
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDisrupting the PCSK9/LDLR protein–protein interaction by an imidazole-based minimalist peptidomimetic†
Mattia
Stucchi
*ab,
Giovanni
Grazioso
 *c,
Carmen
Lammi
*c,
Silvia
Manara
c,
Chiara
Zanoni
c,
Anna
Arnoldi
c,
Giordano
Lesma
a and
Alessandra
Silvani
a
*c,
Carmen
Lammi
*c,
Silvia
Manara
c,
Chiara
Zanoni
c,
Anna
Arnoldi
c,
Giordano
Lesma
a and
Alessandra
Silvani
a
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy. E-mail: mattia.stucchi@unimi.it
bDipartimento di Scienze della Vita, Università di Modena e Reg-gio Emilia, via G. Campi 103, 41125 Modena, Italy
cDipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, Via L. Mangiagalli 25, 20133 Milano, Italy. E-mail: giovanni.grazioso@unimi.it; carmen.lammi@unimi.it
First published on 27th September 2016
Abstract
Herein we report on the multicomponent synthesis of a novel imidazole-based compound, able to act efficiently as a minimalist β-strand mimic. Biological evaluation proved its ability to impair the LDLR–PCSK9 protein–protein interaction, disclosing it as the first small molecule exerting a PCSK9-mediated hypocholesterolemic effect.
Introduction
Protein–protein interactions (PPIs) determine the biological role of the relative proteins. Only in the last decade they have begun to be considered as viable targets for therapeutic intervention, dysregulation of PPIs being the cause of many diseases, such as cancer, diabetes, neurodegeneration and HIV.1 Quite recently, the PPI between the proprotein convertase subtilisin/kexin type 9 (PCSK9) and the hepatic low density lipoprotein receptor (LDLR) has attracted the attention of the scientific community.2 More in depth, PCSK9 is expressed primarily in liver, kidney, and intestine.3 It binds to LDLR, promoting its degradation,4,5 which results in an increased level of plasmatic low-density lipoprotein. A high concentration of plasma LDL cholesterol is a major cause of atherosclerosis, which subsequently promotes the development of cardiovascular diseases.6,7 For this reason, over the past few years PCSK9 has become a sure and potent target for the treatment of hypercholesterolemia.2 Moreover, as most of statin drugs’ patents have expired recently, numerous pharmaceutical industries are devoting efforts to develop new molecules that can be used in patients with hypercholesterolemia, in combination with, or as alternatives to, statins. On the other hand, the only PCSK9 inhibitors approved for clinical use are, at the moment, expensive monoclonal antibodies, such as evolocumab and alirocumab.8In the search for alternatives to the current therapy, the design of small molecules able to efficiently impair the PCSK9/LDLR PPI may become an attractive approach.2 In general, it is well known that the use of small peptidomimetics of the main recognition motifs is a promising way to disrupt PPIs.9 In this context, while α-helix10 and β-turn11 domains have been extensively investigated, the β-strand motif has received less attention,12 even though some β-strand peptidomimetics have been successfully employed to inhibit enzymes’ activity, in order to treat different diseases, ranging from cancer and AIDS to anthrax and Alzheimer's.13
Concerning PCSK9/LDLR PPI, the X-ray crystal structure14 clearly reports evidence of a β-strand-mediated interaction. In particular, PCSK9 comes into contact with the EGF-A domain of LDLR by means of a β-sheet shaped by the residues C378-F379-V380-S381 of PCSK9 and V328-C329-N330-D331 (VCND) of LDLR (Fig. 1).
 | ||
| Fig. 1 Schematic representation of the PCSK9/LDLR PPI, as found in the X-ray structure of the complex. | ||
Going on with our studies on PCSK9/LDLR PPI, we reasoned that small, non-peptidic β-strand foldamers, resembling or not the VCND sequence, could be able to positively interfere with the PCSK9/LDLR reciprocal interaction.
