
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isothiourea-catalysed enantioselective pyrrolizine synthesis: synthetic and computational studies†
Daniel G.
Stark
a,
Patrick
Williamson
a,
Emma R.
Gayner
b,
Stefania F.
Musolino
a,
Ryan W. F.
Kerr
a,
James E.
Taylor
a,
Alexandra M. Z.
Slawin
a,
Timothy J. C.
O'Riordan
c,
Stuart A.
Macgregor
*b and
Andrew D.
Smith
*a
aEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
bInstitute of Chemical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: s.a.macgregor@hw.ac.uk
cSyngenta, Jealott's Hill International Research Centre, Bracknell, RG42 6EY, UK
First published on 21st July 2016
Abstract
The catalytic enantioselective synthesis of a range of cis-pyrrolizine carboxylate derivatives with outstanding stereocontrol (14 examples, >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr, >98:2 er) through an isothiourea-catalyzed intramolecular Michael addition-lactonisation and ring-opening approach from the corresponding enone acid is reported. An optimised and straightforward three-step synthetic route to the enone acid starting materials from readily available pyrrole-2-carboxaldehydes is delineated, with benzotetramisole (5 mol%) proving the optimal catalyst for the enantioselective process. Ring-opening of the pyrrolizine dihydropyranone products with either MeOH or a range of amines leads to the desired products in excellent yield and enantioselectivity. Computation has been used to probe the factors leading to high stereocontrol, with the formation of the observed cis-steroisomer predicted to be kinetically and thermodynamically favoured.
:5 dr, >98:2 er) through an isothiourea-catalyzed intramolecular Michael addition-lactonisation and ring-opening approach from the corresponding enone acid is reported. An optimised and straightforward three-step synthetic route to the enone acid starting materials from readily available pyrrole-2-carboxaldehydes is delineated, with benzotetramisole (5 mol%) proving the optimal catalyst for the enantioselective process. Ring-opening of the pyrrolizine dihydropyranone products with either MeOH or a range of amines leads to the desired products in excellent yield and enantioselectivity. Computation has been used to probe the factors leading to high stereocontrol, with the formation of the observed cis-steroisomer predicted to be kinetically and thermodynamically favoured.
Introduction
The 5,5-bicyclic pyrrolizine and pyrrolizidine structural motifs that contain a bridgehead nitrogen atom are present within the core of many biologically active natural products1 such as that of dehydroretronecine 1 (Fig. 1).2 This natural product, along with many other derivatives, originates from metabolism of pyrrolizidine alkaloids (PAs), a natural alkaloid prevalent in plant life throughout nature.3 Such PA-derived molecules are known to be potent heptatoxins,4 carcinogens,5 teratogens6 and genotoxins.7 Compelled by such levels of biological activity, many of these natural products and their derivatives have become commercial therapeutic agents. For example, the partially reduced pyrrolizine mitomycin C 2, isolated from Streptomyces caespitosus or Streptomyces lavendulae, is a potent antitumour drug with a broad scope of applications.8 The non-steroidal anti-inflammatory drug (NSAID), Licofelone 3 has shown great promise in osteoarthritis treatment through a dual inhibition of 5-LOX/COX.9 Additionally, another pyrrolizine based NSAID, Ketorolac 4, has found success as a commercial analgesic.10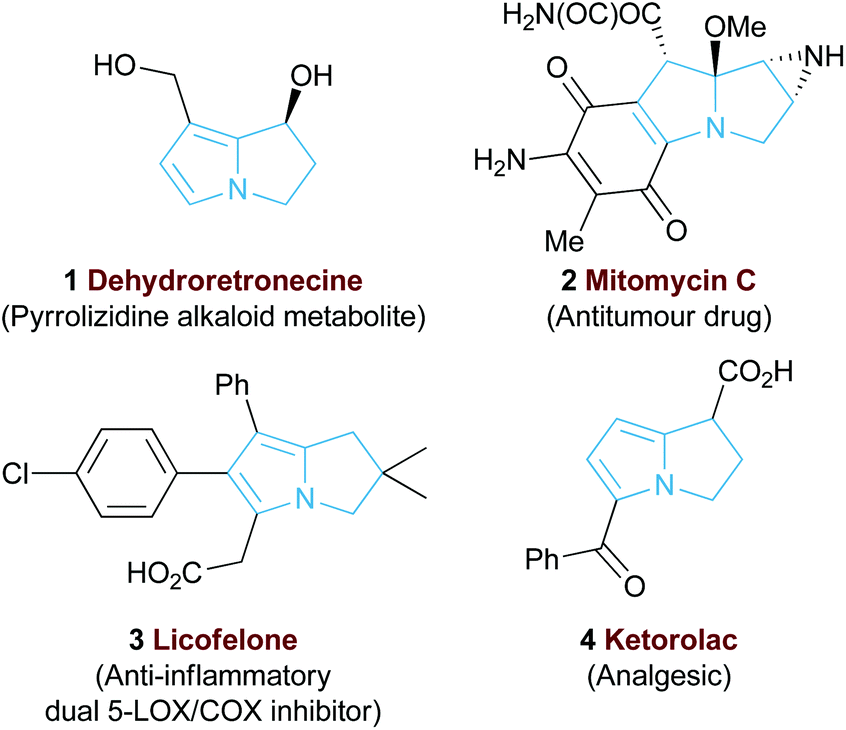 | ||
| Fig. 1 Representative biologically active pyrrolizines. | ||
Given the value and potential of these bicyclic compounds, a variety of synthetic routes towards these motifs have been designed, with many syntheses involving classic total synthesis approaches towards specific target molecules.11 In recent years the state-of-the-art in catalytic pyrrolizine syntheses has involved diastereoselective multi-step reaction processes such as the phosphine-catalysed domino reaction developed by Tong and co-workers13 (Fig. 2a) or the gold-catalysed process by Matsuya and co-workers (Fig. 2b).14 Catalytic enantioselective methodologies that enable efficient access to this desirable structural motif are relatively limited. Within this area, Cho and co-workers showed that an enantioselective organocatalysed Michael addition-aldol approach could generate functionalised pyrrolizines with excellent diastereo- and enantiocontrol (18 examples, >95:5 dr and 95:5 to 99:1 er, Fig. 2c), although relatively high catalyst loadings were employed to promote this process.15 In spite of these advances there is still a clear requirement for easily accessible and reliable catalytic methodologies that can produce stereodefined chiral pyrrolizine derivatives with high levels of efficiency and enantiocontrol.
