Preparation of oxocene terpenes. The first enantiospecific synthesis of cytotoxic arenaran A†
Received
29th July 2016
, Accepted 20th September 2016
First published on 20th September 2016
Abstract
The first syntheses of cytotoxic marine arenarans A and B starting from commercial (−)-sclareol are reported. The oxocene ring of the target compound is formed via ring-closing metathesis, a process that depends on certain structural requirements. The trans-fused structure of the natural product is confirmed by comparison with the cis-fused isomer, which was synthesized. This synthetic strategy is also applicable to the synthesis of other oxocene terpenes.
Introduction
Although terpenes containing eight-membered ether rings are infrequent in nature, their biological activities are of interest. Arenaran A (1) and B (2), two sesquiterpene ethers isolated from the marine sponge Dysidea arenaria, belong to this type of compound. Compound 1 is in vitro-active against several types of cancer cells, with reported IC50 values (μg mL−1) of 9.51 (A-549, human lung carcinoma), 9.11 (HT-29, human colon adenocarcinoma), 5.28 (HCT-29, human colon adenocarcinoma) and 3.17 (P-388 murine leukemia). Another example of oxocane terpene is the labdane type brominated diterpene 3, isolated from the red alga Laurencia obtuse (Fig. 1).2
 |
| | Fig. 1 Natural oxocane terpenes. | |
Despite the rare structure of this type of compound, which is biogenetically related to aplysistatin and other bioactive metabolites, and the relevant biological activity observed in some cases, no syntheses of these oxocane terpenes have yet been reported. The structure of arenaran A (1) has been established in 2D NMR experiments, utilizing mainly H–C COSY correlations (J = 140 and J = 9 Hz); however, this result is insufficient to characterise the trans-fused union assigned. Moreover, the absolute configuration of natural terpene 1 remains unknown.
Results and discussion
These considerations, together with the possibility of preparing large quantities of arenaran A (1) in order to conduct an in-depth study of its biological activity, encouraged us to develop a synthetic route to compound 1 and related terpenes. In this respect, the only relevant antecedent is a study aimed at the synthesis of arenaran A reported by Reggelin et al.3 These authors essayed the construction of the bicyclic oxocene structure of 1 based on the intramolecular attack of an allyl alcohol on an alkenyl sulfoximine. The failure of their attempt highlights the difficulty in forming an eight-membered ring. In view of this outcome, we planned to create the oxocene ring via a ring-closing metathesis (RCM) process, as shown in Scheme 1.
 |
| | Scheme 1 Retrosynthesis of arenaran A (1) via ring-closing metathesis. | |
In order to determine the reaction conditions, we first investigated the use of α-ionone (6) as a starting material. Considering the chemical behaviour of compound 6 previously reported,4 the utilization of this terpene as the synthetic precursor should provide alcohol 10, the epimer of compound 5, and consequently the complete sequence should lead to the corresponding cis-fused epi-arenanan A (13). Scheme 2 shows the synthesis of alcohol 10 from α-ionone (6). Epoxidation of dihydro-α-ionone (7) gave the expected epoxyketone 8, which after methylenation and treatment with LiAlH4 provided alcohol 10.
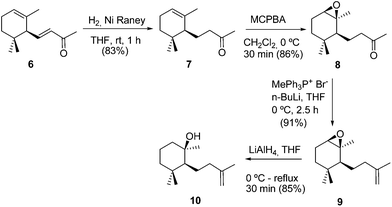 |
| | Scheme 2 Synthesis of alcohol 10 from α-ionone (6). | |
This alcohol was then transformed into epi-arenaran A (13), following the above retrosynthetic plan (Scheme 3). The reaction of alcohol 10 with allyl bromide under basic conditions leads to ether 11, which unexpectedly failed to give the desired RCM after treatment with the second-generation Grubbs catalyst, instead generating alcohol 10. Under these reaction conditions, ether 12, derived from dimethylallyl bromide, underwent the desired ring-closing metathesis, affording compound 13, the cis-fused stereoisomer of arenaran A.
 |
| | Scheme 3 Synthesis of epi-arenaran A (13) from alcohol 10. | |
At this point, it is very important to note that the preparation of alcohol 5 in enantiopure form, the precursor of arenaran A (1), from α-ionone (6) can be achieved via the corresponding diastereoisomer of epoxyketone 8, with the epoxy group on the α side; however, the preparation of this epoxyketone from α-ionone (6), reported by Serra, involves a very long synthetic sequence (10 steps), including a low yield lipase-mediated acetylation reaction.4
Taking into account these difficulties, we investigated the preparation of the enantiopure alcohol 5. The first synthetic proposal for obtaining this alcohol from commercial (+)-sclareolide (16) is depicted in Scheme 4. Alcohol 5 is obtained from ketoester 14, resulting from the Baeyer–Villiger oxidation of the diketone derived from alkene 15, which is easily prepared from lactone 16.
 |
| | Scheme 4 Retrosynthesis of alcohol 5 from (+)-sclareolide (16). | |
Scheme 5 shows the synthetic sequence for the intended synthesis of ketoester 14 from lactone 16. Reduction of iodide 176 with RANEY® Ni, following a procedure developed in our laboratory, gave alkene 15, which was then converted into diketone 18. However, all attempts at obtaining the desired ketoester 14, via Baeyer–Villiger oxidation of diketone utilizing a variety of reaction conditions were unsuccessful, producing instead ketoester 19. These results highlight the difficulty of achieving oxidation of the ketone group linked to the quaternary carbon, probably due to steric hindrance. This outcome contrasts with that of the related, but more rigid 1-decalones;5 however, the possibility of utilizing these ketones as a starting material for preparing alcohol 5 must be disregarded due to the further difficulties arising from the tendency of the subsequent reduction products to dehydrate, affording bicyclic enol ethers.
 |
| | Scheme 5 Attempts at preparing ketoester 14 from (+)-sclareolide (16). | |
The above difficulties were circumvented by utilizing commercial (−)-sclareol (20) as a starting material. Thus, alcohol 5 was prepared from this diterpene, via ketoester 22, resulting from the Baeyer–Villiger oxidation of ketoaldehyde 21, utilizing a procedure developed in our laboratory7 (see Scheme 6). At this point, it is interesting to note the different behaviour of the aldehyde group attached to the quaternary carbon, present in compound 21, from that of the ketone group joined to the same carbon atom, which is in compound 18; the first of these undergoes Baeyer–Villiger oxidation in good yield, under the usual reaction conditions, and so methylenation of ketoester 22 leads to good yield of the desired alcohol 5.
 |
| | Scheme 6 Synthesis of alcohol 5 from (−)-sclareol (20). | |
Alcohol 5 was then transformed into arenaran A (1) following a similar procedure to that utilized for the cis-fused isomer 13. First, the O-allyl ether 4 was prepared, but this dialkene also failed to give the metathesis process. However, the O-dimethylallyl ether 23 underwent the desired reaction, affording arenaran A (1). At this point it is important to note that ether 23 undergoes the RCM much faster than its epimer 12. The further epoxidation of compound 1 gave arenaran B (2) (Scheme 7).
 |
| | Scheme 7 Synthesis of arenaran A (1) and B (2) from alcohol 5. | |
As mentioned above, the structural elucidation performed by Crews et al. for the natural arenaran A, based on 2D NMR experiments, does not allow us to establish unequivocally the trans-fused union proposed by these authors.1 However, after preparing both stereoisomers, we will be able to confirm this proposal. Tables 1 and 2 show the 13C and 1H NMR chemical shifts for epi-arenaran A (13) (with the cis-fused union), for the synthetic arenaran A (1), reported here, and for the natural arenaran A. NMR data for the synthetic and natural arenarans B (2) are shown in Table 3.
Table 1
13C NMR chemical shifts for epi-arenaran A (13) and for the synthetic and natural arenarans A (1)
|
epi-Arenaran A (13)a |
Synthetic arenaran A (1)a |
Synthetic arenaran A (1)b |
Natural arenaran A (1)b (ref. 1) |
|
CD3OD.
