
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetra-porphyrin molecular tweezers: two binding sites linked via a polycyclic scaffold and rotating phenyl diimide core†
R. B.
Murphy
 a,
R. E.
Norman
a,
J. M.
White
b,
M. V.
Perkins
a and
M. R.
Johnston
*a
a,
R. E.
Norman
a,
J. M.
White
b,
M. V.
Perkins
a and
M. R.
Johnston
*a
aFlinders Centre for NanoScale Science and Technology, School of Chemical and Physical Sciences, Flinders University, Bedford Park, Adelaide, Australia. E-mail: martin.johnston@flinders.edu.au
bSchool of Chemistry, The University of Melbourne, Melbourne, Australia
First published on 19th August 2016
Abstract
The synthesis of a tetra-porphyrin molecular tweezer with two binding sites is described. The bis-porphyrin binding sites are aligned by a polycyclic scaffold and linked via a freely rotating phenyl diimide core. Synthesis was achieved using a divergent approach employing a novel coupling method for linking two polycyclic units to construct the core, with a copper(II)-mediated phenyl boronic acid coupling found to extend to our polycyclic imide derivative. We expect this chemistry to be a powerful tool in accessing functional polycyclic supramolecular architectures in applications where north/south reactivity and/or directional interactions between modules are important. Porphyrin receptor functionalisation was undertaken last, by a four-fold ACE coupling reaction on the tetra-epoxide derivative of the core.
Introduction
Molecular tweezers containing two ligand binding sites, where these are remote but interdependent, can benefit from cooperativity during complexation, to positively or negatively influence ligand binding. Several examples of cooperativity in molecular tweezers appear in the literature, from early architectures such as Rebek's 2,2′-bipyridine/crown ether system,1–3 to more recent bis-porphyrin systems with either crown ether,4,5 pyridyl,6 or biindole7 effector sites. Molecular-based stimuli can also be used to effect changes to guest binding in molecular tweezers. For example, a water-soluble polymer-tethered tweezer has shown progress towards the development of controlled drug release devices, using a proton-activated (pH) switching unit to change the conformation of bis-naphthalene receptors and release a substrate.8 Cooperativity has even been shown to arise from electronic rather than steric effects,9,10 by using the direction of charge transfer (CT) between a porphyrin host and an appropriate guest (CT donor or acceptor).We have previously described the synthesis and binding characteristics of single binding site tweezer system 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 (Fig. 1), in which the bis-metalloporphyrin receptor offers strong intramolecular complexation with the diamino ligand 1,4-diazabicyclo[2.2.2]octane (DABCO).
11 (Fig. 1), in which the bis-metalloporphyrin receptor offers strong intramolecular complexation with the diamino ligand 1,4-diazabicyclo[2.2.2]octane (DABCO).
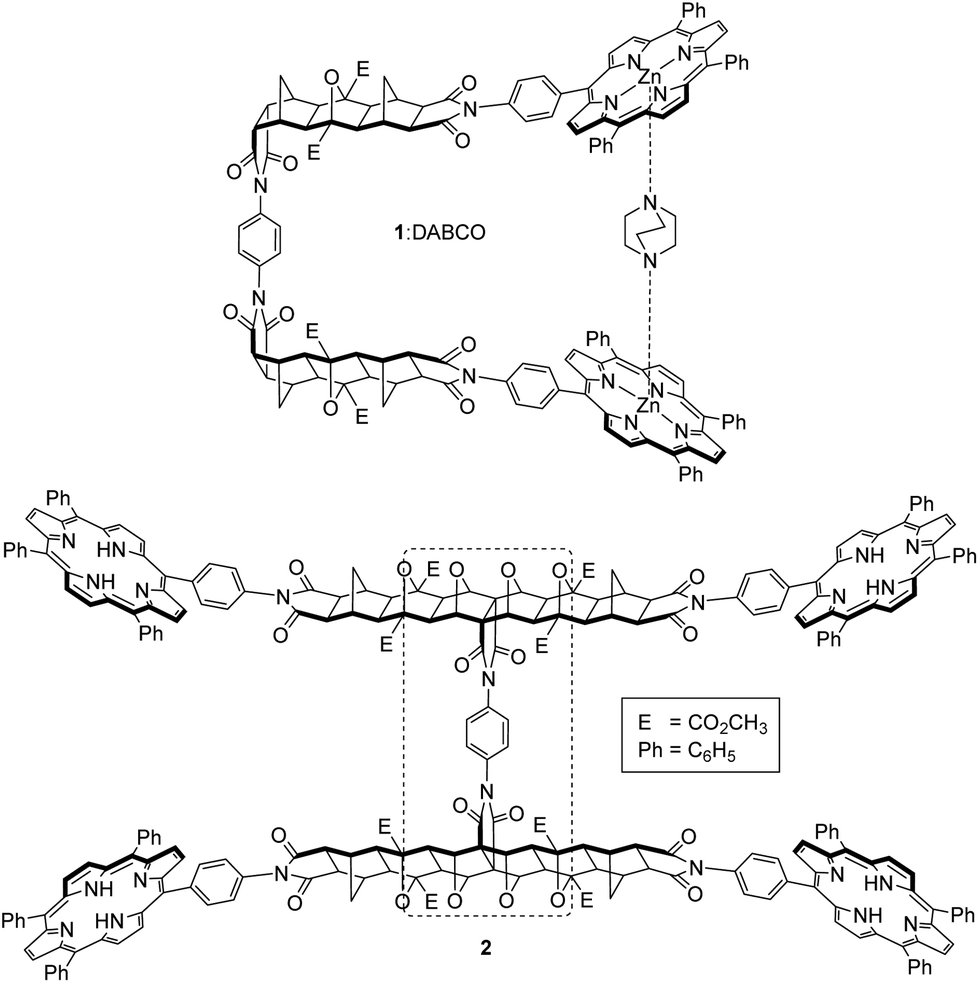 | ||
| Fig. 1 Previously described model tweezer system 111 (shown as complex with DABCO), and the expanded tetra-porphyrin molecular tweezer 2 with two bis-porphyrin binding sites linked via a bridged polycyclic scaffold and freely rotating phenyl diimide core (dotted line). | ||
Polycyclic frameworks, based on norbornyl derivatives, provide several distinct advantages when incorporated into molecular tweezers; namely excellent control of the direction of both lateral and vertical extension and well defined positioning of pendant moieties. These structures are highly synthetically variable using chemistry developed extensively by the research groups of Warrener and Butler12–16 and Paddon-Row,17–20 and is very much modular, enabling the synthesis of supramolecular architectures with customisable dimensions. More recent examples include those by the groups of Johnston,21,22 Pfeffer,23,24 and Margetić.25–27 Molecular modelling by Warrener28,29 and Johnston,30 and experimental evidence by Margetić26 has shown that the inclusion of certain modules provides either linearity or curvature to the polycyclic scaffold. Importantly, a number of studies by several research groups has identified that polycyclic backbones are characterised by a higher degree of rigidity when only consisting of fused bridged polycyclic rings, with non-bridged cyclohexane(ene) rings and fused aromatic rings increasing flexibility.19,20,31–37 However, caution must be exercised, as small degrees of flexibility within rigid modular sections can be amplified across the complete scaffold.38
Our original bis-porphyrin tweezer model architecture 1 has now been expanded to tetra-porphyrin system 2 with two homotropic binding sites (Fig. 1). We reasoned that with the inclusion of a largely rigid polycyclic backbone, the interporphyrin distance from the binding of one guest may be able to be transferred to a second remote site with interannular cooperativity. This type of cooperativity arises in systems when there is interplay between two or more intramolecular binding interactions.39 The two polycyclic backbones would be connected via a centrally located phenyl diimide structural unit, which would be fundamental in conferring the rotational degrees of freedom required by the host to facilitate interannular cooperativity between the two remote but interdependent binding sites. Furthermore, rotation about the porphyrin-polycyclic backbone bonds would allow for further cavity adjustment as required by different sized guests.
Results and discussion
Two overall approaches to synthesising 2 were considered; convergent and divergent (Fig. 2). The convergent approach, whereby p-phenylenediamine is used to link two bis-porphyrin alicyclic systems via their anhydride (Fig. 2a), is synthetically preferable as this route provides increased structural control; functionalisation of each half with porphyrin receptors involves two points of reactivity.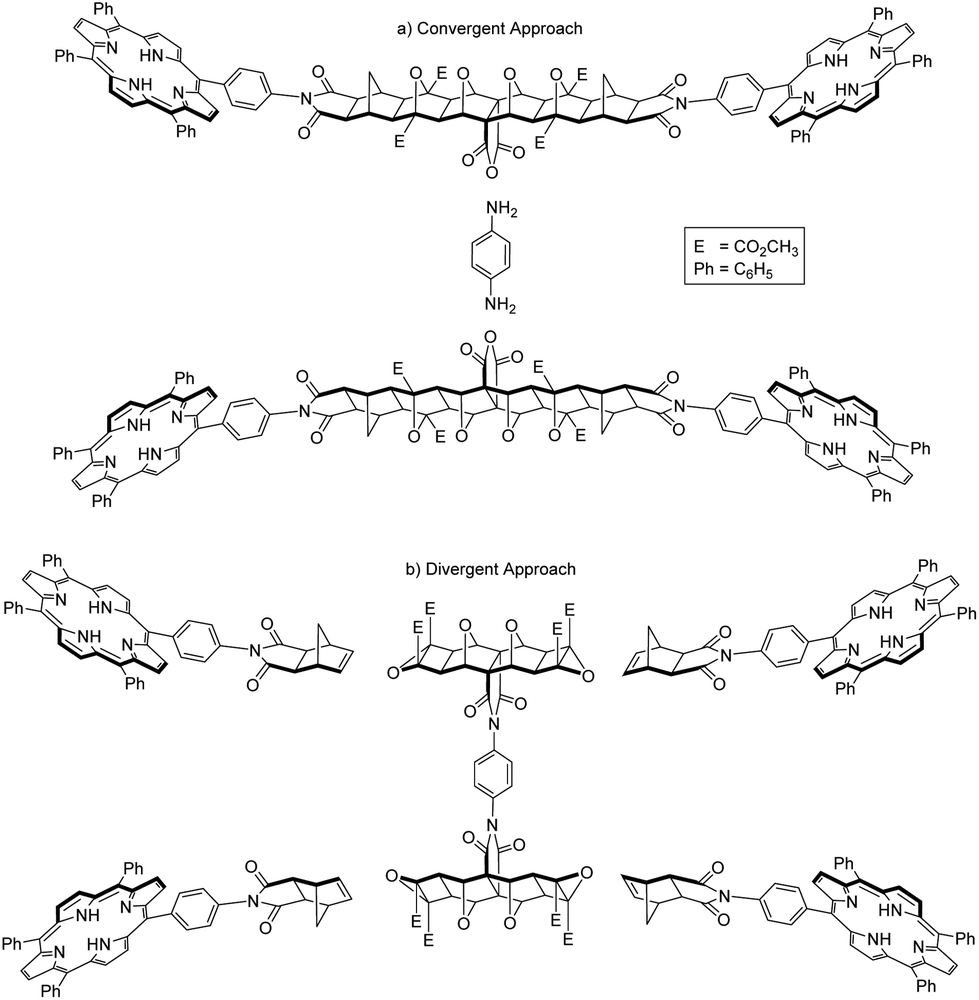 | ||
| Fig. 2 Schematic representations of convergent and divergent synthetic approaches to the synthesis of tetra-porphyrin tweezer 2. | ||
On the contrary, a divergent approach was envisaged in which two alicyclic systems were already linked to create a central core, to which porphyrin receptors could be subsequently attached (Fig. 2b). This avenue provides less structural control, as building out from the core would involve four simultaneous points of reactivity during receptor functionalisation.
Convergent approach towards 2
Initial attempts focused on the convergent approach since this was envisaged to allow the highest degree of control over the final system synthesised. This began with the application of Mitsudo conditions40,41 to anhydride functionalised bis-oxa block 3 (Fig. 3), and which afforded the bis-(cyclobutene methyl ester) adduct 442 in excellent yield. This was subsequently epoxidised in the normal manner,14,16 giving bis-epoxide 5 in good yield. Microwave-accelerated ACE reaction43 with mono-porphyrin receptor 611 successfully generated the bis-porphyrin anhydride 7. The identity of 7 was confirmed by HRMS (S5,† calcd [M + 2H]2+ 1050.3421, found 1050.3432), as poor solubility, presumably due to aggregation, in many organic solvents prohibited characterisation by NMR.
