
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general approach to iridoids by applying a new Julia olefination and a tandem anion-radical-carbocation crossover reaction†
Lucie
Řehová
,
Martin
Dračínský
and
Ullrich
Jahn
*
Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Flemingovo náměstí 2, 16610 Prague 6, Czech Republic. E-mail: jahn@uochb.cas.cz
First published on 13th September 2016
Abstract
A unified, asymmetric approach to the total synthesis of naturally occurring iridoids is presented. The synthesis features a recently discovered ortho → α transmetalation of alkyl aryl sulfone carbanions, thus enabling Julia reactions, by which so far hardly accessible disilylated olefins have been obtained. A subsequent tandem alkoxycarbonylation/oxidative radical cyclization afforded substituted cyclopentane building blocks with high diastereoselectivity. These compounds serve as unique central intermediates for short access to dihydronepetalactone, dolicholactone and potentially other iridoids.
Introduction
Cyclopentanoid monoterpenes (iridoids) have long been recognized as an important class of plant secondary metabolites displaying a wide range of biological activities.1a–e A number of iridoids including iridomyrmecin (1), isoiridomyrmecin (2) and dihydronepetalactone (3) were isolated from the volatile oils present in the cat-attracting plant Actinidia polygama.2 These iridoids, accompanied by nepetalactone (4), were also isolated from Nepeta cataria, commonly known as catnip.3 Catnip is famous for its irresistible action on cats and has been recently used in medical preparations as an antidiaphoretic, antispasmodic and mild sedative. Both, 3 and 4 have been identified as effective insect repellents.4 Dolicholactone (5) was isolated from the wild plant Teucrium marum growing in the Mediterranean area.5 The iridoid scaffold is biosynthesized by the enzyme iridoid synthase using 8-oxocitral as a substrate.6 Recently, other iridoids have been found in nature. However, their isolation from plants with sufficient purity and amount is often complicated. Because of this their absolute or even their relative configuration as well as their biological activities could not be established.Therefore, total synthesis serves best to gain access to the natural products and their analogs. A number of syntheses of iridoids have been published over the years.7–12 Most of them target only individual members of the family, and are not suitable for the preparation of these compounds in larger amounts or provide the desired iridoid in racemic form.8 More divergent approaches include Wolinski's synthesis of four of the eight possible stereoisomers of dihydronepetalactone (3),9 Francke's synthesis of the eight trans-fused diastereomers,10 Hofferberth's intramolecular Michael addition-based synthesis of iridoid lactols or lactones,11 and the synthesis of iridolactones via a stereoselective Favorski rearrangement (Fig. 1).12
 | ||
| Fig. 1 Major naturally occurring iridoids. | ||
We developed previously a short approach to racemic (±)-dihydronepetalactone (3). Keysteps were a telescoped ozonolysis/Wittig reaction, which provided ω-silylated citronellate in only 30% yield as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z mixture, and an oxidative radical cyclization to obtain the cyclopentane unit of 3, which was obtained as an inseparable 2:1 trans/cis-diastereomeric mixture.13 Obviously, the strategy was far from ideal. We hypothesized that the Julia reaction14 may serve as a better option and that a more symmetrical trisubstituted olefin unit would destabilize boat-type transition states in the crucial radical cyclization. However, recent attempts to promote Julia reactions with β,β′-branched alkyl phenyl sulfones have led to a surprising reversal in the metalation selectivity, in that such substrates are initially ortho-metalated despite having a significantly more acidic α-proton.15 Further studies revealed that a rearrangement of the carbanion from the ortho- to the α-position is possible under thermodynamic conditions. Therefore, to successfully apply the Julia reaction in the total synthesis of iridoids, suitable silylated precursors and rearrangement conditions had to be found.
:1 E/Z mixture, and an oxidative radical cyclization to obtain the cyclopentane unit of 3, which was obtained as an inseparable 2:1 trans/cis-diastereomeric mixture.13 Obviously, the strategy was far from ideal. We hypothesized that the Julia reaction14 may serve as a better option and that a more symmetrical trisubstituted olefin unit would destabilize boat-type transition states in the crucial radical cyclization. However, recent attempts to promote Julia reactions with β,β′-branched alkyl phenyl sulfones have led to a surprising reversal in the metalation selectivity, in that such substrates are initially ortho-metalated despite having a significantly more acidic α-proton.15 Further studies revealed that a rearrangement of the carbanion from the ortho- to the α-position is possible under thermodynamic conditions. Therefore, to successfully apply the Julia reaction in the total synthesis of iridoids, suitable silylated precursors and rearrangement conditions had to be found.
Here we describe an efficient integrated, asymmetric synthetic approach to iridoids, exemplified by dihydronepetalactone (3) and dolicholactone (5), by applying a carbanion rearrangement-driven Julia reaction and a highly diastereoselective tandem enolate alkoxycarbonylation-oxidative radical cyclization with cationic termination as the key steps.
Results and discussion
The retrosynthesis of both, dihydronepetalactone (3) and dolicholactone (5), leads to the common cyclopentane building block 7 (Scheme 1). The effect of differently substituted silyl groups on this transformation must be investigated; since 3 will be accessed by protodesilylation, whereas 5 is envisaged to be formed by oxidative desilylation.16 The cyclopentane building block 7 is envisaged to be obtained from bis(allylsilanes) 8. Since the few known methods to access the so far synthetically rarely used allylic bis(silanes)17 are not applicable for the synthesis of 8 because of functional group incompatibilities, the Julia reaction of sulfones 10 with aldehyde 9 seemed to be the most promising approach. | ||
| Scheme 1 Retrosynthetic analysis of dihydronepetalactone (3) and dolicholactone (5). | ||
β,β′-Disilylated sulfones 10a–c were prepared in good yields by a one-pot sequential alkylation of methyl phenyl sulfone with diverse (chloromethyl) silanes.15 Their metalation selectivity was initially studied to enable the Julia olefination (Scheme 2, for full details, see the ESI†). Indeed, the initial deprotonation of all β,β-bis(silyl) sulfones 10a–c by n-BuLi in the presence of TMEDA at −78 °C generated the undesired ortho-aryllithium o-11a–c with moderate to good selectivities. From o-11a the ortho → α transmetalation took place on warming from −60 to 0 °C as determined by quenching the reaction mixture with D2O and isolation of deuterated sulfone α-12a.15a The deprotonation of silylated sulfone 10b was slower and proceeded less selectively. Moreover, α-sulfonyllithium α-11b resulting after the rearrangement was not stable in THF and deteriorated to an appreciable extent. Therefore, the metalation/transmetalation was more conveniently performed in diethyl ether under otherwise similar conditions resulting in the clean formation of α-12b. In contrast ortho-sulfonylphenyllithium o-11c underwent the desired transmetalation to α-11c only very slowly, and did not proceed to completion. Thus, application of 10c in the Julia olefination is not possible.
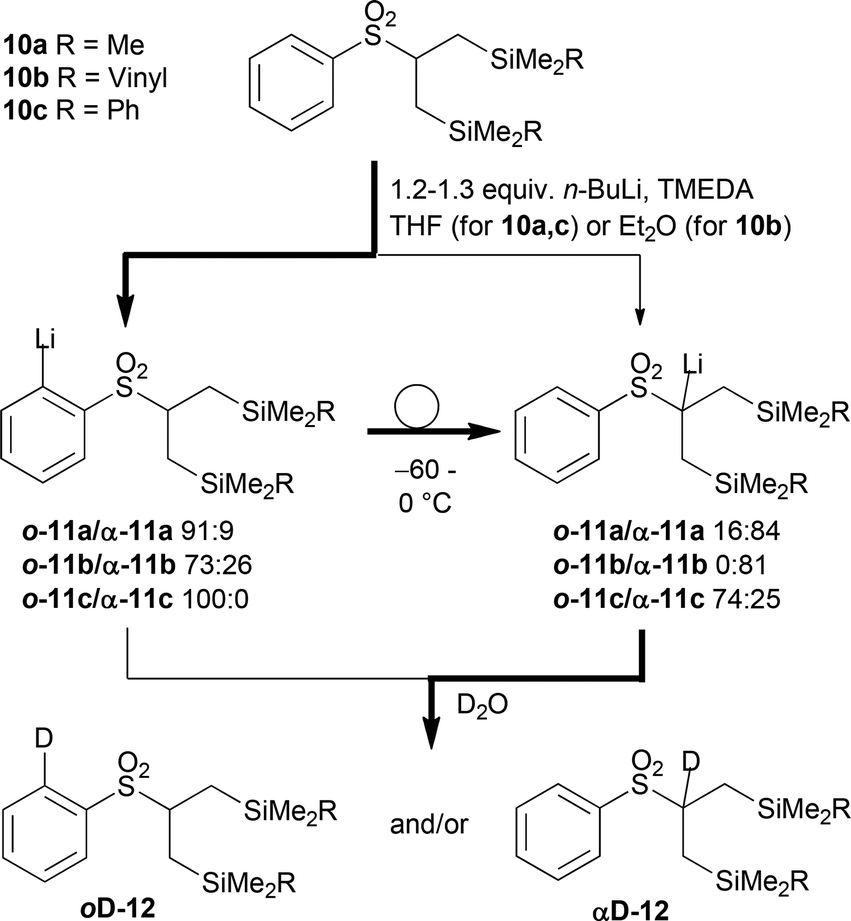 | ||
| Scheme 2 Metalation selectivity of silylated sulfones 10a–c and subsequent ortho → α transmetalation. The ratios were determined by taking aliquots and quenching by D2O. | ||
Aldehyde 9 was synthesized from (S)-citronellol (13) in three steps (Scheme 3). Initial oxidation by pyridinium dichromate (PDC) in dry DMF gave (S)-citronellic acid, which was esterified with sodium hydride and ethyl bromide. Subsequent ozonolysis provided aldehyde 9, which was subsequently added to the reaction mixture of α-phenylsulfonyl lithium intermediates α-11a,b. The resulting alkoxide was acylated in situ with benzoyl chloride affording β-benzoyloxy sulfones 14. In contrast to the reported Julia reactions with similar substrates,15a the olefination step of 14 using SmI2 did not afford the desired olefins 8. However, they were obtained by classical deoxygenation by using sodium amalgam in dry ethanol. It must be mentioned that compounds 14 and 8 are not stable to silica gel chromatography unless NEt3 is added as a coeluent (see the ESI† for details).