C6D6.
|
| 140.6 (C) |
132.9 (C) |
133.1 (C) |
132.8 (C) |
| 123.6 (CH) |
123.3 (CH2) |
124.7 (CH) |
124.5 (CH) |
| 77.4 (C) |
80.2 (C) |
79.5 (C) |
79.3 (C) |
| 56.4 (CH2) |
61.1 (CH) |
61.8 (CH2) |
61.5 (CH2) |
| 55.5 (CH) |
45.4 (CH2) |
46.1 (CH) |
45.9 (CH) |
| 42.8 (CH2) |
41.9 (CH) |
42.6 (CH2) |
42.4 (CH2) |
| 41.9 (CH2) |
35.2 (CH3) |
35.7 (CH2) |
35.5 (CH2) |
| 35.5 (CH2) |
34.0 (C) |
34.5 (C) |
34.3 (C) |
| 34.1 (C) |
32.3(CH2) |
33.4 (CH3) |
33.1 (CH3) |
| 31.4 (CH3) |
28.9 (CH3) |
29.7 (CH2) |
29.5 (CH2) |
| 25.2 (CH2) |
25.0(CH2) |
26.4 (CH3) |
26.1 (CH3) |
| 24.6 (CH3) |
24.1 (CH3) |
24.9 (CH2) |
24.7 (CH2) |
| 21.1 (CH3) |
21.9 (CH2) |
23.2 (CH3) |
23.0 (CH3) |
| 18.0 (CH2) |
21.0 (CH2) |
22.1 (CH3) |
21.9 (CH3) |
| 15.1 (CH3) |
19.7 (CH3) |
20.5 (CH2) |
20.1 (CH2) |
Table 2
1H NMR chemical shifts for epi-arenaran A (13) and for the synthetic and natural arenarans A (1)
|
epi-Arenaran A (13)a |
Synthetic arenaran A (1)a |
Synthetic arenaran A (1)b |
Natural arenaran A (1)b (ref. 1) |
|
CD3OD.
C6D6.
|
| 5.43 (t, J = 6.9) |
5.15 (br s) |
5.07 (br s) |
5.06 (br s) |
| 3.96 (dd, J = 7.0, 13.8) |
4.22 (dd, J = 2.1, 18.4) |
4.11 (br s) |
4.06 (br s) |
| 3.76 (dd, J = 6.9, 13.8) |
4.03 (d, J = 18.4) |
3.68 (ddd, J = 19.5, 11.3) |
3.67 (ddd, J = 19.9, 11.5, 1.3) |
| 2.40–2.03 (m) |
3.37 (m) |
1.68 (m) |
1.68 (m) |
| 1.96 (ddd, J = 3.1, 13.1) |
1.76 (ddd, J = 4.5, 12.7) |
1.68 (s) |
1.67 (s) |
| 1.87 (m) |
1.69 (s) |
1.63 (m) |
1.63 (m) |
| 1.76 (ddd, J = 3.3, 13.4) |
1.65 (m) |
|
|
| 1.71 (s) |
1.61–1.58 (m) |
1.62 (m) |
1.62 (m) |
| 1.45–1.40 (m) |
1.55 (m) |
1.52 (m) |
1.51 (m) |
| 1.34 (d, J = 3.0) |
1.51 (m) |
1.35 (m) |
1.34 (m) |
| 1.20 (m) |
1.46 (dt, J = 3.0, 13.5) |
1.31 (m) |
1.31 (m) |
| 1.13 (s) |
1.35–1.32 (m) |
1.29 (s) |
1.26 (s) |
| 1.08 (dd, J = 4.0, 13.4) |
1.25 (s) |
1.25 (m) |
1.25 (m) |
| 1.03 (s) |
1.16 (ddd, J = 4.1,13.3) |
0.86 (s) |
0.85 (s) |
| 0.89 (s) |
0.91 (s) |
0.78 (s) |
0.79 (s) |
Table 3
1H and 13C NMR data in CDCl3 for the synthetic and natural arenarans B (2)
| Synthetic arenaran B (2) |
Natural arenaran B (2) (ref. 1) |
Synthetic arenaran B (2) |
Natural arenaran B (2) (ref. 1) |
| 4.02 (dd, J = 17.6, 1.7) |
4.02 (dd, J = 15.6, 2.1) |
80.0 (C) |
80.0 (C) |
| 3.90 (d, J = 17.6) |
3.89 (d, J = 15.6) |
64.1 (CH) |
64.1 (CH) |
| 2.79 (s) |
2.79 (d, J = 2.1) |
60.8 (C) |
60.8 (C) |
| 2.42 (dt, J = 13.4, 4.9) |
2.41 (dt, J = 13.5, 5.1) |
58.1 (CH2) |
58.2 (CH2) |
| 1.86 (dt, J = 13.2, 3.9) |
1.86 (dt, J = 13.5, 3.6) |
44.7 (CH) |
45.0 (CH) |
| 1.67 (m) |
1.67 (m) |
42.2 (CH2) |
42.4 (CH2) |
| 1.58 (m) |
1.58 (m) |
35.9 (CH2) |
36.0 (CH2) |
| 1.50 (m) |
1.50 (m) |
34.7 (C) |
35.5 (C) |
| 1.45 (m) |
1.45 (m) |
33.2 (CH3) |
33.3 (CH3) |
| 1.40 (m), 1.21 (m) |
1.40 (m), 1.21 (m) |
31.8 (CH2) |
32.7 (CH2) |
| 1.35 (m) |
1.35 (m) |
22.9 (CH3) |
23.0 (CH3) |
| 1.29 (s) |
1.21 (s) |
22.5 (CH3) |
22.6 (CH3) |
| 1.16 (s) |
1.16 (s) |
22.0 (CH2) |
22.1 (CH2) |
| 0.98 (s) |
0.97 (s) |
21.5 (CH3) |
21.9 (CH3) |
| 0.85 (s) |
0.84 (s) |
19.9 (CH2) |
20.0 (CH2) |
As can be seen in Tables 1 and 2, there is an excellent correlation of spectral data between the synthetic and the natural arenaran A (1), while those of the cis-fused stereoisomer (epi-arenaran A, 13) are very discordant. This finding corroborates the trans-fused stereochemistry proposed for natural arenaran A by Crews et al. On the other hand, the Z configuration of the carbon–carbon double bond of arenaran A has been confirmed on the basis of the observed NOE effect between Me-12 (δ 1.68) and H-2 (δ 5.07) (see the ESI†). The NMR data of the synthetic and natural arenarans B (2), which are placed in order of decreasing δ in Table 3, showed some discrepancies. In order to dispel doubts, a thorough study on the structure of synthetic arenaran B (2) has been conducted. This includes 1D and 2D NMR spectra at 500 MHz (TOCSY, COSY, HSQC, HMBC and NOESY experiments). This allowed us to corroborate unequivocally the proposed structure for this compound, and to assign correctly the 1H and 13C NMR signals (Table 4). The β disposition of the epoxide group has been confirmed on the basis of the observed NOE effect between H-2 (δ 2.79) and H-6 (δ 1.50). All these experiments are included in the ESI.† With respect to the absolute stereochemistry of natural arenaran A, it should be noted that the optical rotation for the natural compound ([α]25D: +154.0; c 0.01, CHCl3) is very discordant from that measured in our laboratory for synthetic arenaran A ([α]25D: −32.1; c 0.01, CHCl3); however, the optical rotation of synthetic arenaran B (2) ([α]25D: −24.9; c 0.2, CHCl3) is very similar to that described for the natural epoxide ([α]25D: −24.4; c 0.23, CHCl3). This finding leads us to believe that the [α]25D value previously reported for natural arenaran A (1) might be mistaken, and that the absolute stereochemistry proposed by Crews et al. for this compound is correct, because of its correlation with (−)-sclareol (20).