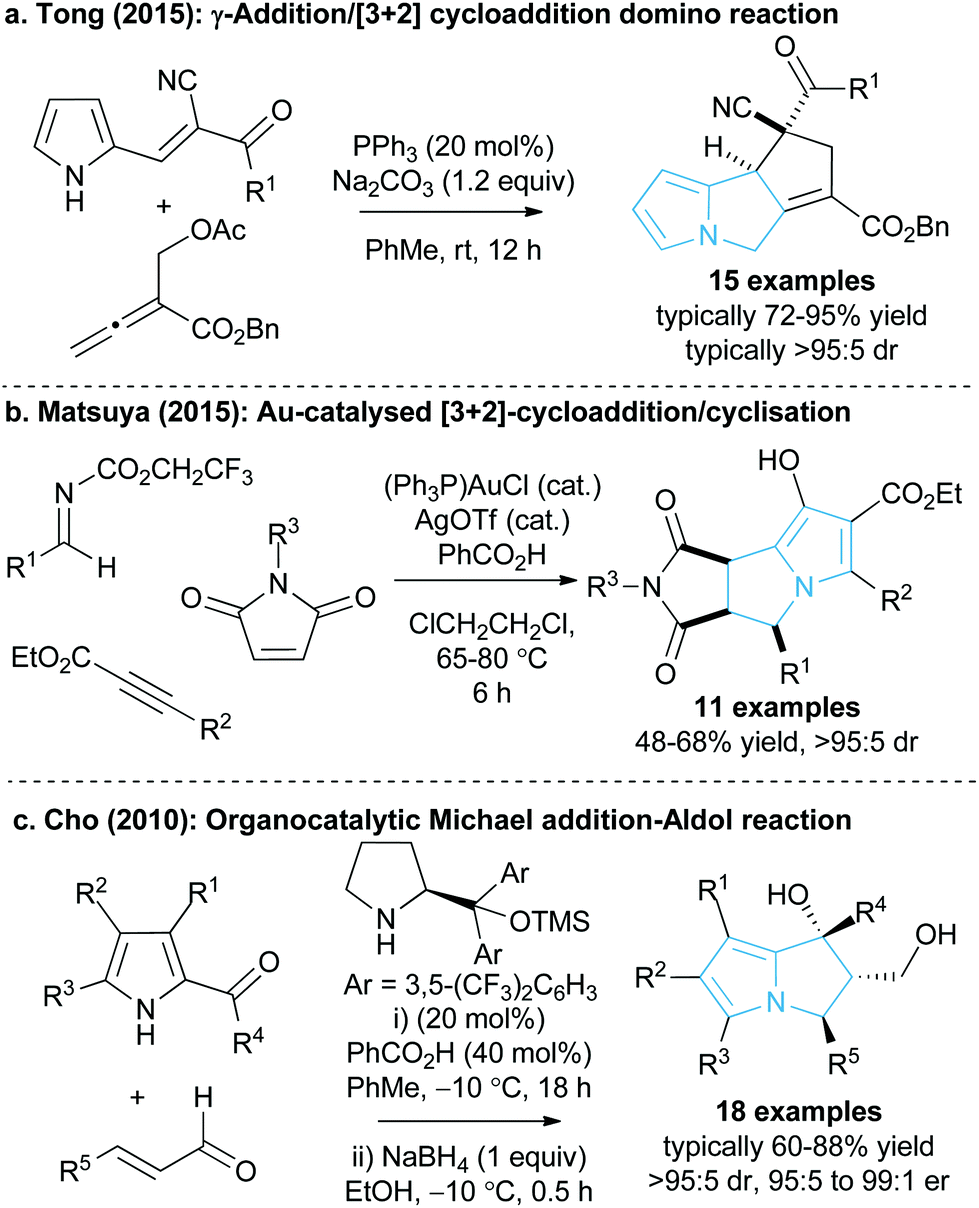 | ||
| Fig. 2 Current catalytic methods for pyrrolizine synthesis. | ||
Following seminal work from Romo and co-workers using ammonium enolates generated from carboxylic acids,16 ourselves and others,17 have used isothioureas18 to catalyse a range of formal [2 + 2],19 [3 + 2]20 and [4 + 2]21 cycloaddition processes that employ an ammonium enolate intermediate.22 Related intramolecular Michael addition-lactonisation cascades from enone-acid substrates have been used to generate simple heterocyclic products such as THFs and pyrrolidines with excellent enantioselectivity.23 Building upon this previous work, the application of this strategy to construct the highly desirable pyrrolizine core in a catalytic enantioselective fashion starting from pyrrole-derived enone acid substrates such as 5 is investigated (Fig. 3). At the onset of these studies the main challenges were envisaged to arise from the incorporation of the electron-rich N-functionalised pyrrole core within the target enone-acid. A robust method to access this structural motif has not been reported previously, while the effect of incorporating this planar electron-rich aromatic structure upon stability and reactivity, as well as the conformational and steric effects upon diastereo- and enantioselectivity were unknown. Furthermore, the potential for competitive intramolecular Friedel–Crafts acylation of the pyrrole via a mixed anhydride or acyl ammonium ion intermediate,24 or alternatively β-elimination from an ammonium enolate, needed to be assessed. To further enhance our understanding of this process, we also wanted to probe the course of the proposed cascade cyclisation process via computation in order to understand the factors leading to stereocontrol. Very limited computational analysis of the use of isothiourea-derived ammonium enolates in catalysis has been reported.25 To date only the single report from Muck-Lichtenfeld and Studer concerning the intermolecular formal 1,3-dipolar cycloaddition of azomethine imines with mixed anhydrides under isothiourea catalysis has incorporated DFT analysis.20a
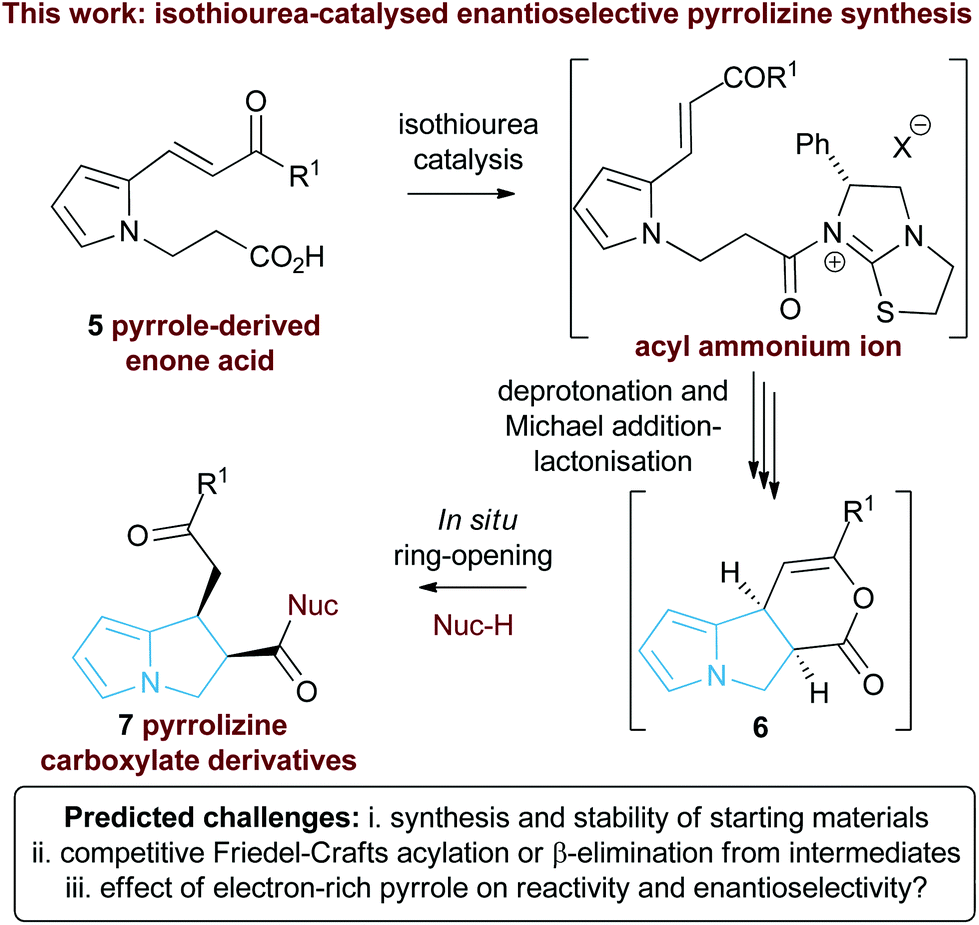 | ||
| Fig. 3 Proposed isothiourea-catalysed Michael addition-lactonisation methodology for pyrrolizine synthesis. | ||
In this manuscript we report the realisation of this strategy to facilitate the catalytic enantioselective synthesis of these valuable heterocyclic products 6 and 7. A straightforward three-step synthetic route to the enone acid starting materials from readily available pyrrole-2-carboxaldehydes is delineated, with commercially available benzotetramisole (BTM) proving the optimal catalyst for the enantioselective process. Furthermore, the use of computational analysis allows insight into the origin of stereocontrol in this intramolecular cascade process.
Results and discussion
Substrate synthesis
Initial studies set out to devise a practical synthetic route towards the target pyrrole-derived enone-acid substrates. Substrate 11 was identified as a model system, and was initially prepared from pyrrole 2-carboxaldehyde by N-alkylation, ester hydrolysis and Wittig olefination. However, attempted synthesis of a range of pyrrole-derived enone-acid substrates or scale-up of this synthetic route proved low yielding and irreproducible when directed towards alternative substrates. After a thorough investigation into alternative synthetic approaches, a reliable three-step sequence starting from the corresponding pyrrole 2-carboxaldehyde was established (Scheme 1). Aldol-condensation with the requisite ketone provides a general and chromatography-free preparation of pyrrole-enones 8. Treatment with sub-stoichiometric t-BuOK and methyl acrylate gives the N-alkylation product 9, with subsequent ester hydrolysis providing the desired enone-acid substrates 10 in good overall yield with a wide scope available. This reliable synthetic sequence allowed the preparation of a range of substrates 11–18 in up to 58% yield over three steps and on multigram scale, and allows for substituent variation within both the pyrrole and enone components.26 Direct chlorination of 11 with N-chlorosuccinimide (NCS) led to substrate 19, while an analogous procedure using acetone for the aldol reaction procedure, followed by alkylation and ester hydrolysis gave methyl enone 20.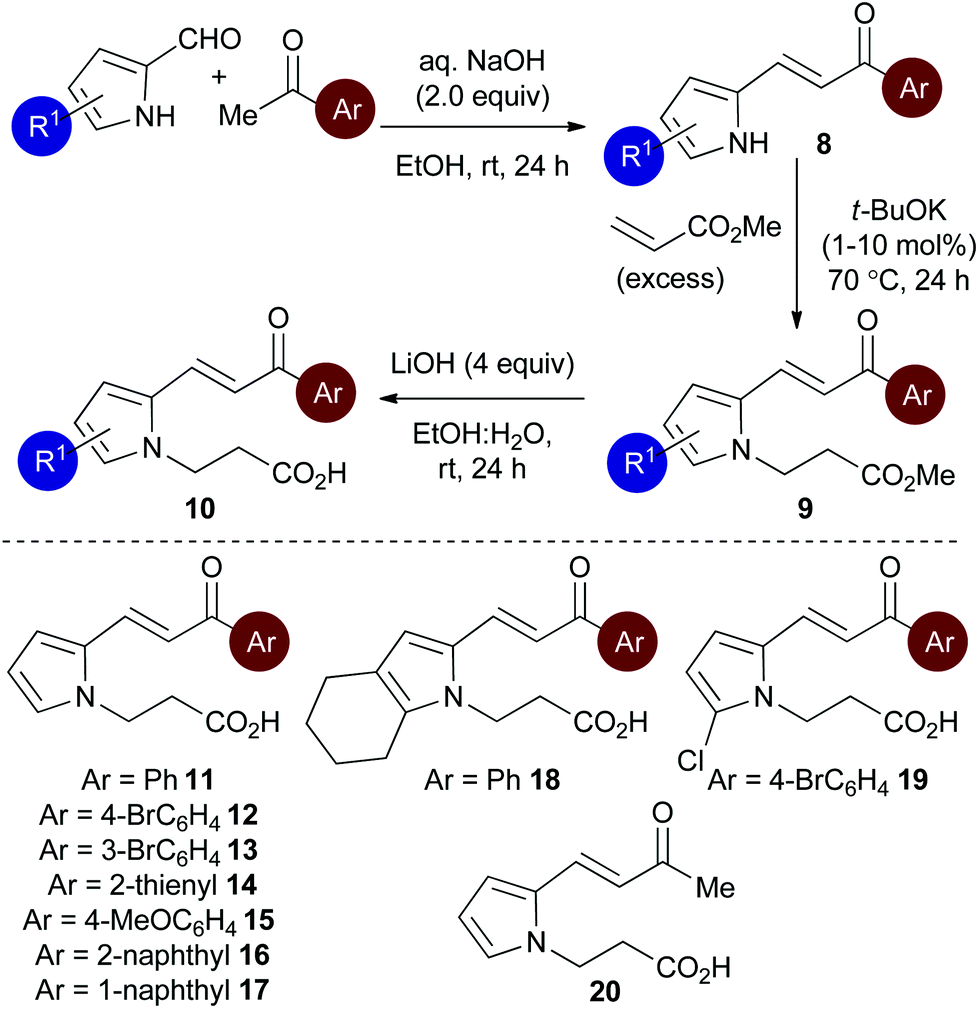 | ||
| Scheme 1 Optimised synthesis of pyrrole-derived enone-acid substrates. | ||
Reaction optimisation
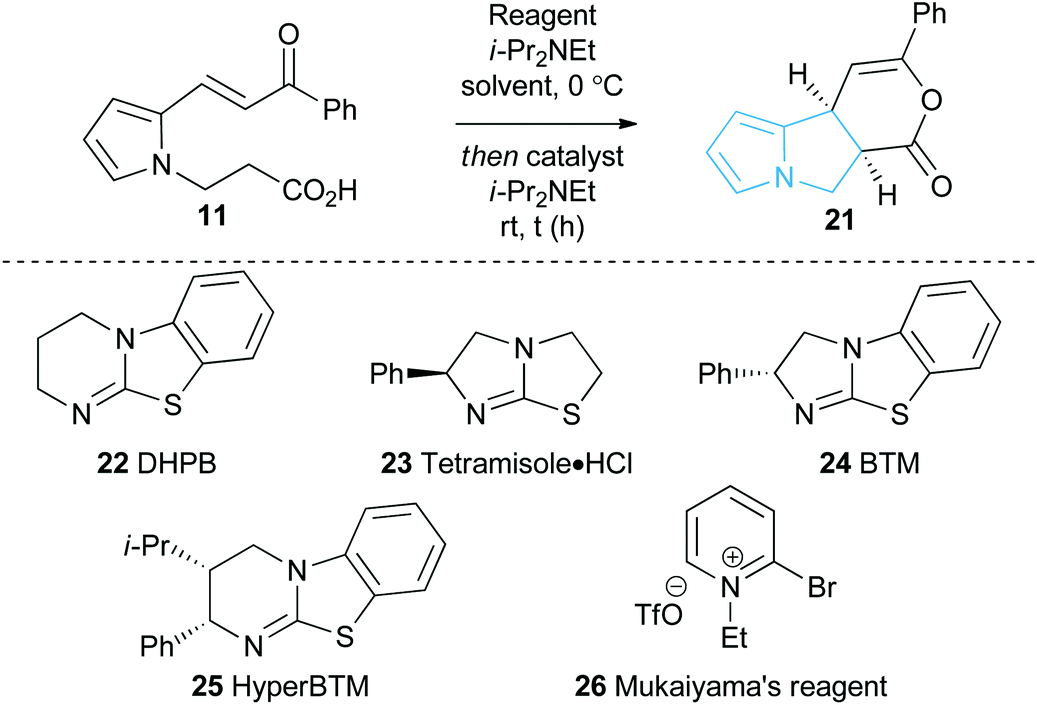
With a reliable synthetic route to pyrrole-derived enone acid substrates in hand, the feasibility and subsequent optimisation of the isothiourea-catalysed Michael addition-lactonisation protocol was conducted (Table 1). Utilising achiral isothiourea DHPB 22, i-Pr2NEt and t-BuCOCl provided the desired pyrrolizine dihydropyranone 21 from enone-acid 11 in a moderate 41% isolated yield but with excellent >95:5 dr (entry 1). Alternative methods for in situ generation of a reactive carboxylate derivative, such as Mukaiyama's reagent 26, did not improve conversion or isolated yield (entry 2). A significant improvement in yield was observed when the equivalents of both t-BuCOCl and i-Pr2NEt were increased from 1.5 to 3.0 equiv., with 21 obtained in 84% yield. Subsequent studies assessed the feasibility of an enantioselective process, with chiral isothiourea catalysts 23–25 examined. Tetramisole·HCl 23 and benzotetramisole (BTM) 24-mediated reactions (entries 4 and 5) provided 21 in excellent yield, >95:5 dr and with >99:1 er. Application of HyperBTM 25 proved marginally less effective, with the product 21 obtained in high yield and dr but with reduced 96.5:3.5 er (entry 6). Notably in all of these catalytic processes no competitive products arising from either Friedel–Crafts acylation or β-elimination were observed, despite the proposed intermediacy of an acyl ammonium ion. The effect of reduced catalyst loading using BTM 24 and tetramisole·HCl 23 was next evaluated. Using 5 mol% BTM 24 maintained excellent levels of diastereo- and enantioselectivity, giving product 21 in 82% yield, while using 5 mol% tetramisole·HCl 23 led to reduced but still acceptable 67% yield (entries 7 and 8). Further reduction of the catalyst loading to 1 mol% showed the same trend in reactivity, with BTM-24 giving 21 in 42% isolated yield, and tetramisole·HCl 23 a lower 34% yield, yet still in high dr and er (entries 9 and 10). All further studies used BTM 24 (5 mol%) as the optimal reaction conditions for this enantioselective process.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagenta (equiv.) | Catalyst (mol%) | Yieldb (%) | drc | erd |
|
a Applied in 1:1 ratio with i-Pr2NEt.
b Isolated yield.
c Determined by 1H NMR of the crude reaction product.
d Determined by chiral HPLC.
|
|||||
| 1 | t-BuCOCl (1.5) | 22 (10) | 41 | >95:5 |
— |
| 2 | 26 (1.5) | 22 (10) | 45 | >95:5 |
— |
| 3 | t-BuCOCl (3.0) | 22 (10) | 84 | >95:5 |
— |
| 4 | t-BuCOCl (3.0) | 23 (10) | 84 | >95:5 |
>99:1(ent) |
| 5 | t-BuCOCl (3.0) | 24 (10) | 81 | >95:5 |
>99:1 |
| 6 | t-BuCOCl (3.0) | 25 (10) | 81 | >95:5 |
96.5:3.5 |
| 7 | t-BuCOCl (3.0) | 24 (5) | 82 | >95:5 |
>99:1 |
| 8 | t-BuCOCl (3.0) | 23 (5) | 67 | >95:5 |
>99:1(ent) |
| 9 | t-BuCOCl (3.0) | 24 (1) | 42 | >95:5 |
>99:1 |
| 10 | t-BuCOCl (3.0) | 23 (1) | 34 | >95:5 |
99:1(ent) |
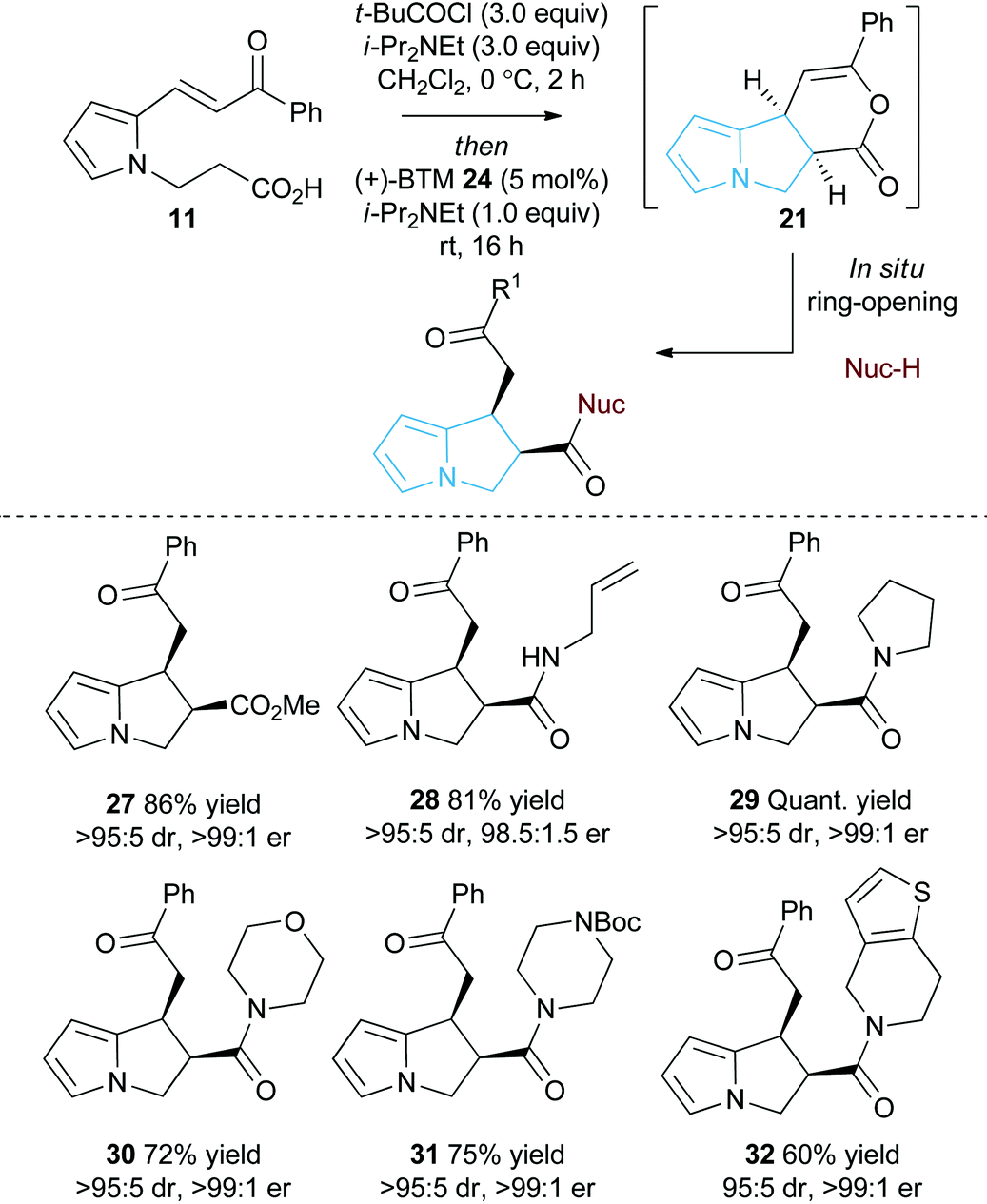
As an alternative strategy to the isolation of dihydropyranone 21in situ ring-opening with a suitable nucleophile was investigated to provide access to pyrrolizine carboxylate derivatives (Table 2). Taking pyrrole-derived enone-acid 11 under the optimum catalysis conditions the resulting dihydropyranone 21 can be readily ring-opened in situ with MeOH giving pyrrolizine methyl ester 27 in excellent 86% yield, >95:5 dr and >99:1 er. Ring-opening using both primary and secondary amines to give pyrrolizine amides also proved successful. For example, use of allylamine provided 28 in 81% yield, >95:5 dr and 98.5:1.5 er, while ring-opening with pyrrolidine gave pyrrolizine amide 29 in quantitative yield, >95:5 dr and >99:1 er. Morpholine, N-Boc piperazine and tetrahydrothieno[3,2-c]pyridine could also be utilised, giving the corresponding pyrrolizine amides 30–32 in good to excellent yield (60–75%), >95:5 dr and >99:1 er. Upon standing the pyrrolizine carboxylate products proved considerably more stable to storage than the dihydropyranone 21, therefore ring-opening with MeOH was used as the general procedure when exploring further substrate scope.
Substrate scope: Michael addition-lactonisation/methanolysis
With a reliable synthetic route to pyrrole-derived enone acid substrates available, the generality of the isothiourea catalysis using BTM 24 as the catalyst and MeOH as the nucleophile for in situ ring-opening was evaluated (Table 3). Variation of the enone-substituent of the substrate was first investigated with a range of aryl groups accommodated. Brominated aryl units can be included to give 33 and 34 in 78% and 74% yield, respectively, with both generated as single diastereoisomers in >99:1 er. Incorporation of the heterocyclic 2-thienyl substituent was readily tolerated giving 35 in 77% yield, >95:5 dr and 99:1 er. Electron-rich groups (4-MeOC6H4) can be installed, giving 36 in excellent 93% yield, >95:5 dr and >99:1 er.
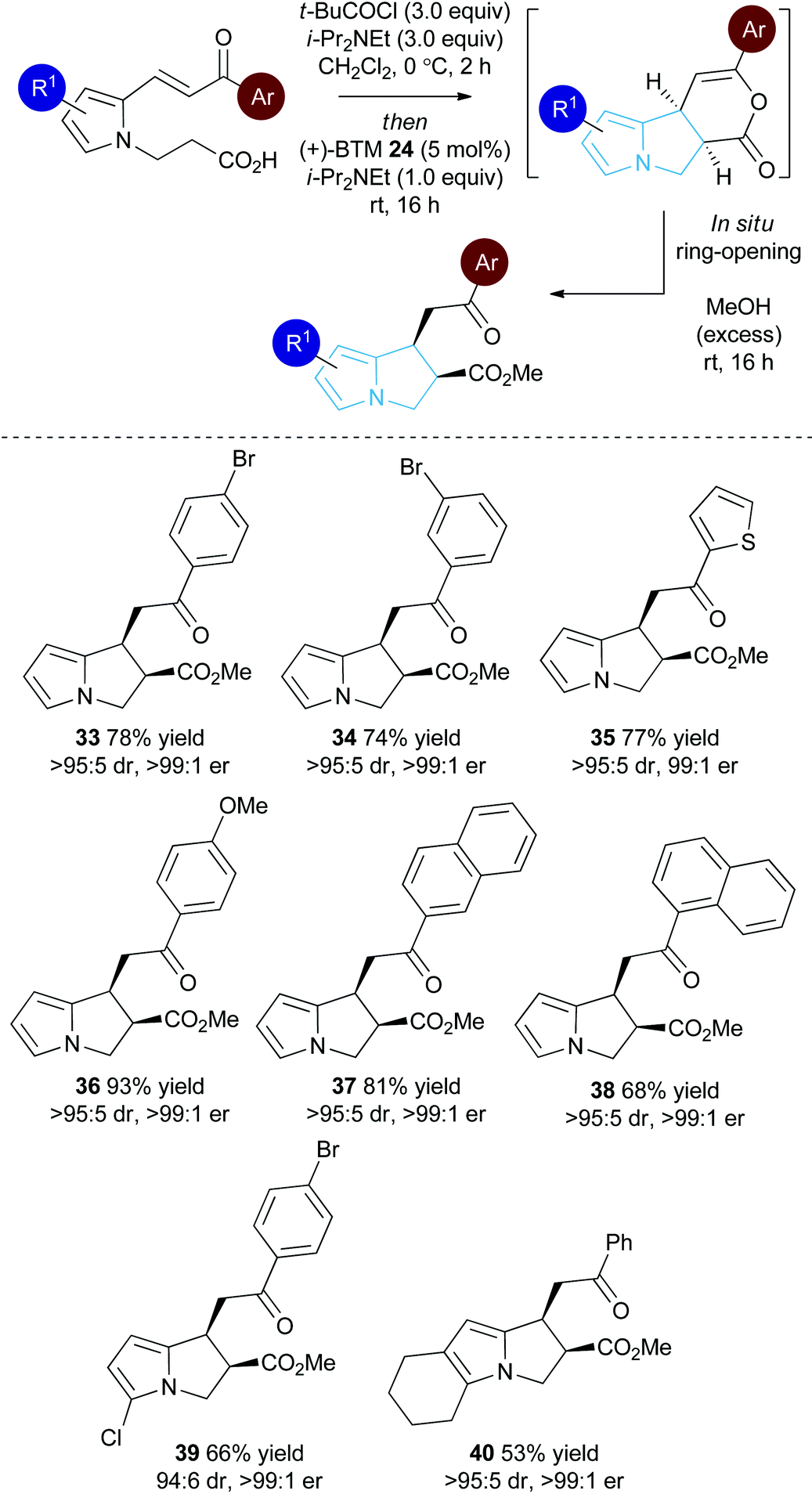
Both 1-naphthyl and 2-naphthyl units can also be incorporated to access the corresponding products 37 and 38 in good yields and excellent enantioselectivity, although attempted catalysis using Me-enone 20 did not result in any conversion to product. Variation within the pyrrole core of the pyrrolizine carboxylate product was also assessed. Chlorinated product 39 was produced in 66% yield with good 94:6 dr and as a single enantiomer (>99:1 er). The core motif can be expanded to the hexahydro-1H-pyrroloindole structure with the corresponding product 40 achieved in 53% yield and excellent stereoselectivity (>95:5 dr and >99:1 er).27 The reaction to form 33 was readily carried out on a 1 gram scale, giving the desired product in 67% yield in >95:5 dr and >99:1 er. The relative and absolute configuration within 33 was assigned by X-ray crystallography analysis, with the configuration within all other products assigned by analogy (Fig. 4).28
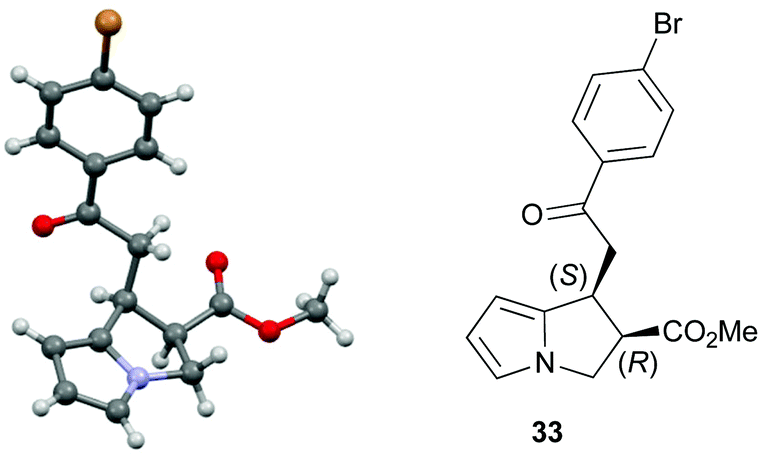 | ||
| Fig. 4 Molecular representation of the X-ray structure of 33. | ||
Product derivatisation
To exemplify that this methodology can potentially be used as a basis for further synthetic elaboration, product derivatisation within both the pyrrole core and aromatic ketone substituent was explored (Scheme 2). For example, the synthesis of product 33 using this cascade methodology gives a product with the bromine functional handle. Through the use of a Suzuki–Miyaura coupling 33 was readily elaborated to access the polyheteroaromatic pyrrolizine 41 in 60% isolated yield and with no loss of stereointegrity. Alternatively, a simple chlorination of the pyrrole core within 27 was conducted using NCS to access chloropyrrolizine 42 in 79% yield.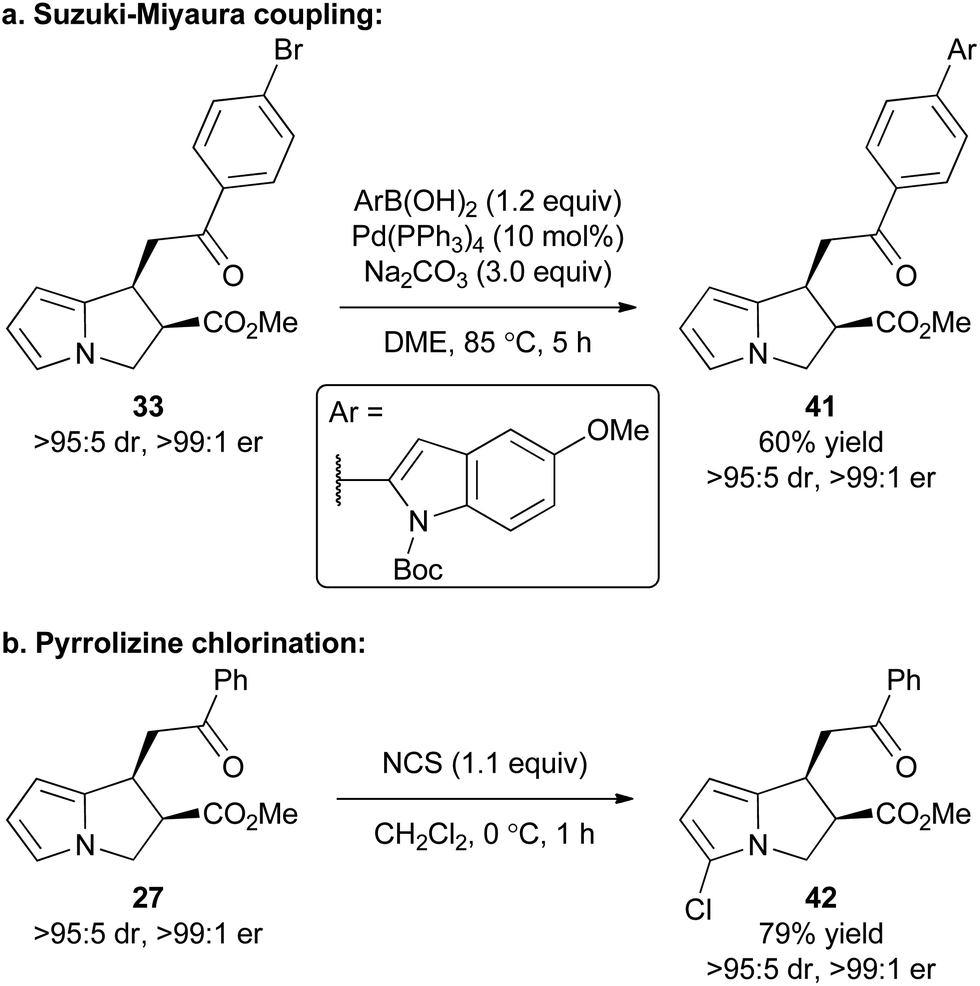 | ||
| Scheme 2 Product derivatisation. | ||
Proposed mechanism and computational studies
A proposed catalytic cycle for the synthesis of pyrrolizine dihydropyranone 21 from enone-acid 11 is shown in Scheme 3. Firstly, pivaloyl chloride reacts with the enone-acid to form mixed anhydride 43, with subsequent nucleophilic attack from the Lewis base catalyst BTM 24 generating acyl ammonium ion 44. Deprotonation to form (Z)-ammonium enolate 45, followed by intramolecular Michael-addition forms the new C–C bond and two stereocentres in the initial cyclisation step. Intermediate 45 is believed to be stabilised by a favourable non-bonding or electrostatic O to S interaction and this has been investigated computationally (vide infra). Subsequent lactonisation releases catalyst 24 and the polycyclic dihydropyranone product 21, which can be ring-opened upon addition of a nucleophilic amine or alcohol to give the isolated pyrrolizine carboxylate derivative.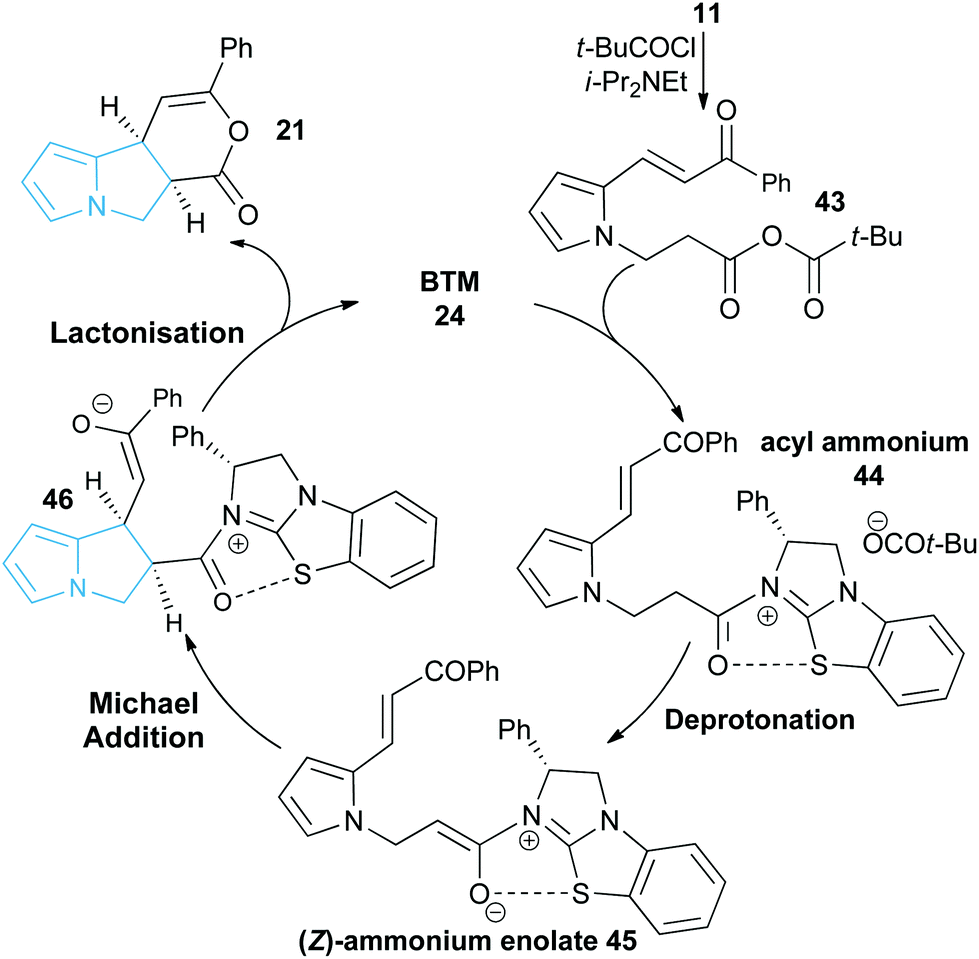 | ||
| Scheme 3 Proposed mechanism of isothiourea-catalysed Michael addition-lactonisation. | ||
On the basis of this mechanistic hypothesis, the origin of the diastereo- and enantioselective formation of the pyrrolizine dihydropyranone products in this BTM 24-catalysed cascade process was further probed through density functional theory (DFT) calculations. Calculations employed the M06-2X functional and investigated the cyclisation of the parent enone-acid 21 reacting in the presence of 24 in dichloromethane solvent from either the (E)- or (Z)-form of the ammonium enolate. Full computational details, including results obtained with the B3LYP functional, are supplied in the ESI.†29
Fig. 5 shows computed profiles for the alternative reactions of the (Z)-ammonium enolate, (Z)-45, to form the cis- and trans-isomers of pyrrolizine dihydropyranone 21. From (Z)-45 two low energy transition states were located for the Michael cyclisation step: TS1cis at +5.2 kcal mol−1 and TS1trans at +10.0 kcal mol−1. The onward reaction viaTS1cis (highlighted in red) leads to 46cis at −0.6 kcal mol−1 in which the newly formed C4–C9 bond is rather long at 1.62 Å. Attack of the enolate oxygen then permits formation of a zwitterionic tetrahedral dihydropyran intermediate 47cisviaTS2cis at +6.3 kcal mol−1. Facile dissociation of BTM 24 from 47cisviaTS3cis forms 21cis at −18.2 kcal mol−1. An analogous series of steps (highlighted in blue) was also characterized for the formation of 21trans at −13.0 kcal mol−1. Formation of 21cis is therefore both thermodynamically and kinetically favoured, as both TS1cis and TS2cis are significantly lower than TS1transen route to 21trans. A third pathway starting from the (E)-ammonium enolate precursor, (E)-45 (+3.7 kcal mol−1) and leading to the enantiomeric-trans product was also characterised, but has a significantly higher barrier of 16.7 kcal mol−1 and can therefore be discounted (see ESI†).
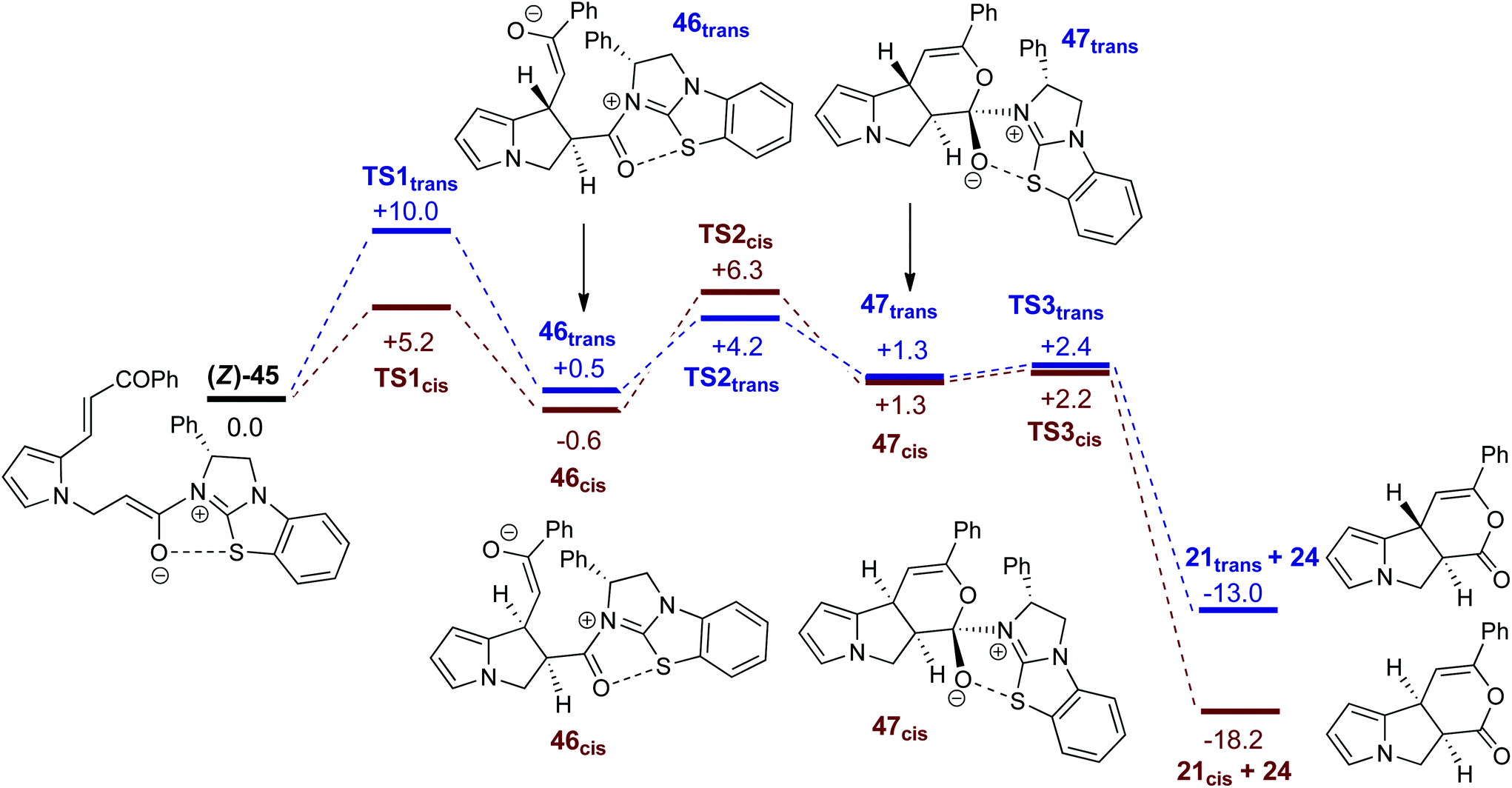 | ||
| Fig. 5 Computed free energy profiles (M06-2X(CH2Cl2), kcal mol−1) for the formation of cis- and trans-pyrrolizine dihydropyranones 21 from (Z)-ammonium enolate (Z)-45. | ||
It is notable that all the computed structures for both reaction pathways in Fig. 5 from enolate (Z)-45 up to the final BTM dissociation steps feature a co-planar [1,5]-S⋯O motif with [1,5]-S⋯O interatom distances ranging from 2.60 Å to 2.82 Å. The importance of non-bonding S⋯O interactions has been widely recognized in structural and medicinal chemistry in the solid state (often described as a stabilising nO to 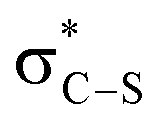 interaction),30 and has been previously recognised as a controlling element in enantioselective isothiourea-catalyzed reaction processes.31 Although the origin of this interaction is still under debate,32 in all calculated structures the [1,5]-S⋯O distance is considerably below the sum of the van der Waals radii. Interestingly, the shortest distances calculated are found within (Z)-45 (2.64 Å) and zwitterionic intermediates (47cis 2.60 Å and 47trans 2.64 Å), presumably reflecting the formal negative charge on the oxygen atom within these structures. However, as this feature appears within all the structures in the computed reaction profiles, and varies in a similar way along those profiles, it does not appear to be a discriminating factor between the two pathways in Fig. 5.
interaction),30 and has been previously recognised as a controlling element in enantioselective isothiourea-catalyzed reaction processes.31 Although the origin of this interaction is still under debate,32 in all calculated structures the [1,5]-S⋯O distance is considerably below the sum of the van der Waals radii. Interestingly, the shortest distances calculated are found within (Z)-45 (2.64 Å) and zwitterionic intermediates (47cis 2.60 Å and 47trans 2.64 Å), presumably reflecting the formal negative charge on the oxygen atom within these structures. However, as this feature appears within all the structures in the computed reaction profiles, and varies in a similar way along those profiles, it does not appear to be a discriminating factor between the two pathways in Fig. 5.
Computed structures for the selectivity-determining transition states TS1cis and TS1trans are compared in Fig. 6. Both structures have similar distances (2.09 and 2.10 Å) for the forming C4⋯C9 bond, with Michael addition occurring anti-to the stereodirecting phenyl substituent and thus accounting for the observed enantioselectivity. In the favoured TS1cis arrangement the prostereogenic centres adopt an approximately eclipsed conformation with a H–C4⋯C9–H dihedral of 3°, while in TS1trans the corresponding H–C4⋯C9–H dihedral is 149.5°. Both transition states also exhibit short and approximately co-planar [1,5]-S⋯O1 contacts of ca. 2.7 Å between the BTM sulfur atom and the enolate oxygen. The near-parallel arrangement of the BTM moiety and the enone aryl substituent in TS1cis (inter-plane angle = 5.7°) appears set up for stabilising π-stacking interactions that should be captured by the M06-2X functional.29 Indeed with the B3LYP functional (where such dispersion effects are not treated) the equivalent inter-plane angle = 28.1° and the preference for TS1cis is reduced to only 1.4 kcal mol−1 (see ESI†). Also noticeable in TS1cis are two short contacts between the forming oxy-anion on the enone (O2) and two C–H hydrogens upon the positively charged isothiouronium core (O1⋯H1 2.19 Å and O1⋯H2 2.37 Å). The enhanced acidity of these hydrogen atoms may facilitate some non-classical H-bonding and so confer greater stability on TS1cis over TS1trans where the analogous contacts are distinctly longer (O1⋯H1 2.72 Å and O1⋯H2 2.65 Å). While not particularly strong individually, these various effects likely combine to stabilise TS1cis over TS1trans and so account for the observed selectivity for the cis product.
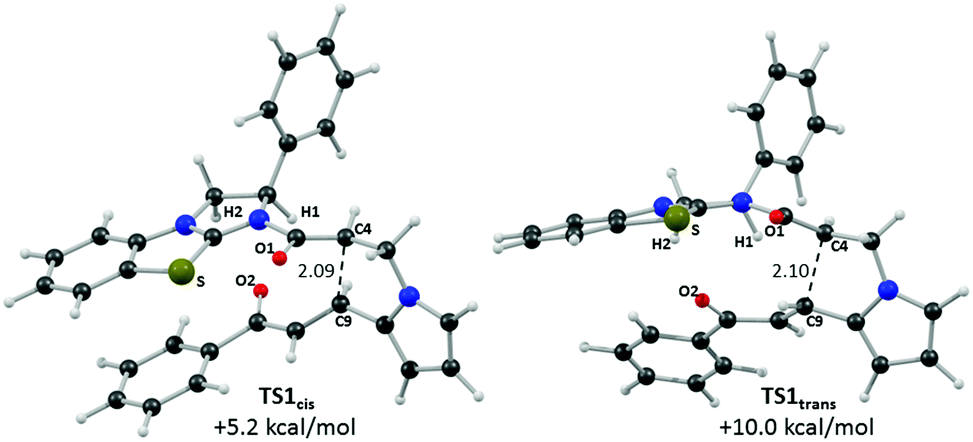 | ||
| Fig. 6 Computed selectivity determining transition states TS1cis and TS1trans with key atom labels and the forming C4⋯C9 distance highlighted in Å. | ||
Conclusions
In conclusion, an optimised and straightforward three-step synthetic route to a range of pyrrole-derived enone acid starting materials from readily available pyrrole-2-carboxaldehydes is delineated. The catalytic enantioselective synthesis of a range of cis-pyrrolizine carboxylate derivatives proceeds with outstanding stereocontrol (14 examples, >95:5 dr, >98:2 er) with benzotetramisole proving the optimal catalyst for this process. In situ ring-opening of the pyrrolizine dihydropyranone products with either MeOH or a range of amines leads to the desired products in excellent yield and enantioselectivity. Computation has been used to probe the factors leading to high stereocontrol in this reaction process, with the formation of the observed cis-stereoisomer predicted to be both kinetically and thermodynamically favoured. Further work from this laboratory will utilise this methodology for the synthesis of target molecules and probe the utility of isothioureas and other Lewis bases in enantioselective catalysis.
Acknowledgements
We thank Syngenta and the EPSRC (grant code EP/K503162/1) (DGS), and the EPSRC Centre for Doctoral Training in Critical Resource Catalysis (CRITICAT, grant code EP/L016419/1) (ERG, SFM, RWFK) for funding. The European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013) ERC Grant Agreement No. 279850 is also acknowledged (JET). ADS thanks the Royal Society for a Wolfson Research Merit Award. We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University.Notes and references
- (a) H. Ulbrich, B. Fiebich and G. Dannhardt, Eur. J. Med. Chem., 2002, 37, 953 CrossRef CAS PubMed; (b) S. E. Abbas, F. M. Awadallah, N. A. Ibrahim and A. M. Gouda, Eur. J. Med. Chem., 2010, 45, 482 CrossRef CAS PubMed; (c) A. M. Gouda, A. H. Abdelazeem, E.-S. A. Arafa and K. R. A. Abdellatif, Bioorg. Chem., 2014, 53, 1 CrossRef CAS PubMed; (d) A. Belal and B. E.-D. M. El-Gendy, Bioorg. Med. Chem., 2014, 22, 46 CrossRef CAS PubMed.
- (a) W. F. Haddon, J. L. Dallas, D. W. Wilson and H. J. Segall, Science, 1985, 229, 472 Search PubMed; (b) R. L. Reed, K. G. Ahern, G. D. Pearson and D. R. Buhler, Carcinogenesis, 1988, 9, 1355 CrossRef CAS PubMed.
- L. W. Smith and C. C. J. Culvenor, J. Nat. Prod., 1981, 44, 129 CrossRef CAS.
- (a) R. Schoental and P. N. Magee, J. Pathol. Bacteriol., 1957, 74, 305 CrossRef CAS; (b) E. K. McLean, Pharmacol. Rev., 1970, 22, 420 Search PubMed.
- K. Kuhara, J. Takanashi, I. Hirono, T. Furuya and Y. Asada, Cancer Lett., 1980, 10, 117 CrossRef CAS PubMed.
- (a) C. R. Green and G. S. Christie, Br. J. Exp. Pathol., 1961, 42, 369 CAS; (b) J. E. Peterson and M. V. Jago, J. Pathol., 1980, 131, 339 CrossRef CAS PubMed.
- (a) C. E. Green, H. J. Segall and J. L. Byard, Toxicol. Appl. Pharmacol., 1980, 60, 176 CrossRef; (b) G. M. Williams, H. Mori, I. Hirono and M. Nagao, Mutat. Res., 1980, 79, 1 CrossRef CAS PubMed.
- (a) M. M. Paz, Antitumour and Anitbiotics, in Anticancer Therapeutics, ed. S. Missailidis, John Wiley and Sons, Chichester, UK, 2008, pp. 112–115 Search PubMed; (b) M. Tomasz, Chem. Biol., 1995, 2, 575 CrossRef CAS PubMed.
- W. Liu, J. Zhou, K. Bensdorf, H. Zhang, H. Liu, Y. Wang, H. Qian, Y. Zhang, A. Wellner, G. Rubner, W. Huang, C. Guo and R. Gust, Eur. J. Med. Chem., 2011, 46, 907 CrossRef CAS PubMed.
- A. Gusman, F. Yuste, R. A. Toscano and J. M. Young, J. Med. Chem., 1986, 29, 589 CrossRef CAS.
- K. Stevens and J. Robertson, Nat. Prod. Rep., 2014, 31, 1721 RSC.
- The research data underpinning this manuscript can be found at DOI:10.17630/2e866671-4a64-401a-8305-438f7dc60464.
- Y. Gu, P. Hu, C. Ni and X. Tong, J. Am. Chem. Soc., 2015, 137, 6400 CrossRef CAS PubMed.
- K. Sugimoto, N. Yamamoto, D. Tominaga and Y. Matsuya, Org. Lett., 2015, 17, 1320 CrossRef CAS PubMed.
- J.-Y. Bae, H.-J. Lee, S.-H. Youn, S.-H. Kwon and C.-W. Cho, Org. Lett., 2010, 12, 4352 CrossRef CAS PubMed.
- (a) G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945 CrossRef CAS PubMed; (b) G. S. Cortez, S. H. Oh and D. Romo, Synthesis, 2001, 173 Search PubMed; (c) S. H. Oh, G. S. Cortez and D. Romo, J. Org. Chem., 2005, 70, 2835 CrossRef CAS PubMed.
- (a) X. Li, K. Gai, Z. Yuan, J. Wu, A. Lin and H. Yao, Adv. Synth. Catal., 2015, 357, 3479–3484 CrossRef CAS; (b) K. Schwartz, J. L. Amos, J. C. Klein, D. T. Do and T. N. Snaddon, J. Am. Chem. Soc., 2016, 138, 5214–5217 CrossRef PubMed.
- For seminal work on isothiourea catalysis in kinetic resolution and acyl transfer reaction processes see: (a) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef CAS PubMed; (b) V. B. Birman, H. Jiang, X. Li, L. Guo and E. W. Uffman, J. Am. Chem. Soc., 2006, 128, 6536–6537 CrossRef CAS PubMed; (c) M. Kobayashi and S. Okamoto, Tetrahedron Lett., 2006, 47, 4347–4350 CrossRef CAS; (d) V. B. Birman and X. Li, Org. Lett., 2008, 10, 1115–1118 CrossRef CAS PubMed; (e) Y. Zhang and V. B. Birman, Adv. Synth. Catal., 2009, 351, 2525–2529 CrossRef CAS PubMed; (f) C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS PubMed. For recent reviews, see: (g) L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC; (h) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC.
- (a) S. R. Smith, J. Douglas, H. Prevet, P. Shapland, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1626–1639 CrossRef CAS PubMed; (b) L. C. Morrill, S. M. Smith, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1640–1655 CrossRef CAS PubMed.
- (a) L. Hesping, A. Biswas, C. G. Daniliuc, C. Muck-Lichtenfeld and A. Studer, Chem. Sci., 2015, 6, 1252–1257 RSC; (b) S. R. Smith, C. Fallan, J. E. Taylor, R. McLennan, D. S. B. Daniels, L. C. Morrill, A. M. Z. Slawin and A. D. Smith, Chem. – Eur. J., 2015, 21, 10530–10536 CrossRef CAS PubMed.
- For select examples see: (a) D. G. Stark, T. J. C. O'Riordan and A. D. Smith, Org. Lett., 2014, 16, 6496–6499 CrossRef CAS PubMed; (b) S. R. Smith, S. M. Leckie, R. Holmes, J. Douglas, C. Fallan, P. Shapland, D. Pryde, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2014, 16, 2506–2509 CrossRef CAS PubMed; (c) P.-P. Yeh, D. S. B. Daniels, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2014, 16, 964–967 CrossRef CAS PubMed; (d) L. C. Morrill, L. A. Ledingham, J.-P. Couturier, J. Bickel, A. D. Harper, C. Fallan and A. D. Smith, Org. Biomol. Chem., 2014, 12, 624–636 RSC; (e) D. G. Stark, L. C. Morrill, P.-P. Yeh, A. M. Z. Slawin, T. J. C. O'Riordan and A. D. Smith, Angew. Chem., Int. Ed., 2013, 52, 11642–11646 CrossRef CAS PubMed; (f) L. C. Morrill, J. Douglas, T. Lebl, A. M. Z. Slawin, D. J. Fox and A. D. Smith, Chem. Sci., 2013, 4, 4146–4155 RSC; (g) L. C. Morrill, T. Lebl, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2012, 3, 2088–2093 RSC; (h) C. Simal, T. Lebl, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2012, 51, 3653–3657 CrossRef CAS PubMed; (i) D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef CAS PubMed.
- For a selection of alternative isothiourea-catalysed processes see below. For reactions utilising α,β-unsaturated acyl ammonium intermediates see: (a) E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193–2200 RSC; (b) S. Vellalath, K. N. Van and D. Romo, Angew. Chem., Int. Ed., 2013, 52, 13688–13693 CrossRef CAS PubMed; (c) G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049–1057 CrossRef CAS PubMed; (d) Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320–5323 CrossRef CAS PubMed . For ammonium ylide intermediates see: T. H. West, D. S. B. Daniels, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476–4479 Search PubMed . For seminal studies utilising acyl ammonium intermediates see ref. 17.
- For pyrrolidines see: (a) D. Belmessieri, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2013, 15, 3472 CrossRef CAS PubMed; For THFs see: (b) D. Belmessieri, A. de la Houpliere, E. D. D. Calder, J. E. Taylor and A. D. Smith, Chem. – Eur. J., 2014, 20, 9762–9769 CrossRef CAS PubMed.
- (a) H. F. Motiwala, R. H. Vekariya and J. Aube, Org. Lett., 2015, 17, 5484–5487 CrossRef CAS PubMed; (b) J. E. Taylor, M. D. Jones, J. M. J. Williams and S. D. Bull, Org. Lett., 2010, 12, 5740–5743 CrossRef CAS PubMed; (c) M. Sechi, A. Mura, L. Sannia, M. Orecchioni and G. Paglietti, ARKIVOC, 2004, 5, 97–106 Search PubMed; (d) B. Sayah, N. Pelloux-Leon and Y. Vallee, J. Org. Chem., 2000, 65, 2824–2826 CrossRef CAS PubMed.
- For select computational analyses of alternative isothiourea-catalysed reaction processes that involve acyl ammonium intermediates for kinetic resolutions see: (a) I. Shiina, K. Nakata, K. Ono, Y.-S. Onda and M. Itagaki, J. Am. Chem. Soc., 2010, 132, 11629–11641 CrossRef CAS PubMed; (b) X. Yang, V. D. Bumbu, P. Liu, X. Li, H. Jiang, E. W. Uffman, L. Guo, W. Zhang, X. Jiang, K. N. Houk and V. B. Birman, J. Am. Chem. Soc., 2012, 134, 17605–17612 CrossRef CAS PubMed; (c) X. Yang, P. Liu, K. N. Houk and V. B. Birman, Angew. Chem., Int. Ed., 2012, 51, 9638–9642 CrossRef CAS PubMed.
- See ESI† for full details.
- The enone-acid substrate 18 proved difficult to purify to homogeneity and so was applied to the catalysis in crude form after ester hydrolysis. This naturally leads to a lower isolated yield of product 40 as this is the yield isolated in 3 steps from the ester substrate.
- The crystallographic data obtained for 33 has been deposited with the Cambridge Crystallographic Data Centre and the supplementary data can be found via CCDC 1483759.
- For recent reviews of the computational modelling of organocatalysis and the importance of dispersion interactions in such systems see: (a) S. E. Wheeler, T. J. Seguin, Y. Guan and A. C. Doney, Acc. Chem. Res., 2016, 49, 1061 CrossRef CAS PubMed; (b) D. M. Walden, O. M. Ogba, R. C. Johnston and P. H.-Y. Cheong, Acc. Chem. Res., 2016, 49, 1279 CrossRef CAS PubMed.
- (a) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed; (b) R. C. Reid, M.-K. Yau, R. Singh, J. Lim and D. P. Fairlie, J. Am. Chem. Soc., 2014, 136, 11914–11917 CrossRef CAS PubMed; (c) F. T. Burling and B. M. Goldstein, J. Am. Chem. Soc., 1992, 114, 2313–2320 CrossRef CAS.
- For the initial postulate of 1,5-S⋯O interactions as a control element in isothiourea catalysis see:
(a) V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS PubMed. For other manuscripts of interest see:
(b) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef CAS PubMed;
(c) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef CAS PubMed . Romo and Tantillo (ref. 31b) have probed the nature of 1,5-S⋯O interactions of α,β-unsaturated acyl ammonium species with NBO and postulate this interaction is due to a number of orbital interactions. In particular, unfavourable
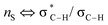 interactions disfavour alternative conformations with an O–C–N–C dihedral angle of 180°.
interactions disfavour alternative conformations with an O–C–N–C dihedral angle of 180°. - For a selection of discussions on the nature and relevance of these types of interaction see: (a) X. Zhang, Z. Gong, J. Li and T. Lu, J. Chem. Inf. Model., 2015, 55, 2138–2153 CrossRef CAS PubMed; (b) J. G. Ángyán, Á. Kucsman, R. A. Poirier and I. G. Csizmadia, J. Mol. Struct.: THEOCHEM, 1985, 123, 189–201 CrossRef; (c) J. S. Murray, P. Lane and P. Politzer, Int. J. Quantum Chem., 2008, 108, 2770–2781 CrossRef CAS; (d) M. Iwaoka, S. Takemoto and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 10613–10620 CrossRef CAS PubMed; (e) K. A. Brameld, B. Kuhn, D. C. Reuter and M. Stahl, J. Chem. Inf. Model., 2008, 48, 1–24 CrossRef CAS PubMed.

Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, HPLC analysis and computational co-ordinates. Data available.12 CCDC 1483759. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01557c |
| This journal is © The Royal Society of Chemistry 2016 |