Initial efforts towards the synthesis of a non-peptidic β-strand foldamer could be traced back to the work of Hirschmann and Smith in 1992.15 Their innovative idea was to replace the peptidic backbone with a heterocyclic scaffold, namely a polypyrrolinone, able to mimic native β-strands with regard to both the side chain orientations and the inter-strand H-bond donating capabilities. Later, various non-peptidic β-strand mimics have been developed on the basis of Hirschmann and Smith's concept.16 A milestone in the peptidomimetic chemistry was then posed by Burgess and coworkers in 2011,17 with the definition of minimalist mimics, as frameworks in which only selected side chains of the original peptide chain are present. In this context, Hamilton and co-workers18 recently reported non-peptidic minimalist frameworks, able to mimic the i, i + 2 and i + 4 residues of a β-strand motif, stabilizing their conformations either by intramolecular hydrogen bonds or by the dipolar repulsion effect (Fig. 2).
 | ||
| Fig. 2 (A) Native β-strand. (B) Hirschmann and Smith's β-strand foldamer.15 (C) Hamilton's β-strand minimalist mimic.18 | ||
Relying on our experience with the synthesis of minimalist peptidomimetics,19 we conceived a multicomponent reaction (MCR)-based approach to a novel class of potential β-strand mimics, which are characterized by the presence of multiple C2–C5′-linked imidazole rings, spanning N–R substituents in place of amino acid side chains (Scheme 1). The synthetic strategy is based on the van Leusen three-component reaction20 (vL-3CR)/C2-formylation iterative protocol. The vL-3CR is able to generate 1,4,5-trisubstituted imidazoles in a single step, by a simple base-induced condensation between an aldehyde, a primary amine and tosylmethyl isocyanide (TosMIC).20
 | ||
| Scheme 1 A non-peptidic, multicomponent approach to the synthesis of β-strand mimics. | ||
As a proof of concept of the goodness of our approach, herein we report the synthesis of the N-methyl tetraimidazole derivative 7 (Scheme 2), demonstrating its ability to mimic a β-strand motif, by means of NMR, computational studies and biological evaluation of PCSK9-LDLR PPI.
 | ||
| Scheme 2 Synthesis of tetraimidazole 7. Reaction conditions: (a) MeNH2 (40 wt% H2O, 2 eq.), DMF (1–0.25 M), 2 h, rt; then K2CO3 (1.5 eq.), TosMIC (1.2 eq.), 24 h, 50 °C; (b) BuLi (1.5 eq.), THF (0.5–0.25 M), 2 h, −78 °C, then DMF (2 eq.), −78 to rt, 24 h; (c) BuLi (1.5 eq.), TMEDA (2 eq.), THF (0.05 M), 2 h, −78 °C; then DMF (2 eq.), −78 to rt, 24 h. | ||
Results and discussion
Although diverse reaction conditions have been reported for the vL-3CR,20 the combination of potassium carbonate as a base and a highly polar solvent such as methanol, ethanol or dimethylformamide (DMF) usually provides best results.21 Therefore, we selected high-boiling DMF as the solvent, and introduced a pre-condensation time of two hours, in order to allow the in situ formation of the corresponding imine, starting from the simple and easy-to-handle benzaldehyde and aqueous methylamine.The subsequent addition of potassium carbonate and TosMIC, followed by a reaction time of 24 hours at 50 °C, smoothly afforded the desired imidazole derivative 1 in high yield. Starting from 1, we applied iteratively the vL-3CR conditions on intermediate formyl derivatives 2, 4 and 6. In this way, we achieved bi- and tri-imidazoles 3 and 5 and finally the desired four residue mimic 7. Intermediates 2, 4 and 6 could be obtained by classical formylation conditions, using n-butyllithium and dimethylformamide at low temperature. In particular, starting from 1 or 3, the corresponding aldehydes 2 and 4 were smoothly achieved in moderate to good yields. Otherwise, under the same conditions, no appreciable formation of the formylated triimidazole 6 could be observed, due to the low solubility of the lithiated derivative of compound 5. However, by adding TMEDA and lowering the reaction concentration, we were able to obtain compound 6 in moderate yield.