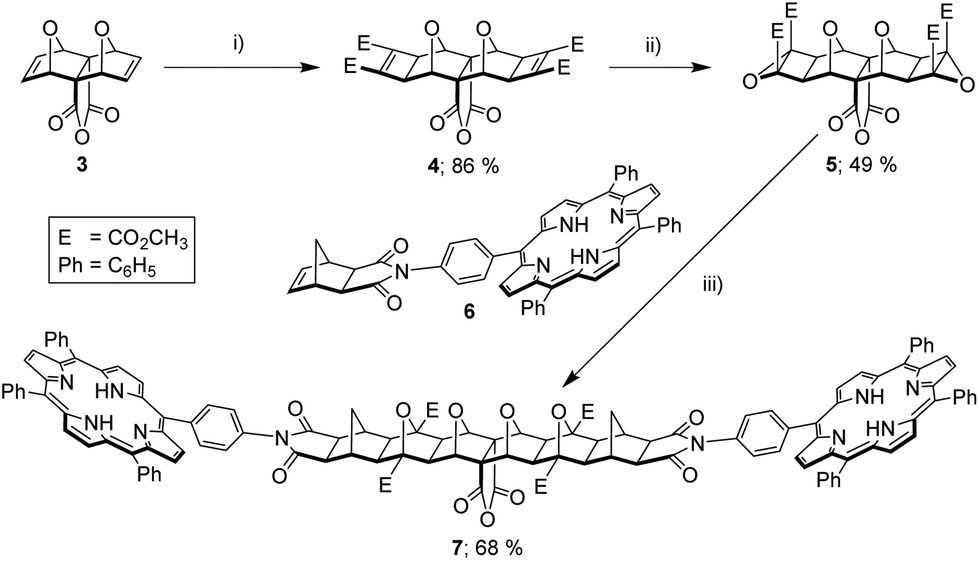 | ||
| Fig. 3 Convergent synthetic approach via bis-porphyrin anhydride 7. (i) DMAD (2 eq.), 5 mol% [RuH2(CO)(PPh3)3], toluene, 80 °C, N2 atmosphere, 4 days, 86%; (ii) anhydrous 3.3 M tBuOOH in toluene (2.5 eq.), dry CH2Cl2, 0 °C, N2 atmosphere, 10 min, tBuOK (1 eq.), r.t., 3.5 h, 49%; (iii) exo-porphyrin receptor 6 (2 eq.), dry THF, microwave 80–220 W, 14–20 bar, 170 °C, 2 h, 68%. | ||
As the bis-porphyrin anhydride 7 displayed poor solubility in common organic solvents, the Mitsudo anhydride 4 was employed as a soluble model compound in an attempt to ascertain whether the convergent synthesis could be completed as represented in Fig. 2a. 4 was frustratingly unable to be successfully reacted with aromatic amines under a variety of conditions. In summary, these problems stem from the exo-bis-oxa-polycyclic moiety, which unfortunately exhibits a combination of thermal instability prior to lateral functionalisation (retro-Diels–Alder),12,15,38,44,45 and poor anhydride reactivity via conventional condensation with aromatic amines,38,44 particularly following lateral functionalisation.45 It has been suggested by others that the anhydride is shielded by endo-hydrogen atoms at the ring junctions in laterally functionalised derivatives.45 Thus even if a highly soluble derivative of bis-porphyrin anhydride 7 could be synthesised, condensation with p-phenylenediamine would not be a viable convergent pathway to the tetra-porphyrin tweezer 2. Hence, our hand was forced to pursue a divergent approach.
Divergent approach towards 2
The divergent approach comprised the synthesis of the phenyl diimide core and subsequent attachment of the porphyrin moieties. Retrosynthetic analysis of the phenyl diimide core (dotted line, Fig. 1) was undertaken and is shown in Fig. 4.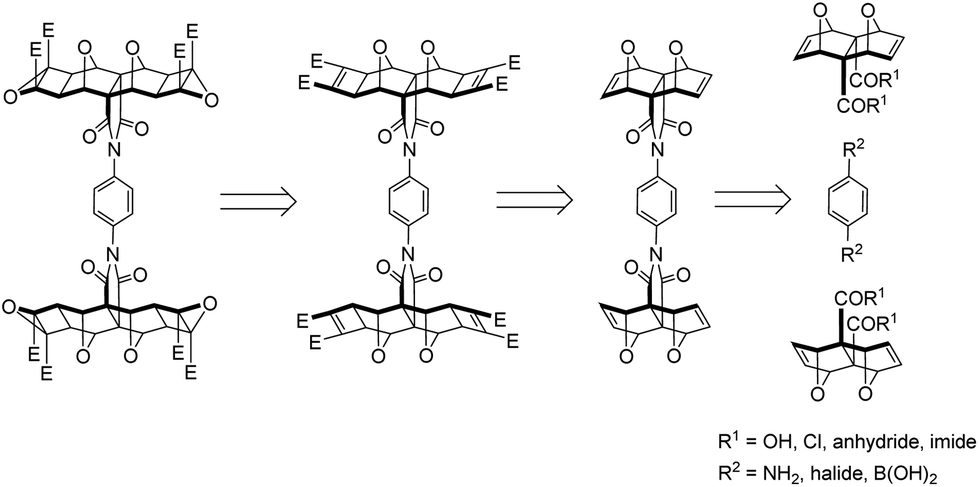 | ||
| Fig. 4 Retrosynthetic analysis of the phenyl diimide core. | ||
The endo-functionality of the polycyclic module (R1 in Fig. 4) can be the dicarboxylic acid,46,47 diacid chloride,38,48 anhydride,49 or imide18,50–53 derivative. The 1,4-substituted phenyl core then needs to possess a functionality (R2 in Fig. 4) suitable for reaction with the aforementioned functional groups. For example, literature precedent exists for the condensation of the diacid chloride derivative 8 with aniline derivatives (Fig. 5).38,44 Alternatively, while the anhydride 3 does not ordinarily react with aniline derivatives, reaction has been reported for non-laterally functionalised anhydride 3 when the aniline derivative is deprotonated by strong base such as n-BuLi.45
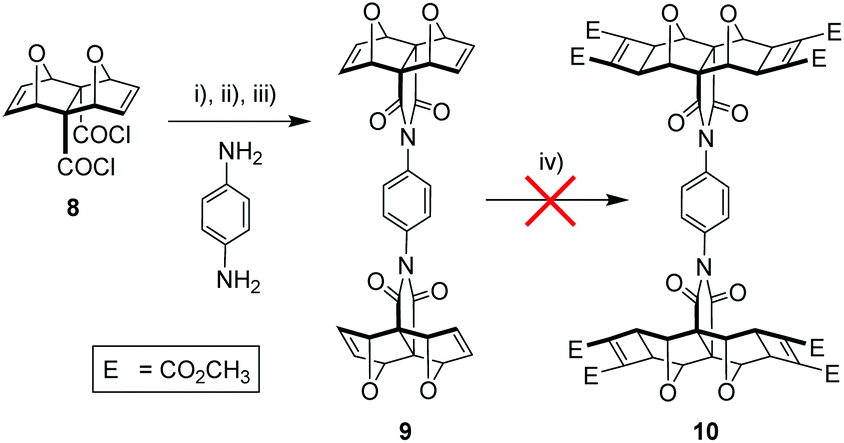 | ||
| Fig. 5 (i) p-phenylenediamine, THF, N2 atmosphere, r.t., slow addition of <1 eq. LiHMDS, stirring 30 min, (ii) diacid chloride (2 eq.), N2 atmosphere, r.t., 1 h, (iii) HOBt/DCC/Et3N, dry THF, N2 atmosphere, r.t., 3 days; (iv) DMAD (8 eq.), 20 mol% [RuH2(CO)(PPh3)3], toluene, 50 °C, N2 atmosphere, 3 days, no reaction. | ||
We chose a combination of these two conditions and utilised the reactivity of acid chloride 8 with the increased nucleophilicity54 of the p-phenylenediamine mono-anion following deprotonation by a strong base, in this case LiHMDS (Fig. 5). This afforded a significant improvement in the reaction time over the aforementioned literature methods, with instant precipitation of the intermediate amic acid from the reaction mixture. Subsequent carbodiimide-mediated ring closing of the amic acid to the imide yielded the tweezer core structure 9, with identical NMR spectra (see S6†) to that reported.44
Conveniently, we obtained a crystal of 9 from DMSO solution suitable for X-ray crystallographic analysis. Solving of the structure revealed the structure shown in Fig. 6. The structure of 9 has a crystallographic centre of inversion and is co-crystallised with two molecules of DMSO which occupy channels extending along the a-direction in the crystal. Additional information on the crystal structure of compound 9 can be found in the ESI (see S8†).
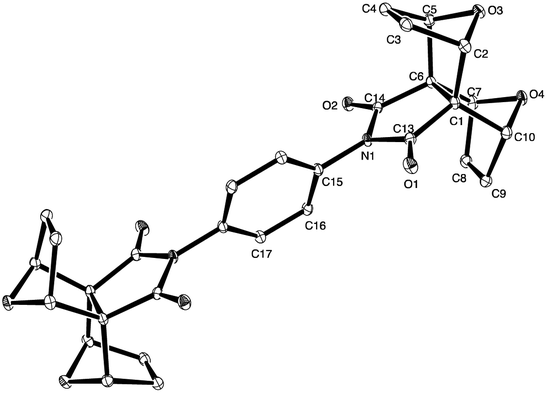 | ||
| Fig. 6 X-ray crystal structure of the centrosymmetric phenyl diimide core 9. The DMSO molecules have been omitted for clarity, H atoms are not shown. | ||
The next step towards the necessary tetra-functional phenyl diimide core 10 was a Mitsudo [2 + 2] cycloaddition reaction using DMAD and a ruthenium catalyst. Unfortunately, core unit 9 was found to be unreactive to Mitsudo conditions (Fig. 5), a problem we had encountered previously during the corresponding linker synthesis for tweezer 1.55 Unfortunately, the retro-Diels–Alder potential of 9 limited the harshness of the conditions that were able to be employed in forcing the Mitsudo reaction, and we were unable to achieve any success in this regard. There are only a few norbornyl based substrates identified in the literature that do not undergo the Mitsudo reaction,56 with no clear trends evident with our compounds, or similar compounds in the literature that allow this result to be rationalised. Further investigations are currently underway in our laboratory in an attempt to rationalise this lack of reactivity of this as well as several other substrates.
It became evident from both our own work and the work of others38,44,45 that the known syntheses currently employed for polycyclic substrates would be unsuitable to generate the tetra-functional core 10 (Fig. 5) required for the tetra-porphyrin tweezer 2. Faced with the competing triad of thermal instability of 3 and 9, poor anhydride reactivity of laterally functionalised 4 with aromatic amines, and the inability of the core unit 9 to undergo the Mitsudo reaction, we required a substrate to circumvent each of these complications. The imide 13 (Fig. 7) alleviated all of these issues; (i) reactivity in the southern direction is moved from the carbonyl to the imide nitrogen avoiding the steric protecting effects of the adjacent endo-hydrogen atoms on anhydride reactivity, and (ii) pre-installed cyclobutene rings (Mitsudo reaction) both prevent thermal degradation and commence lateral functionalisation.
