
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of odoamide, a novel cyclic depsipeptide, from an Okinawan marine cyanobacterium†
Masato
Kaneda
a,
Kosuke
Sueyoshi
b,
Toshiaki
Teruya
b,
Hiroaki
Ohno
a,
Nobutaka
Fujii
a and
Shinya
Oishi
 *a
*a
aGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: soishi@pharm.kyoto-u.ac.jp; Fax: +81-75-753-4570; Tel: +81-75-753-4561
bFaculty of Education, University of the Ryukyus, Nishihara, Okinawa 903-0213, Japan
First published on 1st September 2016
Abstract
Odoamide is a novel cyclic depsipeptide with highly potent cytotoxic activity isolated from the Okinawan marine cyanobacterium Okeania sp. It contains a 26-membered macrocycle composed of a fatty acid moiety, a peptide segment and isoleucic acid. Four possible stereoisomers of the odoamide polyketide substructure were synthesised using a chiral pool approach. The first total synthesis of odoamide was also successfully achieved. The structure of synthetic odoamide was verified by comparing its NMR spectra with those of the natural product.
Introduction
Many peptide secondary metabolites derived from natural resources show attractive biological activities.1 Because of their favourable drug-like properties including good membrane permeability and biostability,2 a number of synthetic and medicinal chemistry studies of macrocyclic peptides and highly N-methylated peptides have been carried out.3 Among them, aurilide-class cyclic depsipeptides exhibit highly potent antiproliferative activity against cancer cell lines (Fig. 1). The first 26-membered cyclic depsipeptide, aurilide (1a), was isolated from the sea hare Dolabella auricularia.4 The related depsipeptides, aurilide B (1b) and C (1c), from Lyngbya majuscula also show potent cytotoxicity.5 Kulokekahilide-2 (2) is a similar cytotoxic depsipeptide from a marine mollusk, Philinopsis speciosa, which has two conformers of the 26-membered macrocycle in dichloromethane.6 Lagunamide A (3a) and B (3b) from Lyngbya majuscula exhibit antimalarial activity against Plasmodium falciparum at submicromolar concentrations.7 Lagunamide C (3c)8 and palau'amide (4)9 exhibit cytotoxicity at nanomolar concentrations comparable to other aurilide-class depsipeptides, although these peptides have unique 27-membered and 24-membered macrocycles, respectively. The configurations of the component amino acids of the depsipeptides were investigated by chiral HPLC, chiral GC-MS, and Marfey's analyses,10 while the stereoselective synthesis and the NMR analysis facilitated the determination of the absolute stereochemistries of the fatty acid substructure. In some cases, the structure was verified or revised through synthetic studies of natural products and their stereoisomers.11 | ||
| Fig. 1 Structures of aurilide-class depsipeptides. | ||
Odoamide (5) is a novel cyclic depsipeptide from the Okinawan marine cyanobacterium Okeania sp. (Fig. 2A), which shows highly potent cytotoxic activity against HeLa S3 cell lines.12 The overall structure of the 26-membered macrocycle is similar to those of aurilide-class depsipeptides, and comprises three substructures: a fatty acid moiety, a peptide segment (Ala-D-MePhe-Sar-Ile-MeAla) and isoleucic acid. At the initial stage of this study, the absolute configurations of the constituent amino acids and isoleucic acid in 5 were determined by chiral HPLC analysis and Marfey's analysis. The absolute configuration of the 5-hydroxy group of the polyketide part was determined by Mosher's method,13 while the remaining configurations of the polyketide were ambiguous. In this study, we carried out a synthetic study of odoamide to verify its structure and complete stereochemistry.
 | ||
| Fig. 2 Structures of odoamide (5) (A) and the polyketide substructures in 5 (B). | ||
The synthetic strategy is illustrated in Scheme 1. During the cyclisation of the linear peptide, epimerisation and dimer formation are often problematic.3e,f,14 To avoid the less reactive process of N-methylated amide (CO–NMe) or ester bond formation compared with standard peptide bond (CO–NH) formation, we chose macrocyclisation of the Ala and D-allo-isoleucic acid residues of the linear precursor 6 for odoamide (5).11d Peptide 6 could be prepared by coupling of alcohol 7, MeAla 8 and tetrapeptide 9, which could be obtained by standard solid-phase peptide synthesis. Alcohol 7 could be synthesised by coupling of D-allo-isoleucic acid ester 1115 with carboxylic acid 10.
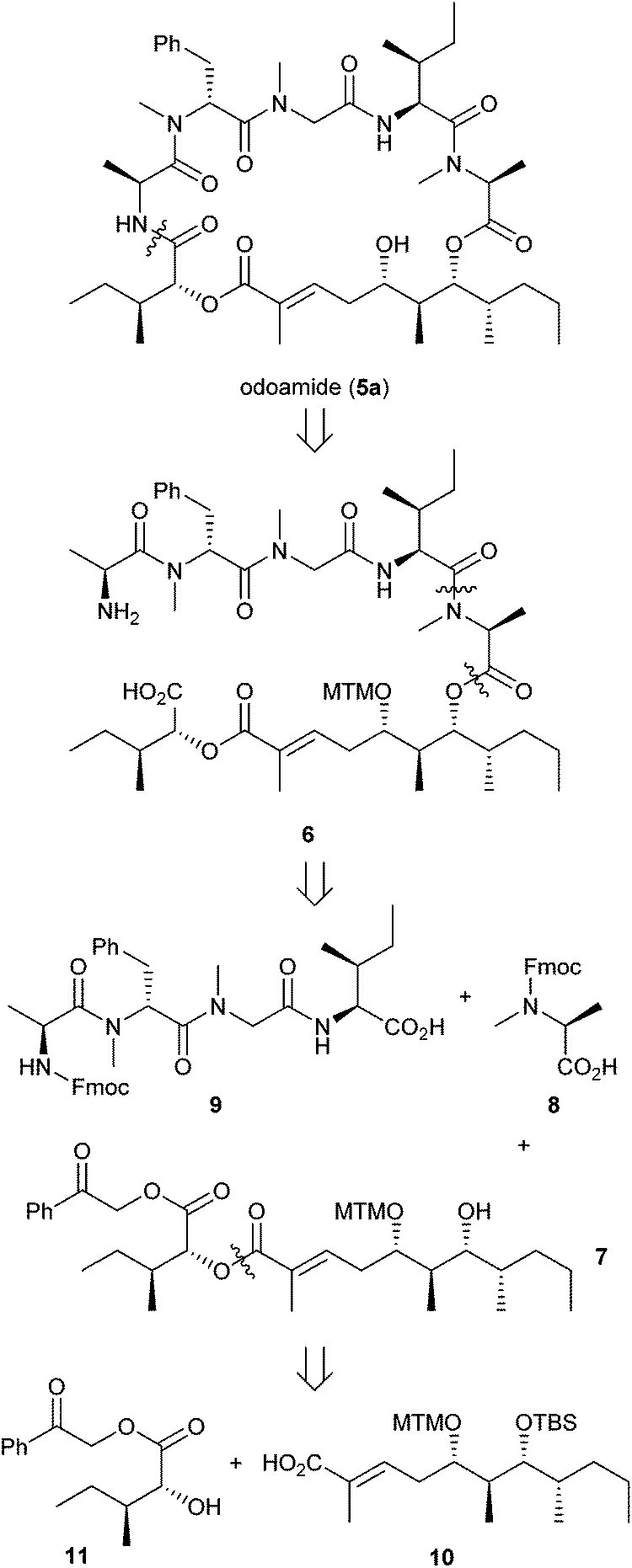 | ||
| Scheme 1 Retrosynthetic analysis of odoamide (5). | ||
Results and discussion
Synthesis of the polyketide substructure of odoamide
The stereochemistries of the polyketide part were unknown when we started this study. Therefore, it was necessary to synthesise all the possible polyketide substructures of odoamide 5. The polyketide substructure in lagunamide A (3a), a closely related structural analogue of 5, has 5S,7R-dihydroxy and 6S,8S-dimethyl groups. Additionally, aurilide-class depsipeptides 1a–c, 2, and 3a,b possess the syn-1,3-diol moiety with a 5S-hydroxy configuration. On the basis of the structures of these related molecules, we expected that the plausible stereochemical configuration of the natural odoamide 5 was 5S,6S,7R,8S. Among these four stereocentres, the configuration at the C8-methyl group was ambiguous because attempts to determine it based on derivatisation and NMR analysis of odoamide (5) were unsuccessful. It was also desirable to confirm the stereochemistry of the C6-methyl group. Therefore, we designed four possible methyl esters 12a–d as polyketide substructure substrates (Fig. 2B).Methyl esters 12a,b were synthesised from (S)-Roche ester 13 according to a similar process described in previous reports by us and others (see the ESI†).11a,12 Preparation of (5S,6R,7R,8S)-ester 12c and (5S,6R,7R,8R)-ester 12d started from the commercially available (R)-Roche ester ent-13 in a similar manner (Scheme 2). (R)-Roche ester ent-13 was converted to alcohol ent-15via benzyl protection16 and LiAlH4-mediated reduction. After Swern oxidation, an n-Bu2BOTf-mediated Evans aldol reaction17 of the resulting aldehyde provided the syn-aldol products 16c and 17d. The requisite stereochemistries at the C8 chiral centre in 12c and 12d were generated at this step by using propionyl- and pentanoyl-oxazolidinones, respectively. TBS protection of the secondary alcohol in 16c and 17d followed by removal of the chiral auxiliary with LiBH4 gave alcohols 20c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 and 21d. Swern oxidation of 20c and the subsequent Wittig reaction of the resulting aldehyde with ethylidene–triphenylphosphorane provided olefin 22c as an E/Z isomeric mixture. Hydrogenation of 22c in the presence of Pd/C afforded the key alcohol 23c with a threo/threo-configuration. Separately, tosylation of 21d followed by LiAlH4-mediated reduction afforded benzyl ether 24d, which was converted to the corresponding alcohol 23d (with a threo/erythro-configuration) by hydrogenation. Swern oxidation of 23c followed by a Mukaiyama aldol reaction19 with 1-methoxy-2-methyl-1-trimethylsiloxy-1,3-butadiene (25)20 produced methyl ester 12c with a (5S)-hydroxy group (dr >99:1).21 Ester 12d was obtained from 23d by using the identical protocol.