With tetraimidazole 7 in hand, we investigated its solution-phase conformational behavior through the NOESY NMR experiment in CDCl3 (Fig. 3). Strong correlations between N-Me groups and the aromatic proton of the next ring (H10 ↔ H3, H10 ↔ H15, H16 ↔ H21, H22 ↔ H27 and H28 ↔ H25) were observed.
 | ||
| Fig. 3 Selected region of the NOESY NMR (CDCl3, 400 MHz, 298 K) spectrum of compound 7, focusing on the cross-peaks between N-Me groups and aromatic protons. Green arrows indicate the observed NOE contacts. | ||
On the other hand, not even weak NOE contacts among N-Me groups (H10 ↔ H16, H16 ↔ H22 and H22 ↔ H28) could be detected, clearly indicating an N-Me alternate, β-strand-like, conformation for compound 7.
In order to properly assess tetraimidazole 7 as a minimalist β-strand mimetic, computational studies were performed. Molecular dynamics (MD) simulations with the GB implicit water solvent model were executed by using the AMBER12 package, and GAFF force field.22,23 In particular, the computational model of 7 was investigated by minimizing, equilibrating and heating up the starting conformation to 300 K, 700 K and 1000 K for a short period (2 ns). Then, after an intermediate equilibration step at 700 K, a production run of 20 ns of MD simulations at 300 K was accomplished,17 acquiring 2800 conformational states. Analyzing the fluctuation of the dihedral angles connecting the imidazole rings, and their distribution percentage (τ1, τ2 and τ3 in Fig. 4A), we were able to confirm that compound 7 assumes the conformation suggested by the NOESY experiment, with the most populated values of dihedral angles in the range from 180° to 150° and from −150° to −180° (for additional details see the ESI†).
 | ||
| Fig. 4 (A) Torsion angle distributions resulting from the MD simulations of compound 7. (B) Superimposition between 7 (yellow) and a hypothetical protein β-strand (cyan). | ||
To determine the robustness of compound 7 as a β-strand mimic, the distances between the N-Me groups were measured on the ab initio optimized (B3LYP/6-31 g(d)/CPCM-water level of theory) lowest energy conformer. By comparing the found values of 5.5, 7.3 and 11.4 Å to those reported by Burgess and coworkers17 for Cβ–Cβ distances in typical secondary structures, we can expect that 7 mimics the parallel β-strand motif almost perfectly (optimal i–i + 1, i–i + 2 and i–i + 3 distances are 5.8, 7.1 and 11.1 Å respectively, see Table S1, ESI†). Moreover, superimposing compound 7 with a hypothetical protein β-strand motif, we observed very good overlapping (Fig. 4B), with distances between the N-Me groups of 7 and the Cβ atoms of β-strand side chains lower than 0.5 Å.
This last observation drove the biological investigation of compound 7's capability to inhibit the therapeutically relevant PPI between PCSK9 and LDLR (Fig. 5).14 Remarkably, the in vitro PCSK9–LDLR binding assay showed that compound 7 induces a concentration dependent inhibition of the LDLR binding on PCSK9, with an IC50 value equal to 11.2 ± 0.2 μM (Fig. 5A). In the same experiments, instead, compound 5 gave an IC50 equal to 116.3 ± 0.16 μM, indicating that the presence of three imidazole rings in the structure are not enough to efficiently impair the PCSK9–LDLR PPI. For comparison, we refer to two known PCSK9 inhibitors. Both are peptides showing IC50 values in a low micromolar range. The first one, recently reported by us, is a natural decapeptide derived from lupin (P5) and showing an IC50 value equal to 1.6 μM.24 The other one, singled out by Zhang and coworkers,25 is Pep2–8 (13 amino acids) displaying an IC50 value of 0.8 μM. The disclosed micro-molar IC50 of compound 7, along with its peptidomimetic nature, is in our opinion particularly relevant and can pave the way for an original, non-peptidic approach to PCSK9 inhibition. With this in mind, we carried out deeper investigation into the activity of compound 7. Thus, the change of the functional capability of HepG2 cells, previously treated with this compound, to uptake extracellular LDL was investigated, by performing fluorescent-LDL uptake experiments. We could observe that the ability of compound 7 to impair the PCSK9–LDLR binding, stabilizing the active LDLR on the cell membrane, leads to an improved ability of hepatic cells to uptake extracellular LDL, with a final hypocholesterolemic effect (Fig. 5B). This evidence indicates that compound 7 does not directly bind the LDLR, localized on the cellular membranes; otherwise the improved capacity of HepG2 cells to absorb extracellular fluorescent LDL would have been severely impaired. MTT experiments have also been performed, in order to exclude any potential cytotoxicity effect after treatment of HepG2 with 1, 10, 50, and 100 μM of compound 7 for 24 h. As clearly shown by the attained results (Fig. S1, see the ESI†), compound 7 is safe for HepG2 cells.