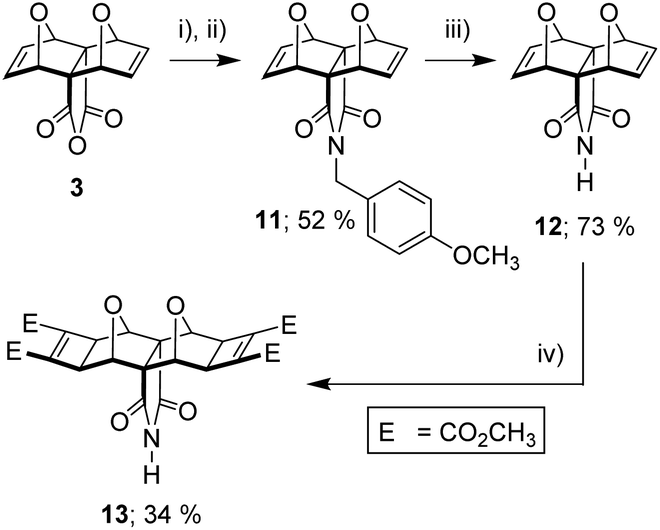 | ||
| Fig. 7 (i) p-methoxybenzylamine (2 eq.), deacidified CHCl3, 50 °C, N2 atmosphere, overnight; (ii) NaOAc/Ac2O, 50 °C, N2 atmosphere, overnight, 52%; (iii) 9:1 CH3CN/H2O, ammonium cerium(IV) nitrate (3.3 eq., 2 h; 2.3 eq. 90 min + 1 eq. 30 min), 40 °C, 73%; (iv) DMAD (4 eq.), 36 mol% [RuH2(CO)(PPh3)3], dry DMF, 60 °C, N2 atmosphere, 24 h, 34%. | ||
Fortunately, there was literature precedent for the non-Mitsudo imide 12,18,50 in two steps from anhydride 3. This method uses p-methoxybenzylamine (PMB) as a nitrogen source, with the intermediate PMB-protected derivative 11 cleaved to yield the free imide 12 (Fig. 7). We prepared 12 using these literature methods and then applied Mitsudo conditions to afford the imide 13.
We were fortunate to obtain X-ray quality crystals for all three compounds in this synthetic series to confidently assign the structures of imides 11, 12 and 13 (Fig. 8). The structures of all three imide block derivatives contain two 7-oxabicycloheptane motifs with a 6-membered ring A and 5-membered ring B approximately perpendicular. The fusion of these rings adopt an envelope conformation in compounds 11–13 with an O atom as the flap. The skeletal framework of these molecules is a strained system (bridgehead O atom bond angles of 96–97° for 11 and 12, and 98° for 13, refer to S15†).
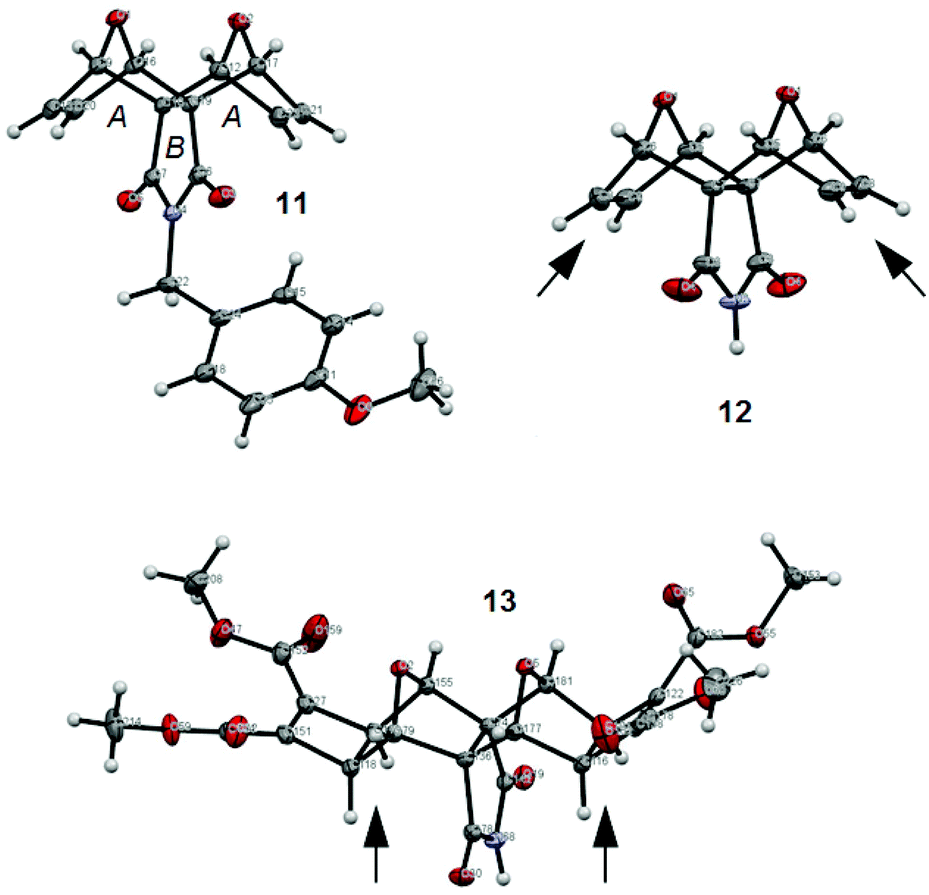 | ||
| Fig. 8 X-ray crystal structures of the imide series 11, 12 and 13. The arrows highlight the change in the position of the hydrogen atoms at the ring junctions following addition of the cyclobutene diester ring. | ||
The non-Mitsudo imide 12 is centrosymmetric and solved as the asymmetric unit with SHELX-97 in a monoclinic unit cell (space group P21/c). In structure 12, the crystal packing features intermolecular N2–H2⋯O1 hydrogen bonding features (see S15†); whereas the PMB protected imide 11 does not exhibit such interactions. Within the crystal lattice of the imide 13 there exists hydrogen bonding contacts between the envelope flap oxygen atoms (O18/O5/O3 and O15/O2/O11) with a molecule of water (see S15†). The imide nitrogen atoms (N36, N48, N68) interact with the same water molecules, as do the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygen atoms from the methyl esters (O203, O87). This creates a pseudo coordination center about each H2O molecule. There are no interactions observed with the CH3CN molecules. Additional information on the crystal structures of compounds 11–13 can be found in the ESI (see S15†).
O oxygen atoms from the methyl esters (O203, O87). This creates a pseudo coordination center about each H2O molecule. There are no interactions observed with the CH3CN molecules. Additional information on the crystal structures of compounds 11–13 can be found in the ESI (see S15†).
Furthermore, the X-ray crystal structures 12 and 13 reveal significant differences in the position of the hydrogen atoms at the ring junctions as the hybridisation of the carbon atom changes from sp2 to sp3 (marked with arrows in Fig. 8). This could support the notion previously reported by others that the anhydride is shielded45 in laterally functionalised derivatives.
The next step in the divergent approach towards 2 was the coupling of two units of imide 13 together through the imide nitrogen atom. On review of the literature, two candidates for coupling nitrogen-containing molecules with aryl species were identified, neither of which appeared to have been demonstrated on norbornyl imides to our knowledge.
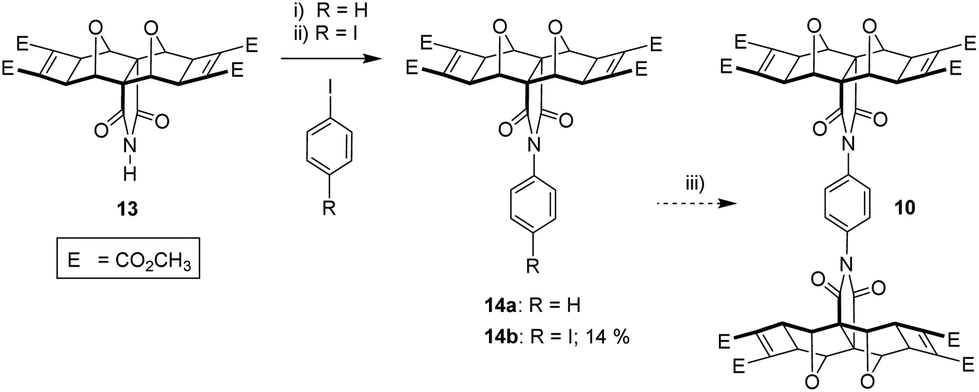 | ||
| Fig. 9 (i) 1-Iodobenzene (excess, as solvent), freshly precipitated Cu(0) (5 eq.), 4 × 10 minute microwave cycles (300 W, high stirring, air cooling, 150 °C); (ii) Initially 1,4-diodobenzene (8 eq.), freshly precipitated Cu(0) (8 eq.), EtOAc (1 mL), microwave settings as for step (i), 120–150 °C, 1 h. Then further addition of Cu(0) (8 eq.), microwave, 120–150 °C, 1.5 h. Addition of toluene (1 mL), microwave, 150 °C, 1 h + 8.5 h. 14% of 14b; (iii) 14b, 13 (1 eq.), not attempted. | ||
A small quantity of the asymmetric phenyl 1-imide-4-iodo adduct 14b was generated by using excess 1,4-diiodobenzene in solvent and subjecting to microwave irradiation in several cycles over approximately 12 hours. However, the inefficiency associated with forming precursor 14b, along with the second coupling of 13 with 14b (not attempted) which would be required to generate diimide 10 led us to explore an alternative imide coupling procedure.
Given the success of this coupling reaction, we employed 1,4-phenyl diboronic acid and polycyclic imide 13, in a 1:2 stoichiometric quantity, to produce phenyl diimide 10 (Fig. 10). The identity of 10 was confirmed by HRMS (calcd [M + Na]+ 1127.2182, found 1127.2166), while the 1H and 13C NMR spectra (see S18 and S19†) both showed the expected number of resonances. A singlet in the 1H NMR at 7.38 ppm was assigned to the phenyl core and suggests that the rotation of both phenyl–imide bonds is rapid on the NMR timescale.
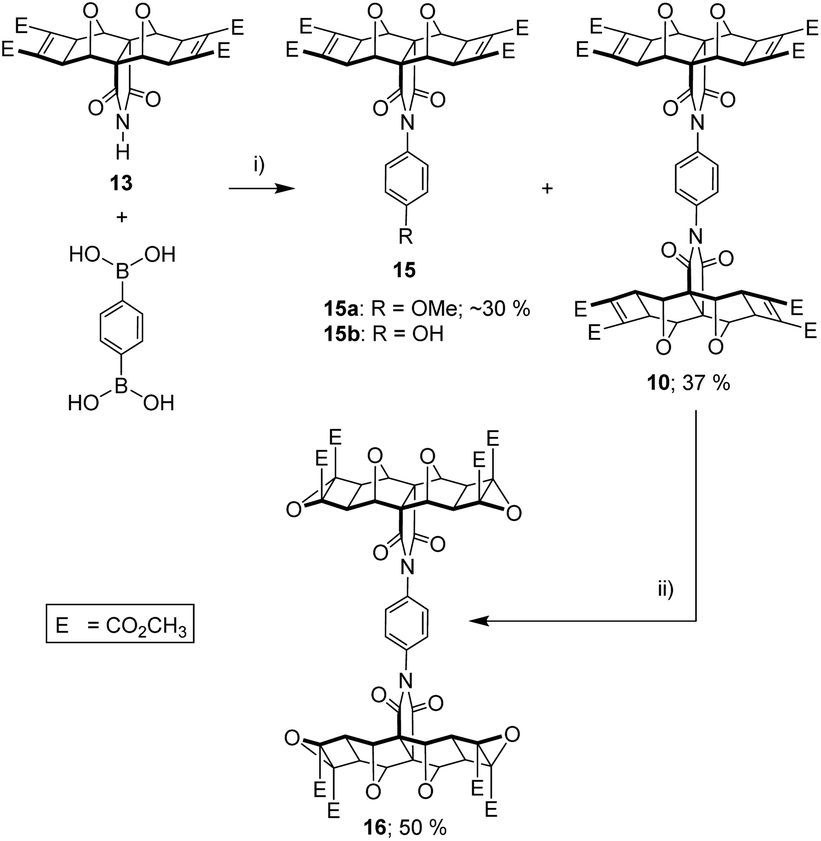 | ||
| Fig. 10 (i) benzene-1,4-diboronic acid, copper(II) acetate (5 mol% per boronic acid), MeOH or 9:1 THF/H2O, reflux, air bubbler, 18 h, ∼30% 15a + 37% 10; (ii) anhydrous 3.3 M tBuOOH in toluene (5 eq.), dry CH2Cl2, 0 °C, N2 atmosphere, 10 min, tBuOK (2 eq.), r.t., 3.5 h, 50%. | ||
The optimum Cu(II) catalyst loading was found to be less than 5%, at which the reaction yielded approximately equal quantities of unreacted starting material imide 13, desired product 10, but also an unexpected side product 15 (Fig. 10). This side product was identified as 15a or 15b depending on the solvent used for the reaction (see next paragraph). These products indicate a competitive reaction between boronic acid, imide, and solvent. At greater than 5% catalyst loadings, side product 15 became the major product. These side products were not reported in the original methods,58,59 which only use aryl mono-boronic acid derivatives, and this led us to briefly explore the effect of solvent on the coupling reaction.