18 and 21d. Swern oxidation of 20c and the subsequent Wittig reaction of the resulting aldehyde with ethylidene–triphenylphosphorane provided olefin 22c as an E/Z isomeric mixture. Hydrogenation of 22c in the presence of Pd/C afforded the key alcohol 23c with a threo/threo-configuration. Separately, tosylation of 21d followed by LiAlH4-mediated reduction afforded benzyl ether 24d, which was converted to the corresponding alcohol 23d (with a threo/erythro-configuration) by hydrogenation. Swern oxidation of 23c followed by a Mukaiyama aldol reaction19 with 1-methoxy-2-methyl-1-trimethylsiloxy-1,3-butadiene (25)20 produced methyl ester 12c with a (5S)-hydroxy group (dr >99:1).21 Ester 12d was obtained from 23d by using the identical protocol.
 | ||
| Scheme 2 Synthesis of esters 12c,d. Reagents and conditions: (a) benzyl 2,2,2-trichloroacetimidate, TfOH, CH2Cl2, cyclohexane, 0 °C to rt, 85%; (b) LiAlH4, THF, 0 °C, 81%; (c) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (d) (R)-4-benzyl-3-propionyl-2-oxazolidinone, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 80% (2 steps); (e) (R)-4-benzyl-3-pentanoyl-2-oxazolidinone, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 72% (2 steps); (f) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C to rt, 89% (18c) and 87% (19d); (g) LiBH4, MeOH, THF, 0 °C to rt, 69% (20c) and 80% (21d); (h) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (i) ethyltriphenylphosphonium bromide, n-BuLi, THF, rt, 69% (2 steps, Z/E = 7:1); (j) TsCl, Et3N, Me3N·HCl, CH2Cl2, rt; (k) LiAlH4, THF, 0 °C to rt, 70% (2 steps) (l) Pd/C, H2, EtOH, rt, 92% (23c) and 85% (23d); (m) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (n) 25, BF3·OEt2, CH2Cl2, Et2O, −78 °C, 54% (12c) and 69% (12d) (2 steps). | ||
The stereochemistry of the 5-hydroxy group in alcohol 12a was confirmed by the NMR analysis of the corresponding acetonide (Scheme 3). TBS deprotection of 26a22 and 12a provided 1,3-diols 27a and 28a, which were treated with 2,2-dimethoxypropane in the presence of PPTS to give acetonides 29a and 30a, respectively. It is known that 13C NMR chemical shifts of the ketal methyl groups in syn- and anti-1,3-diol acetonides are different.23 A syn-acetonide shows different chemical shifts for the two ketal methyl groups (e.g., 19.5 and 30.0 ppm for 30a) because of its predominant chair conformation. In contrast, an anti-acetonide shows close chemical shifts (e.g., 23.5 and 25.2 ppm for 29a), because the anti-isomer exists in a twist-boat conformation to avoid the 1,3-diaxial interaction that would be present in the chair conformation. Accordingly, it was demonstrated that 1,3-diol 28a, the precursor of acetonide 30a has the desired 1,3-syn configuration. Of note, esters 12a–d were employed as the key substrates for the stereochemical assignment of the polyketide substructure in 5 in our previous research.12 Manipulations of esters 12a–d including DIBAL-mediated reductive transformation provided the corresponding triol derivatives. The comparative NMR analysis between the natural product-derived triol and synthetic triols demonstrated that the polyketide substructure had the 5S,6S,7R,8S configuration (see the ESI†).12
 | ||
| Scheme 3 Stereochemical assignment of 1,3-diols. Reagents and conditions: (a) TBAF, THF, rt, 81% (27a) and 81% (28a); (b) 2,2-dimethoxypropane, PPTS, CH2Cl2, rt, 86% (29a) and 90% (30a). | ||
Synthesis of odoamide and its biological evaluation
After the determination of the stereochemistry of the polyketide part,12 we attempted the total synthesis of odoamide using (5S,6S,7R,8S)-ester 12a. Synthesis of odoamide (5) began with methylthiomethyl (MTM) protection of the secondary hydroxy group in 12a to give thioacetal 31 (Scheme 4).11a,24 Hydrolysis of 31 with LiOH followed by coupling with D-allo-isoleucic acid phenacyl ester (11) using 2-methyl-6-nitrobenzoic anhydride (MNBA)25 and DMAP afforded ester 32. The TBS group in 32 was deprotected with HF·pyridine to produce the corresponding alcohol 7. In the coupling of Fmoc-MeAla-OH 8 with 7 using DCC and DMAP, significant epimerisation occurred. The coupling using Fmoc-MeAla-Cl26 with 7 in the presence of DIPEA followed by Fmoc deprotection with Et2NH gave amine 33 in 54% yield (two steps) without epimerisation. Tetrapeptide 9 was conjugated with 33 using EDCI–HOAt to afford 34 as a 1.4:1 epimeric mixture at the α-position of Ile.27 After removal of the phenacyl (with Zn and AcOH) and Fmoc groups (with Et2NH), the epimer mixture of the linear peptides was separated into the desired compound 6a (major, L-Ile) and undesired compound 6b (minor, D-allo-Ile) by HPLC purification. Cyclisation of 6a and 6b with HATU followed by deprotection of the MTM group with AgNO3 and 2,6-lutidine gave the desired odoamide 5a and its diastereomer 5b. Both cyclisations of 6a and 6b proceeded smoothly within five hours without epimerisation. The configurations of L-Ile and D-allo-Ile in peptides 5a and 5b, respectively, were determined by Marfey's analysis and 1H NMR analysis after acid hydrolysis.