Table 4
1H and 13C NMR assignments for synthetic arenaran B (2)
| Carbon or proton |
1H |
13C |
| 1 |
4.03 (dd, J = 15.9, 2.3) |
58.2 (CH2) |
| 3.90 (d, J = 15.9) |
|
| 2 |
2.79 (d, J = 2.3) |
64.2 (CH) |
| 3 |
|
60.9 (C) |
| 4 |
2.43 (ddd, J = 13.4, 4.3, 4.3) |
32.0 (CH2) |
| 1.88 (ddd, J = 13.4, 13.4, 4.3) |
|
| 5 |
1.68 (tt, J = 13.4, 4.3) |
22.2 (CH2) |
| 1.61 (tdd, J = 13.4, 4.3, 4.3) |
|
| 6 |
1.50 (dd, J = 13.4, 4.3) |
44.9 (CH) |
| 7 |
|
80.2 (C) |
| 8 |
1.72 (ddd, J = 13.5, 3.8, 3.8) |
36.1 (CH2) |
| 1.33 (ddd, J = 13.5, 13.5, 3.8) |
|
| 9 |
1.55 (dp, J = 13.5, 3.8) |
20.0 (CH2) |
| 1.45 (ddd, J = 13.5, 3.8, 3.8) |
|
| 10 |
1.39 (ddd, J = 13.5, 3.8, 3.8) |
42.4 (CH2) |
| 1.19 (ddd, J = 13.5, 13.5, 3.8) |
|
| 11 |
|
34.8 (C) |
| 12 |
1.30 (s) |
23.1 (CH3) |
| 13 |
1.16 (s) |
22.7 (CH3) |
| 14 |
0.85 (s) |
21.7 (CH3) |
| 15 |
0.98 (s) |
33.4 (CH3) |
Utilizing a similar strategy, commercial (−)-sclareol (20) was transformed into the oxocene epoxide 29, via alcohol 26 (Scheme 9). This was synthesized in two alternative ways. First, methylenation of ketoester 24, whose efficient preparation in one step from (−)-sclareol (20) has been developed by our group,8 gave in high yield alcohol 26. Alternatively, aldehyde 25, previously synthesised in our laboratory,9 was converted into this alcohol under the Wolff–Kishner conditions; to the best of our knowledge, this transformation of an α-hydroxyaldehyde into an alkene has not yet been described (Scheme 8).
 |
| | Scheme 8 Synthesis of alcohol 26 from (−)-sclareol (20). | |
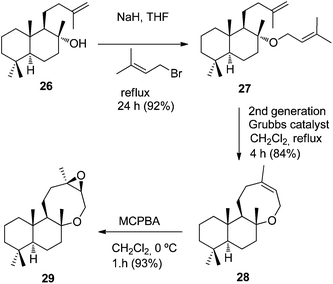 |
| | Scheme 9 Synthesis of oxocene epoxide 29 from alcohol 26. | |
Next, alcohol 26 was transformed into the oxocene epoxide 29, the 3-debromoderivative of the marine metabolite 3. Treatment of dimethylallyl ether 27 with the second-generation Grubbs catalyst afforded in good yield oxocene 28, which underwent stereoselective epoxidation, after reaction with MCPBA at 0 °C, to give epoxide 29, the 3-debromoderivative of natural terpene 3. The 13C NMR chemical shifts of the carbons of the bicyclic ether moiety of epoxide 29 are similar to those reported for natural terpene 3.
Experimental
Materials and methods
Unless stated otherwise, the reactions were performed in oven-dried glassware under an argon atmosphere using dry solvents. The solvents were dried as follows: THF over Na-benzophenone, and DCM and MeOH over CaH2. Thin-layer chromatography (TLC) was performed using F254 precoated plates (0.25 mm) and visualized by UV fluorescence quenching and phosphomolybdic acid solution staining. Flash chromatography was performed on silica gel (230–400 mesh). Chromatography separations were carried out by using a conventional column on silica gel 60 (230–400 mesh), using hexanes–EtOAc mixtures of increasing polarity.1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz, respectively. CDCl3 was treated with K2CO3. Chemical shifts (δH) are quoted in parts per million (ppm) referenced to the appropriate residual solvent peak and tetramethylsilane. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration), with the abbreviations s, br s, d, br d, t, q, and m denoting singlet, broad singlet, doublet, broad doublet, triplet, quartet and multiplet respectively. J = coupling constant in Hertz (Hz). Data for 13C NMR spectra are reported in terms of the chemical shift relative to Me4Si (δ 0.0) and the signals are assigned utilizing DEPT experiments and on the basis of heteronuclear correlations. Infrared spectra (IR) were recorded as thin films or as solids on a FTIR spectrophotometer with samples between sodium chloride plates or as potassium bromide pellets and are reported in the frequency of absorption (cm−1). Only selected absorbances (νmax) are reported. ([α]D) measurements were carried out on a polarimeter, utilizing a 1 dm length cell and CHCl3 as a solvent. The concentration is expressed in mg mL−1. HRMS were recorded on a spectrometer, using FAB with a thioglycerol or a glycerol matrix doped with 1% NaI.
Synthetic procedures
4-(2,6,6-Trimethylcyclohex-2-enyl)butan-2-one (7).
Ni RANEY® (50% in water, 6 mL) was added to a solution of α-ionone (6) (10.0 g, 52 mmol) in THF (120 mL), and the mixture was stirred under an ordinary hydrogen pressure (balloon) at room temperature for 1 h. Then, the reaction mixture was filtered through a silicagel-Na2SO4 pad (100 g), eluting with acetone (100 mL). After evaporation of the solvent under vacuum, ketone 7 (8.39 g, 83%) was obtained, as a colourless oil. Compound 7 showed identical spectroscopic properties to those reported in the literature.10,11
4-((1S,2S,6R)-1,3,3-Trimethyl-7-oxa-bicyclo[4.1.0]heptan-2-yl)butan-2-one (8).
m-Chloroperbenzoic acid (70%, 4.92 g, 20.00 mmol) was added to a solution of dihydro-α-ionone (7) (3.52 g, 18.11 mmol) in dichloromethane (70 mL), cooled at 0 °C, and the reaction mixture was stirred for 30 min. Then, a 10% Na2SO3 solution (10 mL) was added, and the mixture was extracted with EtOAc (3 × 20 mL). The organic phase was successively washed with sat. NaHCO3 (3 × 30 mL) and brine (2 × 30 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under vacuum, compound 8 (3.27 g, 86%) was obtained as a colourless syrup. Compound 8 exhibited identical properties to those reported in the literature.4
(1S,6R)-1,3,3-Trimethyl-2-(3-methylbut-3-en-1-yl)-7-oxabicyclo[4.1.0]heptane (9).
2 M n-BuLi in cyclohexane (7.8 mL, 15.7 mmol) was added to a solution of methyltriphenylphosphonium bromide (5.59 g, 15.7 mmol) in anhydrous THF (75 mL), and the mixture was stirred at 0 °C under an argon atmosphere for 15 min. Then, a solution of ketone 8 (3 g, 14.26 mmol) in anhydrous THF (2 mL) was added, and the resulting mixture was kept stirring for 2.5 h. Then, the reaction was carefully quenched with water (10 mL), and the solvent was evaporated. Then, ether (100 mL) was added and the organic phase was washed with water (3 × 30 mL) and brine (2 × 30 mL), dried over anhydrous Na2SO4 and evaporated to afford a crude product that was purified by column chromatography on silica gel (10% EtOAc/hexane) to yield epoxide 9 (2.67 g, 91%) as a colourless oil. 1H NMR (CDCl3, 400 MHz): δ 0.82 (s, 3H), 0.88 (s, 3H), 1.28 (m, 1H), 1.33 (s, 3H), 1.46–1.58 (m, 2H), 1.37 (m, 1H), 1.75 (s, 3H), 1.83 (m, 1H), 1.92 (dd, J = 15.5, 6.1 Hz, 1H), 2.05 (m, 1H), 2.29 (m, 1H), 2.93 (s, 1H), 4.71 (s, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.3 (CH2), 22.8 (CH3), 25.8 (CH2), 27.1 (CH3), 27.1 (CH2), 27.5 (CH3), 27.9 (CH), 31.6 (C), 37.7 (CH2), 46.9 (CH3), 59.8 (C), 60.3 (CH), 109.7 (CH2), 146.8 (C). IR (film): 756, 884, 1095, 1182, 1216, 1366, 1376, 1449, 1649, 3073 cm−1. HRMS (FAB) m/z calcd for C14H24ONa (M + Na+) 231.1725, found 231.1733.
(1R,2S)-1,3,3-Trimethyl-2-(3-methylbut-3-en-1-yl) cyclohexan-1-ol (10).
LiAlH4 (130 mg, 3.43 mmol) was added to a solution of epoxide 9 (2.5 g, 12.00 mmol) in anhydrous THF (50 mL) at 0 °C. The mixture was stirred at reflux under an argon atmosphere for 30 min, at which time TLC showed no 9 remaining. Then, the mixture was poured into ice and the solvent was evaporated. Ether (100 mL) was added and the phases were shaken. The organic phase was washed with water (3 × 30 mL) and brine (2 × 30 mL), dried over anhydrous Na2SO4, and evaporated to give alcohol 10 (2.15 g, 85%) as a yellow oil. 1H RMN (CDCl3, 400 MHz): δ 0.87 (s, 3H), 0.97 (s, 3H), 1.17 (s, 3H), 1.25 (br s, 2H), 1.33–1.47 (m, 4H), 1.53–1.65 (m, 2H), 1.75 (m, 1H), 1.75 (s, 3H), 2.05 (m, 2H), 4.69 (s, 2H). 13C RMN (CDCl3, 101 MHz): δ 18.3 (CH2), 21.4 (CH3), 22.5 (CH3), 24.3 (CH2), 30.8 (CH3), 32.0 (CH3), 34.7 (C), 41.1 (CH2), 41.7 (CH2), 41.8 (CH2), 54.0 (CH), 73.0 (C) 109.5 (CH2), 146.5 (C). IR (film): 757, 884, 909, 930, 1024, 1042, 1099, 1178, 1214, 1378, 1386, 1454, 1648, 3072, 3400–3600 cm−1. HRMS (FAB) m/z calcd for C14H26ONa (M + Na+) 233.1881, found 233.1876.