As reported in the original methods, we also found that no coupling was obtained in aprotic solvents such as THF, but that successful coupling could be achieved protic solvents such as MeOH, or mixed aprotic/protic solvents such as THF/H2O.58,59 In our case, THF/H2O (9:1) yielded not only the phenyl diimide 10, but side product with hydroxy substitution 15b. When the solvent was MeOH, the side product was methoxy substituted 15a.
Even with this side reaction, a sufficient quantity of the tetra-functional phenyl diimide core 10 was obtained to allow further reactions to be carried out. Thus 10 was subsequently subjected to a four-fold nucleophilic epoxidation at the electron deficient alkene, which gave decent yields of tetra-epoxide 16 (Fig. 10). The identity of tetra-epoxide 16 was confirmed by using HRMS (calcd [M + Na]+ 1191.1978, found 1191.1984, 1191.2006). The successful incorporation of the epoxide functionality set the stage for the ACE coupling to attach the porphyrin moieties.
Tetra-porphyrin tweezer
The tetra-epoxide core 16 was appended with exo-porphyrin receptors 611via the alkene plus cyclobutane epoxide (ACE) reaction (Fig. 11), accelerated by the use of microwave irradiation.43 Although ACE chemistry has been well established for polycyclic scaffolds for some time, there appears to be no examples of tetra-functional substrates undergoing four simultaneous ACE reactions. The tetra-porphyrin two binding site tweezer 2 was found to be the major product of the reaction (by 1H NMR of the crude reaction mixture), however, the high degree of purity required for host–guest experiments resulted in significant loss of product from column chromatography and repeated recrystallisations to remove traces of the other porphyrin stoichiometries. The identity of the freebase tweezer 2 was confirmed by using HRMS (see S25,† calcd [M + 4H]4+ 1068.3540, found 1068.3566, plus [M + 3H]3+ and [M + 2H]2+).
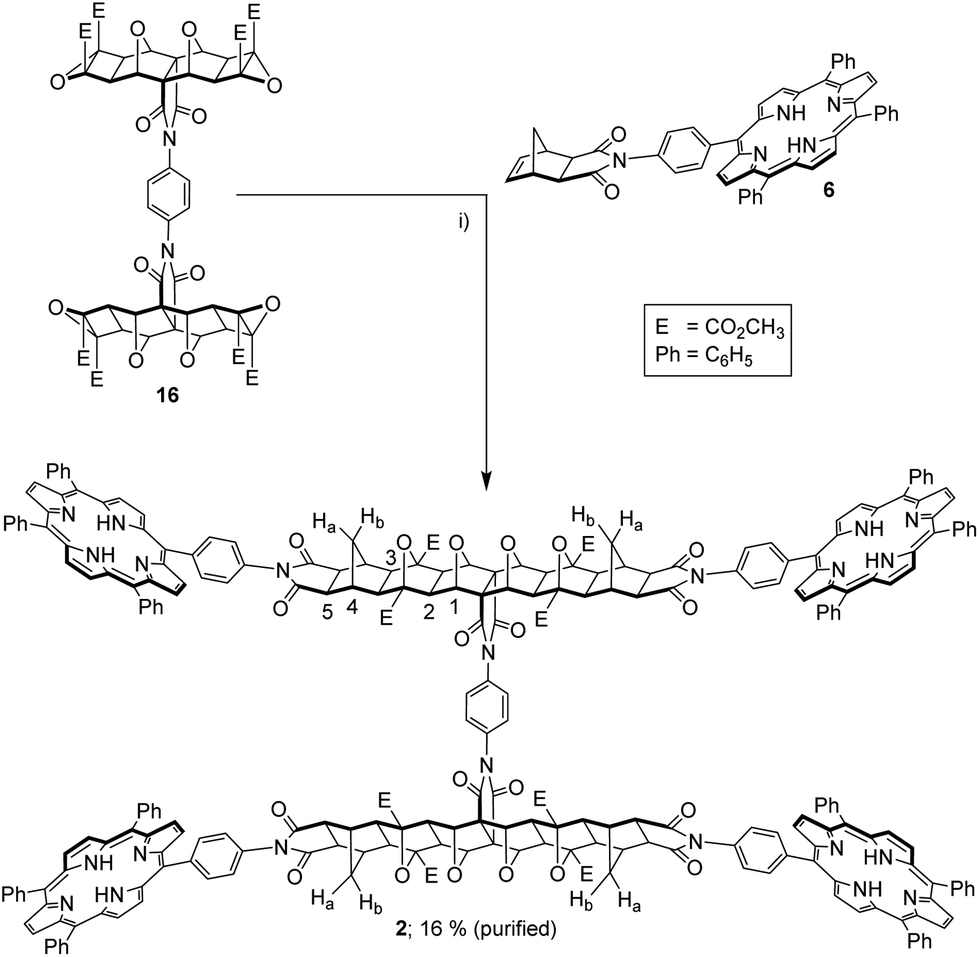 | ||
| Fig. 11 (i) exo-Porphyrin receptor 6 (4 eq.), dry THF, microwave 80–220 W, 14–20 bar, 170 °C, 2 h, 16% after purification. | ||
The 1H NMR spectra reflected the symmetry of the tetra-porphyrin tweezer 2, arising from free rotation about the phenyl diimide core and porphyrin receptors. Several features characteristic of ACE-coupled reaction products could be observed, including a small downfield shift for the methyl ester resonance,43 along with the disappearance of the norbornene proton resonance from the exo-receptor 6 at 6.45 ppm. The resonance at 90 ppm in the 13C NMR spectrum (not shown) is observed in similar polycyclic systems,34 and is assigned to the carbon bridgehead atoms in the newly formed oxanorbornane.34
In the 1H NMR spectrum (see S24†), seven polycyclic resonances can be observed as expected (excluding the methyl ester); five singlets and two doublets. Of the five singlets, two arise from the central 7-oxanorbornane component, labelled 1–2, Fig. 11), while the remaining three signals arise from the exo-component (labelled 3–5, Fig. 11). The doublets, which occur at chemical shifts of 1.27 and 2.62 ppm, correspond to the methylene bridge protons Ha/Hb (Fig. 11), which appear at different chemical shifts due to the influence of the 7-oxanorbornane in these linear systems.25,26,30
There is some evidence of facial differentiation within the porphyrin macrocycles in the 1H NMR spectrum of the freebase tetra-porphyrin tweezer 2 (see S24†). While the β-pyrrole resonance appears as a singlet, there is slight splitting/desymmetrisation within some of the meso-phenyl resonances, and the linking meso-phenyl resonances are somewhat broad (see S24†). This facial differentiation indicates there is some amount of either intramolecular interaction of opposing porphyrins or intermolecular aggregation at NMR concentrations (10−3–10−4 M).
The various porphyrin containing molecules were also examined using UV-Vis spectroscopy. Table 1 summarises the main spectral features of the freebase derivatives of the tetra-porphyrin tweezer 2 (see S26†), bis-porphyrin tweezer 1,11 and mono-porphyrin receptor 6. The Soret peak band width of 12.1 nm for tetra-porphyrin tweezer 2 is barely broadened compared to the value for mono-porphyrin receptor 6 (11.8 nm), indicating that exciton coupling interactions between the porphyrins in the tetra-porphyrin tweezer 2 are largely absent at UV-Vis concentrations (10−6–10−7 M). This suggests either the porphyrin units are able to undergo rotation either about the single bond between the imide and/or porphyrin moiety, and/or the two polycyclic arms can undergo rotation about the central phenyl diimide core.
Molecular modelling
It was envisaged that an unsubstituted central phenyl core would confer the rotational ability required by the polycyclic scaffold to facilitate interannular cooperativity between the two remote binding sites. A significant number of computational and experimental works exploring bond rotation in N-phenyl imide derivatives have been reported.60–76 In summary, the energy barrier to rotation increases as the size of the ortho-phenyl substituent increases due to steric repulsion between the substituent and the oxygen atom of the imide.60,64,65 Additionally, the angle between the aryl and imide rings increases towards perpendicular with increasing size of the ortho-substituent on the phenyl ring, to minimise steric repulsion between the substituent and the oxygen atom of the imide.60–62,64 This competes with the resonance delocalisation and conjugation favoured in the planar conformation77‡. Free rotation is reported for unsubstituted N-phenyl imide derivatives,66,67 with rotation becoming restricted for ortho-substituents other than hydrogen,66–76 while meta- and para-phenyl substitution does not affect rotation in these systems.67To gain an increased understanding of the barrier to rotation about the imide–phenyl bond at the core of tetra-porphyrin tweezer 2, a rotational energy profile was calculated§ for a simplified model of the linker, compound 17¶ (N2–C3, Fig. 12). The relative energy difference between the global maximum and minimum is approximately 9 kJ mol−1 (2.15 kcal mol−1) and supports the notion of free rotation in this system at room temperature, from thermal energy available from the surroundings.65 Not surprisingly, the highest energy conformer occurs at a dihedral angle of 0°, when the phenyl ring proton atoms and imide carbonyl oxygen atoms are in the same plane and interacting sterically. The lowest energy conformation occurs at a dihedral angle of approximately 45°, while a local maximum occurs at approximately 90°.
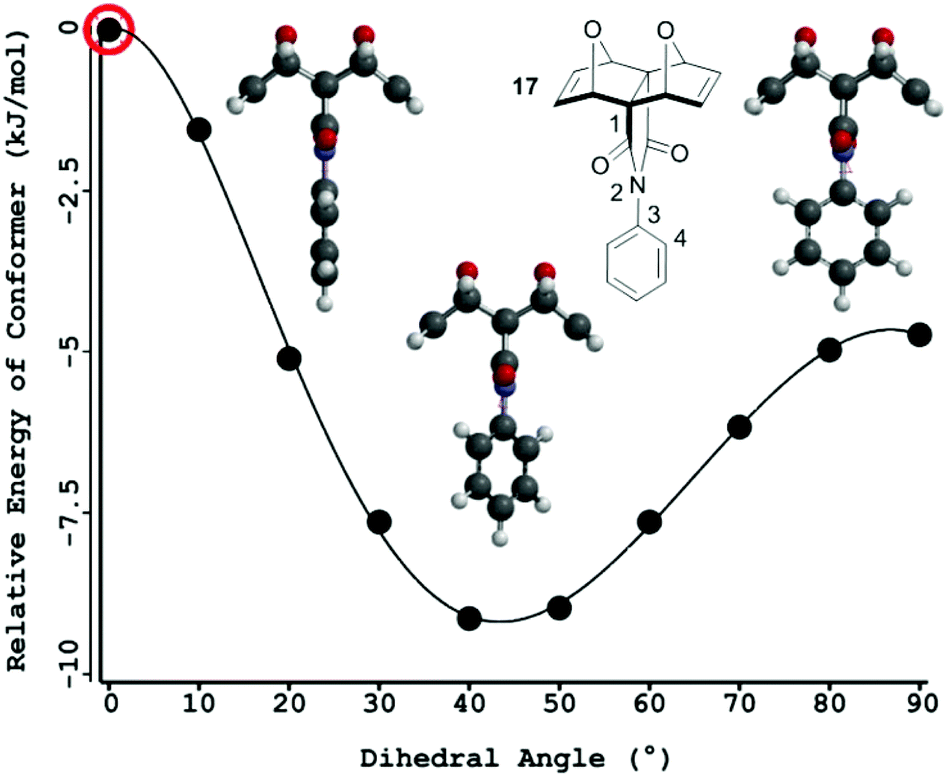 | ||
| Fig. 12 Rotational energy profile and barrier to rotation about the imide–phenyl single bond in a model of the linker, compound 17 (energy profile Hartree–Fock 6-31G*, energy of each conformation recalculated Density Functional B3LYP/6-31G*). | ||
Molecular modelling has been previously used to calculate the ground state optimised geometry in a polycyclic N-phenyl imide system.17,78 In this study, the global energy minimum corresponded to a phenyl–imide angle of 70–71° (RHF/3-21G,78 HF3-21G17), and was 5.65 kJ (1.35 kcal) more stable than the case where the phenyl ring was constrained to the plane of the imide.78 While the phenyl–imide angle for the global energy minimum is different depending on the computational method employed (rotational energy profile in our work, and the ground state optimised geometry in the literature17,78), both calculations confer that the highest energy state occurs when the phenyl ring is in the plane of the imide, where the hydrogen and oxygen atoms are sterically interacting.