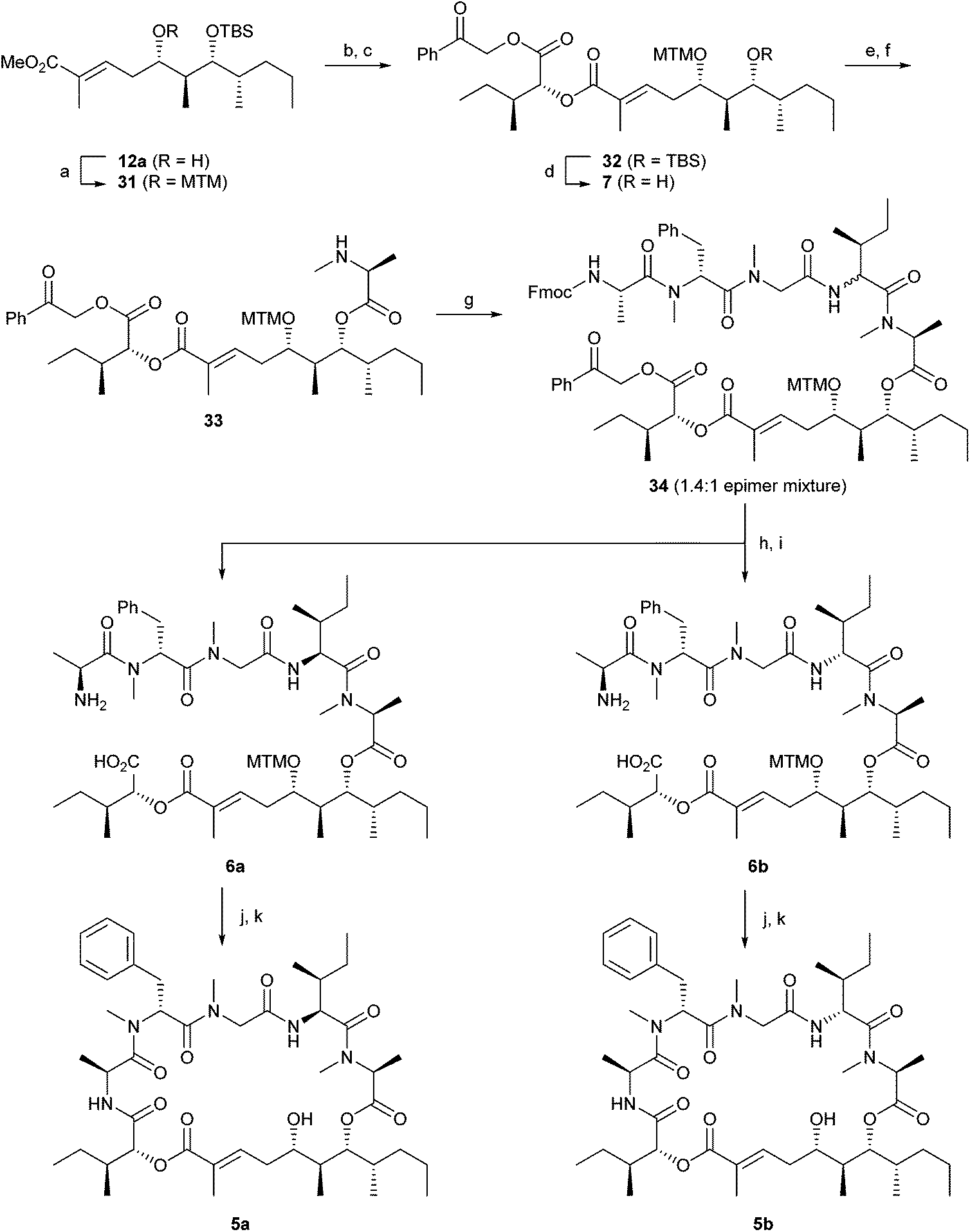 | ||
| Scheme 4 Synthesis of odoamide (5a) and its epimer 5b. Reagents and conditions: (a) Ac2O, DMSO, AcOH, rt, 59%; (b) LiOH, THF, MeOH, H2O, 0 °C to 30 °C; (c) 11, MNBA, DMAP, CH2Cl2, rt, 87% (2 steps); (d) HF·pyridine, THF, pyridine, 0 °C to rt, 74%; (e) Fmoc-MeAla-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (f) Et2NH, MeCN, 0 °C to rt, 54% (2 steps); (g) 9, EDCI·HCl, HOAt, CH2Cl2, 0 °C to rt; (h) Zn, CH3COOH, H2O, EtOAc, rt; (i) Et2NH, MeCN, 0 °C to rt, 30% (6a) and 24% (6b) (3 steps); (j) HATU, HOAt, collidine, DMF, rt; (k) AgNO3, 2,6-lutidine, THF, H2O, rt to 70 °C, 85% (5a) and 62% (5b) (2 steps). | ||
We analysed the 1H NMR and 13C NMR spectra of the natural and synthetic products (Fig. 3 and 4). The NMR spectra of the synthetic odoamide 5a were identical to those of the natural product 5, suggesting that the chemical structure of odoamide was the same as 5a. The cytotoxicity of synthetic odoamides 5a and 5b against A549 cells was also evaluated by the MTS assay. Peptide 5a showed highly potent cytotoxicity (IC50 = 2.1 nM), corroborating our correct structural assignment of odoamide (5). However, the epimer peptide 5b showed significantly less potent antiproliferative activity (IC50 = 0.54 μM), suggesting that the L-Ile configuration is crucial for the cytotoxic activity of odoamide.
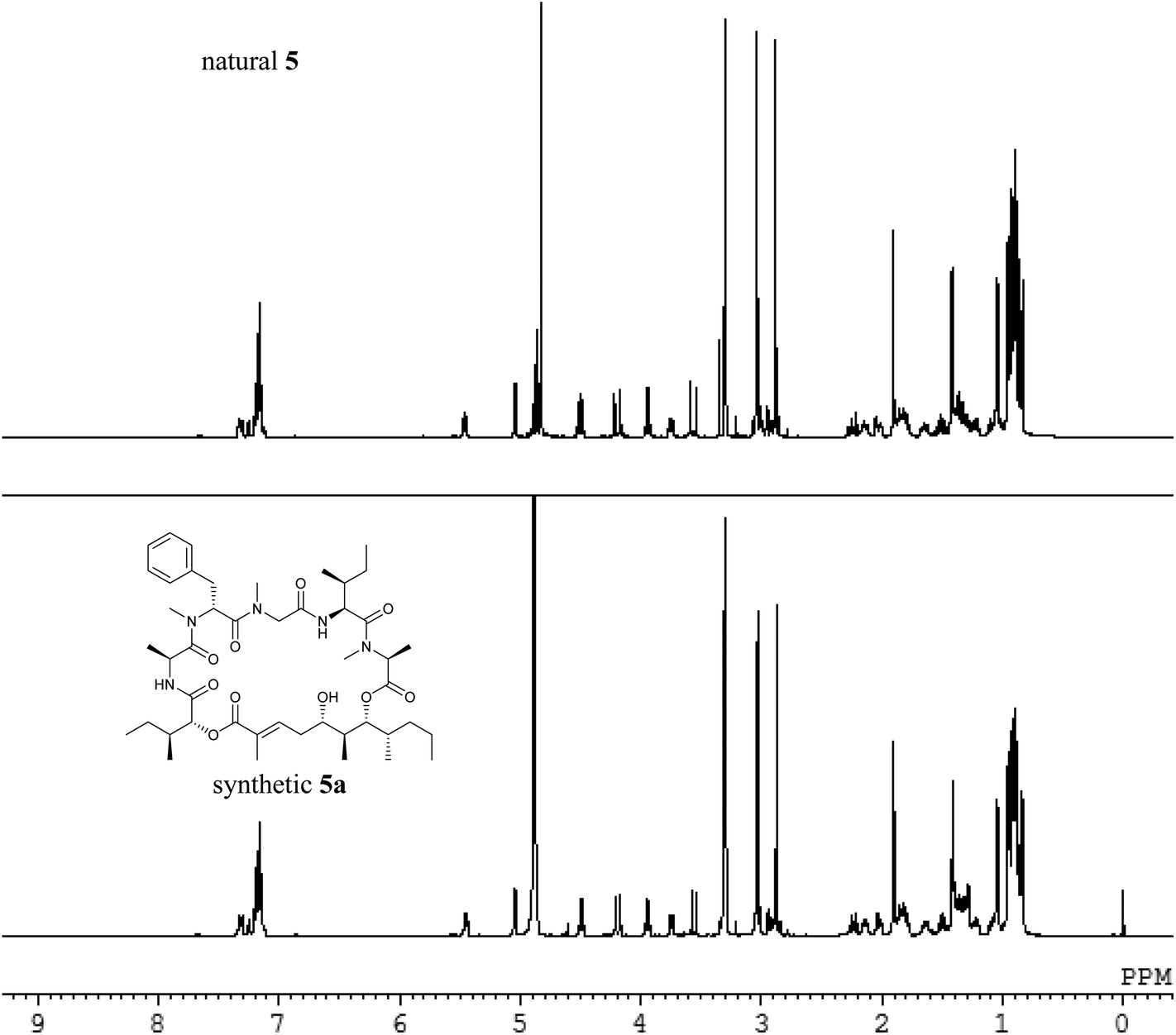 | ||
| Fig. 3 Comparison of the 1H NMR spectra between the natural compound 5 and the synthetic compound 5a (in CD3OD). | ||
 | ||
| Fig. 4 Comparison of the 13C NMR spectra between the natural compound 5 and the synthetic compound 5a (in CD3OD). | ||
Conclusions
In this study, the total synthesis of odoamide was completed via the synthesis of four possible polyketide substructures 12a–d. The NMR spectra of the synthetic peptide 5a were identical to those of the natural odoamide 5. Accordingly, the full structural assignment and first total synthesis of odoamide were achieved.Experimental section
Synthetic general method
NMR spectra were recorded using a JEOL ECA-500 spectrometer. Chemical shifts are reported in δ (ppm) relative to Me4Si (in CDCl3) as an internal standard. 13C NMR spectra were referenced to the residual solvent signal. Melting points were measured by a hot stage melting point apparatus (uncorrected). Exact mass (HRMS) spectra were recorded on a Shimadzu LC-ESI-IT-TOF-MS instrument. IR spectra were recorded on a JASCO FT/IR-4100 spectrometer. Optical rotations were measured with a JASCO P-1020 polarimeter. For flash chromatography, Wakogel C-300E (Wako) was employed. For analytical HPLC, a Cosmosil 5C18-ARII column (4.6 × 250 mm, Nacalai Tesque, Inc.) was employed with a linear gradient of CH3CN (with 0.1% (v/v) TFA, except for the analysis of final products 5a,b using solvents without TFA) in H2O at a flow rate of 1 cm3 min−1, and eluting products were detected by UV at 220 nm. Preparative HPLC was performed using a Cosmosil 5C18-ARII preparative column (20 × 250 mm, Nacalai Tesque, Inc.) at a flow rate of 8 cm3 min−1. The purity of peptides 5a,b was determined by HPLC analysis (>95%). The synthetic procedures for esters 12a,b were described in our previous report.12:1 to 10:1) to give compound ent-14 (14.8 g, 85%) as a colorless oil. The spectral data were in good agreement with those previously reported.28
:1 to 3:1) to give compound ent-15 (10.3 g, 81%) as a colorless oil. The spectral data were in good agreement with those previously reported.28
To a stirred solution of (R)-4-benzyl-3-propionyloxazolidin-2-one (429.2 mg, 1.84 mmol) in CH2Cl2 (9.2 cm3) under argon were added n-Bu2BOTf (1.0 mol dm−3 in CH2Cl2; 2.0 cm3, 2.00 mmol) and i-Pr2NEt (0.38 cm3, 2.17 mmol) at −78 °C. After stirring for 1 h, the reaction mixture was warmed to 0 °C and stirred for 30 min. To this solution was added the above aldehyde in CH2Cl2 (3.9 cm3) at −78 °C. After stirring for 1 h, the mixture was warmed to −10 °C and stirred for 1 h. The mixture was quenched with pH 7.0 phosphate buffer solution (1.8 cm3) and 30% H2O2 in MeOH (1:2, 4.2 cm3) and stirred overnight at room temperature. The whole mixture was concentrated under reduced pressure and extracted with CH2Cl2. The extract was washed with saturated aqueous NaHCO3, and dried over MgSO4. The filtrate was concentrated under reduced pressure and the residue was purified by flash chromatography over silica gel with hexane–EtOAc (5:1 to 3:1) to give compound 16c (610.7 mg, 80%, dr >15:1) as a colorless oil. The minor isomer was removed by column chromatography: [α]29D −43.2 (c 0.72, CHCl3); IR (neat) νmax/cm−1: 3504 (OH), 1779 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (500 MHz, CDCl3) δ: 1.05 (3H, d, J 6.9), 1.33 (3H, d, J 6.3), 1.87–1.93 (1H, m), 2.77 (1H, dd, J1 13.2, J2 9.7), 3.00 (1H, d, J 2.9), 3.25 (1H, dd, J1 13.2, J2 3.2), 3.46–3.52 (2H, m), 3.96–4.03 (2H, m), 4.16–4.21 (2H, m), 4.51 (2H, s), 4.65–4.69 (1H, m), 7.20–7.21 (2H, m), 7.26–7.36 (8H, m); 13C NMR (125 MHz, CDCl3) δ: 12.4, 12.8, 36.2, 37.7, 40.5, 55.1, 66.0, 73.3, 73.9, 74.1, 127.4 (2C), 127.5 (2C), 128.3 (2C), 128.9 (2C), 129.4 (2C), 135.0, 138.1, 152.7, 177.0; HRMS (ESI) calcd for C24H29NNaO5 (MNa+): 434.1938; found: 434.1938.
O); 1H NMR (500 MHz, CDCl3) δ: 1.05 (3H, d, J 6.9), 1.33 (3H, d, J 6.3), 1.87–1.93 (1H, m), 2.77 (1H, dd, J1 13.2, J2 9.7), 3.00 (1H, d, J 2.9), 3.25 (1H, dd, J1 13.2, J2 3.2), 3.46–3.52 (2H, m), 3.96–4.03 (2H, m), 4.16–4.21 (2H, m), 4.51 (2H, s), 4.65–4.69 (1H, m), 7.20–7.21 (2H, m), 7.26–7.36 (8H, m); 13C NMR (125 MHz, CDCl3) δ: 12.4, 12.8, 36.2, 37.7, 40.5, 55.1, 66.0, 73.3, 73.9, 74.1, 127.4 (2C), 127.5 (2C), 128.3 (2C), 128.9 (2C), 129.4 (2C), 135.0, 138.1, 152.7, 177.0; HRMS (ESI) calcd for C24H29NNaO5 (MNa+): 434.1938; found: 434.1938.
:1) to give compound 18c (16.0 g, 89%) as a colorless oil: [α]27D −38.2 (c 1.21, CHCl3); IR (neat) νmax/cm−1: 1780 (CO); 1H NMR (500 MHz, CDCl3) δ: 0.04 (3H, s), 0.06 (3H, s), 0.90 (9H, s), 0.92 (3H, d, J 7.4), 1.25 (3H, d, J 6.9), 1.88–1.93 (1H, m), 2.75 (1H, dd, J1 13.2, J2 9.7), 3.24 (1H, dd, J1 13.2, J2 3.2), 3.28 (1H, dd, J1 8.9, J2 7.2), 3.49 (1H, dd, J1 8.9, J2 6.6), 3.96–4.02 (1H, m), 4.08–4.16 (3H, m), 4.46–4.52 (2H, m), 4.59–4.64 (1H, m), 7.20–7.34 (10H, m); 13C NMR (125 MHz, CDCl3) δ: −4.1, −3.9, 11.9, 15.0, 18.4, 26.1 (3C), 37.6, 38.8, 41.9, 55.4, 65.9, 72.8, 73.0, 73.4, 127.3, 127.4, 127.6 (2C), 128.3 (2C), 128.9 (2C), 129.4 (2C), 135.3, 138.6, 152.8, 175.9; HRMS (ESI) calcd for C30H43NNaO5Si (MNa+): 548.2803; found: 548.2808.
:1) to give compound 20c (10.3 g, 69%) as a colorless oil: [α]29D −0.41 (c 1.23, CHCl3); IR (neat) νmax/cm−1: 3422 (OH); 1H NMR (500 MHz, CDCl3) δ: 0.03 (3H, s), 0.08 (3H, s), 0.85 (3H, d, J 7.4), 0.89 (9H, s), 0.96 (3H, d, J 6.9), 1.93–1.98 (1H, m), 1.99–2.05 (1H, m), 2.32 (1H, br s), 3.26 (1H, dd, J1 9.2, J2 6.3), 3.39 (1H, dd, J1 9.2, J2 7.2), 3.47–3.51 (1H, m), 3.64–3.68 (1H, m), 3.88–3.89 (1H, m), 4.46–4.51 (2H, m), 7.26–7.36 (5H, m); 13C NMR (125 MHz, CDCl3) δ: −4.5, −4.2, 12.8, 12.9, 18.2, 26.0 (3C), 35.9, 40.1, 66.3, 72.9, 73.6, 74.4, 127.5 (3C), 128.3 (2C), 138.5; HRMS (ESI) calcd for C20H36NaO3Si (MNa+): 375.2326; found: 375.2324.
:1). Further purification by flash chromatography over silica gel with hexane–CHCl3 (8:1) gave compound 22c as a diastereomixture (163.7 mg, 69%, Z/E = 7:1): colorless oil; [α]27D +12.5 (c 1.17, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.01 (0.4H, s), 0.02 (2.6H, s), 0.04 (0.4H, s), 0.05 (2.6H, s), 0.84–0.86 (3H, m), 0.89 (1.2H, s), 0.90 (7.8H, s), 0.94–0.97 (3H, m), 1.61 (2.6H, dd, J1 6.9, J2 1.7), 1.63 (0.4H, d, J 4.6), 1.96–2.03 (1H, m), 2.23–2.30 (0.1H, m), 2.60–2.65 (0.9H, m), 3.22 (1H, dd, J1 8.9, J2 6.9), 3.40 (1H, dd, J1 8.9, J2 7.7), 3.56 (1H, dd, J1 8.0, J2 1.7), 4.43–4.52 (2H, m), 5.16–5.21 (1H, m), 5.35–5.42 (1H, m), 7.25–7.34 (5H, m); 13C NMR (125 MHz, CDCl3) δ: −4.1, −4.0, −3.6, −3.5, 10.8, 11.1, 13.0, 17.6, 18.1, 18.3, 18.4, 18.5, 26.2 (6C), 35.9, 36.6, 37.1, 41.5, 72.7, 72.8, 73.6, 73.9, 76.0, 76.2, 122.7, 123.8, 127.4, 127.5 (2C), 128.3 (2C), 134.5, 134.9, 138.7; HRMS (ESI) calcd for C22H38NaO2Si (MNa+): 385.2533; found: 385.2534.
:1) to give compound 23c (938.1 mg, 92%) as a colorless oil: [α]28D −12.7 (c 1.13, CHCl3); IR (neat) νmax/cm−1: 3328 (OH); 1H NMR (500 MHz, CDCl3) δ: 0.06 (3H, s), 0.08 (3H, s), 0.85 (3H, d, J 6.9), 0.88–0.92 (15H, m), 1.09–1.16 (1H, m), 1.19–1.28 (1H, m), 1.31–1.41 (2H, m), 1.60–1.66 (1H, m), 1.90–1.97 (1H, m), 2.07 (1H, dd, J1 6.0, J2 4.3), 3.45–3.50 (1H, m), 3.62–3.67 (2H, m); 13C NMR (125 MHz, CDCl3) δ: −4.2, −4.1, 12.6, 14.3, 15.8, 18.3, 20.8, 26.0 (3C), 35.9, 37.0, 39.5, 66.5, 77.4; HRMS (ESI) calcd for C15H34NaO2Si (MNa+): 297.2220; found: 297.2221.
:1:0.4 v/v, 1.8 cm3) was added to the reaction mixture. The mixture was warmed to room temperature and stirred for 15 min. Then, saturated aqueous NaHCO3 was added to the mixture at 0 °C. The whole mixture was extracted with CH2Cl2 and the extract was washed with brine, and dried over MgSO4. The filtrate was concentrated under reduced pressure and the residue was purified by flash chromatography over silica gel with hexane–EtOAc (20:1 to 10:1) to give compound 12c (73.4 mg, 54%) as a colorless oil: [α]26D −22.4 (c 1.01, CHCl3); IR (neat) νmax/cm−1: 3523 (OH), 1716 (CO); 1H NMR (500 MHz, CDCl3) δ: 0.08 (3H, s), 0.09 (3H, s), 0.85 (3H, d, J 6.9), 0.88–0.91 (12H, m), 0.94 (3H, d, J 6.9), 1.04–1.11 (1H, m), 1.15–1.22 (1H, m), 1.35–1.42 (1H, m), 1.46–1.53 (1H, m), 1.63–1.69 (2H, m), 1.87 (3H, s), 1.96 (1H, d, J 4.0), 2.31–2.42 (2H, m), 3.65–3.66 (1H, m), 3.74 (3H, s), 3.79–3.83 (1H, m), 6.79–6.82 (1H, m); 13C NMR (125 MHz, CDCl3) δ: −4.2, −3.5, 8.9, 12.7, 14.4, 15.4, 18.3, 21.1, 26.0 (3C), 34.7, 35.3, 37.9, 40.1, 51.7, 73.7, 78.8, 129.3, 138.8, 168.4; HRMS (ESI) calcd for C21H42NaO4Si (MNa+): 409.2745; found: 409.2748.
:0 to 70:1) to give compound 24d (4.9 g, 70%) as a colorless oil: [α]27D +1.74 (c 1.16, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.00 (3H, s), 0.03 (3H, s), 0.86–0.89 (18H, m), 1.00–1.05 (1H, m), 1.18–1.26 (1H, m), 1.36–1.42 (2H, m), 1.56–1.60 (1H, m), 1.95–2.00 (1H, m), 3.22 (1H, dd, J1 8.6, J2 6.6), 3.37 (1H, dd, J1 8.6, J2 7.4), 3.62 (1H, dd, J1 5.7, J1 2.3), 4.45–4.52 (2H, m), 7.27–7.34 (5H, m); 13C NMR (125 MHz, CDCl3) δ: −4.3, −3.9, 11.9, 14.4, 15.9, 18.4, 20.7, 26.1 (3C), 35.5, 35.6, 38.3, 72.8, 74.4, 75.4, 127.4, 127.5 (2C), 128.3 (2C), 138.7; HRMS (ESI) calcd for C22H40NaO2Si (MNa+): 387.2690; found: 387.2691.
:1) to give compound 31 (1.6 g, 59%) as a colorless oil: [α]27D −75.4 (c 0.72, CHCl3); IR (neat) νmax/cm−1: 1716 (CO); 1H NMR (500 MHz, CDCl3) δ: 0.05 (6H, s), 0.85–0.92 (18H, m), 1.15–1.28 (2H, m), 1.33–1.42 (2H, m), 1.62–1.66 (1H, m), 1.85 (3H, d, J 1.1), 1.98–2.05 (1H, m), 2.16 (3H, s), 2.26–2.39 (2H, m), 3.47 (1H, dd, J1 6.9, J2 2.9), 3.73 (3H, s), 4.00–4.03 (1H, m), 4.53 (1H, d, J 11.5), 4.63 (1H, d, J 11.5), 6.92–6.95 (1H, m); 13C NMR (125 MHz, CDCl3) δ: −3.7, −3.6, 11.4, 12.8, 14.0, 14.1, 14.3, 18.4, 20.9, 26.2 (3C), 29.1, 36.4, 36.6, 39.0, 51.6, 73.0, 75.9, 77.2, 128.4, 140.3, 168.5; HRMS (ESI) calcd for C23H46NaO4SSi (MNa+): 469.2778; found: 469.2779.
:1) to give 10, which was used without further purification. To a stirred solution of acid 10 in CH2Cl2 (12.8 cm3) were added MNBA (1.32 g, 3.8 mmol), DMAP (935.0 mg, 7.7 mmol) and 11 (959.0 mg, 3.8 mmol) at room temperature. After stirring overnight, the mixture was quenched with 1 N HCl. The whole mixture was extracted with CH2Cl2 and the extract was washed with brine, and dried over MgSO4. The filtrate was concentrated under reduced pressure and the residue was purified by flash chromatography over silica gel with hexane–EtOAc (10:1) to give compound 32 (1.48 g, 87%) as a colorless oil: [α]26D −43.5 (c 0.89, CHCl3); IR (neat) νmax/cm−1: 1710 (CO); 1H NMR (500 MHz, CDCl3) δ: 0.05 (3H, s), 0.07 (3H, s), 0.86–0.91 (18H, m), 0.98 (3H, t, J 7.4), 1.15 (3H, d, J 6.9), 1.18–1.28 (2H, m), 1.34–1.47 (3H, m), 1.52–1.58 (1H, m), 1.62–1.64 (1H, m), 1.89 (3H, s), 1.99–2.03 (1H, m), 2.11 (3H, s), 2.19–2.25 (1H, m), 2.34–2.37 (2H, m), 3.47 (1H, dd, J1 7.4, J2 2.9), 4.01–4.04 (1H, m), 4.52 (1H, d, J 11.5), 4.61 (1H, d, J 11.5), 5.19 (1H, d, J 3.4), 5.25 (1H, d, J 16.6), 5.55 (1H, d, J 16.6), 7.04–7.07 (1H, m), 7.49 (2H, t, J 7.7), 7.60–7.63 (1H, m), 7.89–7.91 (2H, m); 13C NMR (125 MHz, CDCl3) δ: −3.6, −3.5, 11.3, 11.8, 12.7, 14.1, 14.2 (2C), 14.3, 18.4, 20.9, 26.2 (3C), 26.3, 29.4, 36.4, 36.6, 36.9, 39.2, 66.2, 73.0, 74.6, 76.1, 77.2, 127.7 (2C), 128.2, 128.9 (2C), 133.9, 134.1, 141.4, 167.4, 169.7, 191.6; HRMS (ESI) calcd for C36H60NaO7SSi (MNa+): 687.3721; found: 687.3720.
:1) to give compound 7 (49.2 mg, 74%) as a colorless oil: [α]25D −5.29 (c 1.08, CHCl3); IR (neat) νmax/cm−1: 3526 (OH), 1708 (CO); 1H NMR (500 MHz, CDCl3) δ: 0.83–0.85 (6H, m), 0.89–0.91 (3H, m), 0.98 (3H, t, J 7.4), 1.15 (3H, d, J 6.9), 1.24–1.37 (4H, m), 1.38–1.46 (1H, m), 1.50–1.59 (1H, m), 1.61–1.64 (1H, m), 1.90 (3H, s), 1.92–1.99 (1H, m), 2.15–2.16 (4H, m), 2.19–2.26 (1H, m), 2.37–2.49 (2H, m), 3.38–3.41 (1H, m), 4.08–4.12 (1H, m), 4.64 (2H, s), 5.22 (1H, d, J 2.9), 5.26 (1H, d, J 16.6), 5.55 (1H, d, J 16.6), 7.03–7.06 (1H, m), 7.49 (2H, t, J 7.7), 7.60–7.63 (1H, m), 7.89–7.91 (2H, m); 13C NMR (125 MHz, CDCl3) δ: 11.4, 11.7 (2C), 12.6, 14.2 (3C), 20.5, 26.3, 29.6, 34.4, 36.7, 36.9, 38.5, 66.2, 73.5, 74.6, 76.2, 78.3, 127.7 (2C), 128.4, 128.9 (2C), 133.9, 134.1, 140.7, 167.4, 169.7, 191.6; HRMS (ESI) calcd for C30H46NaO7S (MNa+): 573.2856; found: 573.2855.
:1 to 3:1) to give crude Fmoc-protected amine, which was used without further purification. To a stirred solution of the above protected amine in MeCN (7.0 cm3) was added Et2NH (2.3 cm3) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 1.5 h. The mixture was concentrated under reduced pressure and the residue was purified by flash chromatography over silica gel with hexane–EtOAc (3:1 to 1:2) to give compound 33 (94.9 mg, 54%) as a yellow oil: [α]27D −55.4 (c 0.79, CHCl3); IR (neat) νmax/cm−1: 1712 (CO); 1H NMR (500 MHz, CD3CN) δ: 0.78 (3H, t, J 7.2), 0.81–0.84 (6H, m), 0.86 (3H, t, J 7.4), 0.99 (3H, d, J 6.9), 1.02–1.08 (1H, m), 1.09–1.14 (1H, m), 1.16 (3H, d, J 7.1), 1.23–1.34 (3H, m), 1.39–1.47 (1H, m), 1.69–1.77 (4H, m), 1.95 (3H, s), 2.02–2.08 (1H, m), 2.13–2.17 (1H, m), 2.19–2.27 (4H, m), 2.32–2.36 (1H, m), 3.13 (1H, q, J 7.1), 3.63 (1H, dt, J1 10.3, J2 2.6), 4.44 (1H, d, J 11.5), 4.55 (1H, d, J 11.5), 4.79 (1H, dd, J1 10.3, J2 2.3), 5.05 (1H, d, J 3.4), 5.30 (1H, d, J 16.6), 5.42 (1H, d, J 16.6), 6.78–6.81 (1H, m), 7.46 (2H, t, J 8.0), 7.57–7.61 (1H, m), 7.85–7.87 (2H, m); 13C NMR (125 MHz, CD3CN) δ 10.3, 12.0, 12.8, 13.1, 14.1, 14.4, 14.6, 18.7, 20.9, 26.8, 29.6, 34.4, 34.5, 36.7, 37.0, 37.7, 59.1, 67.7, 73.5, 75.3, 76.1, 78.3, 128.7 (2C), 129.0, 129.8 (2C), 134.9, 135.0, 142.4, 167.9, 170.5, 174.9, 193.2; HRMS (FAB) calcd for C34H54NO8S (MH+): 636.3565; found: 636.3569.
:1 to 2:3) to give peptide 34 as a 1.4:1 diastereomixture, which was used without further purification. To a stirred solution of 34 in AcOH/EtOAc/H2O (60:35:5, 4.3 cm3) was added Zn (92.2 mg, 1.4 mmol) at room temperature. After stirring for 8 h, the reaction mixture was filtered through Celite, and 1 N HCl was added to the filtrate. The whole mixture was extracted with EtOAc and the extract was washed with brine, and dried over MgSO4. After the filtrate was concentrated under reduced pressure, AcOH was removed by azeotropic distillation with toluene to give the corresponding carboxylic acid, which was used without further purification. To a stirred solution of the above acid in MeCN (2.4 cm3) was added Et2NH (0.80 cm3) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 1.5 h. The reaction mixture was concentrated under reduced pressure and the residue was purified by reverse-phase preparative HPLC (59% CH3CN in 0.1% TFA solution) to give linear peptides 6a (29.6 mg, 30% from 33) and 6b (24.0 mg, 24% from 33) as a colorless powder.
6a: [α]27D −46.8 (c 0.89, CHCl3); IR (neat) νmax/cm−1: 1645 (CO); 1H NMR (500 MHz, CD3CN, 1:1 mixture of rotamers) δ: 0.77 (1.5H, d, J 7.0), 0.81–0.91 (18H, m), 0.96 (1.5H, d, J 3.1), 0.97 (1.5H, d, J 3.1), 1.06–1.22 (4.5H, m), 1.26–1.33 (3H, m), 1.37 (3H, d, J 7.3), 1.41–1.53 (2H, m), 1.74–1.84 (5H, m), 1.96–2.01 (1H, m), 2.07 (1.5H, s), 2.08 (1.5H, s), 2.20–2.38 (3H, m), 2.75–2.86 (4.5H, m), 3.00–3.12 (6.5H, m), 3.53 (0.5H, d, J 16.4), 3.70–3.71 (1H, m), 4.00 (0.5H, d, J 18.0), 4.05–4.09 (0.5H, m), 4.17 (0.5H, d, J 18.0), 4.24 (0.5H, d, J 16.4), 4.31–4.36 (0.5H, m), 4.48–4.50 (0.5H, m), 4.53 (0.5H, d, J 5.0), 4.55 (0.5H, d, J 5.0), 4.62 (0.5H, d, J 5.0), 4.64 (0.5H, d, J 5.0), 4.73–4.76 (0.5H, m), 4.87–4.89 (1H, m), 4.95 (0.5H, d, J 3.4), 4.96 (0.5H, d, J 3.4), 5.19 (0.5H, q, J 7.3), 5.24 (0.5H, q, J 7.3), 5.43–5.46 (0.5H, m), 5.63 (0.5H, dd, J1 9.9, J2 5.7), 6.82–6.89 (1H, m), 7.15–7.28 (6H, m), 7.64 (2H, br s); 13C NMR (125 MHz, CD3CN) δ: 10.2 (2C), 11.1, 11.3, 11.9 (2C), 12.7 (2C), 13.0, 13.1, 14.2 (2C), 14.3 (2C), 14.8 (3C), 14.9, 15.5, 15.7, 16.0, 16.1, 20.8 (2C), 24.9, 25.1, 26.8 (2C), 29.3, 29.4, 30.5, 31.5, 32.5 (2C), 34.5, 34.6, 35.4, 35.6, 35.7, 36.8 (2C), 37.0 (3C), 37.2, 37.3 (2C), 37.7, 48.0, 48.6, 52.0, 52.3, 53.5 (2C), 53.7, 55.1, 55.3, 56.0, 73.6 (2C), 75.4, 75.6, 76.5 (2C), 78.9 (2C), 127.4, 127.7, 128.9, 129.0, 129.1 (2C), 129.2 (2C), 130.2 (2C), 130.3 (2C), 137.5, 137.8, 142.4 (2C), 168.3, 168.5, 168.9, 169.5, 170.0, 170.4, 171.2, 171.7, 172.2, 172.3, 172.5 (2C), 173.4, 174.0; HRMS (ESI) calcd for C48H80N5O11S (MH+): 934.5570; found: 934.5567.
6b: [α]28D −23.5 (c 1.00, CHCl3); IR (neat) νmax/cm−1: 1648 (CO); 1H NMR (500 MHz, CD3CN, 3:3:3:1 mixture of rotamers) δ: 0.74 (0.9H, d, J 6.9), 0.78–0.98 (21.9H, m), 1.05 (1.2H, d, J 6.9), 1.08–1.51 (11H, m), 1.75–1.90 (5H, m), 1.97–2.02 (1H, m), 2.05–2.06 (2.7H, m), 2.09 (0.3H, s), 2.19–2.45 (3H, m), 2.72 (0.3H, s), 2.81–2.86 (4.8H, m), 2.89–2.91 (2H, m), 2.93 (0.3H, s), 2.95 (0.3H, s), 3.00–3.09 (1.8H, m), 3.12–3.18 (1.5H, m), 3.43 (0.1H, d, J 16.1), 3.55–3.56 (0.1H, m), 3.61–3.76 (1.6H, m), 4.09–4.16 (0.6H, m), 4.25–4.36 (1.3H, m), 4.45–4.48 (0.4H, m), 4.52–4.64 (1.9H, m), 4.70–4.73 (0.3H, m), 4.79–5.00 (3H, m), 5.08–5.12 (0.7H, m), 5.26 (0.3H, dd, J1 9.3, J2 6.0), 5.33 (0.3H, dd, J1 11.1, J2 4.4), 5.46 (0.3H, dd, J1 11.1, J2 4.8), 5.52 (0.1H, dd, J1 9.5, J2 6.4), 6.84–6.91 (1H, m), 6.97 (0.3H, d, J 9.2), 7.07–7.26 (5H, m), 7.42 (0.3H, d, J 7.3), 7.69 (0.4H, d, J 6.7), 8.11 (2H, br s); 13C NMR (125 MHz, CD3CN) δ: 10.3 (3C), 10.6, 12.0 (2C), 12.2 (2C), 12.8, 12.9, 13.2 (3C), 13.7, 14.2 (2C), 14.4 (2C), 14.6 (2C), 14.7, 14.8, 14.9 (2C), 15.1, 15.2, 15.9 (2C), 16.1 (2C), 16.2, 16.3, 20.8, 20.9, 26.8 (2C), 26.9 (2C), 27.0 (2C), 27.4, 29.4, 29.5 (2C), 29.6, 30.4, 30.5, 30.6, 31.1, 31.9, 32.6, 32.7, 33.6, 34.5, 34.6, 34.7, 35.2, 35.3, 35.4, 35.5, 35.6, 35.9, 36.5, 36.8, 37.0 (4C), 37.1, 37.2, 37.4 (2C), 38.1, 38.4, 48.0 (2C), 48.6 (2C), 52.5, 52.6, 53.0, 53.2, 54.0, 54.3 (2C), 54.7, 54.8, 55.0, 56.4, 57.1, 58.1, 73.5, 75.3 (2C), 75.6 (2C), 75.9, 76.1 (2C), 78.6, 79.0, 79.6, 79.7, 127.5 (2C), 127.6, 129.1 (3C), 129.2 (3C), 129.3, 130.2 (2C), 130.3 (3C), 130.4 (2C), 137.5, 137.8, 138.1, 138.3, 141.4, 141.5, 141.9, 142.2, 167.9, 168.1, 168.2, 168.3, 168.7, 169.6, 170.1, 170.2, 170.3 (2C), 170.8, 171.2 (2C), 171.3, 171.4, 171.8, 172.0, 172.2 (2C), 172.5, 173.4, 174.4, 174.7; HRMS (ESI) calcd for C48H80N5O11S (MH+): 934.5570; found: 934.5580.
:1 to 0:1) to give the corresponding cyclic peptide. To a stirred solution of the above cyclic peptide in THF/H2O (4:1, 0.566 cm3) were added 2,6-lutidine (0.0394 cm3, 0.34 mmol) and AgNO3 (115.5 mg, 0.68 mmol) at room temperature. The reaction mixture was warmed to 70 °C and stirred for 4 h. The mixture was filtered through Celite, and 1 N HCl was added to the filtrate. The whole mixture was extracted with EtOAc and the extract was washed with H2O, saturated aqueous NaHCO3 and brine, and dried over MgSO4. The filtrate was concentrated under reduced pressure and the residue was purified by reverse-phase preparative HPLC (72% CH3CN in H2O) to give odoamide (5a) (12.4 mg, 85%) as a colorless powder: [α]28D −15.8 (c 1.14, CH3OH); IR (neat) νmax/cm−1: 3305 (OH), 1645 (CO); 1H NMR (500 MHz, CD3OD) δ: 0.83–0.96 (21H, m), 1.04–1.12 (4H, m), 1.19–1.26 (1H, m), 1.29–1.40 (4H, m), 1.42 (3H, d, J 6.9), 1.45–1.55 (1H, m), 1.57–1.67 (1H, m), 1.78–1.87 (3H, m), 1.90 (3H, s), 2.01–2.04 (1H, m), 2.12–2.16 (1H, m), 2.20–2.28 (1H, m), 2.85–2.95 (4H, m), 3.01–3.06 (4H, m), 3.30 (3H, s), 3.56 (1H, d, J 18.3), 3.74–3.76 (1H, m), 3.94 (1H, q, J 6.9), 4.19 (1H, d, J 18.3), 4.49 (1H, q, J 6.9), 4.86–4.89 (2H, m), 5.05 (1H, d, J 6.3), 5.45 (1H, dd, J1 10.3, J2 5.2), 7.12–7.20 (5H, m), 7.31–7.32 (1H, m); 13C NMR (125 MHz, CD3OD) δ: 10.0, 11.7, 12.0, 12.1, 13.1, 13.8, 14.5, 14.6, 15.6, 16.0, 21.6, 24.7, 27.4, 30.5 (2C), 35.8, 35.9, 36.6, 37.6, 37.8, 38.5, 39.4, 41.3, 46.4, 52.6, 54.7, 55.0, 60.3, 71.5, 77.6, 79.2, 127.4, 128.6, 129.1 (2C), 130.6 (2C), 138.4, 146.8, 170.4, 171.3, 172.5, 172.7, 172.8, 173.0, 174.9; HRMS (ESI) calcd for C46H73N5NaO10 (MNa+): 878.5250; found: 878.5254.
Growth inhibition assay
A549 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma) supplemented with 10% (v/v) fetal bovine serum at 37 °C in a 5% CO2-incubator. Growth inhibition assays using A549 cells were performed in 96-well plates (BD Falcon). A549 cells were seeded at 500 cells per well in 0.050 cm3 of culture media, respectively, and were cultured for 6 h. Chemical compounds in DMSO were diluted 250-fold with the culture medium in advance. Following the addition of 0.040 cm3 of the fresh culture medium to the cell cultures, 0.030 cm3 of the chemical diluents were also added. The final volume of DMSO in the medium was equal to 0.1% (v/v). The cells under chemical treatment were incubated for further 72 h. The wells in the plates were washed twice with the cultured medium without phenol-red. After 1 h incubation with 0.100 cm3 of the medium, the cell culture in each well was supplemented with 0.020 cm3 of the MTS reagent (Promega), followed by incubation for additional 40 min. Absorbance at 490 nm of each well was measured using a Wallac 1420 ARVO SX multilabel counter (Perkin Elmer). Three experiments were performed per condition and the average of inhibition rates in each condition was evaluated to determine IC50 values using the GraphPad Prism software.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research from JSPS, Japan (24659004 and 15J05499); the Platform for Drug Discovery, Informatics, and Structural Life Science from MEXT, Japan; and the Takeda Science Foundation. M. K. is grateful for JSPS Research Fellowships for Young Scientists.References
- For reviews, see: (a) Y. Hamada and T. Shioiri, Chem. Rev., 2005, 105, 4441–4482 CrossRef CAS PubMed; (b) S. Sivanathan and J. Scherkenbeck, Molecules, 2014, 19, 12368–12420 CrossRef PubMed.
- For reviews, see: (a) J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, Acc. Chem. Res., 2008, 41, 1331–1342 CrossRef CAS PubMed; (b) J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
- (a) W. Huang, R. G. Ren, H. Q. Dong, B. G. Wei and G. Q. Lin, J. Org. Chem., 2013, 78, 10747–10762 CrossRef CAS PubMed; (b) J. Tulla-Puche, S. Auriemma, C. Falciani and F. Albericio, J. Med. Chem., 2013, 56, 5587–5600 CrossRef CAS PubMed; (c) M. Pelay-Gimeno, A. Meli, J. Tulla-Puche and F. Albericio, J. Med. Chem., 2013, 56, 9780–9788 CrossRef CAS PubMed; (d) R. Nabika, S. Oishi, R. Misu, H. Ohno and N. Fujii, Bioorg. Med. Chem., 2014, 22, 6156–6162 CrossRef CAS PubMed; (e) W. He, H.-B. Qui, Y.-J. Chen, J. Xi and Z.-J. Yao, Tetrahedron Lett., 2014, 55, 6109–6112 CrossRef CAS; (f) R. Nabika, T. L. Suyama, A. M. Hau, R. Misu, H. Ohno, J. E. Ishmael, K. L. McPhail, S. Oishi and N. Fujii, Bioorg. Med. Chem. Lett., 2015, 25, 302–306 CrossRef CAS PubMed; (g) G. Yao, Z. Pan, C. Wu, W. Wang, L. Fang and W. Su, J. Am. Chem. Soc., 2015, 137, 13488–13491 CrossRef CAS PubMed.
- (a) K. Suenaga, T. Mutou, T. Shibata, T. Itoh, H. Kigoshi and K. Yamada, Tetrahedron Lett., 1996, 37, 6771–6774 CrossRef CAS; (b) K. Suenaga, T. Mutou, T. Shibata, T. Itoh, T. Fujita, N. Takada, K. Hayamizu, M. Takagi, T. Irifune, H. Kigoshi and K. Yamada, Tetrahedron, 2004, 60, 8509–8527 CrossRef CAS.
- B. Han, H. Gross, D. E. Goeger, S. L. Mooberry and W. H. Gerwick, J. Nat. Prod., 2006, 69, 572–575 CrossRef CAS PubMed.
- Y. Nakao, W. Y. Yoshida, Y. Takada, J. Kimura, L. Yang, S. L. Mooberry and P. J. Scheuer, J. Nat. Prod., 2004, 67, 1332–1340 CrossRef CAS PubMed.
- A. Tripathi, J. Puddick, M. R. Prinsep, M. Rottmann and L. T. Tan, J. Nat. Prod., 2010, 73, 1810–1814 CrossRef CAS PubMed.
- A. Tripathi, J. Puddick, M. R. Prinsep, M. Rottmann, K. P. Chan, D. Y. Chen and L. T. Tan, Phytochemistry, 2011, 72, 2369–2375 CrossRef CAS PubMed.
- P. G. Williams, W. Y. Yoshida, M. K. Quon, R. E. Moore and V. J. Paul, J. Nat. Prod., 2003, 66, 1545–1549 CrossRef CAS PubMed.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- (a) T. Mutou, K. Suenaga, T. Fujita, T. Itoh, N. Takada, K. Hayamizu, H. Kigoshi and K. Yamada, Synlett, 1997, 199–201 CrossRef CAS; (b) Y. Takada, E. Mori, M. Umehara, Y. Nakao and J. Kimura, Tetrahedron Lett., 2007, 48, 7653–7656 CrossRef CAS; (c) Y. Takada, M. Umehara, Y. Nakao and J. Kimura, Tetrahedron Lett., 2008, 49, 1163–1165 CrossRef CAS; (d) L. Dai, B. Chen, H. Lei, Z. Wang, Y. Liu, Z. Xu and T. Ye, Chem. Commun., 2012, 48, 8697–8699 RSC.
- The isolation and structural assignment of odoamide were reported in our previous article, see: K. Sueyoshi, M. Kaneda, S. Sumimoto, S. Oishi, N. Fujii, K. Suenaga and T. Teruya, Tetrahedron, 2016, 72, 5472–5478 CrossRef CAS.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096 CrossRef CAS.
- E. Marcucci, J. Tulla-Puche and F. Albericio, Org. Lett., 2012, 14, 612–615 CrossRef CAS PubMed.
- (a) G. C. Stelakatos, A. Paganou and L. Zervas, J. Chem. Soc. C, 1966, 1191–1199 RSC; (b) T. Takahashi, H. Nagamiya, T. Doi, P. G. Griffiths and A. M. Bray, J. Comb. Chem., 2003, 5, 414–428 CrossRef CAS PubMed.
- T. Kawabata, Y. Kimura, Y. Ito, S. Terashima, A. Sasaki and M. Sunagawa, Tetrahedron, 1988, 44, 2149–2165 CrossRef CAS.
- (a) D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS; (b) D. A. Evans, J. V. Nelson, E. Vogel and T. R. Taber, J. Am. Chem. Soc., 1981, 103, 3099–3111 CrossRef CAS.
- Alcohol 20c was synthesised from ent-13 according to a similar process in a previous report, see: A. Zampella, M. Sorgente and M. V. D'Auria, Tetrahedron: Asymmetry, 2002, 13, 681–685 CrossRef CAS.
- (a) T. Mukaiyama, K. Banno and K. Narasaka, J. Am. Chem. Soc., 1974, 96, 7503–7509 CrossRef CAS; (b) D. A. Evans, M. J. Dart, J. L. Duffy and M. G. Yang, J. Am. Chem. Soc., 1996, 118, 4322–4343 CrossRef CAS.
- (a) D. W. Cameron, M. G. Looney and J. A. Pattermann, Tetrahedron Lett., 1995, 36, 7555–7558 CrossRef CAS; (b) G. T. Kim, M. Wenz, J. I. Park, J. Hasserodt and K. D. Janda, Bioorg. Med. Chem., 2002, 10, 1249–1262 CrossRef CAS PubMed.
- Two hydroxy group configurations in 12c and 12d were determined by the NMR analysis of the corresponding acetonides.
- (5R)-Hydroxy ester 26a is the substrate for the synthesis of (5S)-hydroxy ester 12a (see the ESI†).12.
- S. D. Rychnovsky, B. Rogers and G. Yang, J. Org. Chem., 1993, 58, 3511–3515 CrossRef CAS.
- P. M. Pojer and S. J. Angyal, Aust. J. Chem., 1978, 31, 1031–1040 CrossRef CAS.
- (a) I. Shiina, R. Ibuka and M. Kubota, Chem. Lett., 2002, 31, 286–287 CrossRef; (b) I. Shiina, M. Kubota, H. Oshiumi and M. Hashizume, J. Org. Chem., 2004, 69, 1822–1830 CrossRef CAS PubMed.
- L. A. Carpino, B. J. Cohen, K. E. Stephens, Jr., S. Y. Sadat-Aalaee, J. H. Tien and D. C. Langridge, J. Org. Chem., 1986, 51, 3732–3734 CrossRef CAS.
- Among several conditions investigated for coupling of peptide 9, EDCI–HOAt provided the desired compound 6a in higher chemical yield; however, significant epimerization at the C-terminal L-Ile occurred.
- J. D. White and M. Kawasaki, J. Org. Chem., 1992, 57, 5292–5300 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob01583b |
| This journal is © The Royal Society of Chemistry 2016 |