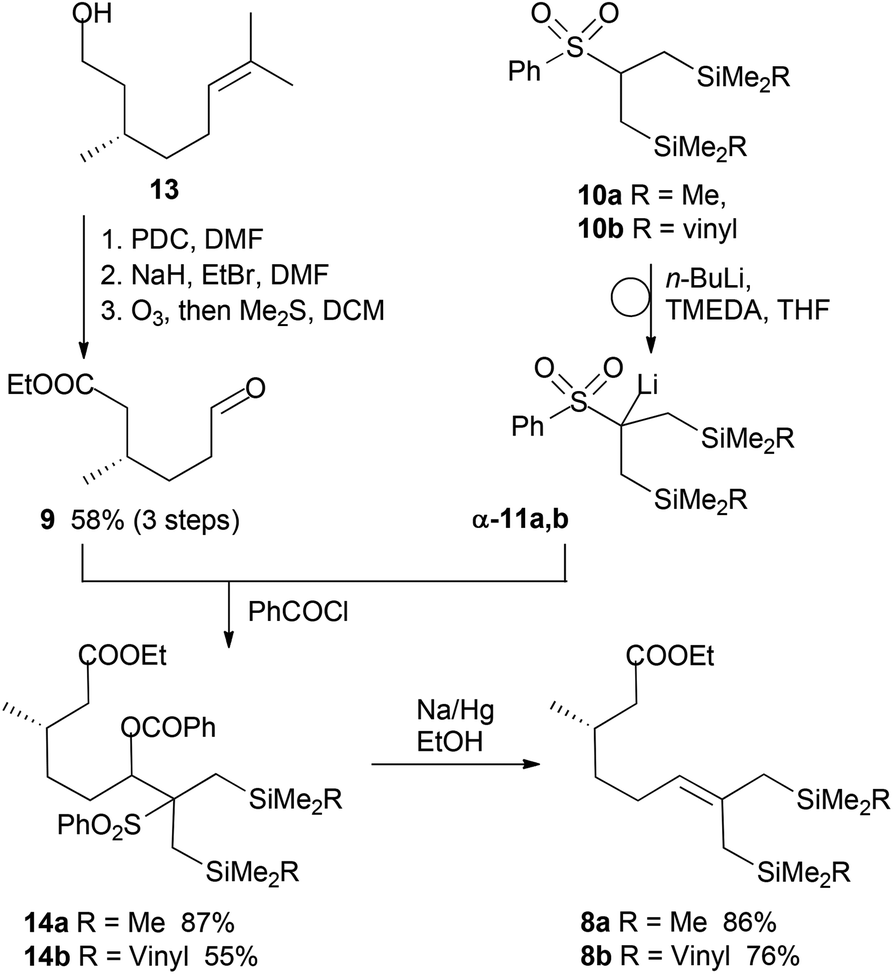 | ||
| Scheme 3 Preparations of olefins 8a,b by Julia reactions. | ||
Disilylated esters 8a and 8b were subjected to tandem alkoxycarbonylation/oxidative radical cyclization by deprotonating with 2.6 equiv. of LiTMP and adding 1.2 equiv. of ethyl chloroformate followed by 2.3 equiv. of ferrocenium hexafluorophosphate (Scheme 4). In contrast to the previous synthesis,13 the desired cyclopentanes 7a and 7b were obtained in good to excellent 10:1 and 20:1 trans/cis selectivities, respectively, as inseparable mixtures. The formation of cyclopentanes 7 is rationalized by deprotonation and alkoxycarbonylation giving malonate enolates 15, which undergo selective oxidative single electron transfer mediated by ferrocenium hexafluorophosphate. Thus generated radicals cyclize efficiently to radicals 18; the high trans-diastereoselectivity of the radical 5-exo cyclization is secured by favoring the chair-like transition state 16. The (Z)-oriented allylic silyl group effectively increases the energy of the competing boat-like transition state 17 because of allylic strain.18 Subsequently, carbocations 19 are generated by a second SET oxidation. Their desilylation affords predominately trans-cyclopentanes 7.
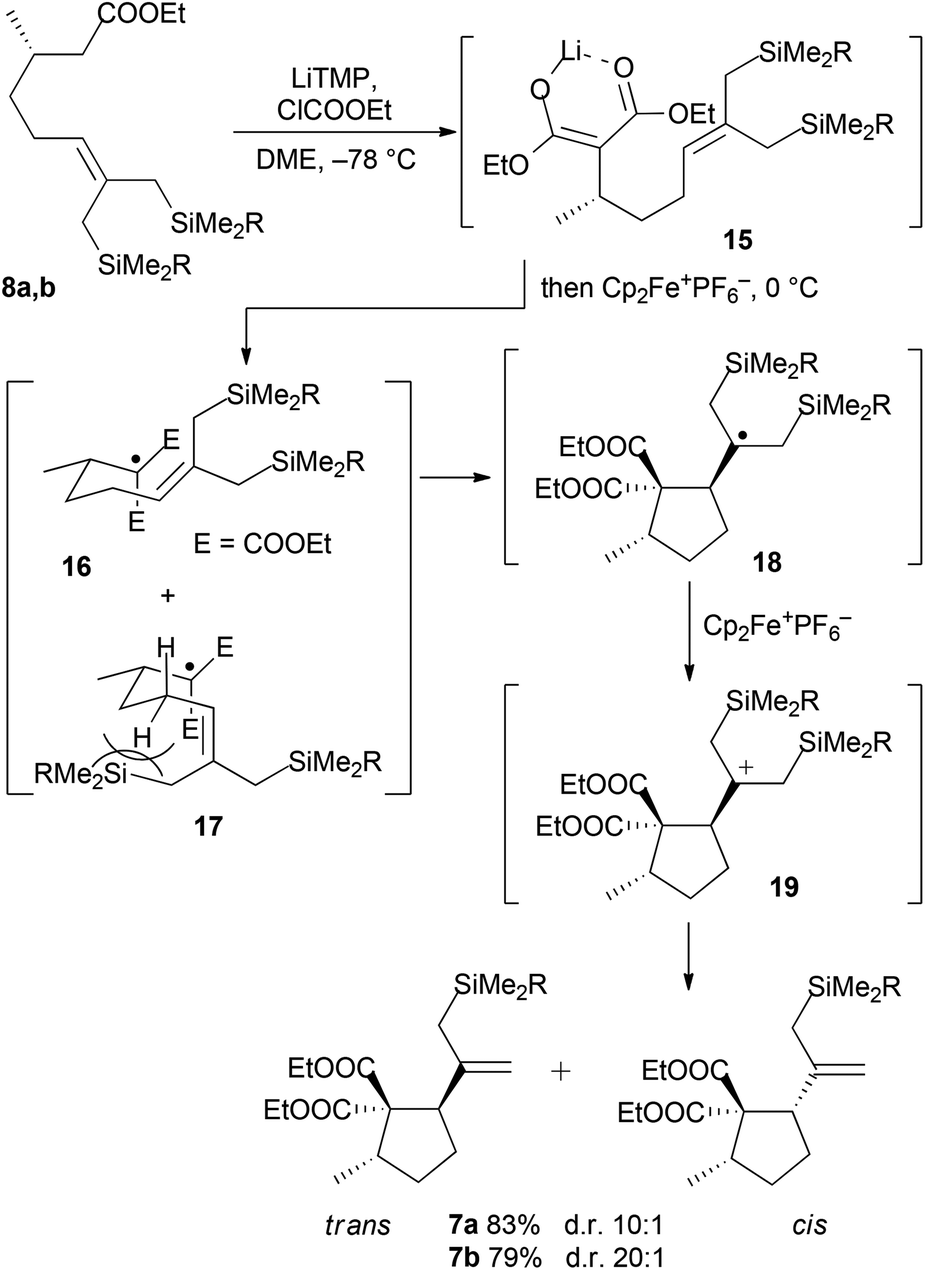 | ||
| Scheme 4 Tandem alkoxycarbonylation/oxidative radical cyclization of 8a,b. | ||
For the synthesis of 3, protodesilylation of the diastereomeric mixture 7a by BF3·OEt2 was carried out affording an inseparable 10:1 diastereomeric mixture of trans- and cis-cyclopentanedicarboxylates 20 in very good yield (Scheme 5). The remaining oxygen atom was introduced by a highly diastereoselective hydroboration/oxidation sequence providing alcohols 22. The rationale for the diastereoselectivity of hydroboration is provided by a strongly preferred conformation of the isopropenyl group in 21 to minimize allylic strain.18 In this conformation the two ester groups effectively shield the α-face, therefore the borane exclusively attacks the double bond from the opposite face.
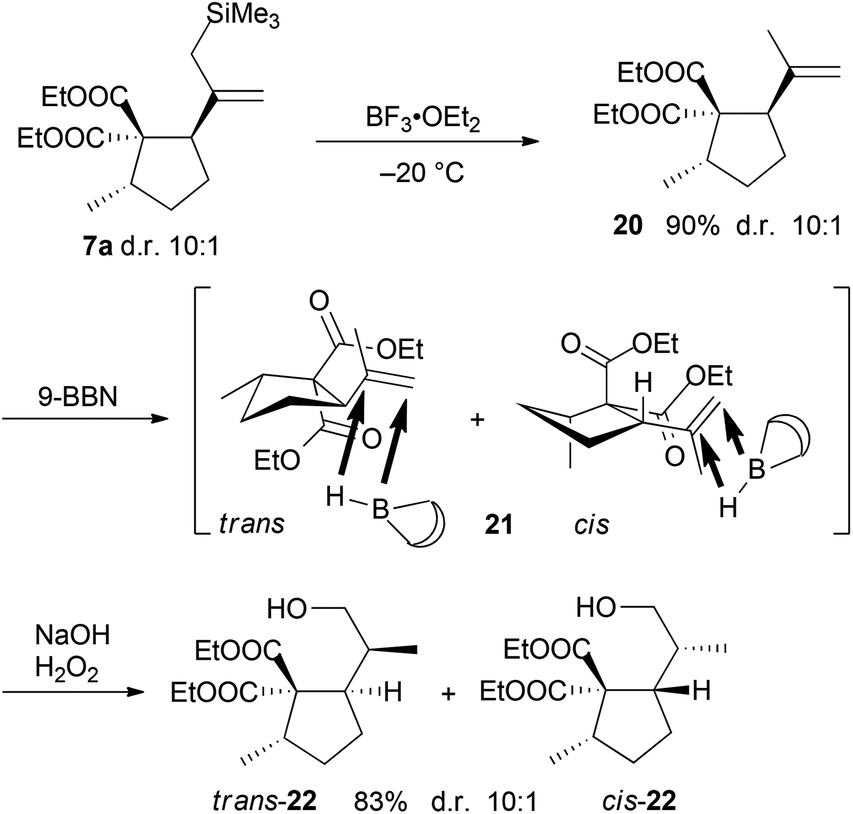 | ||
| Scheme 5 Synthesis of hydroxy esters 22. | ||
The transformation of alcohols 22 into dihydronepetalactone (3) required some experimentation (Scheme 6). Saponification with an excess of potassium hydroxide selectively provided crude carboxylic acid 23 in high yield, but the transformation required seven days to go to completion. The subsequent thermal decarboxylation furnished dihydronepetalactone (3) in 85% yield. The saponification can be performed with the mixture of trans- and cis-22, because the latter did not undergo lactonization and was therefore easily separable from 3 after decarboxylation (not shown). To reduce the rather long reaction time, lactonization and the hydrolysis of the ester function of 22 with 10 equiv. of potassium trimethylsilanolate19 in THF under reflux for two days afforded an approximately 1:1 mixture of the corresponding carboxylic acid and dihydronepetalactone (3) in combined 96% yield, which converged to 3 by heating the crude mixture in DMSO at 130 °C for 5 h. Lactonization of trans-22 was also promoted under acidic conditions using a catalytic amount of p-toluenesulfonic acid at room temperature in 96% yield, whereas unreacted cis-22 was recovered and separated. However, the subsequent Krapcho dealkoxycarbonylation of lactone ester 24 did not proceed as expected, since conversion was slow and dihydronepetalactone formation was accompanied by the formation of the cis,trans-alcohol 25.