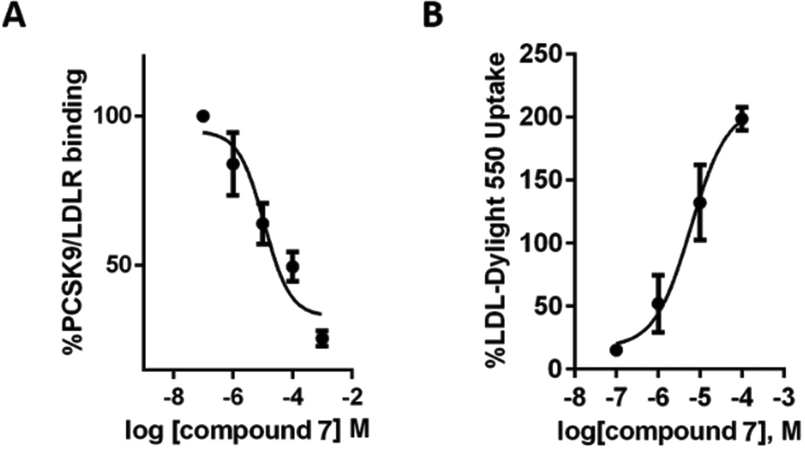 | ||
| Fig. 5 (A) Inhibitory effects of compound 7 on the PCSK9–LDLR PPI in vitro. Data points represent averages ± SEM of three independent experiments in triplicate. (B) Effect of compound 7 on the HepG2 ability to uptake LDL from the extracellular environment. Data points represent averages ± SEM of three independent experiments. | ||
In conclusion, we have developed a novel synthetic route to a C2–C5′-linked tetraimidazole scaffold, by means of an iterative vL-3CR/C2-formylation protocol. Its solution-phase conformational behaviour was investigated through NOESY, NMR and computational studies, demonstrating its ability to mimic the i, i + 1, i + 2 and i + 3 amino acid residues of a β-strand motif. This evidence led us to test the capability of this compound to effectively disrupt a β-strand-mediated PPI. With this aim, the therapeutically relevant PCSK9–LDLR PPI was chosen as the biological target. The attained results confirmed that the tetraimidazole scaffold is able to impair the PCSK9–LDLR reciprocal interaction, with an IC50 value equal to 11.2 μM. Also the LDL-uptake is increased (EC50 = 6.04 μM), while MTT assays assured that such a compound is also safe on liver HepG2 cells.
Undoubtedly, compound 7 constitutes a versatile scaffold for a generation of new molecular entities capable of potentially mimicking any β-strand motif. Furthermore, since there are no reports yet in the literature on small molecules able to inhibit PCSK9, we are also confident that compound 7 will constitute a lead compound for the rational design of a new class of PCSK9 inhibitors. Further work is underway in order to better refine the chemical framework, in particular, through substitution of N-Me groups with different residues, more strictly resembling the actual LDLR β-strand side chains.
We are grateful for financial support from the University of Milan, Grant 2015, Line 2 – Action A (PI GG).
Notes and references
- (a) L. Garner and K. D. Janda, Curr. Top. Med. Chem., 2011, 11, 258 CrossRef PubMed; (b) B. O. Villoutreix, M. A. Kuenemann, J. Poyet, H. Bruzzoni-Giovanelli, C. Labbé, D. Lagorce, O. Sperandio and M. A. Miteva, Mol. Inf., 2014, 33, 414 CrossRef CAS PubMed; (c) T. L. Nero, C. J. Morton, J. K. Holien, J. Wielens and M. W. Parker, Nat. Rev. Cancer, 2014, 14, 248 CrossRef CAS PubMed.