In the absence of X-ray crystallographic data, molecular modelling79 was undertaken to provide information on the equilibrium geometry of the two binding site tweezer 2. Fig. 13 shows the equilibrium geometry (semi-empirical, AM1) of the zinc(II) metallated adduct of tetra-porphyrin tweezer 2 without guest, and indicates that the polycyclic scaffolds do not necessarily adopt a parallel alignment, as per the idealised structures shown in the reaction schemes, with the two polycyclic arms rotated with respect to each other around the central phenyl diimide group. The phenyl–imide angle from this model of 2 was calculated to be 43° or 21° (depending on whether measured from either the top or bottom half of the model), and is within the trough of the rotational energy profile shown earlier for compound 17 in Fig. 12.
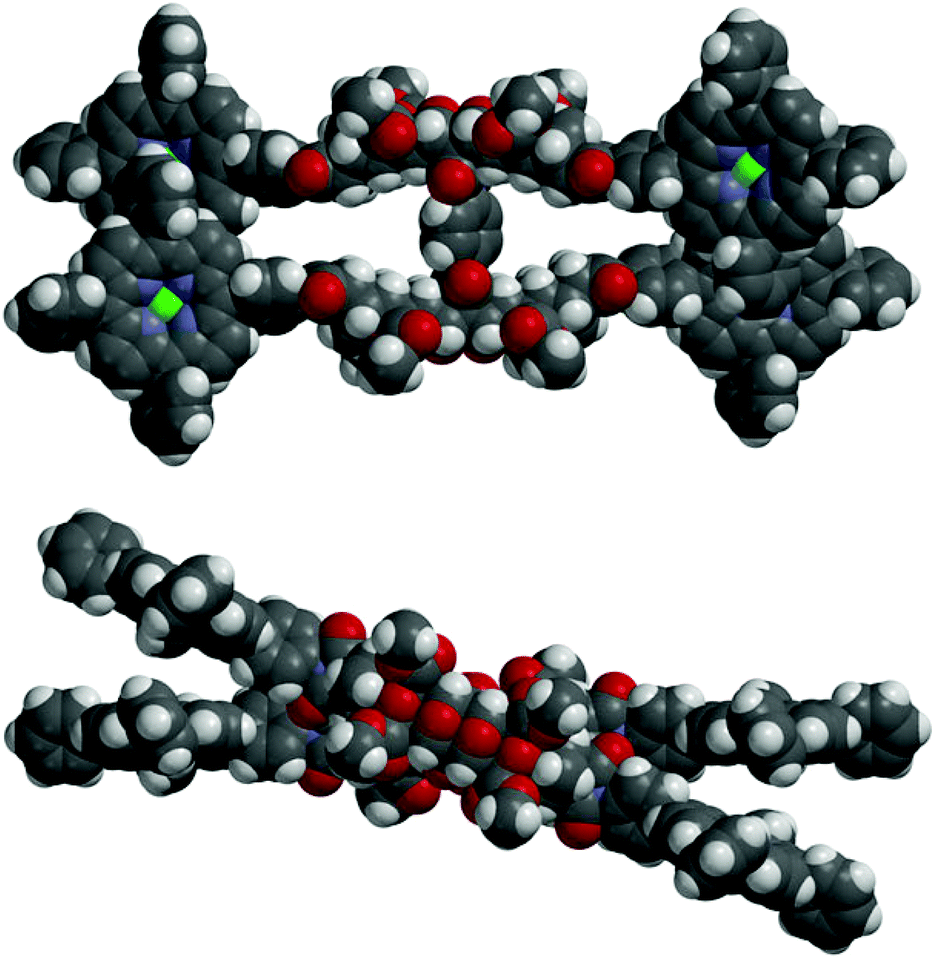 | ||
| Fig. 13 Molecular modelling of the zinc(II) metallated adduct of the two binding site tetra-porphyrin tweezer 2 in the absence of guest (side-on and top-down) (semi-empirical, AM1). | ||
In addition, the idealised parallel arrangement of the porphyrin units has been replaced by a skewed interporphyrin geometry as a result of rotation about the imide–phenyl–porphyrin bond. Rotation of porphyrins about unsubstituted meso-phenyl rings with respect to the porphyrin β-pyrrole region is well known.80–82 Thus the freely rotating porphyrin receptors provides the system with the ability to cofacially align the bis-porphyrins after undergoing defined changes to interporphyrin distance upon guest binding as the polycyclic scaffold is rotated through the central core.
Another structural feature not accounted for by the reaction scheme structures is the high degree of curvature in polycyclic scaffolds with 7-oxanorbornane modules.28,29 This is clearly observable in the molecular models in Fig. 13, and is a crucial design feature in our tweezer which enables the bis-porphyrin binding sites to be coplanar in each half30 and cofacial between the two halves, if an idealised conformation is adopted in the presence of a guest.||
Fig. 14 shows the equilibrium geometry of the zinc(II) metallated adduct of tetra-porphyrin tweezer 2 with diamino ligand; for (a) DABCO and (b) 4,4′-bipy. The system appears able to accommodate diamino ligands of different lengths (N–N distance for DABCO is 2.6 Å, 7.2 Å for 4,4′-bipy, semi-empirical AM1) as eluded to previously, by rotating both the rigid polycyclic arms (via the phenyl diimide core) and the porphyrin receptors, to adjust the interporphyrin distance as required. For example, the polycyclic arms are rotated further with respect to each other for bipy than for DABCO, due to the difference in ligand length. With respect to each guest, approximately equal interporphyrin distance was observed at both binding sites. Neither the DABCO nor bipy complexed structures appear to be significantly strained, and overlay of the polycyclic arms for molecular models with and without guest (not shown) revealed only minimal distortion, providing further evidence that the polycyclic arms are largely rigid. The experimental host–guest complexation of the zinc(II) metallated adduct of tetra-porphyrin tweezer 2 with diamino ligands, and analysis of interannular cooperativity will be described in a future publication.
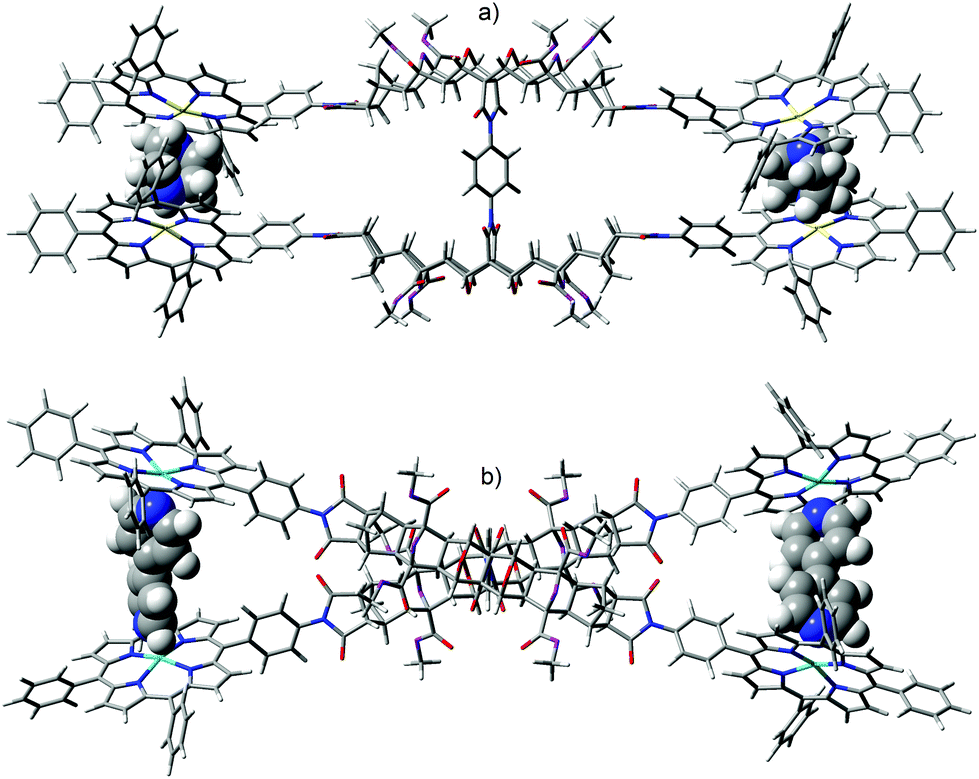 | ||
| Fig. 14 Molecular modelling of the zinc(II) metallated adduct of the two binding site tetra-porphyrin tweezer 2 in the presence of diamino ligands; (a) 2:(DABCO)2 (side-on), (b) 2:(bipy)2 (top-down) (semi-empirical, AM1).Image rendered in GaussView. | ||
Conclusions
Tetra-porphyrin tweezer 2 with two remote but interdependent binding sites was synthesised via a divergent approach from a tetra-functional phenyl diimide core. The key step in obtaining this target was the application of a copper(II)-mediated coupling of benzene-1,4-diboronic acid with a polycyclic imide, and represents a novel use of such coupling in polycyclic scaffolds. We expect that the polycyclic imide coupling methodology presented in this paper, either via the phenyl boronic acid (superior for the synthesis of compound 10) and/or aryl halide route, is easily and widely applicable to other modular polycyclic systems. This will aid in the synthesis of functional supramolecular architectures where control of north/south reactivity or other directional interactions is required. Furthermore, the microwave-accelerated ACE reaction allowed simultaneous attachment of the four porphyrin moieties to afford the tetra-porphyrin tweezer 2.Molecular modelling of both the equilibrium geometry of tetra-porphyrin tweezer 2 and the rotational energy profile of a model of the phenyl–imide linker core suggests the architecture is well suited for the complexation of diamino ligands of varying lengths, such as DABCO and 4,4′-bipy. This appears to be facilitated by bond rotation about both the central phenyl diimide core and the porphyrin receptors.
The absence of reactivity at the anhydride of laterally functionalised systems such as compound 4 continues to complicate efforts towards the convergent synthesis of extended polycyclic architectures. Our laboratory is currently pursuing the imide analogue of bis-porphyrin anhydride 7, which is expected to be able to be coupled using the boronic acid method employed for the synthesis of compound 10. The establishment of a convergent strategy would enable architecture 2 to be expanded to include asymmetric molecular tweezers with two different receptor binding sites.
Experimental
Molecular modelling
Molecular modelling was undertaken using the Wavefunction Spartan ‘10 software package.79 Equilibrium geometry was calculated using a semi-empirical AM1 model. Rotational energy profiles were calculated using a Hartree–Fock 6-31G* model for each conformation about the bond specified by the dihedral angle constraint, with the energy of each conformation recalculated using a B3LYP/6-31G* Density Functional model to improve the fitting function.Characterisation
Melting points were measured using a Barloworld Scientific SMP10 melting point apparatus.NMR spectra were recorded on a Bruker UltraShield Avance III 400 MHz or 600 MHz NMR Spectrometer running the TopSpin software package at 299 K and 293 K respectively. Spectra were calibrated to the residual solvent signal.83 CDCl3 was deacidified by passing through neutral activated aluminium oxide (Scharlau, activity degree 1, 70–290 mesh, grain size 0.05–0.2 mm) and stored under a nitrogen atmosphere over silver foil/molecular sieves in a brown glass bottle.
