can be incorporated into the biosynthetic pathway of 1 to generate its structural analogue. We then fed Streptomyces sp. MA37 with 2-oxobutyrate (50 mM L−1) during the fermentation process. However, no detectable signal of any new compound was observed in the fermentation culture (data not shown), implying that the whole biosynthetic machinery of 1 could not accept substrates other than pyruvate, or perhaps 2-oxobutyrate is degraded by β-oxidation faster than it can be incorporated into the biosynthetic pathway of 1.
To gain insights into the substrate preference of NzsH, we next performed a kinetic assay of NzsH for both pyruvate and 2-oxobutyrate. As shown in Fig. 3, NzsH has a higher affinity for pyruvate (341.6 μM) over 2-oxobutyrate (2180 μM) as measured by KM in the presence of indol-3-pyruvate (2 mM). Moreover, the catalytic efficiency (kcat/KM) of NzsH for pyruvate was ∼8.7-fold higher than that for 2-oxobutyrate, suggesting that pyruvate is a preferred substrate of NzsH.
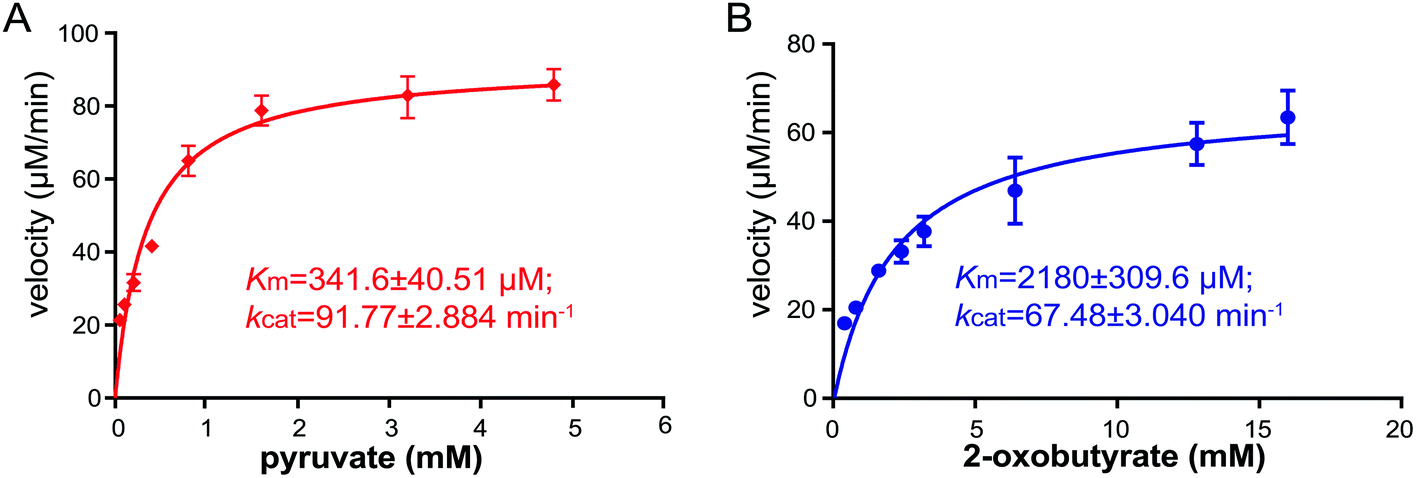 | ||
| Fig. 3 The kinetic analysis of NzsH with pyruvate (A) and 2-oxobutyrate (B) as substrates. The enzyme concentration of NzsH for both assays is 1 μM. Assays were performed in triplicates, and the standard deviations (S.D.) are shown as error bars. | ||
ThDP-dependent enzymes are highly diverse in their sequences, but can be grouped into nine superfamilies based on their structural architectures.17,18 Interestingly, NzsH shares low sequence identity (27%) with ScyA although both enzymes utilize the same substrate, indole-3-pyruvate, which makes it an interesting issue to investigate the phylogenetic relationship of NzsH with ScyA and other ThDP-dependent enzymes, such as YerE. To this end, we set out to construct a Bayesian Markov chain Monte Carlo (MCMC) tree of NzsH and its analogs belonging to the decarboxylases (DC) superfamily, which includes the ALS (Fig. 4). The MCMC tree is consistent with the previous evolutionary analysis of the ThDP-dependent enzyme family that ALS and other pyruvate decarboxylase (PDC)-like enzymes including InPDC (indole-3-pyruvate decarboxylase), BAL (benzaldehyde lyase), BFD (benzoylformate decarboxylase), MenD, and CDH form distinct clades within the DC superfamily.19 Notably, NzsH, YerE, and ScyA form three different branches from ALS enzymes, implying that they may have evolved from a common ancestor of an ALS-like enzyme and diverged from each other by recruiting different mutations in sequence during gene duplications.12,17 Although this is a highly speculative proposal, the above phylogenetic analysis, combined with available protein structure information, may guide us to understand in detail how the evolution of ThDP-dependent enzymes determines their highly diverse catalytic chemistries in the future. Taken together, NzsH represents a unique ThDP-dependent enzyme with respect to its reactivity characteristics and amino acid sequence.