 | ||
| Scheme 6 Completion of the total synthesis of dihydronepetalactone (3). | ||
Cyclopentane 7b was envisaged to provide access to dolicholactone (5) via oxidative Tamao–Fleming desilylation16 (cf.Scheme 2). However, the attempts to implement this strategy failed and resulted in protodesilylation instead. Therefore, allylsilane 7a was alternatively subjected to dihydroxylation20 to obtain a 6:1 diastereomeric mixture of trans-butyrolactones 26a and 26b (Scheme 7). The mixture converged to the optically pure compound 27 by a Peterson olefination using BF3·OEt2.21 Ethyl ester 27 was saponified by potassium hydroxide within 48 hours. The reaction with potassium trimethylsilanolate did in contrast not provide the desired carboxylic acid. The crude carboxylic acid was subjected to final thermal decarboxylation affording dolicholactone (5) in very good yield.
 | ||
| Scheme 7 The successful approach to dolicholactone (5). | ||
Conclusions
In conclusion, an efficient unified approach to iridoid type natural products was developed. The ortho → α-sulfonyl carbanion transmetalation-enabled Julia reaction is a well applicable reaction to provide bis(allylic silanes), whose potential is widely untapped. They undergo highly diastereoselective oxidative tandem polar-radical crossover processes. It was demonstrated that silylated trans-cyclopentanedicarboxylate 7 serves as a unique starting point for general syntheses of iridoids. Dihydronepetalactone and dolicholactone were synthesized in ten steps and 18% and 20% overall yields, respectively. Although not executed, the reported approach may serve as a basis for further diversification to other iridoid natural products, such as 1, 2, 4 or the related monoterpene alkaloid 6 by functional group interconversion as well as to those whose configuration and biological activities are not yet known.Experimental
For general information see the ESI.† Compounds 922 and 10a15a are literature-known and their analytical data agree with those reported in the cited references.
Sequential dialkylation of methyl phenyl sulfone (General procedure)
n-BuLi (8.13 mL, 13 mmol, 1.6 M in hexane) was added dropwise to a stirred solution of methyl phenyl sulfone (10 mmol) and TMEDA (3 mL, 19.5 mmol) in dry THF (50 mL) at −78 °C under a nitrogen atmosphere. After stirring for 15 min, dimethylvinyl(chloromethyl)silane (13 mmol) for 10b and dimethylphenyl(chloromethyl)silane (13 mmol) for 10c were added dropwise at −78 °C. After stirring for 10 min, the reaction mixture was warmed to room temperature and stirred for 0.5 hour for 10b and for two hours for 10c. The reaction mixture was cooled to −78 °C, and TMEDA (3 mL, 19.5 mmol) and n-BuLi (8.13 mL, 13 mmol, 1.6 M in hexane) were added dropwise. After 10 min, the corresponding (chloromethyl)silane (13 mmol) was added, and the solution was stirred at −78 °C for 5 min, warmed to room temperature and stirred until completion as indicated by TLC. The reaction mixture was quenched with saturated NH4Cl solution. The layers were separated and the aqueous layer was extracted with diethyl ether (3 × 50 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and evaporated. Purification by column chromatography (EtOAc/hexane 1:40, gradient to 1:10) gave sulfones 10b–c.
:5) 0.61; IR ν 3051, 3090, 2957, 2928, 2899, 2855, 2801, 1592, 1465, 1406, 1305, 1253, 1143, 1008, 958, 846, 830, 709; MS (ESI+), m/z (%) 727 (15) [2M + Na+], 375 (100) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C17H28O2SSi2Na+ 375.1241; found 375.1243; 1H NMR (400 MHz, CDCl3) δ 0.13 (s, 12H), 0.83 (dd, J = 7.3, 15.1 Hz, 2H), 1.10 (dd, J = 6.3, 15.1 Hz, 2H), 3.23 (tt, J = 6.3, 7.3 Hz, 1H), 5.66 (dd, J = 3.8, 20.1 Hz, 2H), 5.98 (dd, J = 3.8, 14.7 Hz, 2H), 6.14 (dd, J = 14.7, 20.1 Hz, 2H), 7.56 (m, 2H), 7.63 (m, 1H), 7.85 (m, 2H); 13C NMR (100 MHz, CDCl3) δ −2.8 (q), −2.4 (q), 18.1 (t), 59.4 (d), 128.9 (d), 129.4 (d), 132.4 (t), 133.3 (d), 136.9 (s), 138.5 (d).
:5) 0.52; IR ν 3069, 2954, 1427, 1304, 1251, 1143, 1113, 840, 732, 700, 647; MS (ESI+), m/z (%) 475 (100) [M + Na+]; Anal. calcd for C25H32O2SSi2 (452.76) C 66.32, H 7.12, S 7.08; found C 66.12, H 7.15, S 7.20; 1H NMR (400 MHz, CDCl3) δ 0.24 (s, 12H), 0.92 (A part of the ABM system, J = 7.1, 15.3 Hz, 2H), 1.19 (B part of the ABM system, J = 6.3, 15.3 Hz, 2H), 3.10 (m, 1H), 7.31 (m, 10H), 7.45 (m, 2H), 7.62 (m, 3H); 13C NMR (100 MHz, CDCl3) δ −2.4 (q), −1.8 (q), 18.7 (t), 59.3 (d), 128.0 (d), 129.0 (d), 129.28 (d), 129.30 (d), 133.4 (d), 133.9 (d), 137.1 (s), 138.1 (s).
Preparation of 2-benzoyloxy sulfones 14a,b (General procedure)
n-BuLi (1.4 mL, 2.23 mmol or 1.6 mL, 2.57 mmol, 1.6 M in hexane) was added dropwise to a stirred solution of sulfone 10a or 10b (1.88 mmol) and TMEDA (0.35 mL, 2.23 mmol or 0.4 mL, 2.57 mmol) in dry DME (12 mL) for 10a or diethyl ether (12 mL) for 10b, respectively, at −78 °C under a nitrogen atmosphere. The reaction mixture was warmed to room temperature over 2 h for 10a or to 0 °C for 30 min for 10b and aldehyde 9 (357 mg, 2.07 mmol) in DME (1.5 mL) or diethyl ether (1.5 mL), respectively, was added dropwise at −78 °C. The reaction mixture was stirred at this temperature for 15 min until completion as indicated by TLC. Benzoyl chloride (268 μL, 2.26 mmol) was added and the reaction mixture was warmed to room temperature after 20 min. 3-(Dimethylamino)propan-1-ol (292 μL, 2.5 mmol) was added and the reaction was quenched with water after 10 min. The layers were separated and the aqueous layer was extracted with diethyl ether (3 × 40 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and evaporated. Purification by column chromatography (EtOAc/hexane/Et3N 1:20:0.05) gave benzoyloxy sulfones 14a,b.
:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.31; IR ν 3091, 3072, 3065, 2957, 2932, 2907, 2875, 1719, 1602, 1585, 1452, 1447, 1393, 1373, 1315, 1296, 1271, 1265, 1251, 1177, 1162, 1134, 1106, 1095, 1080, 1070, 1026, 843, 711, 690; MS (ESI+), m/z (%) 627 (5) [M + Na+], 485 (100) [M − PhSO2H + Na+], 269 (20) [M+ − PhSO2 − PhCO2 − TMS]; Anal. calcd for C31H48O6SSi (604.95) C 61.55, H 8.00, S 5.30; found C 61.82, H 8.14, S 5.39; 1H NMR (400 MHz, CDCl3) δ 0.20 (s, 36H), 0.82 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H), 1.11 (t, J = 7.1 Hz, 3H), 1.18 (t, J = 7.1 Hz, 3H), 1.19–1.37 (m, 10H), 1.45 (A part of the AB system, J = 14.6 Hz, 1H), 1.47 (A part of the AB system, J = 14.6 Hz, 1H), 1.63 (m, 3H), 1.86 (m, 3H), 1.98 (dd, J = 5.7, 14.7 Hz, 1H), 2.02 (dd, J = 8.2, 14.7 Hz, 1H), 2.15 (dd, J = 7.3, 14.7 Hz, 1H), 2.17 (dd, J = 6.0, 14.7 Hz, 1H), 3.98 (m, 2H), 4.05 (q, J = 7.2 Hz, 2H), 5.43 (dd, J = 2.9, 10.2 Hz, 2H), 7.23 (m, 4H), 7.32 (m, 10H), 7.44 (m, 2H), 7.85 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 1.6 (q), 1.7 (q), 14.3 (q), 14.4 (q), 19.7 (q), 19.8 (q), 20.56 (t), 20.60 (t), 21.57 (t), 21.61 (t), 29.6 (t), 29.7 (t), 30.6 (d), 30.7 (d), 33.7 (t), 33.8 (t), 41.78 (t), 41.81 (t), 60.30 (t), 60.34 (t), 76.78 (s), 76.81 (s), 77.1 (d, 2C), 128.19 (d), 128.20 (d), 128.95 (d), 128.96 (d), 129.4 (s), 129.5 (s), 129.52 (d), 129.54 (d), 130.3 (d, 2C), 132.9 (d), 133.1 (d), 139.3 (s), 139.4 (s), 165.8 (s), 165.9 (s), 172.89 (s), 172.92 (s).
:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.40; IR ν 3059, 2965, 2937, 2865, 1728, 1453, 1409, 1377, 1303, 1270, 1255, 1180, 1139, 1098, 1084, 1073, 1030, 1013, 957, 832, 760, 713, 693; MS (ESI+), m/z (%) 651 (30) [M + Na+], 509 (100) [M − PhSO2H + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C33H48O6SSi2Na+ 651.2602; found 651.2604; 1H NMR (400 MHz, CDCl3) δ 0.27 (s, 6H), 0.29 (s, 6H), 0.30 (s, 6H), 0.31 (s, 6H), 0.85 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H), 1.13 (t, J = 7.0 Hz, 3H), 1.21 (t, J = 7.1 Hz, 3H), 1.19–1.34 (m, 8H), 1.45 (m, 2H), 1.57 (A part of the AB system, J = 14.6 Hz, 1H), 1.59 (A part of the AB system, J = 14.7 Hz, 1H), 1.72 (m, 3H), 1.87 (m, 3H), 2.00 (dd, J = 4.1, 8.2 Hz, 1H), 2.04 (dd, J = 4.2, 8.2 Hz, 1H), 2.17 (dd, J = 6.0, 8.8 Hz, 1H), 2.22 (m, 1H), 4.01 (m, 2H), 4.08 (q, J = 7.1 Hz, 2H), 5.45 (m, 2H), 5.76 (dd, J = 3.7, 20.2 Hz, 2H), 5.78 (dd, J = 3.6, 20.5 Hz, 2H), 6.02 (m, 4H), 6.28 (dd, J = 14.4, 20.2 Hz, 1H), 6.30 (dd, J = 14.5, 20.3 Hz, 1H), 6.42 (dd, J = 14.3, 20.3 Hz, 1H), 6.44 (dd, J = 14.4, 20.2 Hz, 1H), 7.26 (m, 4H), 7.34 (m, 10H), 7.47 (m, 2H), 7.89 (m, 4H); 13C NMR (100 MHz, CDCl3) δ −0.64 (q), −0.62 (q), −0.55 (q), −0.54 (q), −0.27 (q), −0.26 (q), −0.25 (q), −0.24 (q), 14.3 (q), 14.4 (q), 19.7 (q), 19.8 (q), 19.96 (t), 19.97 (t), 21.1 (t), 21.2 (t), 29.5 (t), 29.7 (t), 30.6 (d), 30.8 (d), 33.5 (t), 33.6 (t), 41.8 (t), 41.9 (t), 60.29 (t), 60.33 (t), 76.51 (s), 76.54 (s), 77.1 (d), 77.4 (d), 128.17 (d), 128.19 (d), 128.96 (d), 128.97 (d), 129.42 (s), 129.45 (s), 129.5 (d), 129.6 (d), 130.4 (d), 132.0 (t), 133.0 (d), 133.1 (d), 139.2 (s), 139.3 (s), 140.32 (d), 140.33 (d), 165.8 (s), 165.9 (s), 172.9 (s), 173.0 (s).
Julia olefination with sodium amalgam (General procedure)
Benzoyloxy sulfones 14a,b (0.739 mmol) were dissolved in dry THF (5 mL) and dry ethanol (10 mL) under a nitrogen atmosphere. Sodium amalgam (369 mg, 1.7 mmol) was added at −20 °C. After 3 h at this temperature, the reaction mixture was diluted with diethyl ether (15 mL) and decanted from mercury. The organic layer was washed with brine and the aqueous layer was extracted with diethyl ether (3 × 40 mL). The combined organic extracts were dried over Na2SO4, filtered and evaporated. Purification by column chromatography (EtOAc/hexane/NEt3 1:50:1) afforded olefins 8a,b. NEt3 is mandatory as a coeluent, since partial decomposition occurs during chromatography in its absence.
:5) 0.89; [α]20D −1.1 (c 1.116 in CHCl3); IR ν 2986, 2957, 2927, 2876, 2854, 1726, 1645, 1478, 1462, 1447, 1415, 1394, 1382, 1371, 1352, 1259, 1248, 1115, 1095, 1032, 855, 840, 699; MS (ESI+), m/z (%) 365 (100) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C18H38O2Si2Na+ 365.2303; found 365.2303; 1H NMR (400 MHz, CDCl3) δ −0.01 (s, 9H), 0.02 (s, 9H), 0.94 (d, J = 6.6 Hz, 3H), 1.18–1.34 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H), 1.38 (s, 2H), 1.44 (AB system, J = 13.5 Hz, 2H), 1.81–2.02 (m, 3H), 2.09 (dd, J = 8.6, 14.4 Hz, 1H), 2.30 (dd, J = 5.8, 14.4 Hz, 1H), 4.12 (q, J = 7.2 Hz, 2H), 4.75 (t, J = 7.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ −1.0 (q), −0.5 (q), 14.5 (q), 19.8 (q), 23.9 (t), 26.3 (t), 29.5 (t), 30.3 (d), 37.5 (t), 42.1 (t), 60.2 (t), 119.4 (d), 134.4 (s), 173.4 (s).
:5) 0.77; [α]20D −3.2 (c 0.437 in CHCl3); IR ν 3058, 2967, 2936, 2864, 1743, 1467, 1409, 1375, 1291, 1252, 1194, 1159, 1100, 1071, 1038, 1012, 953, 835, 759, 619; MS (ESI+), m/z (%) 389 (100) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C20H38O2Si2Na+ 389.2303; found 389.2303; 1H NMR (400 MHz, CDCl3) δ 0.06 (s, 6H), 0.09 (s, 6H), 0.93 (d, J = 6.5 Hz, 3H), 1.16–1.34 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H), 1.44 (s, 2H), 1.50 (AB system, J = 13.6 Hz, 2H), 1.81–2.02 (m, 3H), 2.10 (dd, J = 8.4, 14.5 Hz, 1H), 2.30 (dd, J = 5.7, 14.6 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 4.80 (t, J = 7.0 Hz, 1H), 5.65 (dd, J = 3.9, 20.3 Hz, 1H), 5.67 (dd, J = 3.9, 20.2 Hz, 1H), 5.92 (dd, J = 3.8, 14.6 Hz, 1H), 5.94 (dd, J = 3.8, 14.7 Hz, 1H), 6.13 (dd, J = 14.8, 20.4 Hz, 1H), 6.17 (dd, J = 14.7, 20.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ −2.9 (q), −2.5 (q), 14.5 (q), 19.8 (q), 22.8 (t), 26.3 (t), 28.3 (t), 30.2 (d), 37.4 (t), 42.1 (t), 60.2 (t), 120.4 (d), 131.5 (t), 133.5 (s), 139.6 (d), 173.4 (s).
Alkoxycarbonylation/oxidative radical cyclization/carbocation desilylation (General procedure)
n-BuLi (1.14 mL, 1.82 mmol, 1.6 M in hexane) was added dropwise to a stirred solution of 2,2,6,6-tetramethylpiperidine (0.31 mL, 1.82 mmol) in dry DME (20 mL) at −78 °C under a nitrogen atmosphere. After stirring for 30 min, olefin 8a or 8b (0.70 mmol) in DME (0.5 mL) was added dropwise at −78 °C. After stirring for 30 min, ethyl chloroformate (0.08 mL, 0.84 mmol) was added. After completion of the carboxylation, ferrocenium hexafluorophosphate (534 mg, 1.61 mmol) was added in portions at 0 °C until a blue color of the mixture persisted. The reaction mixture was stirred for additional 30 min and quenched with a few drops of saturated NH4Cl solution. The mixture was diluted with diethyl ether (25 mL) and filtered through a pad of silica gel. The solvent was evaporated and purification of the residue by column chromatography (EtOAc/hexane 1:200) afforded cyclopentanes 7a,b.
:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.56; IR ν 3083, 2982, 2958, 2931, 2906, 2875, 2856, 1741, 1716, 1629, 1475, 1463, 1447, 1420, 1390, 1380, 1368, 1258, 1249, 1115, 1095, 1044, 1023, 880, 857, 841, 695; MS (CI+), m/z (%) 341 (95) [M + H+], 325 (100) [M+ − CH3], 295 (30) [M+ − OEt]; Anal. calcd for C18H32O4Si (340.54) C 63.49, H 9.47; found C 63.80, H 9.75; 1H NMR (400 MHz, CDCl3) major trans-diastereomer: δ 0.01 (s, 9H), 0.95 (d, J = 7.0 Hz, 3H), 1.20 (t, J = 7.2 Hz, 3H), 1.23 (m, 1H), 1.25 (t, J = 7.2 Hz, 3H), 1.51 (dd, J = 13.3, 0.9 Hz, 1H), 1.61 (dd, J = 13.3, 1.0 Hz, 1H), 1.67 (m, 1H), 1.94 (m, 2H), 2.91 (m, 1H), 3.29 (dd, J = 7.5, 9.6 Hz, 1H), 3.93 (dq, J = 10.8, 7.2 Hz, 1H), 4.12 (dq, J = 10.8, 7.1 Hz, 1H), 4.13 (dq, J = 10.8, 7.1 Hz, 1H), 4.23 (dq, J = 10.7, 7.1 Hz, 1H), 4.57 (q, J = 1.1 Hz, 1H), 4.60 (t, J = 1.0 Hz, 1H); minor cis-diastereomer, detectable resonances: δ 2.29 (m, 1H), 3.20 (dd, J = 10.6, 8.8 Hz, 1H), 4.61 (s), 4.71 (d, J = 1.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) major trans-diastereomer: δ −1.3 (q), 14.1 (q), 14.3 (q), 17.1 (q), 29.1 (t), 31.3 (t), 33.1 (t), 41.2 (d), 52.2 (d), 60.7 (t), 60.9 (t), 68.1 (s), 108.3 (t), 148.9 (s), 171.1 (s), 172.0 (s); minor cis-diastereomer: δ −2.3 (q), 14.20 (q), 14.25 (q), 15.6 (q), 27.6 (t), 29.8 (t), 31.5 (t), 45.1 (d), 53.0 (d), 60.3 (t), 61.1 (t), 67.8 (s), 108.7 (t), 146.6 (s), 169.3 (s), 172.6 (s).