(1R,2S)-1-(Allyloxy)-1,3,3-trimethyl-2-(3-methylbut-3-en-1-yl)cyclohexane (11).
NaH (208 mg, 5.20 mmol, 60% dispersion in mineral oil) was added to a solution of alcohol 10 (470 mg, 2.238 mmol) in anhydrous THF (20 mL) at 0 °C under an argon atmosphere, and allyl bromide (0.4 mL, 4.62 mmol) was added, and the reaction mixture was kept stirring at reflux for 24 h, at which time TLC showed no 10 remaining. The mixture was poured into ice and the solvent was evaporated under vacuum. Then ether (50 mL) was added and the organic phase was washed with water (3 × 15 mL) and brine (2 × 15 mL), dried over anhydrous Na2SO4, and evaporated to give a crude residue, which, after column chromatography on silica gel (5% EtOAc/hexane), afforded ether 11 (520 mg, 93%) as a colorless oil. 1H NMR (CDCl3, 400 MHz): δ 0.86 (s, 3H), 0.99 (s, 3H), 1.07 (dd, J = 14.1, 3.8 Hz, 1H), 1.12 (s, 3H), 1.16 (dd, J = 13.4, 3.6 Hz, 1H), 1.27–1.34 (m, 2H), 1.36–1.45 (m, 2H), 1.53–1.64 (m, 2H), 1.75 (s, 3H), 1.93–2.12 (m, 3H), 3.82 (d, J = 4.8 Hz, 2H), 4.68 (d, J = 5.0 Hz, 2H), 5.9 (m, 1H), 5.27 (d, J = 17.2 Hz, 1H), 5.05 (d, J = 10.5 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 18.4 (CH2), 22.5 (CH3), 24.1 (CH2), 24.5 (CH3), 32.2 (CH3), 34.9 (C), 34.93 (CH), 41.8 (CH2), 42.2 (CH2), 56.0 (CH), 61.6 (CH2), 22.1 (CH3), 76.7 (C), 109.3 (CH2), 114.1 (CH2), 136.6 (CH), 146.9 (C). IR (film): 917, 1065, 1075, 1154, 1171, 1264, 1372, 1454, 1647 cm−1. HRMS (FAB) m/z calcd for C17H30ONa (M + Na+) 273.2194, found 273.2201.
Treatment of ether 11 with the 2nd generation Grubbs catalyst. Obtention of alcohol 10
The 2nd Generation Grubbs catalyst (20 mg) was added to a solution of ether 11 (170 mg, 0.68 mmol) in anhydrous CH2Cl2 (30 mL), and the mixture was kept stirring at reflux under an argon atmosphere for 48 h. Then, the solvent was evaporated and the crude product was purified by column chromatography (20% EtOAc/hexane) to yield alcohol 10 (124 mg, 87%).
(2S,3R)-1,1,3-Trimethyl-3-((3-methylbut-2-en-1-yl)oxy)-2-(3-methylbut-3-en-1-yl)cyclohexane (12).
NaH (208 mg, 5.2 mmol, 60% dispersion in mineral oil) was added to a solution of alcohol 10 (0.5 g, 2.38 mmol) in anhydrous THF (20 mL) at 0 °C under an argon atmosphere, and 3,3-dimethylallyl bromide (0.4 mL, 3.46 mmol) was added, and the reaction mixture was kept stirring at reflux for 24 h, at which time TLC showed no 10 remaining. The mixture was poured into ice and the solvent was evaporated under vacuum. The aqueous phase was extracted with ether (2 × 30 mL) and the organic phase was washed with water (3 × 15 mL) and brine (2 × 15 mL), dried over anhydrous Na2SO4, and evaporated to give a crude residue, which, after column chromatography on silica gel (5% EtOAc/hexane), afforded ether 12 (0.61 g, 92%) as a colorless oil. 1H NMR (CDCl3, 400 MHz): δ 0.85 (s, 3H), 0.97 (s, 3H), 1.05 (dd, J = 13.9, 3.8 Hz, 1H), 1.12 (s, 3H), 1.15 (dd, J = 13.3, 3.6 Hz, 1H), 1.27–1.33 (m, 2H), 1.34–1.42 (m, 2H), 1.63 (s, 3H), 1.69 (s, 1H), 1.71 (s, 3H), 1.75 (s, 3H), 1.96–2.01 (m, 2H), 2.04 (m, 2H), 3.79 (d, J = 6.2 Hz, 2H), 4.68 (d, J = 10.0 Hz, 2H), 5.28 (tt, J = 6.3, 1.4 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 18.0 (CH3) 18.5 (CH2), 22.0 (CH3), 22.5 (CH3), 24.2 (CH2), 24.6 (CH3), 25.7 (CH3), 32.2 (CH3), 34.9 (C), 35.0 (CH2), 41.9 (CH2), 42.4 (CH2), 56.0 (CH), 57.7 (CH2), 76.5 (C), 109.3 (CH2), 123.3 (CH), 133.3 (C), 147.0 (C). IR (film): 909, 1029, 1106, 1374, 1450, 1648 cm−1. HRMS (FAB) m/z calcd for C19H34ONa (M + Na+) 301.2507, found 301.2498.
(6aS,10aR,Z)-4,7,7,10a-Tetramethyl-5,6,6a,7,8,9,10,10a-octahydro-2H-benzo[b]oxocine (epi-arenaran A) (13).
The 2nd Generation Grubbs catalyst (20 mg) was added to a solution of ether 12 (200 mg, 0.72 mmol) in anhydrous CH2Cl2 (40 mL), and the reaction mixture was kept stirring at reflux under an argon atmosphere for 48 h. Then, the solvent was evaporated and the crude product was purified by column chromatography (3% EtOAc/hexane) to yield ether 13 (0.13 g, 84%). 1H NMR (CD3OD, 400 MHz): δ 0.89 (s, 3H), 1.03 (s, 3H), 1.08 (dd, J = 13.4, 4.0 Hz, 1H), 1.13 (s, 3H), 1.20 (m, 1H), 1.34 (d, J = 3.0 Hz, 2H), 1.40–1.45 (m, 2H), 1.71 (s, 3H), 1.76 (dd, J = 13.4, 3.3 Hz, 1H), 1.87 (m, 1H), 1.96 (dd, J = 13.1, 3.1 Hz, 1H), 2.03–2.40 (m, 2H), 3.76 (dd, J = 13.8, 6.9 Hz, 1H), 3.96 (dd, J = 13.8, 7.0 Hz, 1H), 5.43 (t, J = 6.9 Hz, 1H). 13C NMR (CD3OD, 100 MHz): δ 15.1 (CH3), 18.0 (CH2), 21.1 (CH3), 24.6 (CH3), 25.2 (CH2), 31.4 (CH3), 34.1 (C), 35.5 (CH2), 41.9 (CH2), 42.8 (CH2), 55.5 (CH), 56.4 (CH2), 77.4 (C), 123.6 (CH), 140.6 (C). IR (film): 1052, 1071, 1095, 1153, 1176, 1212, 1364, 1386, 1454, 1475, 1671 cm−1. HRMS (FAB) m/z calcd for C15H26ONa (M + Na+) 245.1881, found 245.1876.
(4aS,8aS)-8-Ethyl-4,4,7,8a-tetramethyl-1,2,3,4,4a,5,6,8a-octahydronaphthalene (15).