Theoretical mass spectra were generated by using the mMass software package.84
Single crystals of compounds 11–13 suitable for X-ray diffraction experiments were covered in Paratone-N oil and mounted on a glass fibre. Data (2θmax 55°) were collected at 150 K using an Oxford Diffraction X-Calibur X-ray diffractometer and MoKα (λ 0.71073 Å) radiation. After integration and scaling, the datasets were merged into N unique reflections (Rint). The structures were solved using conventional methods and refined by using full-matrix least-squares using the SHELX-97 software85 in conjunction with the X-Seed interface.86 Non-hydrogen atoms were refined with anisotropic thermal parameters and hydrogen atoms were placed in calculated positions. Early refinement of compound 13 in an alternate monoclinic space group (P21/n) was attempted; however, structural solution in this space group failed. The structure was solved and refined in the more orthogonal space group P21/c. Structure solution and refinement succeeded with three crystallographically independent but structurally identical molecules in the asymmetric unit. Intensity data for compound 9 was collected with an Oxford Diffraction SuperNova CCD diffractometer using Cu-Kα radiation. The temperature during data collection was maintained at 100.0(1) using an Oxford Cryosystems cooling device. The structure was solved by direct methods and difference Fourier synthesis.85 Thermal ellipsoid plots were generated using the program ORTEP-387 integrated within the WINGX88 suite of programs. Additional information on the crystal structures of compounds 9 and 11–13 can be found in the ESI (see S8 and S15†). CCDC 1486427–1486430 contain the supplementary crystallographic data for this paper.
Where necessary, solvents and reagents for synthesis were purified according to the methods published.89 Dry THF was freshly distilled from sodium/benzophenone, dry CH2Cl2 freshly distilled from CaH2, and dry DMF and DMSO were distilled under reduced pressure onto fresh molecular sieves after stirring on molecular sieves overnight. Potassium tert-butoxide was purified by sublimation under high vacuum at 0.17 mmHg at 160 °C, stored under a nitrogen atmosphere, protected from light, and in a desiccator at room temperature. Benzene-1,4-diboronic acid (Sigma-Aldrich) was recrystallised from THF/H2O and dried in vacuo at 50 °C for 3 hours. Zinc dust was treated with 2% hydrochloric acid to remove zinc oxide. LiHMDS and 4-methoxybenzylamine are available from Sigma-Aldrich.
Silica gel (Davisil, 60 Å, 40–63 μm, Grace Davison Discovery Sciences) was used for column chromatography. Kieselgel silica gel 60 F254 aluminium sheets (Merck) was used for TLC. Colourless compounds were visualised using a UV lamp or permanganate dip stain.
General procedure for microwave-accelerated ACE reactions
Microwave reactions were performed in a CEM Discover S-Class microwave in 10 mL reaction vessels loaded with 0.25 g combined starting materials suspended in no more than 2–3 mL of dry THF. The ACE reaction conditions are based on those prescribed.43 The microwave was operated in variable power (dynamic) mode with the following parameters: temperature 170–180 °C, power 300 W, stirring high, compressed air cooling (PowerMAX) off. Ramp time to reach temperature set point was approximately 10–30 minutes, with the pressure reaching between 8–16.5 bar (maximum pressure cut-off 20 bar). The sample was held at the set temperature for a further 1–2 hours, with the microwave input power automatically modulated between 80–220 W. The pressure tapered off during the course of the reaction.Synthesis of compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 42.
Using a similar method to the literature,90 a solution of anhydride 349 (1.0 g, 4.31 mmol), DMAD91,92 (1.3 g, approx. 2 eq., 9.15 mmol), and [RuH2(CO)(PPh3)3]93 (0.2 g, 0.22 mmol, 5 mol%) in toluene (20 mL) was heated at 80 °C for 4 days under a nitrogen atmosphere and protected from light, forming a white precipitate. The mixture was cooled, filtered, and the precipitate washed with EtOAc to afford a white powder (1.92 g, 86%), used for subsequent synthesis without further purification. M.p. decomposition 272–276 °C. 1H NMR (400 MHz, CDCl3, 26 °C): 4.83 (s, 4H), 3.81 (s, 12H), 3.27 (s, 4H). 13C NMR (100 MHz, CDCl3): 166.69, 160.04, 140.00, 77.74, 72.87, 52.48, 44.57. HRMS (ESI-TOF-MS) for C24H20O13Na+ [M + Na]+: Calc: 539.0802. Found: 539.0801.
38,48 (1.0 g, 3.48 mmol, 2 eq.) was added in portions as a solid, immediately forming a precipitate. Although no change was observed after 1 hour, the mixture was allowed to stir overnight for good measure, the precipitate filtered and washed with Et2O to afford a red/brown powder (0.46 g, 0.78 mmol, 86%, NMR consistent with amic acid). The powder was subjected to ring closing conditions38 using the following procedure. The powder was suspended in dry THF (50 mL) under an argon atmosphere, and HOBt (at least 0.32 g, 2.33 mmol, at least 3 eq.) added with stirring. After 5 minutes, DCC (at least 0.48 g, 2.33 mmol, at least 3 eq.**) and Et3N (1.1 mL, 7.8 mmol, ∼10 eq.) were added in quick succession and the mixture stirred at room temperature for 4 days (ring closing halfway), then at 50 °C for a further 2 days. The mixture was cooled and filtered, and the solids washed with hexane (100 mL), acetone (100 mL), CH3CN (100 mL), EtOH (100 mL), and hexane (100 mL) to remove impurity (product sparingly soluble). The remaining solid was dissolved in CH2Cl2 (200 mL) and washed with HCl (2 M, 100 mL) NaOH (2 M, 100 mL), H2O (100 mL), dried with Na2SO4, filtered, and the solvent removed in vacuo to afford a beige powder. M.p. >300 °C. 1H NMR (600 MHz, CDCl3, 20 °C): 7.12 (s, 4H), 6.73 (s, 8H), 5.35 (s, 8H). 13C NMR (150 MHz, CDCl3): 172.91, 139.37, 131.28, 126.69, 81.73, 69.35. HRMS (ESI-TOF-MS) for C30H20N2O8Na+ [M + Na]+: Calc: 559.1117. Found: 559.1123. Single crystal grown from DMSO by slow evaporation for X-ray diffraction. CCDC 1486430 contains the supplementary crystallographic data for this paper.
49 (21 g, 0.090 mol) was dissolved in CHCl3 (300 mL, deacidified by passing through neutral activated aluminium oxide) under a nitrogen atmosphere. 4-Methoxybenzylamine (2 eq., 25 g per 23.6 mL, 0.18 mol, Sigma-Aldrich) was added and the solution stirred at 50 °C overnight under a nitrogen atmosphere, during which a white precipitate formed. The mixture was cooled, filtered, and the precipitate washed with CHCl3 (3 × 100 mL). The precipitate was dissolved in Ac2O (350 mL), NaOAc (2 eq., 24.6 g, 0.181 mol) added, and the mixture stirred overnight at 50 °C under a nitrogen atmosphere, after which the Ac2O was removed by distillation under reduced pressure at 50 °C. The solids were redissolved into CHCl3 (300 mL), washed with H2O (2 × 200 mL), HCl (2 M, 200 mL), NaOH†† (2 M, 5 × 200 mL), H2O (200 mL), dried with Na2SO4, filtered, and the solvent removed in vacuo. The solid was washed with hexane (2 × 100 mL), Et2O (2 × 100 mL), and EtOH (5 × 100 mL, to remove significant brown byproduct) to afford a white powder (16.5 g, 52%). A sample was recrystallised from CHCl3/hexane for MS and melting point analysis. M.p. 231–233 °C. 1H NMR (600 MHz, CDCl3, 20 °C): 7.15 (d, J = 8.8 Hz, 2H), 6.79 (d, 8.8 Hz, 2H), 6.37 (m, 4H), 5.20 (m, 4H), 4.28 (s, 2H), 3.77 (s, 3H). 13C NMR (150 MHz, CDCl3): 173.86, 159.31, 138.90, 130.51, 127.28, 113.74, 81.39, 69.21, 55.30, 41.79. HRMS (ESI-TOF-MS) for C20H17NO5Na+ [M + Na]+: Calc: 374.1004. Found: 374.1000. Single crystals for X-ray analysis were grown from CH3CN by slow evaporation. CCDC 1486427 contains the supplementary crystallographic data for this paper.
:1 CH3CN/H2O (1 L) at 40 °C for 1 h. Ammonium cerium(IV) nitrate (82 g, 3.3 eq.) was added in two portions; 2.3 eq. for 90 min, followed by 1 eq. for a further 30 minutes; with reaction progress monitored by NMR. The volume of the solution was reduced by approximately two thirds in vacuo at 50 °C, or until a precipitate formed. The precipitate was filtered and put aside to later be combined with additional product. The filtrate volume was then further reduced in vacuo at 50 °C until only H2O remained. The mixture was diluted with further H2O, extracted with EtOAc (10 × 200 mL), and the EtOAc extracts combined. The volume of EtOAc was reduced in vacuo at 50 °C until a precipitate formed. The precipitate was filtered, and combined with the first crop of product. The filtrate volume was further reduced, and the precipitate collection cycle repeated until only an orange/brown oily sludge remained (p-anisaldehyde byproduct). The combined precipitate was washed with H2O (2 × 100 mL) and EtOH (10 × 100 mL) to afford a white powder (7.64 g, 73%). M.p. partial decomposition (darkens) >200 °C, violent decomposition (boiling) at 293–295 °C. Single crystals for X-ray analysis were grown from CH3CN/DMSO by slow evaporation. 1H NMR (600 MHz, DMSO-d6, 20 °C): 10.79 (s, 1H), 6.71 (s, 4H), 5.24 (s, 4H). 13C NMR (150 MHz, DMSO-d6): 175.34, 139.36, 80.39, 70.03. HRMS (ESI-TOF-MS) for C12H8NO4− [M − H]−: Calc: 230.0453. Found: 230.0457. CCDC 1486428 contains the supplementary crystallographic data for this paper.
42.
Using a similar method to the literature,90 a solution of anhydride 349 (1.0 g, 4.31 mmol), DMAD91,92 (1.3 g, approx. 2 eq., 9.15 mmol), and [RuH2(CO)(PPh3)3]93 (0.2 g, 0.22 mmol, 5 mol%) in toluene (20 mL) was heated at 80 °C for 4 days under a nitrogen atmosphere and protected from light, forming a white precipitate. The mixture was cooled, filtered, and the precipitate washed with EtOAc to afford a white powder (1.92 g, 86%), used for subsequent synthesis without further purification. M.p. decomposition 272–276 °C. 1H NMR (400 MHz, CDCl3, 26 °C): 4.83 (s, 4H), 3.81 (s, 12H), 3.27 (s, 4H). 13C NMR (100 MHz, CDCl3): 166.69, 160.04, 140.00, 77.74, 72.87, 52.48, 44.57. HRMS (ESI-TOF-MS) for C24H20O13Na+ [M + Na]+: Calc: 539.0802. Found: 539.0801.
38,48 (1.0 g, 3.48 mmol, 2 eq.) was added in portions as a solid, immediately forming a precipitate. Although no change was observed after 1 hour, the mixture was allowed to stir overnight for good measure, the precipitate filtered and washed with Et2O to afford a red/brown powder (0.46 g, 0.78 mmol, 86%, NMR consistent with amic acid). The powder was subjected to ring closing conditions38 using the following procedure. The powder was suspended in dry THF (50 mL) under an argon atmosphere, and HOBt (at least 0.32 g, 2.33 mmol, at least 3 eq.) added with stirring. After 5 minutes, DCC (at least 0.48 g, 2.33 mmol, at least 3 eq.**) and Et3N (1.1 mL, 7.8 mmol, ∼10 eq.) were added in quick succession and the mixture stirred at room temperature for 4 days (ring closing halfway), then at 50 °C for a further 2 days. The mixture was cooled and filtered, and the solids washed with hexane (100 mL), acetone (100 mL), CH3CN (100 mL), EtOH (100 mL), and hexane (100 mL) to remove impurity (product sparingly soluble). The remaining solid was dissolved in CH2Cl2 (200 mL) and washed with HCl (2 M, 100 mL) NaOH (2 M, 100 mL), H2O (100 mL), dried with Na2SO4, filtered, and the solvent removed in vacuo to afford a beige powder. M.p. >300 °C. 1H NMR (600 MHz, CDCl3, 20 °C): 7.12 (s, 4H), 6.73 (s, 8H), 5.35 (s, 8H). 13C NMR (150 MHz, CDCl3): 172.91, 139.37, 131.28, 126.69, 81.73, 69.35. HRMS (ESI-TOF-MS) for C30H20N2O8Na+ [M + Na]+: Calc: 559.1117. Found: 559.1123. Single crystal grown from DMSO by slow evaporation for X-ray diffraction. CCDC 1486430 contains the supplementary crystallographic data for this paper.
49 (21 g, 0.090 mol) was dissolved in CHCl3 (300 mL, deacidified by passing through neutral activated aluminium oxide) under a nitrogen atmosphere. 4-Methoxybenzylamine (2 eq., 25 g per 23.6 mL, 0.18 mol, Sigma-Aldrich) was added and the solution stirred at 50 °C overnight under a nitrogen atmosphere, during which a white precipitate formed. The mixture was cooled, filtered, and the precipitate washed with CHCl3 (3 × 100 mL). The precipitate was dissolved in Ac2O (350 mL), NaOAc (2 eq., 24.6 g, 0.181 mol) added, and the mixture stirred overnight at 50 °C under a nitrogen atmosphere, after which the Ac2O was removed by distillation under reduced pressure at 50 °C. The solids were redissolved into CHCl3 (300 mL), washed with H2O (2 × 200 mL), HCl (2 M, 200 mL), NaOH†† (2 M, 5 × 200 mL), H2O (200 mL), dried with Na2SO4, filtered, and the solvent removed in vacuo. The solid was washed with hexane (2 × 100 mL), Et2O (2 × 100 mL), and EtOH (5 × 100 mL, to remove significant brown byproduct) to afford a white powder (16.5 g, 52%). A sample was recrystallised from CHCl3/hexane for MS and melting point analysis. M.p. 231–233 °C. 1H NMR (600 MHz, CDCl3, 20 °C): 7.15 (d, J = 8.8 Hz, 2H), 6.79 (d, 8.8 Hz, 2H), 6.37 (m, 4H), 5.20 (m, 4H), 4.28 (s, 2H), 3.77 (s, 3H). 13C NMR (150 MHz, CDCl3): 173.86, 159.31, 138.90, 130.51, 127.28, 113.74, 81.39, 69.21, 55.30, 41.79. HRMS (ESI-TOF-MS) for C20H17NO5Na+ [M + Na]+: Calc: 374.1004. Found: 374.1000. Single crystals for X-ray analysis were grown from CH3CN by slow evaporation. CCDC 1486427 contains the supplementary crystallographic data for this paper.
:1 CH3CN/H2O (1 L) at 40 °C for 1 h. Ammonium cerium(IV) nitrate (82 g, 3.3 eq.) was added in two portions; 2.3 eq. for 90 min, followed by 1 eq. for a further 30 minutes; with reaction progress monitored by NMR. The volume of the solution was reduced by approximately two thirds in vacuo at 50 °C, or until a precipitate formed. The precipitate was filtered and put aside to later be combined with additional product. The filtrate volume was then further reduced in vacuo at 50 °C until only H2O remained. The mixture was diluted with further H2O, extracted with EtOAc (10 × 200 mL), and the EtOAc extracts combined. The volume of EtOAc was reduced in vacuo at 50 °C until a precipitate formed. The precipitate was filtered, and combined with the first crop of product. The filtrate volume was further reduced, and the precipitate collection cycle repeated until only an orange/brown oily sludge remained (p-anisaldehyde byproduct). The combined precipitate was washed with H2O (2 × 100 mL) and EtOH (10 × 100 mL) to afford a white powder (7.64 g, 73%). M.p. partial decomposition (darkens) >200 °C, violent decomposition (boiling) at 293–295 °C. Single crystals for X-ray analysis were grown from CH3CN/DMSO by slow evaporation. 1H NMR (600 MHz, DMSO-d6, 20 °C): 10.79 (s, 1H), 6.71 (s, 4H), 5.24 (s, 4H). 13C NMR (150 MHz, DMSO-d6): 175.34, 139.36, 80.39, 70.03. HRMS (ESI-TOF-MS) for C12H8NO4− [M − H]−: Calc: 230.0453. Found: 230.0457. CCDC 1486428 contains the supplementary crystallographic data for this paper.
Method A – microwave/aryl halide/Cu(0). Using a modified procedure (N-aryl imides from succinimide/phthalimide and mono-halo aryl57), Mitsudo imide 13 (100 mg, 0.19 mmol), activated elemental copper95
‡‡ (100 mg, 1.57 mmol, at least 5 eq.), and iodobenzene (500 μL, 0.44 mmol, solvent/excess) were suspended in a 10 mL microwave vessel. The microwave was operated in variable power (dynamic) mode with the following parameters: temperature 150 °C, power 300 W (automatically modulated around the set temperature), stirring high, compressed air cooling (PowerMAX) on. The sample was held at this temperature 10 minutes, and this repeated for 3–4 cycles, removing the sample in between runs to check the vessel integrity, ensure thorough mixing, and was recharged with additional iodobenzene (500 μL) as necessary if the contents had thickened to a paste or solidified. The mixture was diluted with EtOAc (100 mL), filtered to remove Cu(0), and any solids washed with CHCl3 (100 mL). The combined EtOAc/CHCl3 solution was washed with KOH (2 M, 2 × 100 mL), H2O (2 × 100 mL), dried with Na2SO4, and the solvents removed in vacuo. The residual iodobenzene was diluted with hexane to precipitate the product, which was filtered and washed with hexane to afford a white powder. 1H NMR (600 MHz, CDCl3, 20 °C): 7.53–7.48 (m, 2H), 7.48–7.44 (m, 1H), 7.20–7.16 (m, 2H), 4.87 (s, 4H), 3.80 (s, 12H), 3.28 (s, 4H). HRMS (ESI-TOF-MS) for C30H25NO12Na+ [M + Na]+: Calc. 614.1274. Found. 614.1279.
Method B – phenyl boronic acid/Cu(II)/air. Using the method developed from ref. 59 and used for Mitsudo phenyl diimide 10 (described later), the reaction was undertaken on Mitsudo imide 13 using phenyl boronic acid. The crude sample mixture contained the same 1H NMR resonances assigned to product from Method A.
The mixture was cooled, the precipitate removed by filtration, and the solids washed with MeOH (100 mL). The solid was dissolved in CH2Cl2 (100 mL), washed with H2O (100 mL), dried with Na2SO4, the solvent removed in vacuo, and washed again with MeOH (100 mL) and hexane (100 mL) to afford a white powder (150 mg, 37%). This procedure was repeated several times on a similar scale to generate additional material. A sample was recrystallised from CHCl3/hexane for MS and melting point analysis. M.p. >300 °C (partial decomposition). 1H NMR (600 MHz, CDCl3, 20 °C): 7.38 (s, 4H), 4.87 (s, 8H), 3.80 (s, 24H), 3.23 (s, 8H). 13C NMR (150 MHz, CDCl3): 171.50, 160.17, 140.11, 131.21, 127.03, 77.61, 70.37, 52.49, 44.82. HRMS (ESI-TOF-MS) for C54H44N2O24Na+ [M + Na]+: Calc: 1127.2182. Found: 1127.2166.
Side product with solvent MeOH, 15a. 1H NMR (600 MHz, CDCl3, 20 °C): 7.08 (d, 2H, J = 9.0 Hz), 6.99 (d, 2H, J = 9.0 Hz), 4.86 (s, 4H), 3.84 (s, 3H), 3.80 (s, 12H), 3.26 (s, 4H). HRMS (ESI-TOF-MS) for C31H27NO13Na+ [M + Na]+: Calc. 644.1380. Found. 644.1362.
Side product with solvent 9
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :1 THF/H2O, 15b.
1H NMR (600 MHz, CDCl3, 20 °C): 7.04 (d, 2H, J = 8.9 Hz), 6.93 (d, 2H, J = 8.9 Hz), 5.31 (s, 1H), 4.86 (s, 4H), 3.80 (s, 12H), 3.25 (s, 4H). HRMS (ESI-TOF-MS) for C30H25NO13Na+ [M + Na]+: Calc. 630.1224. Found. 630.1203.
:1 THF/H2O, 15b.
1H NMR (600 MHz, CDCl3, 20 °C): 7.04 (d, 2H, J = 8.9 Hz), 6.93 (d, 2H, J = 8.9 Hz), 5.31 (s, 1H), 4.86 (s, 4H), 3.80 (s, 12H), 3.25 (s, 4H). HRMS (ESI-TOF-MS) for C30H25NO13Na+ [M + Na]+: Calc. 630.1224. Found. 630.1203.
Acknowledgements
R. B. M. thanks Flinders University for the provision of an Australian Postgraduate Award and the Playford Memorial Trust for a PhD top-up scholarship. R. B. M. also thanks Dr Sally Duck (Monash University) for efforts and expertise in accurate mass spectrometry determination of the porphyrin compounds 2 and 7.Notes and references
- J. Rebek Jr., J. E. Trend, R. V. Wattley and S. Chakravorti, J. Am. Chem. Soc., 1979, 101, 4333–4337 CrossRef.
- J. Rebek Jr. and R. V. Wattley, J. Am. Chem. Soc., 1980, 102, 4853–4854 CrossRef.
- J. Rebek Jr. and L. Marshall, J. Am. Chem. Soc., 1983, 105, 6668–6670 CrossRef.
- Y. Kubo, Y. Murai, J.-i. Yamanaka, S. Tokita and Y. Ishimaru, Tetrahedron Lett., 1999, 40, 6019–6023 CrossRef CAS.
- Y. Kubo, T. Ohno, J.-i. Yamanaka, S. Tokita, T. Iida and Y. Ishimaru, J. Am. Chem. Soc., 2001, 123, 12700–12701 CrossRef CAS PubMed.
- M. Ayabe, A. Ikeda, S. Shinkai, S. Sakamoto and K. Yamaguchi, Chem. Commun., 2002, 1032–1033 RSC.
- C.-H. Lee, H. Yoon and W.-D. Jang, Chem. – Eur. J., 2009, 15, 9972–9976 CrossRef CAS PubMed.
- J. Leblond, H. Gao, A. Petitjean and J.-C. Leroux, J. Am. Chem. Soc., 2010, 132, 8544–8545 CrossRef CAS PubMed.
- H. Sato, K. Tashiro, H. Shinmori, A. Osuka, Y. Murata, K. Komatsu and T. Aida, J. Am. Chem. Soc., 2005, 127, 13086–13087 CrossRef CAS PubMed.
- H. Sato, K. Tashiro, H. Shinmori, A. Osuka and T. Aida, Chem. Commun., 2005, 2324–2326 RSC.
- R. B. Murphy, D.-T. Pham, S. F. Lincoln and M. R. Johnston, Eur. J. Org. Chem., 2013, 2985–2993 CrossRef.
- R. N. Warrener, D. N. Butler, W. Y. Liao, I. G. Pitt and R. A. Russell, Tetrahedron Lett., 1991, 32, 1889–1892 CrossRef CAS.
- R. N. Warrener, I. G. Pitt and D. N. Butler, J. Chem. Soc., Chem. Commun., 1983, 1340–1342 RSC.
- R. N. Warrener, A. C. Schultz, D. N. Butler, S. Wang, I. B. Mahadevan and R. A. Russel, Chem. Commun., 1997, 1023–1024 RSC.
- R. N. Warrener, S. Wang and R. A. Russell, Tetrahedron, 1997, 53, 3975–3990 CrossRef CAS.
- R. N. Warrener, D. N. Butler, D. Margetic, F. M. Pfeffer and R. A. Russell, Tetrahedron Lett., 2000, 41, 4671–4675 CrossRef CAS.
- A. M. Napper, I. Read, N. J. Head, A. M. Oliver and M. N. Paddon-Row, J. Am. Chem. Soc., 2000, 122, 5220–5221 CrossRef CAS.
- N. J. Head, A. M. Oliver, K. Look, N. R. Lokan, G. A. Jones and M. N. Paddon-Row, Angew. Chem., Int. Ed., 1999, 38, 3219–3222 CrossRef CAS.
- M. J. Shephard and M. N. Paddon-Row, J. Phys. Chem. A, 2000, 104, 11628–11635 CrossRef CAS.
- M. J. Shephard and M. N. Paddon-Row, J. Phys. Chem. A, 1999, 103, 3347–3350 CrossRef CAS.
- M. R. Johnston and D. M. Lyons, Supramol. Chem., 2005, 17, 503–511 CrossRef CAS.
- M. R. Johnston, M. J. Latter and R. N. Warrener, Org. Lett., 2002, 4, 2165–2168 CrossRef CAS PubMed.
- M. D. Johnstone, E. K. Schwarze, J. Ahrens, D. Schwarzer, J. J. Holstein, B. Dittrich, F. M. Pfeffer and G. H. Clever, Chem. – Eur. J., 2016, 22, 10791–10795 CrossRef CAS PubMed.
- M. D. Johnstone, M. Frank, G. H. Clever and F. M. Pfeffer, Eur. J. Org. Chem., 2013, 5848–5853 CrossRef CAS.
- M. Shang, R. N. Warrener, D. N. Butler, Y. Murata and D. Margetić, Mol. Diversity, 2011, 15, 541–560 CrossRef CAS PubMed.
- P. Trošelj, A. Briš, Y. Murata and D. Margetić, Struct. Chem., 2012, 23, 791–799 CrossRef.
- P. Trošelj, I. Đilović, D. Matković-Čalogović and D. Margetić, J. Heterocycl. Chem., 2013, 50, 83–90 CrossRef.
- M. Golić, M. R. Johnston, D. Margetić, A. C. Schultz and R. N. Warrener, Aust. J. Chem., 2006, 59, 899–914 CrossRef.
- D. Margetic, M. R. Johnston, E. R. T. Tiekink and R. N. Warrener, Tetrahedron Lett., 1998, 39, 5277–5280 CrossRef CAS.
- M. Johnston, Molecules, 2001, 6, 406–416 CrossRef CAS.
- D. Margetic, R. N. Warrener, D. N. Butler and D. Officer, Theor. Chem. Acc., 2007, 117, 239–245 CrossRef CAS.
- J. M. Lawson, A. M. Oliver, D. F. Rothenfluh, Y.-Z. An, G. A. Ellis, M. G. Ranasinghe, S. I. Khan, A. G. Franz, P. S. Ganapathi, M. J. Shephard, M. N. Paddon-Row and Y. Rubin, J. Org. Chem., 1996, 61, 5032–5054 CrossRef CAS.
- T. D. M. Bell, K. A. Jolliffe, K. P. Ghiggino, A. M. Oliver, M. J. Shephard, S. J. Langford and M. N. Paddon-Row, J. Am. Chem. Soc., 2000, 122, 10661–10666 CrossRef CAS.
- R. N. Warrener, D. N. Butler, L. Liu, D. Margetic and R. A. Russell, Chem. – Eur. J., 2001, 7, 3406–3414 CrossRef CAS.
- M. J. Gunter, H. Tang and R. N. Warrener, J. Porphyrins Phthalocyanines, 2002, 6, 673–684 CrossRef CAS.
- M. R. Johnston, M. J. Latter and R. N. Warrener, Aust. J. Chem., 2001, 54, 633–636 CrossRef CAS.
- H. Tang, Z. Dong, Z. Merican, D. Margetić, Ž. Marinić, M. J. Gunter, D. Officer, D. N. Butler and R. N. Warrener, Tetrahedron Lett., 2009, 50, 667–670 CrossRef CAS.
- S. P. Gaynor, M. J. Gunter, M. R. Johnston and R. N. Warrener, Org. Biomol. Chem., 2006, 4, 2253–2266 CAS.
- G. Ercolani and L. Schiaffino, Angew. Chem., Int. Ed., 2011, 50, 1762–1768 CrossRef CAS PubMed.
- T.-a. Mitsudo, K. Kokuryo, T. Shinsugi, Y. Nakagawa, Y. Watanabe and Y. Takegami, J. Org. Chem., 1979, 44, 4492–4496 CrossRef CAS.
- T.-a. Mitsudo, H. Naruse, T. Kondo, Y. Ozaki and Y. Watanabe, Angew. Chem., Int. Ed. Engl., 1994, 33, 580–581 CrossRef.
- G. Dennison, unpublished work, 2004.
- R. C. Foitzik, A. Lowe and F. M. Pfeffer, Tetrahedron Lett., 2009, 50, 2583–2584 CrossRef CAS.
- S. Gaynor, PhD thesis, University of New England, Armidale, New South Wales, 2003.
- T.-C. Chou, K.-C. Lin and C.-A. Wu, Tetrahedron, 2009, 65, 10243–10257 CrossRef CAS.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1931, 490, 243–257 CrossRef.
- J. Kallos and P. Deslongchamps, Can. J. Chem., 1966, 44, 1239–1245 CrossRef CAS.
- G. Maier and W. A. Jung, Chem. Ber., 1982, 115, 804–807 CrossRef CAS.
- D. Margetic, R. N. Warrener, G. Sun and D. N. Butler, Tetrahedron, 2007, 63, 4338–4346 CrossRef CAS.
- D. Mink and G. Deslongchamps, Tetrahedron Lett., 1996, 37, 7035–7038 CrossRef CAS.
- M. Yamaura, T. Suzuki, H. Hashimoto, J. Yoshimura and T. Okamoto, Bull. Chem. Soc. Jpn., 1985, 58, 1413–1420 CrossRef CAS.
- J. Yoshimura, M. Yamaura, T. Suzuki and H. Hashimoto, Chem. Lett., 1983, 1001–1002 CrossRef CAS.
- D. G. Lonergan, PhD, The University of New Brunswick, 1998.
- L. Barriault and P. A. Evans, Personal Communication while visiting Flinders University, 2011.
- R. B. Murphy, PhD thesis, Flinders University, Adelaide, South Australia, 2016.
- R. N. Warrener, D. Margetic, A. S. Amarasekara, D. N. Butler, I. B. Mahadevan and R. A. Russell, Org. Lett., 1999, 1, 199–202 CrossRef CAS.
- L. D. S. Yadav, B. S. Yadav and V. K. Rai, Synthesis, 2006, 1868–1872 CrossRef CAS.
- J.-B. Lan, G.-L. Zhang, X.-Q. Yu, J.-S. You, L. Chen, M. Yan and R.-G. Xie, Synlett, 2004, 1095–1097 CAS.
- J.-B. Lan, L. Chen, X.-Q. Yu, J.-S. You and R.-G. Xie, Chem. Commun., 2004, 188–189 RSC.
- C. W. Miller, C. E. Hoyle, E. J. Valente, D. H. Magers and E. S. Jönsson, J. Phys. Chem. A, 1999, 103, 6406–6412 CrossRef CAS.
- C. W. Miller, E. S. Jönsson, C. E. Hoyle, K. Viswanathan and E. J. Valente, J. Phys. Chem. B, 2001, 105, 2707–2717 CrossRef CAS.
- C. Miller, C. Hoyle, E. Valente, J. Zubkowski and E. S. Jönsson, J. Chem. Crystallogr., 2000, 30, 563–571 CrossRef CAS.
- B. A. Langowski, R. Rothchild and A.-M. Sapse, Spectrosc. Lett., 2001, 34, 235–251 CrossRef CAS.
- K. Kondo, H. Fujita, T. Suzuki and Y. Murakami, Tetrahedron Lett., 1999, 40, 5577–5580 CrossRef CAS.
- M. Mao, J. England and S. R. Turner, Polymer, 2011, 52, 4498–4502 CrossRef CAS.
- Y. Yu, L. Shi, D. Yang and L. Gan, Chem. Sci., 2013, 4, 814–818 RSC.
- S. Verma and N. Singh, Aust. J. Chem., 1976, 29, 295–300 CrossRef CAS.
- D. P. Curran, S. Geib and N. DeMello, Tetrahedron, 1999, 55, 5681–5704 CrossRef CAS.
- K. Tanaka, M. Okano, H. Toshino, H. Kita and K.-I. Okamoto, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 907–914 CrossRef CAS.
- Y. Zhang, J. M. Lavin and K. D. Shimizu, J. Am. Chem. Soc., 2009, 131, 12062–12063 CrossRef CAS PubMed.
- D.-S. Choi, Y. S. Chong, D. Whitehead and K. D. Shimizu, Org. Lett., 2001, 3, 3757–3760 CrossRef CAS PubMed.
- Y. Chen, M. D. Smith and K. D. Shimizu, Tetrahedron Lett., 2001, 42, 7185–7187 CrossRef CAS.
- R. D. Rasberry, X. Wu, B. N. Bullock, M. D. Smith and K. D. Shimizu, Org. Lett., 2009, 11, 2599–2602 CrossRef CAS PubMed.
- C. F. Degenhardt, J. M. Lavin, M. D. Smith and K. D. Shimizu, Org. Lett., 2005, 7, 4079–4081 CrossRef CAS PubMed.
- J. M. Lavin and K. D. Shimizu, Chem. Commun., 2007, 228–230 RSC.
- W. R. Carroll, P. Pellechia and K. D. Shimizu, Org. Lett., 2008, 10, 3547–3550 CrossRef CAS PubMed.
- D. P. Curran and N. C. DeMello, J. Chem. Soc., Chem. Commun., 1993, 1314–1317 RSC.
- A. M. Napper, N. J. Head, A. M. Oliver, M. J. Shephard, M. N. Paddon-Row, I. Read and D. H. Waldeck, J. Am. Chem. Soc., 2002, 124, 10171–10181 CrossRef CAS PubMed.
- Spartan ‘10 for Windows, Wavefunction, Inc., 18401 Von Karman Avenue, Suite 370, Irvine, CA 92612, USA, http://www.wavefun.com/index.html.
- S. S. Eaton and G. R. Eaton, J. Chem. Soc., Chem. Commun., 1974, 576–577 RSC.
- R. W. Wagner, T. E. Johnson and J. S. Lindsey, J. Am. Chem. Soc., 1996, 118, 11166–11180 CrossRef CAS.
- S. S. Eaton and G. R. Eaton, J. Am. Chem. Soc., 1975, 97, 3660–3666 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- mMass 5.4.1, Martin Strohalm, http://www.mmass.org/, 2012.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- D. D. Perrin, L. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, Ltd, Oxford, 1966 Search PubMed.
- M. R. Johnston , personal communication.
- E. H. Huntress, T. E. Lesslie and J. Bornstein, Org. Syn. Coll., 1952, 32, 55 CrossRef CAS.
- E. H. Huntress, T. E. Lesslie and J. Bornstein, Org. Syn. Coll., 1963, 4, 329 Search PubMed.
- S. D. Robinson, Inorg. Synth., 1974, 15, 48–50 Search PubMed.
- J. G. Hill, B. E. Rossiter and K. B. Sharpless, J. Org. Chem., 1983, 48, 3607–3608 CrossRef CAS.
- P. H. Gore and G. K. Hughes, J. Chem. Soc., 1959, 1615–1616 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, X-ray crystallographic data, and UV-Vis and HRMS for porphyrin compounds. CCDC 1486427–1486430. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01588c |
| ‡ Ref. 77 refers to anilide derivatives rather than imides, however, the principle of balancing steric and electronic factors is the same here. |
| § Refer to the molecular modelling paragraph in the Experimental section for computational information regarding this calculation. |
| ¶ This simplified model of the linker does not account for possible additional steric crowding on the underside of the polycyclic arms when additional polycycles are fused to the linker, or the possible electronic effect on rotation due to the second para-imide (as in compound 10). The potential stabilising π–π interactions between the opposing bis-porphyrins are also not accounted for by model compound 17. |
| || R. Dennington, T. Keith and J. Millam, GaussView, Version 5.0.9, Semichem Inc., Shawnee Mission, KS, 2009. |
| ** An excess of warm DCC was added as a liquid. |
| †† Caution: NaOH reacts exothermically with residual Ac2O, temperature should remain below 50 °C for optimum product stability. |
| ‡‡ Prior to precipitation of copper, the zinc dust was pre-treated with 2% hydrochloric acid to ensure it met the oxide-free requirement. Freshly precipitated elemental copper was dried in vacuo, and not at 100 °C. The elemental copper appeared as more of a brown powder (not metallic/lustrous in appearance). |
| This journal is © The Royal Society of Chemistry 2016 |