:1 mixture of diastereomers as colorless oil. Rf (EtOAc/hexane 1:5) 0.63; IR ν 2967, 2939, 2883, 1728, 1636, 1467, 1409, 1299, 1252, 1212, 1184, 1118, 1099, 1075, 1048, 1013, 953, 879, 837, 760; MS (CI+), m/z (%) 353 (100) [M + H+], 337 (50) [M+ − CH3], 325 (40) [M+ − CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2], 307 (40) [M+ − OEt], 279 (30) [M+ − COOEt]; HRMS (CI) m/z [M + H+] calcd for C19H33O4Si+ 353.2148; found 353.2136; 1H NMR (400 MHz, CDCl3) major trans-diastereomer: δ 0.09 (s, 3H), 0.10 (s, 3H), 0.95 (d, J = 7.0 Hz, 3H), 1.20 (t, J = 7.2 Hz, 3H), 1.22 (m, 1H), 1.25 (t, J = 7.2 Hz, 3H), 1.61 (d, J = 13.3 Hz, 1H), 1.64 (m, 1H), 1.68 (d, J = 13.3 Hz, 1H), 1.92 (m, 2H), 2.91 (dquint, J = 10.8, 6.9 Hz, 1H), 3.29 (dd, J = 7.4, 9.7 Hz, 1H), 3.94 (dq, J = 10.8, 7.1 Hz, 1H), 4.12 (dq, J = 10.8, 7.0 Hz, 1H), 4.13 (dq, J = 10.8, 7.0 Hz, 1H), 4.23 (dq, J = 10.7, 7.1 Hz, 1H), 4.60 (d, J = 1.1 Hz, 1H), 4.62 (d, J = 0.9 Hz, 1H), 5.67 (dd, J = 3.9, 20.3 Hz, 1H), 5.95 (dd, J = 3.9, 14.6 Hz, 1H), 6.17 (dd, J = 14.6, 20.3 Hz, 1H); minor cis-diastereomer, detectable resonances: δ 1.05 (d, J = 6.9 Hz, 3H), 2.28 (m, 1H), 3.29 (dd, J = 10.6, 8.8 Hz, 1H), 4.64 (s, 1H), 4.74 (d, J = 1.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) major trans-diastereomer: δ −3.4 (q), −3.2 (q), 14.0 (q), 14.2 (q), 17.0 (q), 27.8 (t), 31.1 (t), 32.9 (t), 41.1 (d), 52.0 (d), 60.6 (t), 60.8 (t), 67.9 (s), 108.8 (t), 131.7 (t), 138.8 (d), 148.4 (s), 171.0 (s), 171.8 (s); minor cis-diastereomer: δ −2.5 (q), −2.3 (q), 14.08 (q), 14.14 (q), 15.5 (q), 26.3 (t), 28.6 (t), 31.5 (t), 44.9 (d), 52.8 (d), 60.2 (t), 61.0 (t), 67.6 (s), 109.1 (t), 131.6 (t), 138.9 (d), 146.0 (s), 169.1 (s), 172.5 (s).
:100) afforded 227 mg (90%) of 20 as an inseparable 10:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.41; IR ν 2982, 2959, 2873, 2855, 1717, 1646, 1636, 1465, 1457, 1448, 1437, 1419, 1394, 1374, 1369, 1252, 1113, 1094, 1032, 894, 849, 840; MS (ESI+), m/z (%) 291 (100) [M + Na+]; Anal. calcd for C15H24O4 (268.35) C 67.14, H 9.01; found C 67.39, H 9.20; 1H NMR (400 MHz, CDCl3) major trans-diastereomer: δ 0.90 (d, J = 7.0 Hz, 3H), 1.13 (t, J = 7.2 Hz, 3H), 1.19 (t, J = 7.1 Hz, 3H), 1.22 (m, 1H), 1.62 (m, 1H), 1.65 (d, J = 0.7 Hz, 3H), 1.81–1.99 (m, 2H), 2.85 (dquint, J = 10.3, 7.0 Hz, 1H), 3.33 (dd, J = 8.7, 8.1 Hz, 1H), 3.93 (dq, J = 10.7, 7.1 Hz, 1H), 4.07 (dq, J = 10.8, 7.1 Hz, 1H), 4.08 (dq, J = 10.8, 7.1 Hz, 1H), 4.18 (dq, J = 10.7, 7.1 Hz, 1H), 4.67 (br s, 1H), 4.70 (br s, 1H); minor cis-diastereomer, detectable resonances: δ 1.09 (d, J = 6.9 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H), 1.67 (m, 1H), 1.69 (s, 3H), 1.80 (m, 1H), 1.87 (m, 1H), 1.95 (m, 1H), 2.30 (m, 1H), 3.17 (dd, J = 10.7, 8.7 Hz, 1H), 3.98–4.22 (m, 4H), 4.73 (br s, 1H), 4.74 (br s, 1H); 13C NMR (100 MHz, CDCl3) major trans-diastereomer: δ 14.0 (q), 14.24 (q), 17.3 (q), 23.1 (q), 29.9 (t), 32.9 (t), 40.9 (d), 52.2 (d), 60.8 (t), 60.9 (t), 67.6 (s), 112.5 (t), 146.2 (s), 171.1 (s), 171.5 (s); minor cis-diastereomer: δ 14.1 (q), 14.16 (q), 15.5 (q), 23.3 (q), 27.9 (t), 30.9 (t), 44.7 (d), 53.5 (d), 60.3 (t), 61.0 (t), 66.9 (s), 112.4 (t), 144.8 (s), 169.3 (s), 172.2 (s).
:30, gradient to 1:10) gave 111 mg (83%) of alcohol 22 as an in principle separable 10:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.39; IR ν 3440, 2972, 2944, 2885, 1723, 1467, 1372, 1302, 1254, 1188, 1117, 1099, 1071, 1037, 865, 763; MS (ESI+), m/z (%) 309 (100) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C15H26O5Na+ 309.1673; found 309.1673; 1H NMR (400 MHz, CDCl3) major trans-diastereomer: δ 0.85 (d, J = 7.2 Hz, 3H), 0.99 (d, J = 6.8 Hz, 3H), 1.10–1.32 (m, 7H), 1.36 (m, 1H), 1.63 (m, 1H), 1.86 (m, 1H), 2.09 (m, 1H), 2.65 (dt, J = 7.3, 11.7 Hz, 1H), 2.77 (sext, J = 7.2 Hz, 1H), 3.36 (dd, J = 6.8, 10.8 Hz, 1H), 3.54 (dd, J = 4.4, 10.8 Hz, 1H), 4.08 (m, 2H), 4.20 (m, 2H); minor cis-diastereomer, detectable resonances: δ 0.97 (d, J = 7.0 Hz, 3H), 1.08 (d, J = 6.8 Hz, 3H), 2.27 (m, 1H), 2.57 (m, 1H), 3.37 (m, 1H), 3.56 (m, 1H); 13C NMR (100 MHz, CDCl3) major trans-diastereomer: δ 14.1 (q), 14.23 (q), 16.8 (q), 18.8 (q), 27.5 (t), 32.5 (t), 37.6 (d), 41.1 (d), 47.7 (d), 61.07 (t), 61.09 (t), 66.5 (t), 66.7 (s), 171.7 (s), 172.0 (s); minor cis-diastereomer: δ 14.15 (q), 14.20 (q), 15.3 (q), 16.6 (q), 27.8 (t), 31.0 (t), 38.6 (d), 45.3 (d), 50.2 (d), 60.5 (t), 61.10 (t), 65.9 (t), 66.9 (s), 169.9 (s), 172.6 (s).
:1 mixture of acid 23 and 12 mg of dihydronepetalactone (3) (overall 96%). MS (ESI+), m/z (%) 235 (40) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C11H16O4Na+ 235.0941; found 235.0941; 1H NMR (400 MHz, CDCl3) δ 0.95 (d, J = 7.0 Hz, 3H), 1.15 (d, J = 6.9 Hz, 3H), 1.56 (m, 1H), 1.69 (m, 1H), 1.85 (m, 1H), 1.94 (m, 1H), 2.21 (m, 1H), 2.25 (m, 1H), 3.33 (m, 1H), 4.20 (ddd, J = 1.9, 4.2, 11.2 Hz, 1H), 4.27 (dd, J = 11.2, 11.3 Hz, 1H), 12.04 (broad s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.0 (q), 15.2 (q), 25.4 (t), 31.2 (d), 32.8 (t), 44.6 (d), 48.7 (d), 60.9 (s), 71.4 (t), 170.8 (s), 177.5 (s).
:30, gradient to 1:1) affording 10 mg (85%) of dihydronepetalactone (3) as a colorless oil. Rf (EtOAc/hexane 1:5) 0.30; [α]20D +66.9 (c 0.121 in CHCl3); IR ν 2966, 2935, 2864, 1742, 1467, 1383, 1251, 1209, 1175, 1122, 1085, 1062, 968, 851, 830, 807, 670; MS (ESI+), m/z (%) 191 (100) [M + Na+]; HRMS (EI) m/z [M+] calcd for C10H16O2+ 168.1150; found 168.1151; 1H NMR (400 MHz, CDCl3) δ 0.90 (d, J = 7.1 Hz, 3H), 1.14–1.25 (m, 1H), 1.20 (d, J = 6.4 Hz, 3H), 1.43 (m, 1H), 1.75 (m, 1H), 1.93 (m, 1H), 2.00 (m, 1H), 2.23 (m, 1H), 2.43 (dd, J = 9.3, 10.8 Hz, 1H), 2.52 (m, 1H), 4.02 (ddd, J = 1.6, 3.9, 11.1 Hz, 1H), 4.08 (dd, J = 10.4, 11.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.3 (q), 19.5 (q), 26.5 (t), 31.2 (d), 35.2 (t), 40.6 (d), 41.7 (d), 50.7 (d), 70.1 (t), 174.5 (s).
:30, gradient to 1:10) giving 23 mg (96%) of lactone 24 as a colorless oil. Rf (EtOAc/hexane 1:5) 0.31; [α]20D +23.3 (c 0.300 in CHCl3); IR ν 2966, 2933, 2863, 1749, 1732, 1467, 1382, 1257, 1237, 1208, 1176, 1126, 1043, 1017; MS (ESI+), m/z (%) 263 (100) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C13H20O4Na+ 263.1254; found 263.1254; 1H NMR (400 MHz, CDCl3) δ 0.92 (d, J = 7.1 Hz, 3H), 1.12 (d, J = 7.0 Hz, 3H), 1.25 (t, J = 7.1 Hz, 3H), 1.40 (m, 1H), 1.47 (m, 1H), 1.78 (m, 2H), 2.32 (m, 1H), 2.47 (m, 1H), 2.96 (dt, J = 7.2, 10.9 Hz, 1H), 4.08 (d, J = 5.1 Hz, 2H), 4.17 (dq, J = 7.1, 10.9 Hz, 1H), 4.19 (dq, J = 7.0, 10.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9 (q), 14.3 (q), 15.7 (q), 26.5 (t), 29.7 (d), 33.4 (t), 44.2 (d), 47.3 (d), 61.5 (s), 61.8 (t), 72.0 (t), 171.1 (s), 172.0 (s).
:1:1 (3 mL) at room temperature. The reaction mixture was stirred for 16 h, concentrated and diluted with water. The mixture was extracted with DCM, and the combined organic extracts were dried over MgSO4 and evaporated under reduced pressure. The crude product was purified by column chromatography (EtOAc/hexane 1:30, gradient to 1:2.5) giving 121 mg (84%) of alcohol 26 as an in principle separable 6:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.34 (major diastereomer), 0.30 (minor diastereomer); IR ν 3523, 3489, 2964, 2934, 2863, 1776, 1734, 1466, 1373, 1303, 1253, 1189, 1140, 1082, 1033, 996, 919, 846, 695; MS (ESI+), m/z (%) 679 (20) [2M + Na+], 351 (100) [M + Na+]; Anal. calcd for C16H28O5Si (328.48) C 58.50, H 8.59; found C 58.71, H 8.69; 1H NMR (400 MHz, CDCl3) major diastereomer: δ 0.11 (s, 9H), 0.98 (d, J = 14.8 Hz, 1H), 1.08 (d, J = 7.0 Hz, 3H), 1.23 (d, J = 14.9 Hz, 1H), 1.30 (t, J = 7.1 Hz, 3H), 1.48 (m, 1H), 1.59 (broad s, 1H), 1.72 (m, 1H), 1.82 (m, 1H), 1.94 (m, 1H), 2.52 (m, 1H), 3.26 (t, J = 8.5 Hz, 1H), 3.53 (d, J = 11.9 Hz, 1H), 3.62 (d, J = 11.9 Hz, 1H), 4.23 (q, J = 7.1 Hz, 2H); minor diastereomer: δ 0.09 (s, 9H), 1.05 (d, J = 14.7 Hz, 1H), 1.06 (d, J = 7.1 Hz, 3H), 1.25 (d, J = 14.7 Hz, 1H), 1.33 (t, J = 7.1 Hz, 3H), 1.48 (m, 1H), 1.59 (broad s, 1H), 1.72 (m, 1H), 1.82 (m, 1H), 1.94 (m, 1H), 2.65 (m, 1H), 3.23 (t, J = 7.9 Hz, 1H), 3.60 (d, J = 12.0 Hz, 1H), 3.80 (d, J = 12.0 Hz, 1H), 4.24 (q, J = 7.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) major diastereomer: δ 0.4 (q), 14.36 (q), 16.1 (q), 21.7 (t), 29.0 (t), 35.0 (t), 45.6 (d), 51.9 (d), 61.8 (t), 66.8 (s), 70.1 (t), 88.73 (s), 170.0 (s), 175.6 (s); detectable resonances of minor diastereomer: δ 0.1 (q), 14.38 (q), 15.9 (q), 26.4 (t), 28.5 (t), 35.6 (t), 44.0 (d), 53.6 (d), 62.2 (t), 66.1 (t), 88.75 (s).
:5, gradient to 1:1) gave 13 mg (86%) of lactone ester 27 as a colorless oil. Rf (EtOAc/hexane 1:5) 0.35; [α]20D −15.9 (c 0.270 in CHCl3); IR ν 2967, 2933, 2863, 1734, 1467, 1456, 1383, 1250, 1166, 671; MS (ESI+), m/z (%) 261 (100) [M + Na+]; Anal. calcd for C13H18O4 (238.28) C 65.53, H 7.61; found C 65.69, H 7.69; 1H NMR (400 MHz, CDCl3) δ 1.00 (d, J = 7.1 Hz, 3H), 1.23 (t, J = 7.1 Hz, 3H), 1.38 (m, 2H), 1.85 (m, 1H), 2.14 (m, 1H), 2.84 (m, 1H), 3.62 (m, 1H), 4.18 (dq, J = 7.1, 10.7 Hz, 1H), 4.20 (dq, J = 7.2, 10.7 Hz, 1H), 4.45 (dt, J = 0.7, 12.2 Hz, 1H), 4.47 (dtd, J = 0.5, 1.3, 12.3 Hz, 1H), 4.98 (d, J = 2.6 Hz, 1H), 5.08 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 14.3 (q), 16.5 (q), 31.2 (t), 33.5 (t), 42.6 (d), 47.4 (d), 62.3 (t), 63.2 (s), 71.8 (t), 113.7 (t), 141.6 (s), 170.5 (s), 171.5 (s).
:30, gradient to 10:1) affording 7.9 mg (91%) of dolicholactone (5) as a colorless oil. Rf (EtOAc/hexane 1:5) 0.29; [α]20D +44.0 (c 0.270 in CHCl3); IR ν 2969, 2934, 2862, 1738, 1466, 1382, 1297, 1268, 1188, 1024, 972, 801; MS (EI), m/z (%) 166 (10) [M+], 138 (20) [M+ − CO], 121 (20) [M+ − COOH], 82 (70) [M+ − COOCH2CCH2], 67 (10) [M+ − COOCH2CCH2 − Me]; HRMS (EI) m/z [M+] calcd for C10H14O2+ 166.0993; found 166.0994; 1H NMR (400 MHz, CDCl3) δ 1.13 (d, J = 6.6 Hz, 3H), 1.16 (m, 1H), 1.45 (m, 1H), 1.89 (m, 1H), 2.05 (m, 1H), 2.29 (m, 1H), 2.45 (dd, J = 8.5, 10.7 Hz, 1H), 3.05 (m, 1H), 4.53 (dd, J = 1.0, 12.0 Hz, 1H), 4.61 (dd, J = 0.6, 12.0 Hz, 1H), 4.97 (m, 1H), 5.04 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 20.1 (q), 32.5 (t), 34.7 (t), 39.4 (d), 42.1 (d), 51.2 (d), 71.0 (t), 113.5 (t), 142.3 (s), 174.0 (s).
CH2], 307 (40) [M+ − OEt], 279 (30) [M+ − COOEt]; HRMS (CI) m/z [M + H+] calcd for C19H33O4Si+ 353.2148; found 353.2136; 1H NMR (400 MHz, CDCl3) major trans-diastereomer: δ 0.09 (s, 3H), 0.10 (s, 3H), 0.95 (d, J = 7.0 Hz, 3H), 1.20 (t, J = 7.2 Hz, 3H), 1.22 (m, 1H), 1.25 (t, J = 7.2 Hz, 3H), 1.61 (d, J = 13.3 Hz, 1H), 1.64 (m, 1H), 1.68 (d, J = 13.3 Hz, 1H), 1.92 (m, 2H), 2.91 (dquint, J = 10.8, 6.9 Hz, 1H), 3.29 (dd, J = 7.4, 9.7 Hz, 1H), 3.94 (dq, J = 10.8, 7.1 Hz, 1H), 4.12 (dq, J = 10.8, 7.0 Hz, 1H), 4.13 (dq, J = 10.8, 7.0 Hz, 1H), 4.23 (dq, J = 10.7, 7.1 Hz, 1H), 4.60 (d, J = 1.1 Hz, 1H), 4.62 (d, J = 0.9 Hz, 1H), 5.67 (dd, J = 3.9, 20.3 Hz, 1H), 5.95 (dd, J = 3.9, 14.6 Hz, 1H), 6.17 (dd, J = 14.6, 20.3 Hz, 1H); minor cis-diastereomer, detectable resonances: δ 1.05 (d, J = 6.9 Hz, 3H), 2.28 (m, 1H), 3.29 (dd, J = 10.6, 8.8 Hz, 1H), 4.64 (s, 1H), 4.74 (d, J = 1.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) major trans-diastereomer: δ −3.4 (q), −3.2 (q), 14.0 (q), 14.2 (q), 17.0 (q), 27.8 (t), 31.1 (t), 32.9 (t), 41.1 (d), 52.0 (d), 60.6 (t), 60.8 (t), 67.9 (s), 108.8 (t), 131.7 (t), 138.8 (d), 148.4 (s), 171.0 (s), 171.8 (s); minor cis-diastereomer: δ −2.5 (q), −2.3 (q), 14.08 (q), 14.14 (q), 15.5 (q), 26.3 (t), 28.6 (t), 31.5 (t), 44.9 (d), 52.8 (d), 60.2 (t), 61.0 (t), 67.6 (s), 109.1 (t), 131.6 (t), 138.9 (d), 146.0 (s), 169.1 (s), 172.5 (s).
:100) afforded 227 mg (90%) of 20 as an inseparable 10:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.41; IR ν 2982, 2959, 2873, 2855, 1717, 1646, 1636, 1465, 1457, 1448, 1437, 1419, 1394, 1374, 1369, 1252, 1113, 1094, 1032, 894, 849, 840; MS (ESI+), m/z (%) 291 (100) [M + Na+]; Anal. calcd for C15H24O4 (268.35) C 67.14, H 9.01; found C 67.39, H 9.20; 1H NMR (400 MHz, CDCl3) major trans-diastereomer: δ 0.90 (d, J = 7.0 Hz, 3H), 1.13 (t, J = 7.2 Hz, 3H), 1.19 (t, J = 7.1 Hz, 3H), 1.22 (m, 1H), 1.62 (m, 1H), 1.65 (d, J = 0.7 Hz, 3H), 1.81–1.99 (m, 2H), 2.85 (dquint, J = 10.3, 7.0 Hz, 1H), 3.33 (dd, J = 8.7, 8.1 Hz, 1H), 3.93 (dq, J = 10.7, 7.1 Hz, 1H), 4.07 (dq, J = 10.8, 7.1 Hz, 1H), 4.08 (dq, J = 10.8, 7.1 Hz, 1H), 4.18 (dq, J = 10.7, 7.1 Hz, 1H), 4.67 (br s, 1H), 4.70 (br s, 1H); minor cis-diastereomer, detectable resonances: δ 1.09 (d, J = 6.9 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H), 1.67 (m, 1H), 1.69 (s, 3H), 1.80 (m, 1H), 1.87 (m, 1H), 1.95 (m, 1H), 2.30 (m, 1H), 3.17 (dd, J = 10.7, 8.7 Hz, 1H), 3.98–4.22 (m, 4H), 4.73 (br s, 1H), 4.74 (br s, 1H); 13C NMR (100 MHz, CDCl3) major trans-diastereomer: δ 14.0 (q), 14.24 (q), 17.3 (q), 23.1 (q), 29.9 (t), 32.9 (t), 40.9 (d), 52.2 (d), 60.8 (t), 60.9 (t), 67.6 (s), 112.5 (t), 146.2 (s), 171.1 (s), 171.5 (s); minor cis-diastereomer: δ 14.1 (q), 14.16 (q), 15.5 (q), 23.3 (q), 27.9 (t), 30.9 (t), 44.7 (d), 53.5 (d), 60.3 (t), 61.0 (t), 66.9 (s), 112.4 (t), 144.8 (s), 169.3 (s), 172.2 (s).
:30, gradient to 1:10) gave 111 mg (83%) of alcohol 22 as an in principle separable 10:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.39; IR ν 3440, 2972, 2944, 2885, 1723, 1467, 1372, 1302, 1254, 1188, 1117, 1099, 1071, 1037, 865, 763; MS (ESI+), m/z (%) 309 (100) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C15H26O5Na+ 309.1673; found 309.1673; 1H NMR (400 MHz, CDCl3) major trans-diastereomer: δ 0.85 (d, J = 7.2 Hz, 3H), 0.99 (d, J = 6.8 Hz, 3H), 1.10–1.32 (m, 7H), 1.36 (m, 1H), 1.63 (m, 1H), 1.86 (m, 1H), 2.09 (m, 1H), 2.65 (dt, J = 7.3, 11.7 Hz, 1H), 2.77 (sext, J = 7.2 Hz, 1H), 3.36 (dd, J = 6.8, 10.8 Hz, 1H), 3.54 (dd, J = 4.4, 10.8 Hz, 1H), 4.08 (m, 2H), 4.20 (m, 2H); minor cis-diastereomer, detectable resonances: δ 0.97 (d, J = 7.0 Hz, 3H), 1.08 (d, J = 6.8 Hz, 3H), 2.27 (m, 1H), 2.57 (m, 1H), 3.37 (m, 1H), 3.56 (m, 1H); 13C NMR (100 MHz, CDCl3) major trans-diastereomer: δ 14.1 (q), 14.23 (q), 16.8 (q), 18.8 (q), 27.5 (t), 32.5 (t), 37.6 (d), 41.1 (d), 47.7 (d), 61.07 (t), 61.09 (t), 66.5 (t), 66.7 (s), 171.7 (s), 172.0 (s); minor cis-diastereomer: δ 14.15 (q), 14.20 (q), 15.3 (q), 16.6 (q), 27.8 (t), 31.0 (t), 38.6 (d), 45.3 (d), 50.2 (d), 60.5 (t), 61.10 (t), 65.9 (t), 66.9 (s), 169.9 (s), 172.6 (s).
:1 mixture of acid 23 and 12 mg of dihydronepetalactone (3) (overall 96%). MS (ESI+), m/z (%) 235 (40) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C11H16O4Na+ 235.0941; found 235.0941; 1H NMR (400 MHz, CDCl3) δ 0.95 (d, J = 7.0 Hz, 3H), 1.15 (d, J = 6.9 Hz, 3H), 1.56 (m, 1H), 1.69 (m, 1H), 1.85 (m, 1H), 1.94 (m, 1H), 2.21 (m, 1H), 2.25 (m, 1H), 3.33 (m, 1H), 4.20 (ddd, J = 1.9, 4.2, 11.2 Hz, 1H), 4.27 (dd, J = 11.2, 11.3 Hz, 1H), 12.04 (broad s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.0 (q), 15.2 (q), 25.4 (t), 31.2 (d), 32.8 (t), 44.6 (d), 48.7 (d), 60.9 (s), 71.4 (t), 170.8 (s), 177.5 (s).
:30, gradient to 1:1) affording 10 mg (85%) of dihydronepetalactone (3) as a colorless oil. Rf (EtOAc/hexane 1:5) 0.30; [α]20D +66.9 (c 0.121 in CHCl3); IR ν 2966, 2935, 2864, 1742, 1467, 1383, 1251, 1209, 1175, 1122, 1085, 1062, 968, 851, 830, 807, 670; MS (ESI+), m/z (%) 191 (100) [M + Na+]; HRMS (EI) m/z [M+] calcd for C10H16O2+ 168.1150; found 168.1151; 1H NMR (400 MHz, CDCl3) δ 0.90 (d, J = 7.1 Hz, 3H), 1.14–1.25 (m, 1H), 1.20 (d, J = 6.4 Hz, 3H), 1.43 (m, 1H), 1.75 (m, 1H), 1.93 (m, 1H), 2.00 (m, 1H), 2.23 (m, 1H), 2.43 (dd, J = 9.3, 10.8 Hz, 1H), 2.52 (m, 1H), 4.02 (ddd, J = 1.6, 3.9, 11.1 Hz, 1H), 4.08 (dd, J = 10.4, 11.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.3 (q), 19.5 (q), 26.5 (t), 31.2 (d), 35.2 (t), 40.6 (d), 41.7 (d), 50.7 (d), 70.1 (t), 174.5 (s).
:30, gradient to 1:10) giving 23 mg (96%) of lactone 24 as a colorless oil. Rf (EtOAc/hexane 1:5) 0.31; [α]20D +23.3 (c 0.300 in CHCl3); IR ν 2966, 2933, 2863, 1749, 1732, 1467, 1382, 1257, 1237, 1208, 1176, 1126, 1043, 1017; MS (ESI+), m/z (%) 263 (100) [M + Na+]; HRMS (ESI) m/z [M + Na+] calcd for C13H20O4Na+ 263.1254; found 263.1254; 1H NMR (400 MHz, CDCl3) δ 0.92 (d, J = 7.1 Hz, 3H), 1.12 (d, J = 7.0 Hz, 3H), 1.25 (t, J = 7.1 Hz, 3H), 1.40 (m, 1H), 1.47 (m, 1H), 1.78 (m, 2H), 2.32 (m, 1H), 2.47 (m, 1H), 2.96 (dt, J = 7.2, 10.9 Hz, 1H), 4.08 (d, J = 5.1 Hz, 2H), 4.17 (dq, J = 7.1, 10.9 Hz, 1H), 4.19 (dq, J = 7.0, 10.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9 (q), 14.3 (q), 15.7 (q), 26.5 (t), 29.7 (d), 33.4 (t), 44.2 (d), 47.3 (d), 61.5 (s), 61.8 (t), 72.0 (t), 171.1 (s), 172.0 (s).
:1:1 (3 mL) at room temperature. The reaction mixture was stirred for 16 h, concentrated and diluted with water. The mixture was extracted with DCM, and the combined organic extracts were dried over MgSO4 and evaporated under reduced pressure. The crude product was purified by column chromatography (EtOAc/hexane 1:30, gradient to 1:2.5) giving 121 mg (84%) of alcohol 26 as an in principle separable 6:1 mixture of diastereomers as a colorless oil. Rf (EtOAc/hexane 1:5) 0.34 (major diastereomer), 0.30 (minor diastereomer); IR ν 3523, 3489, 2964, 2934, 2863, 1776, 1734, 1466, 1373, 1303, 1253, 1189, 1140, 1082, 1033, 996, 919, 846, 695; MS (ESI+), m/z (%) 679 (20) [2M + Na+], 351 (100) [M + Na+]; Anal. calcd for C16H28O5Si (328.48) C 58.50, H 8.59; found C 58.71, H 8.69; 1H NMR (400 MHz, CDCl3) major diastereomer: δ 0.11 (s, 9H), 0.98 (d, J = 14.8 Hz, 1H), 1.08 (d, J = 7.0 Hz, 3H), 1.23 (d, J = 14.9 Hz, 1H), 1.30 (t, J = 7.1 Hz, 3H), 1.48 (m, 1H), 1.59 (broad s, 1H), 1.72 (m, 1H), 1.82 (m, 1H), 1.94 (m, 1H), 2.52 (m, 1H), 3.26 (t, J = 8.5 Hz, 1H), 3.53 (d, J = 11.9 Hz, 1H), 3.62 (d, J = 11.9 Hz, 1H), 4.23 (q, J = 7.1 Hz, 2H); minor diastereomer: δ 0.09 (s, 9H), 1.05 (d, J = 14.7 Hz, 1H), 1.06 (d, J = 7.1 Hz, 3H), 1.25 (d, J = 14.7 Hz, 1H), 1.33 (t, J = 7.1 Hz, 3H), 1.48 (m, 1H), 1.59 (broad s, 1H), 1.72 (m, 1H), 1.82 (m, 1H), 1.94 (m, 1H), 2.65 (m, 1H), 3.23 (t, J = 7.9 Hz, 1H), 3.60 (d, J = 12.0 Hz, 1H), 3.80 (d, J = 12.0 Hz, 1H), 4.24 (q, J = 7.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) major diastereomer: δ 0.4 (q), 14.36 (q), 16.1 (q), 21.7 (t), 29.0 (t), 35.0 (t), 45.6 (d), 51.9 (d), 61.8 (t), 66.8 (s), 70.1 (t), 88.73 (s), 170.0 (s), 175.6 (s); detectable resonances of minor diastereomer: δ 0.1 (q), 14.38 (q), 15.9 (q), 26.4 (t), 28.5 (t), 35.6 (t), 44.0 (d), 53.6 (d), 62.2 (t), 66.1 (t), 88.75 (s).
:5, gradient to 1:1) gave 13 mg (86%) of lactone ester 27 as a colorless oil. Rf (EtOAc/hexane 1:5) 0.35; [α]20D −15.9 (c 0.270 in CHCl3); IR ν 2967, 2933, 2863, 1734, 1467, 1456, 1383, 1250, 1166, 671; MS (ESI+), m/z (%) 261 (100) [M + Na+]; Anal. calcd for C13H18O4 (238.28) C 65.53, H 7.61; found C 65.69, H 7.69; 1H NMR (400 MHz, CDCl3) δ 1.00 (d, J = 7.1 Hz, 3H), 1.23 (t, J = 7.1 Hz, 3H), 1.38 (m, 2H), 1.85 (m, 1H), 2.14 (m, 1H), 2.84 (m, 1H), 3.62 (m, 1H), 4.18 (dq, J = 7.1, 10.7 Hz, 1H), 4.20 (dq, J = 7.2, 10.7 Hz, 1H), 4.45 (dt, J = 0.7, 12.2 Hz, 1H), 4.47 (dtd, J = 0.5, 1.3, 12.3 Hz, 1H), 4.98 (d, J = 2.6 Hz, 1H), 5.08 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 14.3 (q), 16.5 (q), 31.2 (t), 33.5 (t), 42.6 (d), 47.4 (d), 62.3 (t), 63.2 (s), 71.8 (t), 113.7 (t), 141.6 (s), 170.5 (s), 171.5 (s).
:30, gradient to 10:1) affording 7.9 mg (91%) of dolicholactone (5) as a colorless oil. Rf (EtOAc/hexane 1:5) 0.29; [α]20D +44.0 (c 0.270 in CHCl3); IR ν 2969, 2934, 2862, 1738, 1466, 1382, 1297, 1268, 1188, 1024, 972, 801; MS (EI), m/z (%) 166 (10) [M+], 138 (20) [M+ − CO], 121 (20) [M+ − COOH], 82 (70) [M+ − COOCH2CCH2], 67 (10) [M+ − COOCH2CCH2 − Me]; HRMS (EI) m/z [M+] calcd for C10H14O2+ 166.0993; found 166.0994; 1H NMR (400 MHz, CDCl3) δ 1.13 (d, J = 6.6 Hz, 3H), 1.16 (m, 1H), 1.45 (m, 1H), 1.89 (m, 1H), 2.05 (m, 1H), 2.29 (m, 1H), 2.45 (dd, J = 8.5, 10.7 Hz, 1H), 3.05 (m, 1H), 4.53 (dd, J = 1.0, 12.0 Hz, 1H), 4.61 (dd, J = 0.6, 12.0 Hz, 1H), 4.97 (m, 1H), 5.04 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 20.1 (q), 32.5 (t), 34.7 (t), 39.4 (d), 42.1 (d), 51.2 (d), 71.0 (t), 113.5 (t), 142.3 (s), 174.0 (s).
Acknowledgements
This work was supported by the Grant Agency of the Czech Republic (P207/11/1598), the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences (RVO: 61388963) and the COST action CM1201 “Biomimetic Radical Chemistry”.Notes and references
- (a) B. Dinda, D. R. Chowdhury and B. C. Mohanta, Chem. Pharm. Bull., 2009, 57, 765–796 CrossRef CAS PubMed; (b) R. Tundis, M. R. Loizzo, F. Menichini, G. A. Statti and F. Menichini, Mini-Rev. Med. Chem., 2008, 8, 399–420 CrossRef CAS PubMed; (c) B. Dinda, S. Debnath and Y. Harigaya, Chem. Pharm. Bull., 2007, 55, 159–222 CrossRef CAS PubMed; (d) E. L. Ghisalberti, Phytomedicine, 1998, 5, 147–163 CrossRef CAS PubMed; (e) D. H. Grayson, Nat. Prod. Rep., 1996, 13, 195–225 RSC.
- T. Sakan, S. Isoe, S. Be Hyeon, R. Katsumura, T. Maeda, J. Wolinsky, D. Dickerson, M. Slabaugh and D. Nelson, Tetrahedron Lett., 1965, 6, 4097–4102 CrossRef.
- R. B. Bates, E. J. Eisenbraun and S. M. McElvain, J. Am. Chem. Soc., 1958, 80, 3420–3424 CrossRef CAS.
- (a) K. R. Chauhan, J. R. Aldrich, P. W. McCardle, G. B. White and R. E. Webb, J. Am. Mosq. Control Assoc., 2012, 28, 301–306 CrossRef CAS PubMed; (b) J. J. Zhu, C. A. Dunlap, R. W. Behle, D. R. Berkebile and B. Wienhold, J. Agric. Food Chem., 2010, 58, 12320–12326 CrossRef CAS PubMed.
- U. Pagnoni, A. Pinetti, R. Trave and L. Garanti, Aust. J. Chem., 1976, 29, 1375–1381 CrossRef CAS.
- F. Geu-Flores, N. H. Sherden, V. Courdavault, V. Burlat, W. S. Glenn, C. Wu, E. Nims, Y. Cui and S. E. O'Connor, Nature, 2012, 492, 138–142 CrossRef CAS PubMed.
- (a) A. F. Thomas and Y. Bessiere, in The Total Synthesis of Natural Products, ed. J. ApSimon, Wiley, New York, 1988, vol. 7, pp. 275–454 Search PubMed; (b) W. Oppolzer and E. J. Jacobsen, Tetrahedron Lett., 1986, 27, 1141–1144 CrossRef CAS; (c) E. J. Eisenbraun, D. W. Sullins, C. E. Browne and J. N. Shoolery, J. Org. Chem., 1988, 53, 3968–3972 CrossRef CAS; (d) E. Lee and C. H. Yoon, J. Chem. Soc., Chem. Commun., 1994, 479–481 RSC; (e) D. E. Chavez and E. N. Jacobsen, Org. Lett., 2003, 5, 2563–2565 CrossRef CAS PubMed; (f) B. Schöllhorn and J. Mulzer, Eur. J. Org. Chem., 2006, 901–908 CrossRef; (g) N. Robert, C. Hoarau and F. Marsais, Tetrahedron, 2007, 63, 3702–3706 CrossRef CAS; (h) E. M. Santangelo, I. Liblikas, A. Mudalige, K. W. Törnroos, P.-O. Norrby and C. R. Unelius, Eur. J. Org. Chem., 2008, 5915–5921 CrossRef CAS; (i) R. A. Jones and M. J. Krische, Org. Lett., 2009, 11, 1849–1851 CrossRef CAS PubMed; (j) L. Candish and D. W. Lupton, Org. Lett., 2010, 12, 4836–4839 CrossRef CAS PubMed; (k) S. Lee, S.-M. Paek, H. Yun, N.-J. Kim and Y.-G. Suh, Org. Lett., 2011, 13, 3344–3347 CrossRef CAS PubMed.
- (a) I. Fleming and N. K. Terrett, Tetrahedron Lett., 1984, 25, 5103–5104 CrossRef CAS; (b) I. Fleming and N. K. Terrett, J. Chem. Soc., Perkin Trans. 1, 1998, 2645–2650 RSC.
- J. Wolinsky and E. J. Eustace, J. Org. Chem., 1972, 37, 3376–3378 CrossRef CAS.
- N. Zimmermann, R. Hilgraf, L. Lehmann, D. Ibarra and W. Francke, Beilstein J. Org. Chem., 2012, 8, 1246–1255 CrossRef CAS PubMed.
- (a) J. S. Beckett, J. D. Beckett and J. E. Hofferberth, Org. Lett., 2010, 12, 1408–1411 CrossRef CAS PubMed; (b) C. J. Fischman, S. Adler and J. E. Hofferberth, J. Org. Chem., 2013, 78, 7318–7323 CrossRef CAS PubMed.
- E. Lee, C. H. Yoon, Y. J. Lee and H. J. Kim, Bull. Korean Chem. Soc., 1997, 18, 1247–1248 CAS.
- U. Jahn, P. Hartmann and E. Kaasalainen, Org. Lett., 2004, 6, 257–260 CrossRef CAS PubMed.
- (a) R. Dumeunier and I. E. Markó, in Modern Carbonyl Olefination, ed. T. Takeda, Wiley-VCH, Weinheim, 2004, pp. 104–150 Search PubMed; (b) I. E. Markó and J. Pospísil, in Science of Synthesis, ed. A. De Meijere, Thieme, Stuttgart, 2009, vol. 47, pp. 105–160 Search PubMed; (c) M. Julia and J.-M. Paris, Tetrahedron Lett., 1973, 4833–4836 CrossRef CAS; (d) P. R. Blakemore, J. Chem. Soc., Perkin Trans. 1, 2002, 2563–2585 RSC; (e) K. Plesniak, A. Zarecki and J. Wicha, Top. Curr. Chem., 2007, 275, 163–250 CrossRef CAS PubMed; (f) C. Aïssa, Eur. J. Org. Chem., 2009, 1831–1844 CrossRef; (g) B. Zajc and R. Kumar, Synthesis, 2010, 1822–1836 CrossRef CAS PubMed.
- (a) B. Puget and U. Jahn, Synlett, 2010, 2579–2582 CAS; (b) L. Řehová, I. Císařová and U. Jahn, Eur. J. Org. Chem., 2014, 1461–1476 CrossRef; (c) L. Řehová and U. Jahn, Eur. J. Org. Chem., 2014, 4610–4623 CrossRef; (d) For similar rearrangement of sulfoximines, see: M. Wessels, V. Mahajan, S. Boßhammer, G. Raabe and H.-J. Gais, Eur. J. Org. Chem., 2011, 2431–2449 CrossRef CAS.
- G. R. Jones and Y. Landais, Tetrahedron, 1996, 52, 7599–7662 CrossRef CAS.
- (a) K. K. Wang and S. Dhumrongvaraporn, Tetrahedron Lett., 1987, 28, 1007–1010 CrossRef CAS; (b) T. Kercher and T. Livinghouse, J. Am. Chem. Soc., 1996, 118, 4200–4201 CrossRef CAS; (c) T. Kercher and T. Livinghouse, J. Org. Chem., 1997, 62, 805–812 CrossRef CAS; (d) J. Uenishi and M. Ohmi, Angew. Chem., Int. Ed., 2005, 44, 2756–2760 CrossRef CAS PubMed; (e) J. Uenishi, T. Iwamoto and M. Ohmi, Tetrahedron Lett., 2007, 48, 1237–1240 CrossRef CAS.
- R. W. Hoffmann, Chem. Rev., 1989, 89, 1841–1860 CrossRef CAS.
- (a) M. Lovrić, I. Cepanec, M. Litvić, A. Bartolinčić and V. Vinković, Croat. Chem. Acta, 2007, 80, 109–115 Search PubMed; (b) E. D. Laganis and B. L. Chenard, Tetrahedron Lett., 1984, 25, 5831–5834 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- D. J. Ager, Org. React., 1990, 38, 1–223 CrossRef CAS.
- (a) P. Heretsch, S. Rabe and A. Giannis, Org. Lett., 2009, 11, 5410–5412 CrossRef CAS PubMed; (b) S. Muto, M. Bando and K. Mori, Eur. J. Org. Chem., 2004, 1946–1952 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical characterization and copies of 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c6ob01599a |
| This journal is © The Royal Society of Chemistry 2016 |