5 mL of an aqueous suspension of RANEY® nickel (Aldrich, cat. 221678) was added to a stirred solution of 17 (2.5 g, 7.22 mmol) in THF (30 mL) and the mixture was further stirred at room temperature for 1 h, under an ordinary hydrogen pressure (balloon). Then the mixture was diluted with diethyl ether (50 mL) and filtered on a silica gel–Na2SO4 mixture (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 g) column, washed with diethyl ether (10 mL) to yield 15 as a colorless oil (1.42 g, 89%). [α]20D +67.1 (c 1.1, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.82 (s, 3H), 0.87 (s, 3H), 0.92 (s, 3H), 0.96 (t, J = 7.5 Hz, 3H), 1.56 (s, 3H), 0.99–1.70 (m, 8H), 1.75–2.08 (m, 5H). 13C NMR (CDCl3, 100 MHz): δ 15.1 (CH3), 19.07 (CH3), 19.09 (CH3), 19.3 (CH2), 20.0 (CH2), 20.5 (CH3), 21.7 (CH2), 33.3 (CH2), 33.61 (C), 33.63 (CH3), 36.9 (CH2), 39.1 (C), 41.8 (CH2), 51.9 (CH), 125.0 (C), 142.3 (C). IR (film): 1374, 1457, 1644 cm−1. HRMS (FAB) m/z calcd for C16H28Na (M + Na+) 243.2089, found 243.2093.
:16 g) column, washed with diethyl ether (10 mL) to yield 15 as a colorless oil (1.42 g, 89%). [α]20D +67.1 (c 1.1, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.82 (s, 3H), 0.87 (s, 3H), 0.92 (s, 3H), 0.96 (t, J = 7.5 Hz, 3H), 1.56 (s, 3H), 0.99–1.70 (m, 8H), 1.75–2.08 (m, 5H). 13C NMR (CDCl3, 100 MHz): δ 15.1 (CH3), 19.07 (CH3), 19.09 (CH3), 19.3 (CH2), 20.0 (CH2), 20.5 (CH3), 21.7 (CH2), 33.3 (CH2), 33.61 (C), 33.63 (CH3), 36.9 (CH2), 39.1 (C), 41.8 (CH2), 51.9 (CH), 125.0 (C), 142.3 (C). IR (film): 1374, 1457, 1644 cm−1. HRMS (FAB) m/z calcd for C16H28Na (M + Na+) 243.2089, found 243.2093.
4-((1S,6S)-2,2,6-Trimethyl-6-propionylcyclohexyl)butan-2-one (18).
O3/O2 was bubbled through a solution of compound 15 (2.0 g, 9.09 mmol) in CH2Cl2 (60 mL) cooled at −78 °C for 1 h, after which time TLC showed no remaining starting material. Then, argon was bubbled through the solution for 5 min, and triphenylphosphine (2.6 g, 9.9 mmol) was added, and the mixture was further stirred at room temperature for 5 h. After evaporation of the solvent under vacuum, the resulting crude product was purified by column chromatography on silica gel (25% EtOAc/hexane) giving diketone 18 (1.95 g, 85%), as a colourless syrup. [α]20D −16.8 (c 0.9, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.87 (s, 3H), 0.88 (s, 3H), 0.98 (t, J = 7.2 Hz, 3H), 1.18 (s, 3H), 1.18 (m, 1H), 1.28–1.75 (m, 8H), 2.05 (s, 3H), 2.25–2.68 (m, 4H). 13C NMR (CDCl3, 100 MHz): δ 8.5 (CH3), 17.2 (CH3), 18.2 (CH2), 22.1 (CH2), 22.5 (CH3), 29.8 (CH3), 30.6 (CH2), 33.4 (CH3), 34.3 (C), 37.1 (CH2), 41.2 (CH2), 45.5 (CH2), 47.7 (CH), 52.8 (C), 209.0 (C), 217.6 (C). IR (film): 772, 957, 1161, 1355, 1460, 1697, 1715 cm−1. HRMS (FAB) m/z calcd for C16H28O2Na (M + Na+) 275.1987, found 275.1979.
2-((1S,6S)-2,2,6-Trimethyl-6-propionylcyclohexyl)ethyl acetate (19).
m-Chloroperbenzoic acid (70%, 493 mg, 2.158 mmol) was added to a solution of compound 18 (272 mg; 1.079 mmol) in chloroform (10 mL), and the reaction mixture was stirred at reflux for 3 days, at which TLC showed no remaining starting material. Then, a 10% Na2SO3 solution (1 mL) was added, and the mixture was stirred for an additional 15 min. Then the reaction was extracted with EtOAc (3 × 10 mL) and the organic phase was successively washed with sat. NaHCO3 (5 × 10 mL) and brine (2 × 10 mL), and dried over anhydrous Na2SO4 and evaporated to give a crude residue, which, after column chromatography on silica gel (10% EtOAc/hexane), afforded 19 (234 mg, 81%) as a colourless syrup. [α]20D −13.3 (c 1.0, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.89 (s, 3H), 0.90 (s, 3H), 1.00 (t, J = 7.2 Hz, 3H), 1.18 (s, 3H), 1.18–1.74 (m, 9H), 1.99 (s, 3H), 2.40–2.56 (m, 2H), 3.79–3.97 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 8.5 (CH3), 17.5 (CH3), 18.2 (CH2), 21.0 (CH3), 22.7 (CH2), 27.2 (CH2), 30.5 (CH2), 33.1 (CH3), 34.0 (C), 36.3 (CH2), 40.8 (CH2), 44.7 (CH), 52.5 (C), 65.1 (CH2), 170.9 (C), 216.9 (C). IR (film): 957, 1096, 1355, 1459, 1697, 1714, 3072 cm−1. HRMS (FAB) m/z calcd for C16H28O3Na (M + Na+) 291.1936, found 291.1944.
(1S,2S)-1,3,3-Trimethyl-2-(3-oxobutyl) cyclohexyl formate (22).
m-Chloroperbenzoic acid (70%, 1.37 g, 5.58 mmol) and NaHCO3 (0.56 g, 6.69 mmol) were added to a solution of ketoaldehyde 21 (0.5 g, 2.23 mmol) in CH2Cl2 (50 mL) and the mixture was stirred under reflux for 1.5 h. Then, 10% aq Na2SO3 (5 mL) was added and the mixture was further stirred at room temperature for 15 min. Then, EtOAc (20 mL) was added and the organic phase was washed with water (3 × 20 mL) and brine (2 × 20 mL), dried over anhydrous Na2SO4, and evaporated to give formate 22 (0.49 g, 93%). 1H NMR (CDCl3, 400 MHz): δ 0.88 (s, 3H), 0.98 (s, 3H), 1.14–1.28 (m, 1H), 1.30–1.40 (m, 1H), 1.43–1.50 (m, 1H), 1.50–1.60 (m, 3H), 1.55 (s, 3H), 1.60–1.75 (m, 3H), 2.16 (s, 3H), 2.48 (dt, J = 12.0, 3.0 Hz, 1H), 2.51–2.59 (m, 1H), 2.65–2.73 (m, 1H), 8.05 (s, 1H). 13C NMR (CDCl3, 100 MHz): δ 19.8 (CH2), 20.2 (CH2), 20.9 (CH3), 21.7 (CH3), 30.0 (CH3), 32.7 (CH3), 35.8 (C), 38.6 (CH2), 40.6 (CH2), 45.9 (CH2), 53.2 (CH), 88.9 (C), 209.3 (C), 160.5 (CH). HRMS (FAB) m/z calcd for C14H24O3Na (M + Na+) 263.1623, found 263.1619.
(1S,2S)-1,3,3-Trimethyl-2-(3-methylbut-3-en-1-yl) cyclohexan-1-ol (5).
2 M n-BuLi in cyclohexane (1.25 mL, 2.5 mmol) was added to a solution of methyltriphenylphosphonium bromide (0.91 g, 2.5 mmol, 98%) in anhydrous THF (25 mL), and the mixture was stirred at 0 °C under an argon atmosphere for 15 min. Then, a solution of ketoester 22 (0.3 g, 1.25 mmol) in anhydrous THF (0.3 mL) was added, and the resulting mixture was kept stirring for 3 h. Then, the reaction was carefully quenched with water (0.5 mL). The solvent was evaporated and ether was added (25 mL), and the organic phase was washed with water (3 × 10 mL) and brine (2 × 10 mL), dried over anhydrous Na2SO4 and evaporated to afford a crude product that was purified by column chromatography on silica gel (20% EtOAc/hexane) to yield alcohol 5 (0.23 g, 86%) as a yellow oil. [α]20D +6.5 (c 1.2, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.83 (s, 3H), 0.95 (s, 3H), 1.11 (t, J = 4.5 Hz, 1H), 1.17 (s, 3H), 1.21 (dd, J = 13.0, 4.0 Hz, 1H), 1.25 (s, 1H), 1.37 (m, 1H), 1.31 (dd, J = 12.3, 4.1 Hz, 1H), 1.40–1.50 (m, 2H), 1.52–1.64 (m, 2H), 1.75 (s, H), 2.11 (m, 1H), 2.20 (m, 1H), 4.70 (s, 2H). 13C NMR (CDCl3, 100 MHz): δ 20.5 (CH2), 21.4 (CH3), 22.6 (CH3), 23.2 (CH3), 24.4 (CH2), 32.8 (CH3), 35.6 (C), 40.9 (CH2), 41.5 (CH2), 43.6 (CH2), 56.8 (CH), 74.1 (C), 109.7 (CH2), 147.1 (C). IR (film): 883, 911, 1063, 1100, 1161, 1373, 1388, 1459, 1648, 1714, 3072, 3300–3600 cm−1. HRMS (FAB) m/z calcd for C14H26ONa (M + Na+) 233.1881, found 233.1890.