- N. Bergeron, B. A. P. Phan, Y. Ding, A. Fong and R. M. Krauss, Circulation, 2015, 132, 1648 CrossRef CAS PubMed.
- N. G. Seidah and A. Prat, Nat. Rev. Drug Discovery, 2012, 11, 367 CrossRef CAS PubMed.
- D. W. Zhang, T. A. Lagace, R. Garuti, Z. Zhao, M. McDonald, J. D. Horton, J. C. Cohen and H. H. Hobbs, J. Biol. Chem., 2007, 282, 18602 CrossRef CAS PubMed.
- B. Gencer, G. Lambert and F. Mach, Swiss Med. Wkly., 2015, 145, w14094 Search PubMed.
- J. G. Canto and A. E. Iskandrian, JAMA, J. Am. Med. Assoc., 2003, 290, 947 CrossRef PubMed.
- E. G. Nabel, N. Engl. J. Med., 2003, 349, 60 CrossRef CAS PubMed.
- B. M. Everett, R. J. Smith and W. R. Hiatt, N. Engl. J. Med., 2015, 373, 1588 CrossRef PubMed.
- (a) S. Jones and J. M. Thornton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13 CrossRef CAS PubMed; (b) I. S. Moreira, P. A. Fernandes and M. J. Ramos, Proteins: Struct., Funct., Bioinf., 2007, 68, 803 CrossRef CAS PubMed; (c) L. Garner and K. D. Janda, Curr. Top. Med. Chem., 2011, 11, 258 CrossRef PubMed.
- (a) C. G. Cummings and A. D. Hamilton, Curr. Opin. Chem. Biol., 2010, 14, 341 CrossRef CAS PubMed; (b) R. S. Harrison, N. E. Shepherd, H. N. Hoang, G. Ruiz-Gomez, T. A. Hill, R. W. Driver, V. S. Desai, P. R. Young, G. Abbenante and D. P. Fairlie, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11686 CrossRef CAS PubMed; (c) G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3 CAS; (d) M. D. Boersma, H. S. Haase, K. J. Peterson-Kaufman, E. F. Lee, O. B. Clarke, P. M. Colman, B. J. Smith, W. S. Horne, W. D. Fairlie and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 315 CrossRef CAS PubMed; (e) V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161 CrossRef CAS PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020 CrossRef CAS PubMed.
- H. Remaut and G. Waksman, Trends Biochem. Sci., 2006, 31, 436 CrossRef CAS PubMed.
- (a) S. Maitra and J. Norwick, The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Material Science, Wiley, New York, 2000, pp. 495–518 Search PubMed; (b) W. A. Loughlin, J. D. A. Tyndall, M. P. Glenn, T. A. Hill and D. P. Fairlie, Chem. Rev., 2010, 110, PR32 CrossRef CAS PubMed.
- P. Lo Surdo, M. J. Bottomley, A. Calzetta, E. C. Settembre, A. Cirillo, S. Pandit, Y. G. Ni, B. Hubbard, A. Sitlani and A. Carfi, EMBO Rep., 2011, 12, 1300 CrossRef CAS PubMed.
- A. B. Smith III, T. P. Keenan, R. C. Holcomb, P. A. Sprengeler, M. C. Guzman, J. L. Wood, P. J. Carroll and R. Hirschmann, J. Am. Chem. Soc., 1992, 114, 10672 CrossRef.