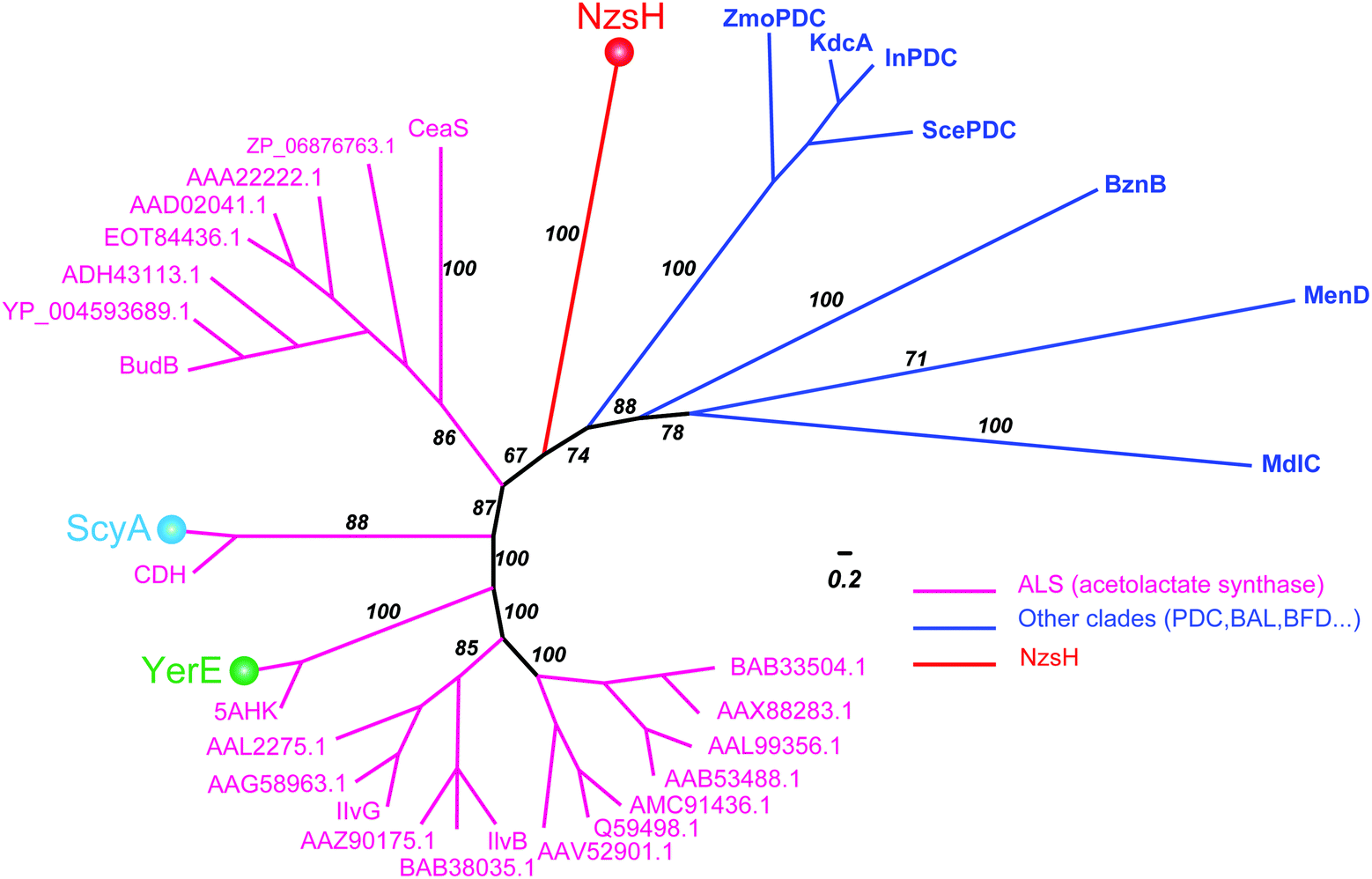 | ||
| Fig. 4 Bayesian phylogenetic tree for NzsH homologs and other groups of ThDP-dependent enzymes. The Bayesian posterior probability is indicated along the branches, which are colored to represent different clades of ThDP-dependent enzymes. Magenta represents the ALS (acetolactate synthase) clade, blue represents other clades containing PDC (pyruvate decarboxylase), InPDC (indole-3-pyruvate decarboxylase), BAL (benzaldehyde lyase), and BFD (benzoylformate decarboxylase). NzsH, YerE, and ScyA are highlighted with balls in different colors, respectively. The scale bar measures the average number of substitutions per site. | ||
Conclusion
The bacterial tricyclic carbazole alkaloid 1 has attracted much interest from researchers not only for its promising bioactivity against free radicals, but also for the intriguing enzymatic reactions underlying the assembly of the carbazole ring. In this study, our in vivo genetic experiments and RT-PCR analysis demonstrate that the gene nzsH is essential for the biosynthesis of 1, and the biotransformation mediated by NzsH is likely to be the rate-limiting step in the process. We also present biochemical evidence that the ThDP-dependent enzyme NzsH catalyzes the acyloin condensation reaction between indole-3-pyruvate and pyruvate, possibly the first enzymatic step to initiate the assembly of the carbazole “A” ring in the biosynthesis of 1. To our knowledge, no other ThDP-dependent enzyme that can utilize pyruvate as the C2 unit donor and indole-3-pyruvate as the acceptor in a C–C bond formation reaction has been reported. Additionally, NzsH displays a narrow substrate tolerance towards other acyl donors except 2-oxobutyrate. Moreover, evolutionary analysis indicates that NzsH has diverged from its ALS-like homologs, including YerE and ScyA, which catalyze similar carboligation reactions in natural product biosynthesis, suggesting that NzsH may represent a new branch of ThDP-dependent enzymes in the DC superfamily.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31570033 to Y. Y.) and the Leverhulme Trust-Royal Society Africa Award (AA090088 to K. K and H. D.).References