(1S,2S)-1-(Allyloxy)-1,3,3-trimethyl-2-(3-methylbut-3-en-1-yl)cyclohexane (4).
NaH (250 mg, 6.24 mmol, 60% dispersion in mineral oil) was added to a solution of alcohol 5 (564 mg, 2.70 mmol) in anhydrous THF (20 mL) at 0 °C under an argon atmosphere, and allyl bromide (0.5 mL, 5.54 mmol) was added, and the reaction mixture was kept stirring at reflux for 20 h, at which time TLC showed no 5 remaining. The mixture was poured into ice and the solvent was evaporated under vacuum. Then ether (50 mL) was added and the organic phase was washed with water (3 × 20 mL) and brine (2 × 20 mL), dried over anhydrous Na2SO4, and evaporated to give a crude residue, which, after column chromatography on silica gel (5% EtOAc/hexane), afforded ether 4 (618 mg, 90%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.87 (m, 1H), 5.25 (dd, J = 1.7, 17.2 Hz, 1H), 5.05 (dd, J = 1.7, 10.4 Hz, 1H), 4.65 (s, 2H), 3.92–3.83 (m, 2H), 2.22 (m, 1H), 2.01 (m, 1H), 1.72 (s, 3H), 1.65–1.52 (m, 2H), 1.41–1.28 (m, 5H), 1.24 (s, 1H), 1.19 (m, 1H), 1.14 (s, 3H), 0.95 (s, 3H), 0.84 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 147.4 (C), 136.5 (CH), 114.6 (CH2), 108.9 (CH2), 78.5 (C), 60.9 (CH2), 53.5 (CH), 41.2 (CH2), 40.7 (CH2), 37.7 (CH2), 35.4 (C), 32.9 (CH3), 25.2 (CH2), 22.6 (CH3), 22.1 (CH3), 19.9 (CH2), 19.8 (CH3). IR (film): 1074, 1155, 1264, 1374, 1455, 1648 cm−1. HRMS (FAB) m/z calcd for C17H30ONa (M + Na+) 273.2194, found 273.2185.
Treatment of ether 4 with the 2nd generation Grubbs catalyst
The 2nd Generation Grubbs catalyst (22 mg) was added to a solution of ether 4 (187 mg, 0.75 mmol) in anhydrous CH2Cl2 (30 mL), and the mixture was kept stirring at reflux under an argon atmosphere for 2 h. Then, the solvent was evaporated affording a crude product, which consists of a complex mixture and the starting material.
(2S,3S)-1,1,3-Trimethyl-3-((3-methylbut-2-en-1-yl)oxy)-2-(3-methylbut-3-en-1-yl)cyclohexane (23).
NaH (34 mg, 0.86 mmol, 60% dispersion in mineral oil) was added to a solution of alcohol 5 (147 mg, 0.7 mmol) in anhydrous THF (12.5 mL) at 0 °C under an argon atmosphere, and 3,3-dimethylallyl bromide (0.24 mL, 20.77 mmol) was added, and the reaction mixture was kept stirring at reflux for 24 h, at which time TLC showed no 5 remaining. The mixture was poured into ice and the solvent was evaporated under vacuum. Then ether (25 mL) was added and the organic phase was washed with water (3 × 10 mL) and brine (2 × 10 mL), dried over anhydrous Na2SO4, and evaporated to give a crude residue, which, after column chromatography on silica gel (3% EtOAc/hexane), afforded ether 23 (179 mg, 92%) as a colorless oil. [α]20D +21.2 (c 0.9, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.86 (s, 3H), 0.97 (s, 3H), 1.17 (s, 3H),), 1.26 (s, 1H), 1.31–1.33 (m, 2H), 1.36 (m, 1H), 1.43–1.46 (m, 2H), 1.56–1.59 (m, 3H), 1.64 (s, 3H), 1.71 (s, 3H), 1.73 (s, 3H), 2.03 (m, 1H), 2.24 (m, 1H), 3.82–3.90 (m, 2H), 4.67 (s, 2H), 5.27 (t, J = 6.4 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 18.02 (CH3), 20.0 (CH2), 20.3 (CH3), 22.3 (CH3), 22.6 (CH3), 25.2 (CH2), 25.8 (CH3), 32.9 (CH3), 35.4 (C), 37.3 (CH2), 40.5 (CH2), 41.2 (CH2), 53.0 (CH), 56.8 (CH2), 78.2 (C), 108.9 (CH2), 122.8 (CH), 134.4 (C), 147.6 (C). IR (film): 754, 960, 1074, 1124, 1276, 1721 cm−1. HRMS (FAB) m/z calcd for C19H34ONa (M + Na+) 301.2507, found 301.2509.
Arenaran A (1).
The 2nd Generation Grubbs catalyst (10 mg) was added to a solution of ether 23 (62 mg, 0.223 mmol) in anhydrous CH2Cl2 (30 mL), and the mixture was kept stirring at reflux under an argon atmosphere for 3 h. Then, the solvent was evaporated and the crude product was purified by column chromatography (3% AcOEt/hexane) to yield 1 (45 mg, 91%). [α]20D −32.1 (c 0.01, CHCl3). 1H NMR (CD3OD, 400 MHz): δ 0.91 (s, 3H), 1.16 (ddd, J = 13.3, 4.1 Hz, 1H), 1.25 (s, 3H), 1.35–1.32 (m, 2H), 1.46 (dt, J = 13.5, 3.0 Hz, 1H), 1.51 (m, 2H), 1.55 (m, 2H), 1.61–1.58 (m, 2 H), 1.65 (m, 2H), 1.69 (s, 3H), 1.76 (ddd, J = 12.7, 4.5 Hz), 3.37 (m, 2H), 4.03 (d, J = 18.4 Hz), 4.22 (dd, J = 18.4, 2.1 Hz), 5.15 (br s). 13C NMR (CD3OD, 100 MHz): δ 19.7 (CH3), 21.0 (CH2), 21.9 (CH2), 24.1 (CH3), 25.0 (CH2), 28.9 (CH3), 32.3 (CH2), 34.0 (C), 35.2 (CH3), 41.9 (CH), 45.4 (CH2), 61.1 (CH), 80.2 (C), 123.3 (CH2), 132.9 (C). 1H NMR (CDCl3, 400 MHz): δ 0.86 (s, 3H), 0.91 (s, 3H), 1.16 (dd, J = 13.3, 4.1 Hz, 1H), 1.25 (s, 3H), 1.32–1.35 (m, 2H), 1.46 (dt, J = 13.5, 3.0 Hz, 1H), 1.51 (m, 1H), 1.55 (m, 1H), 1.58–1.61 (m, 2H), 1.65 (m, 1H), 1.69 (s, 3H), 1.76 (dd, J = 12.7, 4.5 Hz, 1H), 3.37 (m, 1H), 4.03 (d, J = 18.4 Hz, 1H), 4.22 (dd, J = 18.4, 2.1 Hz, 1H), 5.15 (br s, 1H). 13C NMR (CDCl3, 100 MHz): δ 20.1 (CH2), 22.0 (CH3), 22.9 (CH3), 24.5 (CH2), 26.3 (CH3), 29.4 (CH2), 33.3 (CH3), 34.5 (C), 35.5 (CH2), 42.3 (CH2), 45.7 (CH), 61.5 (CH2), 79.8 (C), 123.7 (CH), 133.3 (C). HRMS (FAB) m/z calcd for C15H26ONa (M + Na+) 245.1881, found 245.1893.
Arenaran B (2).
m-Chloroperbenzoic acid (70%, 50.0 mg, 0.20 mmol) was added to a solution of compound 1 (28 mg, 0.126 mmol) in CH2Cl2 (12.5 mL) at 0 °C, and the mixture was stirred for 1 h. Then a 10% Na2SO3 solution (5 mL) was added and the mixture was further stirred for 15 min. Then, EtOAc (10 mL) was added, and the organic phase was washed with sat NaHCO3 (3 × 10 mL) and brine (2 × 10 mL), and dried over anhydrous Na2SO4. Evaporation of the solvent under vacuum gave finally epoxide 2 (28 mg, 93%) as a low m.p. solid. [α]25D: −24.9 (c 0.2, CHCl3) lit.1: −24.4 (c 0.23, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.85 (s, 3H), 0.98 (s, 3H), 1.16 (s, 3H), 1.21 (m, 1H), 1.29 (s, 3H), 1.35 (m, 1H), 1.40 (m, 1H), 1.45 (m, 1H), 1.50 (m, 1H), 1.58 (m, 1H), 1.67 (m, 1H), 1.86 (dt, J = 13.2, 3.9 Hz, 1H), 2.42 (dt, J = 13.4, 4.9 Hz, 1H), 2.79 (s 1H), 3.90 (d, J = 17.6 Hz, 1H), 4.02 (dd, J = 17.6, 1.7 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 19.9 (CH2), 21.5 (CH3), 22.0 (CH2), 22.5 (CH3), 22.9 (CH3), 31.8 (CH2), 33.2 (CH3), 34.7 (C), 35.9 (CH2), 42.2 (CH2), 44.7 (CH), 58.1 (CH2), 60.8 (C), 64.1 (CH), 80.0 (C). 1H NMR (CDCl3, 500 MHz): δ 0.85 (s, 3H), 0.98 (s, 3H), 1.16 (s, 3H), 1.19 (ddd, J = 13.5, 13.5, 3.8 Hz, 1H), 1.30 (s, 3H), 1.33 (ddd, J = 13.5, 13.5, 3.8 Hz, 1H), 1.39 (ddd, J = 13.5, 3.8, 3.8 Hz, 1H), 1.45 (ddd, J = 13.5, 3.8, 3.8 Hz, 1H), 1.50 (dd, J = 13.4, 4.3 Hz, 1H), 1.55 (dp, J = 13.5, 3.8 Hz, 1H), 1.61 (tdd, J = 13.4, 4.3, 4.3 Hz, 1H), 1.68 (tt, J = 13.4, 4.3 Hz, 1H), 1.72 (ddd, J = 13.5, 3.8, 3.8 Hz, 1H), 1.88 (ddd, J = 13.4, 13.4, 4.3 Hz, 1H), 2.43 (ddd, J = 13.4, 4.3, 4.3 Hz, 1H), 2.79 (d, J = 2.3 Hz, 1H), 3.90 (d, J = 15.9 Hz, 1H), 4.03 (dd, J = 15.9, 2.3 Hz, 1H). 13C NMR (CDCl3, 125 MHz): δ 20.0 (CH2), 21.7 (CH3), 22.2 (CH2), 22.7 (CH3), 23.1 (CH3), 32.0 (CH2), 33.4 (CH3), 34.8 (C), 36.1 (CH2), 42.4 (CH2), 44.9 (CH), 58.2 (CH2), 60.9 (C), 64.2 (CH), 80.2 (C).
(1R,2R,4aS,8aS)-2,5,5,8a-Tetramethyl-1-(3-methylbut-3-en-1-yl)decahydronaphthalen-2-ol (26).
2 M n-BuLi in cyclohexane (1.7 mL, 3.3 mmol) was added to a solution of methyltriphenylphosphonium bromide (15.75 g, 60 mmol, 98%) in anhydrous THF (75 mL), and the mixture was stirred at −78 °C under an argon atmosphere for 15 min. Then, a solution of ketoester 24 (3.8 g, 12 mmol) in anhydrous THF (2 mL) was added, and the resulting mixture was kept stirring for 45 min. Then, the reaction was carefully quenched with water (5 mL), and the solvent was evaporated. Then, ether (100 mL) was added, and the organic phase was washed with water (3 × 30 mL) and brine (2 × 30 mL), dried over anhydrous Na2SO4 and evaporated to afford a crude product that was purified by column chromatography on silica gel (20% EtOAc/hexane) to yield alcohol 26 (3.4 g, 91%). [α]20D +8.5 (c 0.8, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.79 (s, 3H), 0.80 (s, 3H), 0.86 (s, 3H), 0.92 (dd, J = 12.1, 2.3 Hz, 1H), 0.97 (dd, J = 12.7, 3.8 Hz, 1H), 1.05 (t, J = 4.0 Hz, 1H), 1.14 (s, 3H), 1.25 (br s, 1H), 1.28 (s, 1H), 1.35–1.40 (m, 2H), 1.41 (s, 1H), 1.43 (m, 1H), 1.53–1.61 (m, 2H), 1.62–1.67 (m, 3H), 1.73 (s, 3H), 1.86 (dt, J = 12.2, 3.1 Hz, 1H), 2.04–2.14 (m, 2H), 4.69 (br s, 2H). 13C NMR (CDCl3, 100 MHz): δ 15.5 (CH3), 18.5 (CH2), 20.6 (CH2), 21.5 (CH3), 22.6 (CH3), 23.6 (CH2), 23.9 (CH3), 33.3 (C), 33.4 (CH3), 39.2 (C), 39.7 (CH2), 41.3 (CH2), 42.0 (CH2), 44.6 (CH2), 56.2 (CH), 61.5 (CH), 74.1 (C), 109.6 (CH2), 147.1 (C). IR (film): 882, 968, 1083, 1103, 1386, 1455, 1648, 1727, 3300–3500 cm−1. HRMS (FAB) m/z calcd for C19H34ONa (M + Na+) 301.2507, found 301.2499.
Treatment of aldehyde 25 with N2H4–KOH. Obtention of alcohol 26
Hydrazine (2 mL, 41.2 mmol) was added to a solution of aldehyde 25 (2.0 g, 6.5 mmol) in triethyleneglycol dimethyl ether (20 mL) and the mixture was stirred under reflux for 1 h, then KOH (2.31 g, 41.25 mol) was added and the mixture was stirred at reflux for an additional 11 h. Then, the mixture was kept at room temperature and H2O (10 mL) was added. EtOAc (50 mL) was added and the organic phase was washed with H2O (10 × 20 mL) and brine (3 × 20 mL), dried over anhydrous Na2SO4 and evaporated to give a crude product which after column chromatography on silica gel (10% EtOAc/hexane), afforded alcohol 26 (1.6 g, 81%) as a colourless oil.
(4aS,5R,6R,8aS)-1,1,4a,6-Tetramethyl-6-((3-methylbut-2-en-1-yl)oxy)-5-(3-methylbut-3-en-1-yl)decahydronaphthalene (27).
NaH (100 mg, 2.5 mmol, 60% dispersion in mineral oil) was added to a solution of alcohol 26 (180 mg, 0.647 mmol) in anhydrous THF (100 mL) at 0 °C under an argon atmosphere, and 3,3-dimethylallyl bromide (0.2 mL, 1.73 mmol) was added, and the reaction mixture was kept stirring at reflux for 24 h, at which time TLC showed no 26 remaining. The mixture was poured into ice and the solvent was evaporated under vacuum. Then, ether (100 mL) was added and the organic phase was washed with water (3 × 30 mL) and brine (2 × 30 mL), dried over anhydrous Na2SO4, and evaporated to give a crude residue, which, after column chromatography on silica gel (5% EtOAc/hexane), afforded ether 27 (206 mg, 92%). [α]20D −7.1 (c 1.0, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 0.79 (s, 3H), 0.83 (s, 3H), 0.85 (s, 3H), 0.90 (dd, J = 12.3, 2.2 Hz, 1H), 0.97 (dd, J = 12.9, 3.6 Hz, 1H), 1.14 (s, 3H), 1.25 (s, 2H), 1.35–1.44 (m, 2H), 1.54–1.61 (m, 4H), 1.63 (s, 3H), 1.65–1.68 (m, 3H), 1.71 (s, 3H), 1.72 (s, 3H), 1.84 (dt, J = 12.2, 3.3 Hz, 1H), 2.01 (dd, J = 13.6, 4.6 Hz, 1H), 2.17 (dd, J = 13.3′ 4.6 Hz, 1H), 3.81–3.90 (m, 2H), 4.65 (br s, 2H), 5.25 (tt, J = 6.4, 1.4 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ 15.9 (CH3), 18.1 (CH3), 18.5 (CH2), 20.1 (CH2), 20.8 (CH3), 21.5 (CH3), 22.6 (CH3), 24.3 (CH2), 25.8 (CH3), 33.2 (C), 33.4 (CH3), 38.5 (CH3), 39.2 (C), 40.1 (CH2), 41.0 (CH2), 42.1 (CH2), 56.1 (CH), 56.8 (CH2), 58.1 (CH), 78.3 (C), 108.9 (CH2), 122.9 (CH), 134.3 (C), 147.6 (C). IR (film): 973, 1035, 1058, 1079, 1131, 1386, 1446, 1647 cm−1. HRMS (FAB) m/z calcd for C24H42ONa (M + Na+) 369.3133, found 369.3141.