- (a) A. B. Smith III, M. C. Guzman, P. A. Sprengeler, T. P. Keenan, R. C. Holcomb, J. L. Wood, P. J. Carroll and R. Hirschmann, J. Am. Chem. Soc., 1994, 116, 9947 CrossRef; (b) W. A. Loughlin, J. D. A. Tyndall, M. P. Glenn and D. P. Fairlie, Chem. Rev., 2004, 104, 6085 CrossRef CAS PubMed; (c) N. G. Angelo and P. S. Arora, J. Am. Chem. Soc., 2005, 127, 17134 CrossRef CAS PubMed; (d) S. Chandrasekhar, B. N. Babu, A. Prabhakar, A. Sudhakar, M. S. Reddy, M. U. Kiran and B. Jagadeesh, Chem. Commun., 2006, 1548 RSC; (e) M. C. Hammond, B. Z. Harris, W. A. Lim and P. A. Bartlett, Chem. Biol., 2006, 13, 1247 CrossRef CAS PubMed; (f) M. C. Hammond and P. A. Bartlett, J. Org. Chem., 2007, 72, 3104 CrossRef CAS PubMed; (g) J. L. Watson and E. R. Gillies, J. Org. Chem., 2009, 74, 5953 CrossRef CAS PubMed; (h) N. Ross, W. P. Katt and A. D. Hamilton, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 989 CrossRef CAS PubMed; (i) A. Raghuraman, E. Ko, L. M. Perez, T. R. Ioerger and K. Burgess, J. Am. Chem. Soc., 2011, 133, 12350 CrossRef CAS PubMed; (j) A. B. Smith III, A. K. Charnley and R. Hirschmann, Acc. Chem. Res., 2011, 44, 180 CrossRef PubMed; (k) C. W. Kang, Y. Sun and J. R. Del Valle, Org. Lett., 2012, 14, 6162 CrossRef CAS PubMed.
- (a) E. Ko, J. Liu and K. Burgess, Chem. Soc. Rev., 2011, 40, 4411 RSC; (b) E. Ko, J. Liu, L. M. Perez, G. Lu, A. Schaefer and K. Burgess, J. Am. Chem. Soc., 2011, 133, 462 CrossRef CAS PubMed.
- (a) P. N. Wyrembak and A. D. Hamilton, J. Am. Chem. Soc., 2009, 131, 4566 CrossRef CAS PubMed; (b) A. G. Jamieson, D. Russell and A. D. Hamilton, Chem. Commun., 2012, 48, 3709 RSC; (c) C. L. Sutherell, S. Thompson, R. T. W. Scott and A. D. Hamilton, Chem. Commun., 2012, 48, 9834 RSC; (d) E. A. German, J. E. Ross, P. C. Knipe, M. F. Don, S. Thompson and A. D. Hamilton, Angew. Chem., Int. Ed., 2015, 54, 2649 CrossRef CAS PubMed; (e) T. Yamashita, P. C. Knipe, N. Busschaert, S. Thompson and A. D. Hamilton, Chem. – Eur. J., 2015, 21, 14699 CrossRef CAS PubMed.
- M. Stucchi, S. Cairati, R. Cetin-Atalay, M. S. Christodoulou, G. Grazioso, G. Pescitelli, A. Silvani, D. C. Yildirim and G. Lesma, Org. Biomol. Chem., 2015, 13, 4993 CAS.
- (a) A. M. van Leusen and J. Strating, Q. Rep. Sulfur Chem., 1970, 5, 67 CAS; (b) A. M. van Leusen, J. Wildeman and O. Oldenziel, J. Org. Chem., 1977, 42, 1977 CrossRef; (c) D. van Leusen and A. M. van Leusen, Org. React., 2003, 57, 419 Search PubMed.
- (a) H. Che, T. N. Tuyen, H. P. Kim and H. Park, Bioorg. Med. Chem. Lett., 2010, 20, 4035 CrossRef CAS PubMed; (b) C. Lamberth, R. Dumeunier, S. Trah, S. Wendeborn, J. Godwin, P. Schneiter and A. Corran, Bioorg. Med. Chem., 2013, 21, 127 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- C. Lammi, C. Zanoni, G. Aiello, A. Arnoldi and G. Grazioso, Sci. Rep., 2016, 6, 29931 CrossRef PubMed.
- Y. Zhang, C. Eigenbrot, L. Zhou, S. Shia, W. Li, C. Quan, J. Tom, P. Moran, P. Di Lello, N. J. Skelton, M. Kong-Beltran, A. Peterson and D. Kirchhofer, J. Biol. Chem., 2014, 289, 942 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, computational and biological experimental procedures, and spectroscopic data for all synthesized compounds, Fig. S1 and S2, Table S1. See DOI: 10.1039/c6ob01642a |
| This journal is © The Royal Society of Chemistry 2016 |