- A. W. Schmidt, K. R. Reddy and H. J. Knolker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed.
- S. Kato, K. Shindo, Y. Kataoka, Y. Yamagishi and J. Mochizuki, J. Antibiot., 1991, 44, 903–907 CrossRef CAS PubMed.
- S. Huang, S. S. Elsayed, M. Lv, J. Tabudravu, M. E. Rateb, R. Gyampoh, K. Kyeremeh, R. Ebel, M. Jaspars, Z. Deng, Y. Yu and H. Deng, Chem. Biol., 2015, 22, 1633–1642 CrossRef CAS PubMed.
- M. Kaneda, T. Kitahara, K. Yamasaki and S. Nakamura, J. Antibiot., 1990, 43, 1623–1626 CrossRef CAS PubMed.
- M. Müller, D. Gocke and M. Pohl, FEBS J., 2009, 276, 2894–2904 CrossRef PubMed.
- R. Kluger and K. Tittmann, Chem. Rev., 2008, 108, 1797–1833 CrossRef CAS PubMed.
- B. Shaanan and D. M. Chipman, FEBS J., 2009, 276, 2447–2453 CrossRef CAS PubMed.
- A. Schellenberger, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1998, 1385, 177–186 CrossRef CAS.
- H. W. Chen, Z. H. Guo and H. W. Liu, J. Am. Chem. Soc., 1998, 120, 11796–11797 CrossRef CAS.
- P. Lehwald, M. Richter, C. Rohr, H. W. Liu and M. Müller, Angew. Chem., Int. Ed., 2010, 49, 2389–2392 CrossRef CAS PubMed.
- E. P. Balskus and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 15260–15261 CrossRef CAS PubMed.
- V. Gedi and M. Y. Yoon, FEBS J., 2012, 279, 946–963 CrossRef CAS PubMed.
- K. Gokhale and B. Tilak, Curr. Drug Targets, 2015, 16, 689–699 CrossRef CAS PubMed.
- D. Chipman, Z. A. Barak and J. V. Schloss, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1998, 1385, 401–419 CrossRef CAS.
- M. Beigi, S. Loschonsky, P. Lehwald, V. Brecht, S. L. A. Andrade, F. J. Leeper, W. Hummel and M. Müller, Org. Biomol. Chem., 2013, 11, 252–256 CAS.
- S. Loschonsky, S. Waltzer, S. Fraas, T. Wacker, S. L. Andrade, P. M. Kroneck and M. Müller, ChemBioChem, 2014, 15, 389–392 CrossRef CAS PubMed.
- C. Vogel and J. Pleiss, Proteins, 2014, 82, 2523–2537 CrossRef CAS PubMed.
- R. G. Duggleby, Acc. Chem. Res., 2006, 39, 550–557 CrossRef CAS PubMed.
- S. J. Costelloe, J. M. Ward and P. A. Dalby, J. Mol. Evol., 2008, 66, 36–49 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob01651k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |