
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective isothiourea-catalysed trans-dihydropyridinone synthesis using saccharin-derived ketimines: scope and limitations†
Daniel G.
Stark
a,
Claire M.
Young
a,
Timothy J. C.
O'Riordan
b,
Alexandra. M. Z.
Slawin
a and
Andrew D.
Smith
*a
aEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
bSyngenta, Jealott's Hill International Research Centre, Bracknell, RG42 6EY, UK
First published on 26th July 2016
Abstract
The catalytic enantioselective synthesis of a range of trans-dihydropyridinones from aryl-, heteroaryl- and alkenylacetic acids and saccharin-derived ketimines with good to excellent stereocontrol (15 examples, up to >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr, up to >99:1 er) is reported. After extensive optimisation, HyperBTM proved the optimal isothiourea catalyst for this transformation at −78 °C, giving trans-dihydropyridones with generally excellent levels of diastereo- and enantioselectivity.
:5 dr, up to >99:1 er) is reported. After extensive optimisation, HyperBTM proved the optimal isothiourea catalyst for this transformation at −78 °C, giving trans-dihydropyridones with generally excellent levels of diastereo- and enantioselectivity.
Introduction
Saccharin (1,2-benzisothiazol-3-one-1,1-dioxide) 1 is a synthetic calorie-free additive, widely used as a sugar substitute in many food products and has proven an important discovery in the fight against diabetes.1 The cyclic sulfonamide core motif embedded within saccharin has attracted much interest in recent decades from the medicinal chemistry community, with this motif a key constituent in many biologically active drugs (Fig. 1a). For example, saccharin-based sultams such as Ipsaspirone 2 are active agonists of 5-HT1A receptors and have been applied as neuroprotectants and anxiolytics.2 Current research within this area has led to the development of saccharin derivatives as inhibitors of carbonic anhydrase enzymes.3 Similarly, related cyclic sulfonamides such as Ampiroxicam 3 are bioactive.4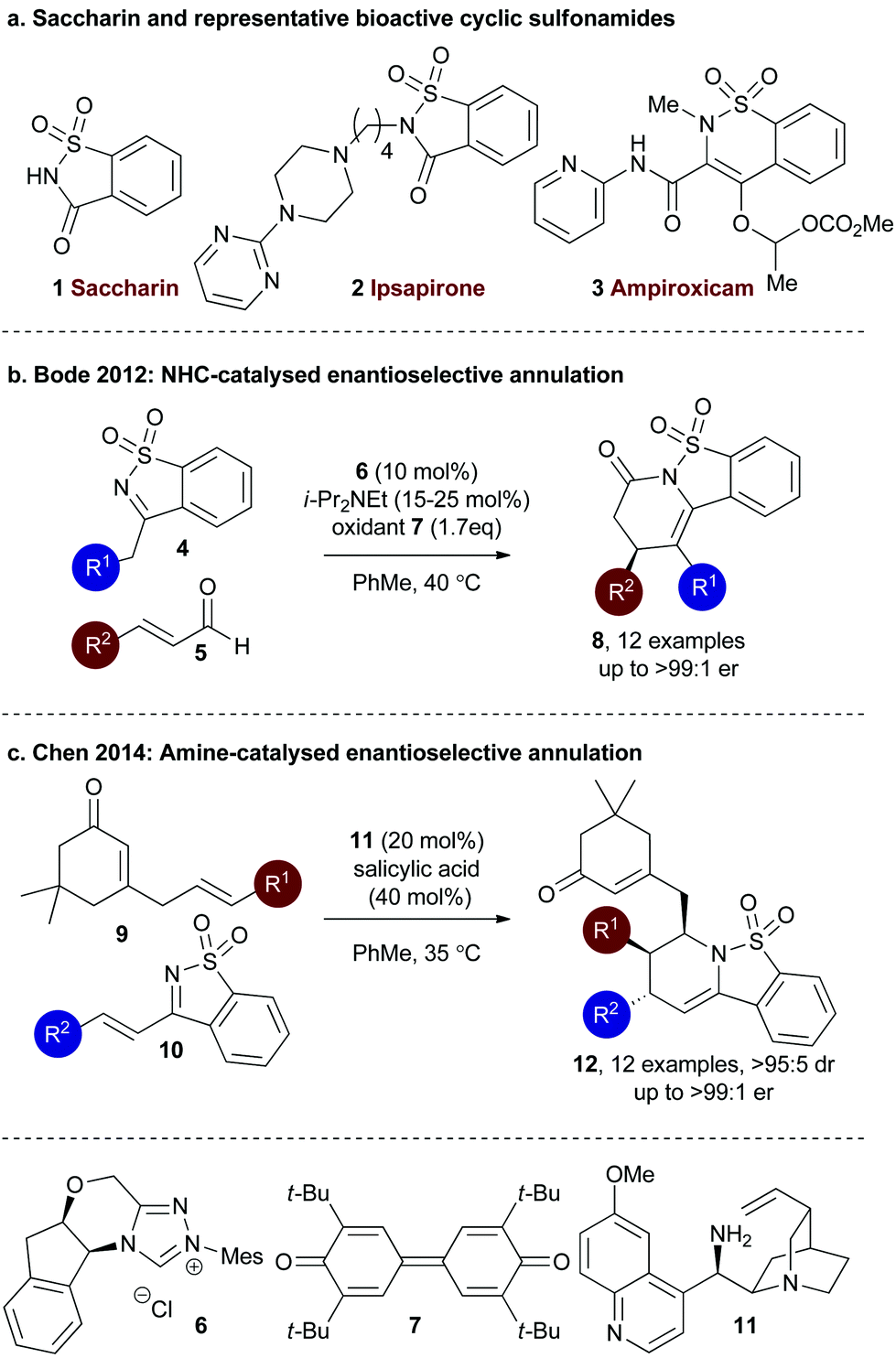 | ||
| Fig. 1 Representative bioactive sultams and enantioselective organocatalytic strategies using saccharin derivatives to prepare cyclic sulfonamides. | ||
A number of enantioselective organocatalytic strategies have been explored to access chiral sultam products that incorporate the saccharin motif. For example, in 2012 Bode and co-workers developed an NHC-catalysed enantioselective annulation process utilising sulfonyl imine 4 and enals 5, giving tricylic sultams 7 in good to excellent yield (67–94%) and excellent enantioselectivity (90:10 to >99:1 er) using monosubstituted enals (Fig. 1b).5 Alternatively, Chen and co-workers have investigated an aza Diels–Alder reaction using organocatalytically-generated trienamines. Cyclic sulfonyl imine 10 and cinchona alkaloid 11 (20 mol%) in the presence of salicylic acid generates a trienamine intermediate that can react through the δ,ε-alkene in an inverse electron demand Diels–Alder reaction with cyclic sulfonyl imines 9 to give products 12 in excellent diastereo- and enantioselectivity (>95:5 dr and 98:2 to >99:1 er, Fig. 1c).6
Following the pioneering nucleophile catalysed aldol lactonisation (NCAL) work of Romo and co-workers using carboxylic acids as ammonium enolate precursors,8 we developed the use of isothioureas9 for enantioselective Michael addition lactonisation processes directly from carboxylic acids.10 The generality of this concept has been extended to a range of formal intermolecular [4 + 2],11 [3 + 2]12 and [2 + 2]13 cycloaddition processes from carboxylic acids or anhydride starting materials (Fig. 2a).14 Of particular relevance to this manuscript we have previously accessed the dihydropyridinone motif from arylacetic acids through enantioselective Michael addition lactamisation using acyclic ketimines derived from chalcones15 and α,β-unsaturated γ-ketoesters.16 Based upon this work, in this manuscript the use of saccharin-derived cyclic ketimines as suitable substrates for the enantioselective preparation of polycyclic dihydropyridinones from aryl-, heteroaryl-, and alkenylacetic acids is investigated (Fig. 2b).
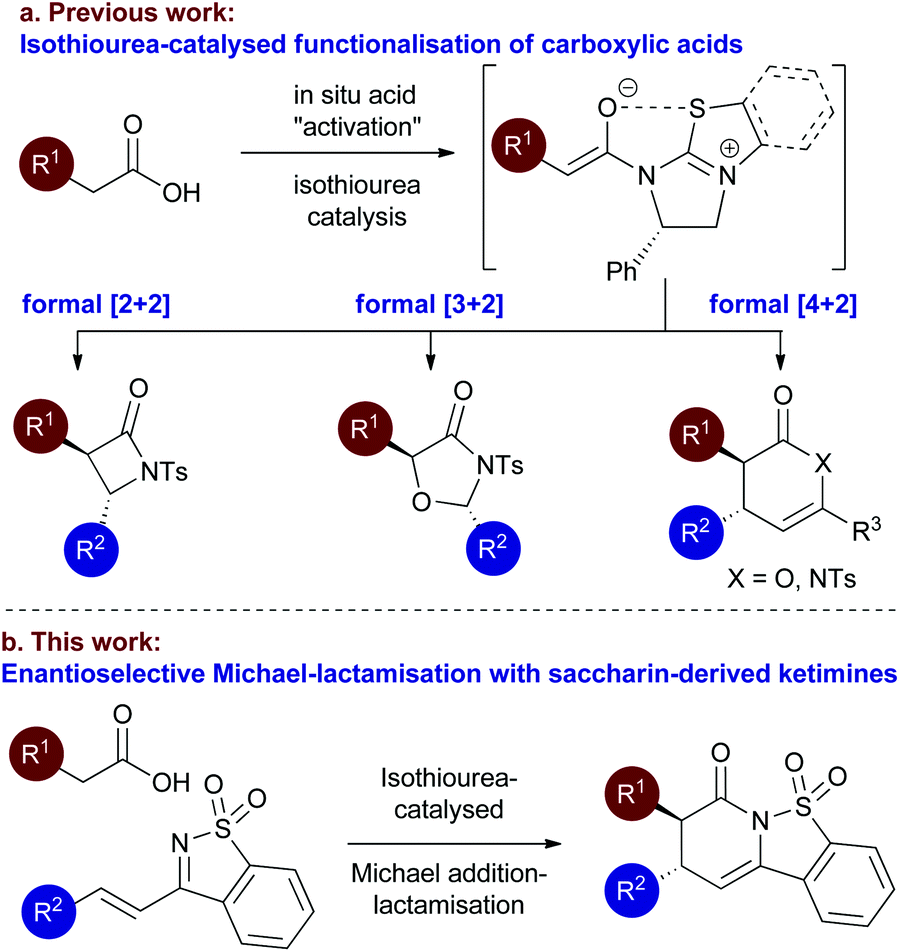 | ||
| Fig. 2 Proposed Isothiourea-catalysed Michael addition-lactamisation using saccharin-derived ketimines. | ||
During the course of this work elegant studies from Pericàs and co-workers reported a very similar reaction process. Using a polymer supported isothiourea catalyst (15 mol%), enantioselective annulation of a limited range of arylacetic acids as enolate precursors and saccharin-derived ketimines gave trans-dihydropyridinones in 86:14 to 96:4 dr and up to >99:1 er.17 Notably, no heteroaryl or alkenyl acetic acids were evaluated as ammonium enolate precursors within this process, and only limited substitution patterns within the arylacetic acid component were included. Similarly, Ye and co-workers have recently reported a related NHC-catalysed process, utilising α-chloroaldehydes as azolium enolate precursors, giving cis-dihydropyridinones in >95:5 dr and >99:1 er upon reaction with saccharin-derived ketimines.18 This effective methodology is however limited to the use of alkyl-α-chloroaldehydes.
Results and discussion
Reaction optimisation
Optimisation studies began with evaluating a small range of isothioureas as catalysts for the synthesis of 15 using phenylacetic acid 13 and ketimine 14 as a model system. Using pivaloyl chloride to make an in situ mixed anhydride and (R)-BTM 16 (10 mol%) gave the desired product 15 in 71% yield, 85:15 dr and 92:8 er. Using (2S,3R)-HyperBTM 17 (10 mol%) at rt gave the desired product 15 in 64% yield, 84:16 dr and 90:10 er. The optimum catalyst, however, was (S)-tetramisole·HCl 18 (10 mol%) giving tricyclic sultam 15 in 73% yield, 85:15 dr and excellent 97.5:2.5 er. Attempts to lower the catalyst loading of (S)-tetramisole·HCl 18 to 5 mol% led to a reduced 56% isolated yield of 15 with 83:17 dr and 94:6 er. Alternative solvents such as EtOAc, THF and toluene were tested but gave poorer dr and er (entries 5–7), with poor solubility in toluene leading to a low product conversion (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Yielda (%) | drb | erc |
| a Isolated following column chromatography using Biotage® Isolera™ 4. b Determined by 1H NMR spectroscopic analysis of crude reaction mixture. c Determined by chiral HPLC analysis. | |||||
| 1 | 16 (10) | CH2Cl2 | 71 | 85:15 |
92:8 (ent) |
| 2 | 17 (10) | CH2Cl2 | 64 | 84:16 |
90:10 (ent) |
| 3 | 18 (10) | CH2Cl2 | 75 | 86:14 |
97.5:2.5 |
| 4 | 18 (5) | CH2Cl2 | 65 | 85:15 |
97.5:2.5 |
| 5 | 18 (2) | CH2Cl2 | 56 | 86:14 |
94:6 |
| 5 | 18 (10) | EtOAc | 65 | 85:15 |
92:8 |
| 6 | 18 (10) | THF | 61 | 85:15 |
92.5:7.5 |
| 7 | 18 (10) | PhMe | 16 | 85:15 |
92:8 |
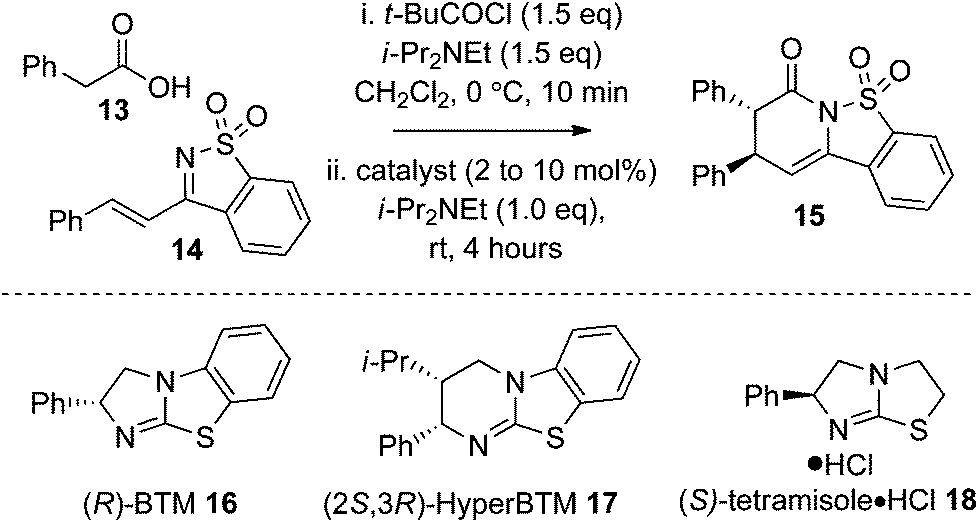
Further studies probed the generality of this enantioselective protocol using (S)-tetramisole·HCl 18 at rt through variation within the acid component, with arylacetic acids bearing both electron donating and withdrawing substituents, as well as heteroarylacetic acids targeted. Although good conversion to product was observed in all cases, significant variation in product diastereo- and enantioselectivity was observed using (S)-tetramisole·HCl 18 at rt (conditions A, Table 2). For example, reaction with 4-bromophenyl acetic acid and m-tolyl acetic acid gave sultams 19 and 22 in 90:10 dr but moderate 80:20 and 75:25 er respectively. Use of 3-thiophenylacetic acid yielded the thienyl sultam product 23 in good 95:5 er but in moderate 74:26 dr. While 4-methoxyphenyl acetic acid gave 20 in acceptable dr and er, incorporating an electron withdrawing substituent in 4-trifluoromethylphenyl acetic acid led to reduced enantioselectivity (21, 90.5:9.5 er). These moderate and variable results indicated that the initial conditions identified in the catalyst screen using (S)-tetramisole·HCl 18 at rt were not general and that further optimisation was required.
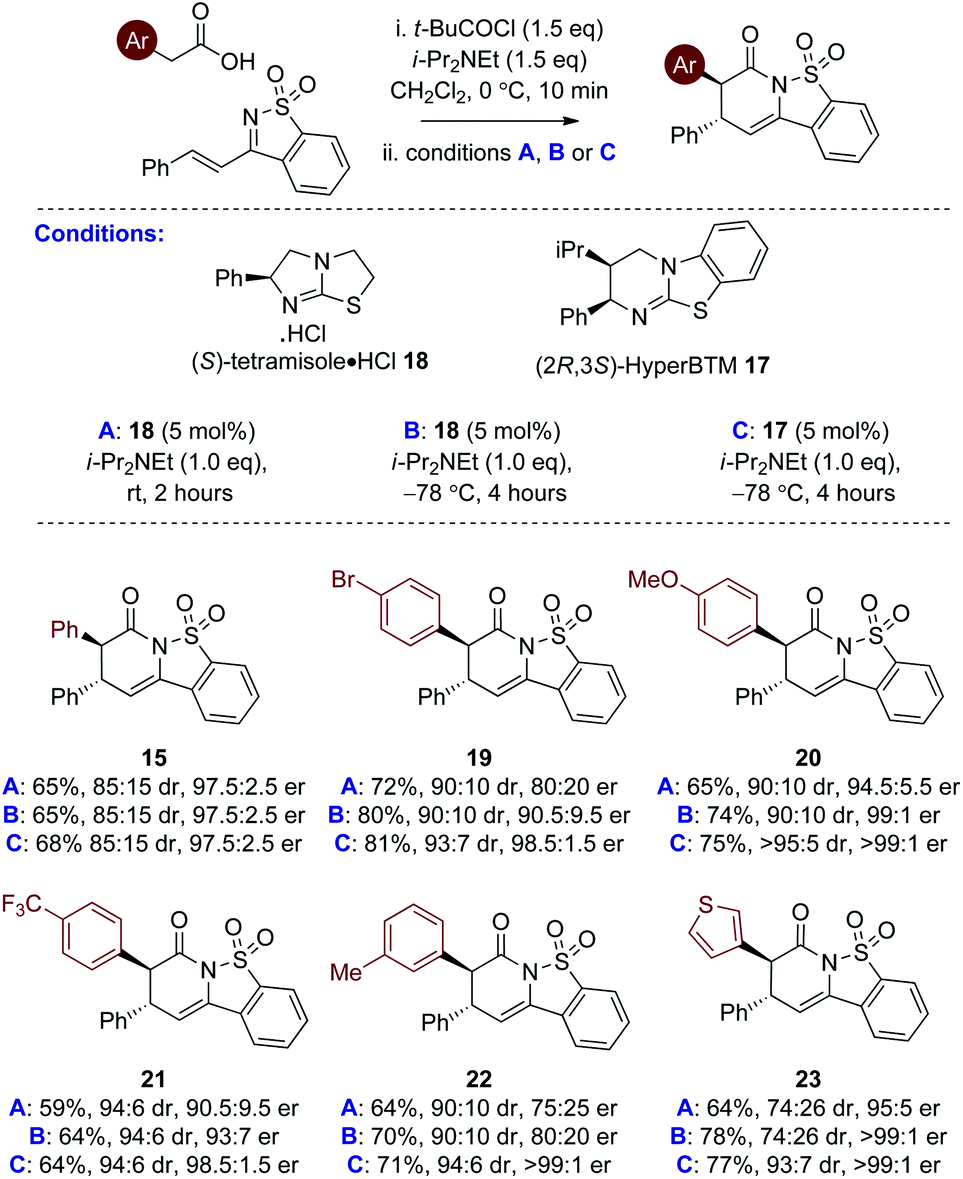
Further optimisation probed the effect of lowering the reaction temperature to −78 °C as this was predicted to minimise any competitive racemic background reaction over the range of substrates.19 Using (S)-tetramisole·HCl 18 (5 mol%) at −78 °C (conditions B, Table 2) led to generally improved enantioselectivity. However, moderate er was observed for the formation of 3-MeC6H4-substituted derivative 22 (80:20 er), and poor diastereoselectivity for 3-thiophenyl substituted 23 (74:26 dr). Pleasingly, however, (2R,3S)-HyperBTM 17 (5 mol%) proved significantly more successful. 4-BrC6H4 substituted sultam 19 was produced in 81% yield, 93:7 dr and excellent 98.5:1.5 er. Sultams 20 and 21 incorporating the electron rich 4-MeOC6H4 and the electron withdrawing 4-CF3C6H4 substituents were isolated in good yield, and excellent diastereo- and enantioselectivity. A dramatic improvement in enantioselectivity was observed for 3-MeC6H4-substituted derivative 22 (>99:1 er), while improved diastereoselectivity was observed for 3-thiophenyl derivative 23 (93:7 dr, >99:1 er).
Further substrate scope
With a reliable enantioselective process in hand using (2R,3S)-HyperBTM 17 at −78 °C, the scope and limitations of this protocol was further investigated, with the extension to alternative heteroaryl and alkenylacetic acids targeted (Table 3). Variation of the carboxylic acid group showed that extended aromatic substituents are readily tolerated, with the 1-naphthyl unit incorporated to give 24 in 82% yield and 99:1 er. The incorporation of alkenyl substituents from the corresponding acids worked well, giving 25 and 26 in excellent dr. Consistent with our previous work the incorporation of the styrenyl unit within 25 led to reduced enantioselectivity (86:14 er) in comparison to 26 (>99:1 er).12b The 3-indolyl unit was also readily included albeit with reduced diastereoselectivity (27, 80:20 dr) but excellent er (>99:1 er). Variation within the β-substituent of the saccharin-derived ketimine was next evaluated (products 28–32). The 1-naphthyl unit was readily incorporated, as were electron-donating and -withdrawing 4-substituents, as well as the 2-furyl motif with good to excellent diastereo- and enantioselectivity.20
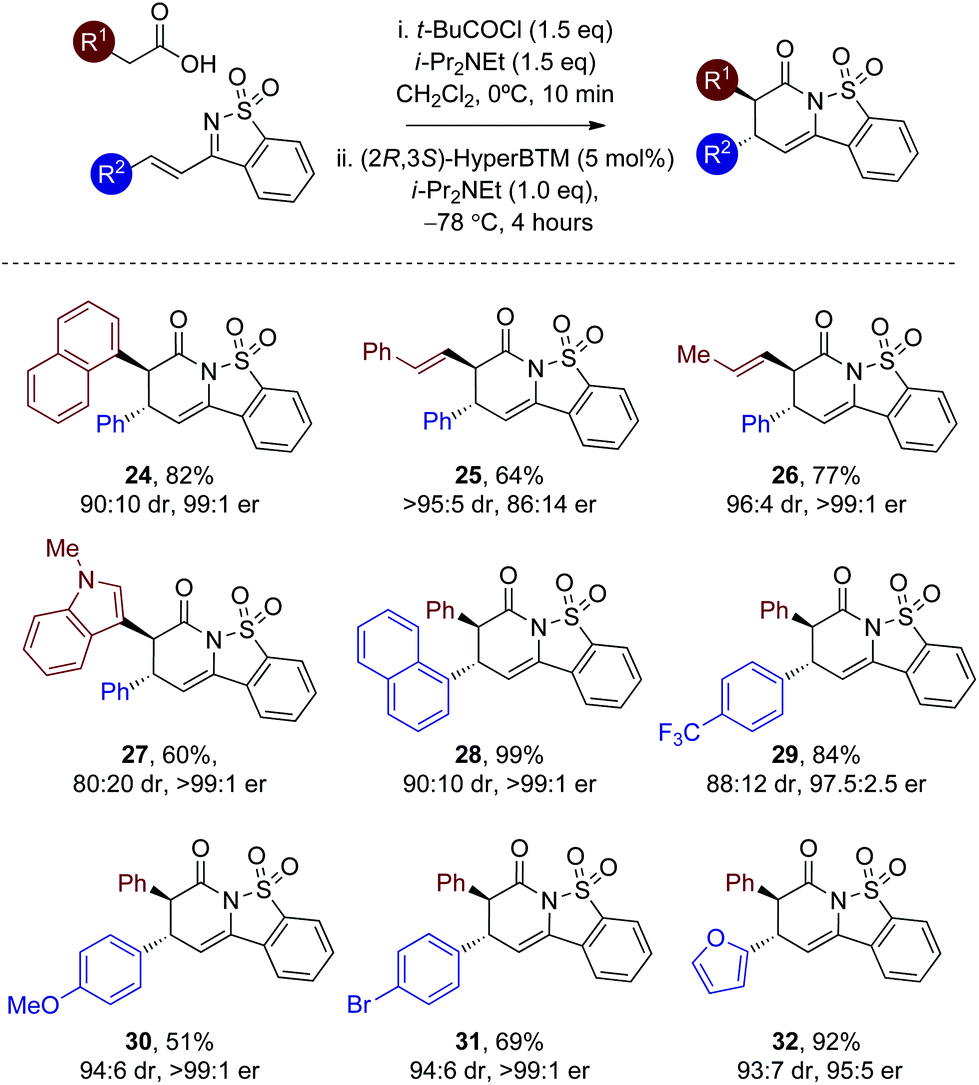
The relative and absolute configuration within 31 was assigned by X-ray crystallography analysis, with the configuration within all other products assigned by analogy (Fig. 3).21
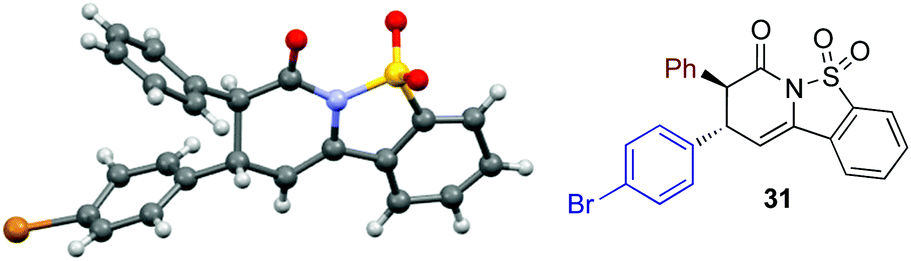 | ||
| Fig. 3 Molecular representation of the X-ray structure of 31. | ||
Proposed mechanism
Consistent with our previous studies, a proposed catalytic cycle for the synthesis of these saccharin-derived dihydropyridones is shown in Scheme 1. Initial in situ formation of the mixed anhydride 33 from pivaloyl chloride and the carboxylic acid, followed by subsequent nucleophilic attack from the (2R,2S)-HyperBTM catalyst 17 generates acyl ammonium ion 34. Deprotonation to form the corresponding (Z)-ammonium enolate 35, followed by stereoselective Michael addition gives 36, with lactonisation releasing catalyst 17 and the polycyclic dihydropyranone product 37. A stabilising n0 to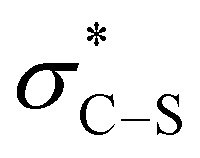 interaction between the enolate oxygen and the sulfur of the isothiouronium ion is proposed to lock the conformation of the enolate species,22 forcing the adjacent stereodirecting phenyl substituent to adopt a pseudoaxial orientation to minimise 1,2-strain.23 Subsequent Michael addition occurs preferentially anti- to this stereodirecting group, with the two prostereogenic centres adopting an approximately staggered array to minimise unfavourable non-bonding interactions. By analogy to Heathcock's model24 a pre-transition state assembly 38 is consistent with the observed sense of diastereo- and enantioselectivity.
interaction between the enolate oxygen and the sulfur of the isothiouronium ion is proposed to lock the conformation of the enolate species,22 forcing the adjacent stereodirecting phenyl substituent to adopt a pseudoaxial orientation to minimise 1,2-strain.23 Subsequent Michael addition occurs preferentially anti- to this stereodirecting group, with the two prostereogenic centres adopting an approximately staggered array to minimise unfavourable non-bonding interactions. By analogy to Heathcock's model24 a pre-transition state assembly 38 is consistent with the observed sense of diastereo- and enantioselectivity.
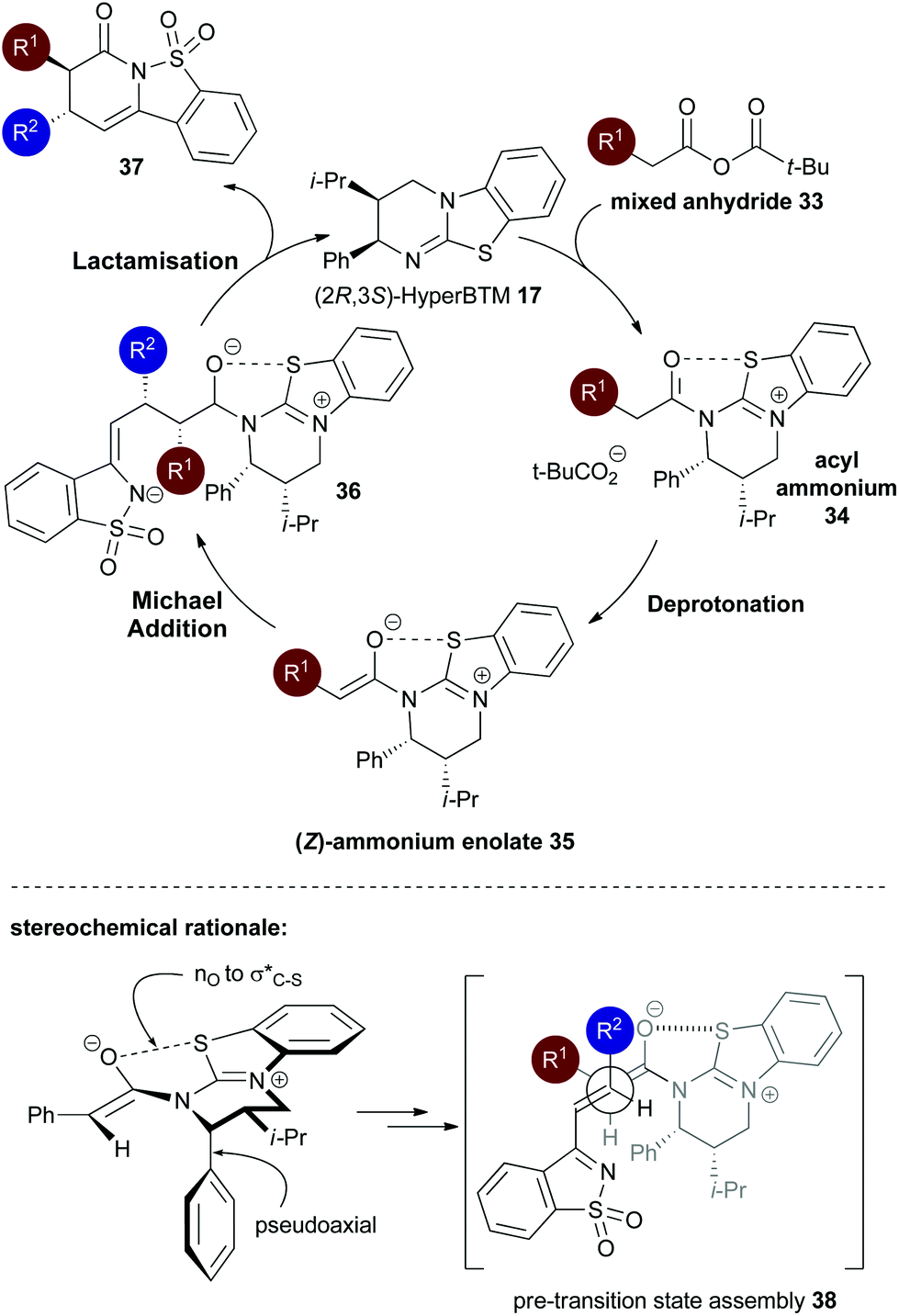 | ||
| Scheme 1 Proposed mechanism and stereochemical rationale of the isothiourea-catalysed Michael addition–lactamisation. | ||
Conclusions
In conclusion, the catalytic enantioselective synthesis of a range of saccharin-derived trans-dihydropyridinones (15 examples, up to >95:5 dr, up to >99:1 er) using both aryl-, heteroaryl-, and alkenylacetic acids as ammonium enolate precursors using (2R,3S)-HyperBTM has been developed. Further work from this laboratory is directed toward developing alternative uses of isothioureas and other Lewis bases in enantioselective catalysis.
Acknowledgements
We thank Syngenta and the EPSRC (grant code EP/K503162/1) (DGS) for funding. The European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013) ERC Grant Agreement No. 279850 is also acknowledged (CMY). ADS thanks the Royal Society for a Wolfson Research Merit Award. We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University.Notes and references
- (a) K. Köhler, A. Hillebrecht, J. Schulze Wischeler, A. Innocenti, A. Heine, C. T. Supuran and G. Klebe, Angew. Chem., Int. Ed., 2007, 46, 7697–7699 CrossRef PubMed; (b) J. Moeker, T. S. Peat, L. F. Bornaghi, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2014, 57, 3522–3531 CrossRef CAS PubMed.
- R. J. Fanelli, T. Schuurman, T. Glaser and J. Traber, Prog. Clin. Biol. Res., 1990, 361, 461–467 CAS.
- (a) M. D'Ascenzio, S. Carradori, C. De Monte, D. Secci, M. Ceruso and C. T. Supuran, Bioorg. Med. Chem., 2014, 22, 1821–1831 CrossRef PubMed; (b) S. Carradori, D. Secci, C. De Monte, A. Mollica, M. Ceruso, A. Akdemir, A. P. Sobolev, R. Codispoti, F. De Cosmi, P. Guglielmi and C. T. Supuran, Bioorg. Med. Chem., 2016, 24, 1095–1105 CrossRef CAS PubMed.
- L. Levy, Drugs Future, 1992, 17, 451–454 CrossRef.
- A. G. Kravina, J. Mahatthananchai and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9433–9436 CrossRef CAS PubMed.
- X. Feng, Z. Zhou, C. Ma, X. Yin, R. Li, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 14173–14176 CrossRef CAS PubMed.
- The research data underpinning this publication can be accessed at DOI:10.17630/12aeb23b-e4ae-402c-a9da-f7fc0b17d374.
- (a) G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945–7946 CrossRef CAS PubMed; (b) S. H. Oh, G. S. Cortez and D. Romo, J. Org. Chem., 2005, 70, 2835–2838 CrossRef CAS PubMed.
- For seminal work on isothiourea catalysis in kinetic resolution and acyl transfer reaction processes see: (a) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef CAS PubMed; (b) V. B. Birman, H. Jiang, X. Li, L. Guo and E. W. Uffman, J. Am. Chem. Soc., 2006, 128, 6536–6537 CrossRef CAS PubMed; (c) M. Kobayashi and S. Okamoto, Tetrahedron Lett., 2006, 47, 4347–4350 CrossRef CAS; (d) V. B. Birman and X. Li, Org. Lett., 2008, 10, 1115–1118 CrossRef CAS PubMed; (e) Y. Zhang and V. B. Birman, Adv. Synth. Catal., 2009, 351, 2525–2529 CrossRef CAS PubMed; (f) C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS PubMed. For recent reviews, see: (g) L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC; (h) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC. For a selection of alternative isothiourea-catalysed processes see below. For reactions utilising α,β-unsaturated acyl ammonium intermediates see: (i) E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193–2200 RSC; (j) S. Vellalath, K. N. Van and D. Romo, Angew. Chem., Int. Ed., 2013, 52, 13688–13693 CrossRef CAS PubMed; (k) G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049–1057 CrossRef CAS PubMed; (l) Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320–5323 CrossRef CAS PubMed. For ammonium ylide intermediates see: (m) T. H. West, D. S. B. Daniels, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476–4479 CrossRef CAS PubMed.
- For select examples see: (a) D. G. Stark, T. J. C. O'Riordan and A. D. Smith, Org. Lett., 2014, 16, 6496–6499 CrossRef CAS PubMed; (b) S. R. Smith, S. M. Leckie, R. Holmes, J. Douglas, C. Fallan, P. Shapland, D. Pryde, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2014, 16, 2506–2509 CrossRef CAS PubMed; (c) P.-P. Yeh, D. S. B. Daniels, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2014, 16, 964–967 CrossRef CAS PubMed; (d) D. G. Stark, L. C. Morrill, P.-P. Yeh, A. M. Z. Slawin, T. J. C. O'Riordan and A. D. Smith, Angew. Chem., Int. Ed., 2013, 52, 11642–11646 CrossRef CAS PubMed; (e) L. C. Morrill, J. Douglas, T. Lebl, A. M. Z. Slawin, D. J. Fox and A. D. Smith, Chem. Sci., 2013, 4, 4146–4155 RSC; (f) L. C. Morrill, T. Lebl, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2012, 3, 2088–2093 RSC; (g) D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef CAS PubMed.
- (a) L. Hesping, A. Biswas, C. G. Daniliuc, C. Muck-Lichtenfeld and A. Studer, Chem. Sci., 2015, 6, 1252–1257 RSC; (b) S. R. Smith, C. Fallan, J. E. Taylor, R. McLennan, D. S. B. Daniels, L. C. Morrill, A. M. Z. Slawin and A. D. Smith, Chem. – Eur. J., 2015, 21, 10530–10536 CrossRef CAS PubMed.
- (a) S. R. Smith, J. Douglas, H. Prevet, P. Shapland, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1626–1639 CrossRef CAS PubMed; (b) L. C. Morrill, S. M. Smith, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1640–1655 CrossRef CAS PubMed.
- J.-Y. Bae, H.-J. Lee, S.-H. Youn, S.-H. Kwon and C.-W. Cho, Org. Lett., 2010, 12, 4352 CrossRef CAS PubMed.
- L. C. Morrill, L. A. Ledingham, J.-P. Couturier, J. Bickel, A. D. Harper, C. Fallan and A. D. Smith, Org. Biomol. Chem., 2014, 12, 624–636 CAS.
- C. Simal, T. Lebl, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2012, 51, 3653–3657 CrossRef CAS PubMed.
- P.-P. Yeh, D. S. B. Daniels, C. Fallan, E. Gould, C. Simal, J. E. Taylor, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2015, 13, 2177–2191 CAS.
- J. Izquierdo and M. A. Pericàs, ACS Catal., 2016, 6, 348–356 CrossRef CAS.
- Z.-Q. Liang, D.-L. Wang, C.-L. Zhang and S. Ye, Org. Biomol. Chem., 2016 10.1039/c6ob01040g.
- Treatment of 4-bromophenyl acetic acid with pivalolyl chloride, i-Pr2NEt and ketimine 14, gave 24% conversion into corresponding product 19 (as determined by 1H NMR spectroscopic analysis). This confirms the presence of a base-catalysed racemic background reaction at room temperature.
- No reaction was observed in this process using alkyl substituted carboxylic acids. We were unable to prepare alkyl substituted ketimines for their use in this transformation.
- The crystallographic data obtained for 31 has been deposited with the Cambridge Crystallographic Data Centre and the supplementary data can be found via CCDC 1491707.
- For the initial postulate of 1,5-S⋯O interactions as a control element in isothiourea catalysis see:
(a) V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS PubMed. For other manuscripts of interest see:
(b) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef CAS PubMed;
(c) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef CAS PubMed . Romo and Tantillo (ref. 22b) have probed the nature of 1,5-S⋯O interactions of α,β-unsaturated acyl ammonium species with NBO and postulate this interaction is due to a number of orbital interactions. In particular, unfavourable
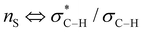 interactions disfavour alternative conformations with an O–C–N–C dihedral angle of 180°.
interactions disfavour alternative conformations with an O–C–N–C dihedral angle of 180°. - For representative examples that demonstrate the preference of substituents adjacent to an N-acyl group in heterocyclic compounds to adopt a pseudo-axial position see: (a) P. J. Sinclair, D. Zhai, J. Reibenspies and R. M. J. Williams, J. Am. Chem. Soc., 1986, 108, 1103 CrossRef CAS; (b) J. F. Dellaria and B. D. Santarsiero, J. Org. Chem., 1989, 54, 3916 CrossRef CAS; (c) M. G. B. Drew, L. M. Harwood, G. Park, D. W. Price, S. N. G. Tyler, C. R. Park and S. G. Cho, Tetrahedron, 2001, 57, 5641 CrossRef CAS.
- For an excellent overview of this area see: (a) D. A. Oare and C. H. Heathcock, Top. Stereochem., 1989, 19, 227–407 CAS. For select representative examples of enolate additions to Michael acceptors see: (b) C. H. Heathcock, M. A. Henderson, D. A. Oare and M. A. Sanner, J. Org. Chem., 1985, 50, 3019–3022 CrossRef CAS; (c) D. A. Oare, M. A. Henderson, M. A. Sanner and C. H. Heathcock, J. Org. Chem., 1990, 55, 132–157 CrossRef CAS; (d) M. Yamaguchi, M. Tsukamoto, S. Tanaka and I. Hirao, Tetrahedron Lett., 1984, 25, 5661–5664 CrossRef CAS. For a computational investigation of intermolecular Michael reactions see: (e) E. E. Kwan and D. A. Evans, Org. Lett., 2010, 12, 5124–5127 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of NMR spectra, HPLC analysis and characterisation. Data available in ref. 7. CCDC 1491707. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01473a |
| This journal is © The Royal Society of Chemistry 2016 |