(6aR,8aS,12aS,12bR,Z)-4,6a,9,9,12a-Pentamethyl-2,3,6a,7,8,8a,9,10,11,12,12a,12b-dodecahydro-1H-naphtho[2,1-b]oxocine (28).
The 2nd Generation Grubbs catalyst (30 mg) was added to a solution of ether 27 (310 mg, 0.895 mmol) in anhydrous CH2Cl2 (40 mL). The mixture was kept stirring at reflux under an argon atmosphere for 4 h. Then, the solvent was evaporated and the crude product was purified by column chromatography (3% AcOEt/hexane) to yield 28 (218 mg, 84%). [α]20D +40.4 (c 0.9, CHCl3). 1H NMR (CD3OD, 400 MHz): δ 0.80 (s, 3H), 0.82 (dd, J = 12.2, 2.5 Hz, 1H), 0.87 (s, 3H), 0.88 (s, 3H), 1.13 (dd, J = 13.3, 3.8 Hz, 1H), 1.24 (s, 3H), 1.30 (dd, J = 12.6, 3.5 Hz, 1H), 1.34–1.43 (m, 3H), 1.50 (dd, J = 12.0, 3.8 Hz, 1H), 1.54–1.58 (m, 2H), 1.60–1.66 (m, 3H), 1.68 (br d, J = 1.5 Hz, 3H), 1.70–1.73 (m, 2H), 1.82 (dd, J = 13.1, 4.5 Hz, 1H), 3.37 (dd, J = 12.7, 4.4 Hz, 1H), 4.03 (d, J = 18.4 Hz, 1H), 4.23 (dd, J = 18.4, 2.0 Hz, 1H), 5.15 (s, 1H). 13C NMR (CD3OD, 100 MHz): δ 15.8 (CH3), 18.8 (CH2), 20.1 (CH2), 21.8 (CH3), 23.4 (CH2), 23.9 (CH3), 26.2 (CH3), 29.2 (CH2), 33.3 (C), 33.6 (CH3), 36.3 (CH2), 38.0 (C), 40.6 (CH), 41.8 (CH), 50.3 (CH), 55.9 (CH), 61.7 (CH2), 79.9 (C), 123.5 (CH), 133.7 (C). IR (film): 1052, 1111, 1127, 1218, 1384, 1450 cm−1. HRMS (FAB) m/z calcd for C20H34ONa (M + Na+) 313.2507, found 313.2516.
4aS,6aR,8aS,9aR,11bS)-4,4,6a,9a,11b-Pentamethyltetradecahydro-1H-naphtho[2,1-b]oxiren[2,3-f]oxocane (29).
m-Chloroperbenzoic acid (70%, 147 mg, 0.6 mmol) was added to a solution of compound 28 (125 mg, 0.431 mmol) in dichloromethane (10 mL), cooled at 0 °C, and the reaction mixture was stirred for 1 h, at which TLC showed no remaining starting material. Then, a 10% Na2SO3 solution (10 mL) was added, and the mixture was stirred for an additional 15 min. Then the reaction was extracted with EtOAc (3 × 20 mL). The organic phase was successively washed with sat. NaHCO3 (3 × 30 mL) and brine (2 × 30 mL), and dried over anhydrous Na2SO4 and evaporated to give a crude residue, which, after column chromatography on silica gel (5% EtOAc/hexane), afforded epoxide 29 (2.3 g, 93%) as a colourless oil. 1H NMR (CDCl3, 400 MHz): δ 0.81 (s, 3H), 0.86 (s, 3H), 0.90 (s, 3H), 1.16 (s, 3H), 1.29 (s, 3H), 1.90–1.21 (m, 15H), 2.44 (dd, J = 13.3, 5.5 Hz, 1H), 2.80 (d, J = 2.0 Hz, 1H), 3.89 (d, J = 15.8 Hz, 1H), 4.03 (dd, J = 15.8, 2.2 Hz, 1H). 13C NMR (CDCl3, 125 MHz): δ 15.33 (CH3), 18.96 (CH2), 20.02 (CH2), 20.82 (CH2), 21.95 (CH3), 23.09 (CH3), 23.68 (CH3), 31.72 (CH2), 33.47 (C), 33.77 (CH3), 36.93 (CH2), 38.35 (C), 40.88 (CH2), 41.84 (CH2), 49.51 (CH), 56.17 (CH), 58.31 (CH2), 61.10 (C), 64.22 (CH), 80.33 (C). HRMS (FAB) m/z calcd for C20H34O2Na (M + Na+) 329.2457, found 329.2461.
Conclusions
In summary, the first synthesis of arenaran A (1) and B (2), utilizing a ring-closing metathesis (RCM) process, starting from commercial (−)-sclareol (20) is reported. For the RCM process to be successfully applied, some structural requirements must be met. The trans-fused structure of the natural products is corroborated by comparison of their spectroscopic data with those of the cis-fused isomer (epi-arenaran A, 13), which was also synthesized. This strategy can also be utilized for preparing other natural oxocene terpenes. Thus, epoxide 29, the 3-debromoderivative of the natural terpene 3, has been synthesized from the diterpene 20.
Acknowledgements
The authors thank the Spanish Ministry of Economy and Competitiveness (Project CTQ2014-56611-R/BQU) and the Regional Government of Andalucia (Project P11-CTS-7651) for financial support and assistance provided to the FQM-348 group. This research is a part of the Doctoral Thesis of P. Gutierrez.
Notes and references
- P. A. Horton and P. Crews, J. Nat. Prod., 1995, 58, 44 CrossRef CAS.
- D. Iliopoulou, N. Mihopoulou, V. Roussis and C. Vagias, J. Nat. Prod., 2003, 66, 1225 CrossRef CAS PubMed.
- M. Reggelin, M. Gerlach and M. Vogt, Eur. J. Org. Chem., 1999, 1011 CrossRef CAS.
- S. Serra and V. Lissoni, Eur. J. Org. Chem., 2015, 2226–2234 CrossRef CAS.
- F. W. J. Demnitz, S. Freiberg and H.-P. Weber, Helv. Chim. Acta, 1995, 78, 887 CrossRef CAS.
- E. Alvarez-Manzaneda, R. Chahboun, E. Cabrera, E. Alvarez, A. Haidour, J. M. Ramos, R. Alvarez-Manzaneda, M. Hmamouchi and H. Es-Samti, Chem. Commun., 2009, 592 RSC.
-
(a) A. F. Barrero, E. Alvarez-Manzaneda, R. Chahboun and M. C. Páiz, Tetrahedron Lett., 1998, 39, 9543 CrossRef CAS;
(b) A. F. Barrero, E. Alvarez-Manzaneda, R. Alvarez-Manzaneda, R. Chahboun, R. Menenes and M. Aparicio, Synlett, 1999, 713 CrossRef CAS.
- E. Alvarez-Manzaneda, R. Chaboun, E. Alvarez, A. Fernández, R. Alvarez-Manzaneda, A. Haidour, J. M. Ramos and A. Akhaouzan, Chem. Commun., 2012, 48, 606 RSC.
- A. F. Barrero, E. Alvarez-Manzaneda, R. Chahboun and A. F. Arteaga, Synth. Commun., 2004, 34, 3631 CrossRef CAS.
- Y. X. Tang and T. Suga, Phytochemistry, 1994, 37, 737 CrossRef CAS PubMed.
- B. A. Baker, Z. V. Boskovic and B. H. Lipshutz, Org. Lett., 2008, 10, 289 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copy of 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c6ob01640e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence








