
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis of a pyridine-N-oxide isophthalamide rotaxane utilizing supplementary amide hydrogen bond interactions†
Nicholas H.
Evans
*,
Charles E.
Gell
and
Michael J. G.
Peach
Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: n.h.evans@lancaster.ac.uk
First published on 1st August 2016
Abstract
The synthesis of a pyridine-N-oxide containing rotaxane, not requiring an additional ionic template, has been achieved in 32% yield. Successful rotaxane formation is dependent upon the structure of the isophthalamide macrocycle used, an observation which has been rationalised by a combination of NMR spectroscopy, X-ray crystallography and computational modelling.
Introduction
Rotaxanes,1 interlocked molecules consisting of macrocyclic ring(s) trapped on a stoppered axle, are increasingly being used in a range of functional applications.2,3 Underpinning this work has been the development of template methodologies to overcome the otherwise entropically unfavourable association of molecular components required for the preparation of a rotaxane. Popular choices of templates and templating interactions that have been used to prepare rotaxanes include: metal cations,4 anions,5 π–π stacking6 and hydrogen bonding.7 While a significant number of template methodologies for the production of rotaxanes have now been reported, the establishment of further versatile synthetic pathways has the potential to aid the supramolecular chemistry community in the preparation of new, functionally useful rotaxanes.The hydrogen bond templated threading of simple 3,5-bisamides of pyridine-N-oxides through isophthalamide macrocycles to form pseudo-rotaxanes has been reported by Jiang and co-workers (Fig. 1).8 Considering the synthetic ease in the generation of 3,5-bisamides of pyridine-N-oxides (including the possibility of installing different substituents on each amide) and isophthalamide macrocycles, such a self-assembled array looks attractive to be incorporated into the synthesis of a fully interlocked rotaxane. However, to date there is a lack of rotaxane syntheses using the system as described by Jiang et al. Beer and co-workers have reported on a number of rotaxanes containing 3,5-bisamide pyridine-N-oxide motifs, but appreciable yields of formation are only observed in the presence of an additional cationic9–11 or anionic12 template. To the best of our knowledge, only one synthesis of a fully interlocked rotaxane using the simple self-assembly motif as described by Jiang et al. without recourse to further ionic templation has been disclosed, but the rotaxane was only isolated in the somewhat modest yield of 15%.13
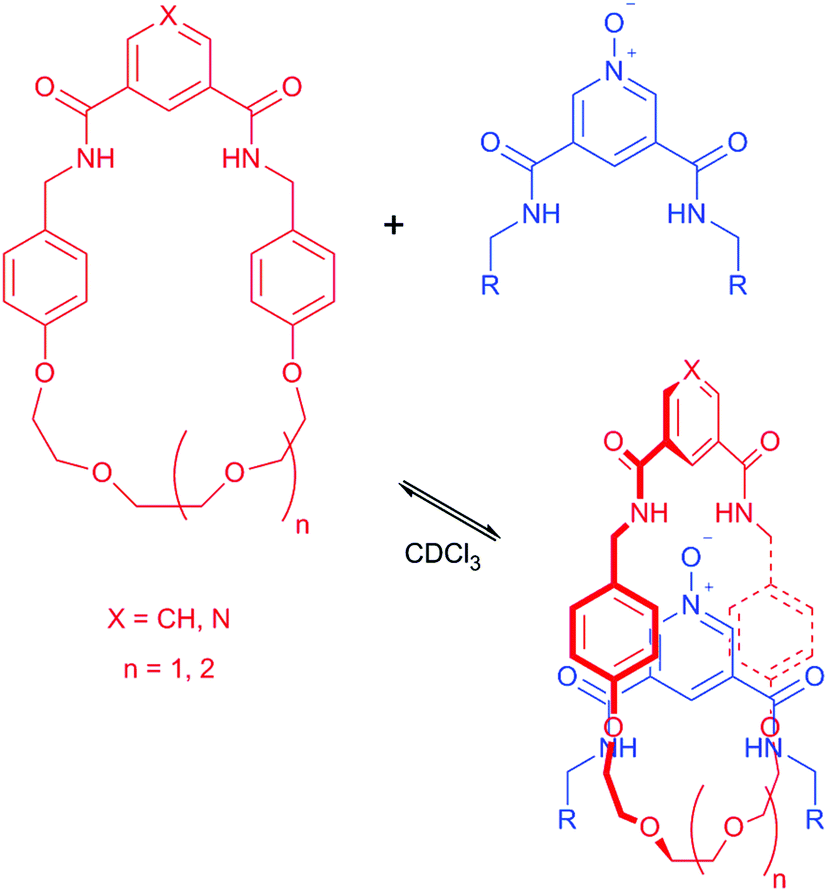 | ||
| Fig. 1 Formation of a hydrogen bond templated pseudo-rotaxane consisting of an isophthalamide macrocycle and unstoppered pyridine-N-oxide axle, as reported by Jiang and co-workers, ref. 8. | ||
Herein we report the synthesis of a fully interlocked rotaxane in a 32% yield, by use of the CuAAC click reaction to stopper a 3,5-bisamide pyridine-N-oxide axle threaded through a polyether containing isophthalamide macrocycle, without the use of additional templation from either a cation or anion to support the self-assembly of the pseudo-rotaxane intermediate. An attempted synthesis with a second isophthalamide macrocycle, containing a benzene ring in place of the polyether chain, yielded no rotaxane. Solution NMR spectroscopy, solid state crystallography and computational modelling have been used to explain the different synthetic outcomes in these two systems.
Results and discussion
Macrocycle synthesis
The two macrocycles, 1 and 2, investigated in this study are depicted in Fig. 1. Each contains an isophthalamide motif that may hydrogen bond to a suitable hydrogen bond acceptor, such as the oxygen atom of a pyridine-N-oxide. With a view to preparing more sophisticated rotaxanes in the future, there is the possibility of incorporating functionality on the isophthalamide ring of both macrocycles by use of 5-substituted isophthalamides. In addition, the inclusion of an additional aromatic ring in the structure of 2 allows for similar opportunities at the other end of this macrocycle.The synthesis of macrocycle 1 has been reported previously.14 Macrocycle 2 was prepared by an analogous synthetic route (Scheme 1). Alkylation of (benzene-1,3-diyl)dimethanol with 2.1 equivalents of 4-(bromomethyl)benzonitrile afforded compound 3 in 73% yield. Bis-nitrile 3 was then reduced to bis-amine 4 using borane–THF in an isolated 40% yield. Bis-amine 4 was then reacted, under high dilution conditions, with one equivalent of isophthaloyl chloride in the presence of excess triethylamine, in dichloromethane. After aqueous work-up and silica gel chromatography, macrocycle 2 was isolated in 38% yield. In addition to characterisation by NMR and IR spectroscopy and mass spectrometry, crystals of 2 suitable for single crystal X-ray diffraction were grown by slow evaporation of a chloroform sample confirming the macrocyclic nature of 2 (see ESI†).
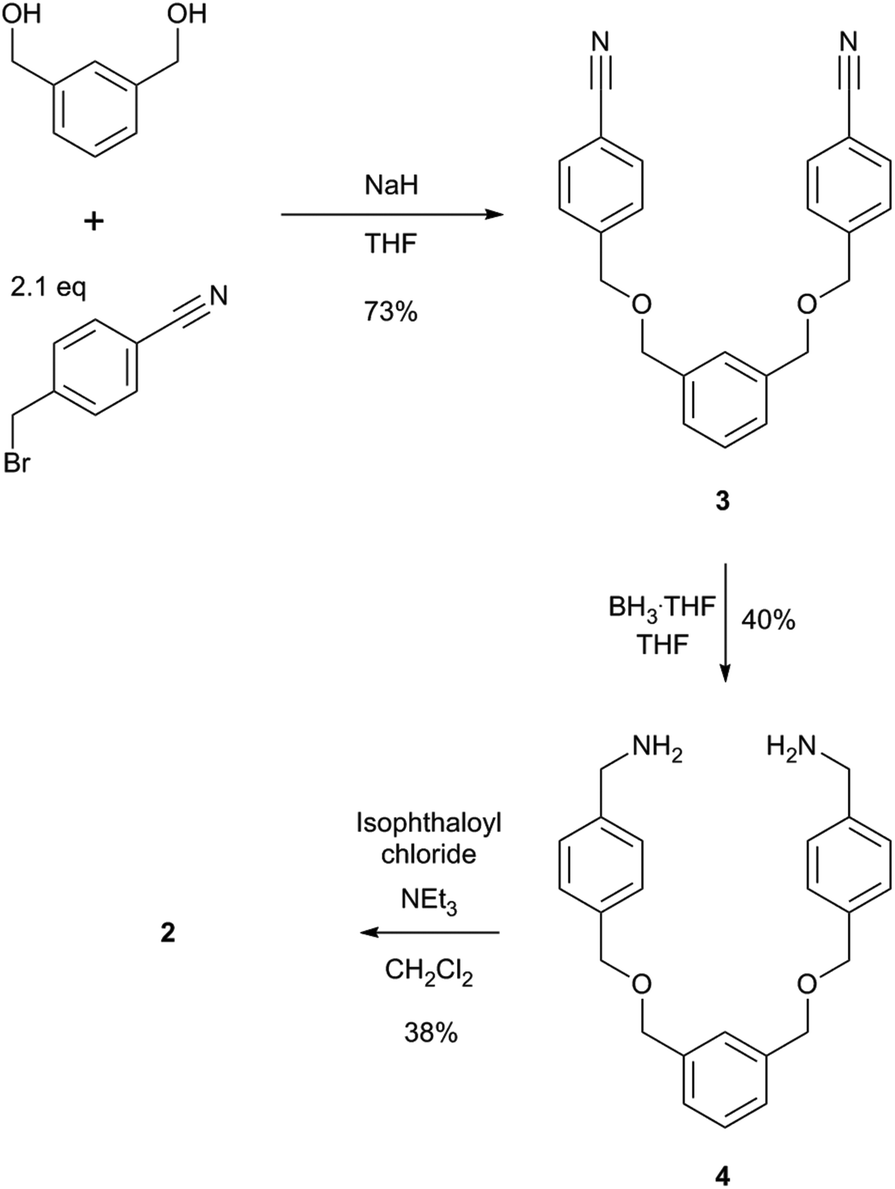 | ||
| Scheme 1 Synthesis of macrocycle 2. | ||
Experimental pseudo-rotaxane investigations
With 1 and 2 in hand, 1H NMR spectroscopy was used to investigate the ability of a 3,5-bisamide pyridine-N-oxide motif to thread through the macrocycles. Equimolar amounts of macrocycle (1 or 2) and unstoppered pyridine-N-oxide thread 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 were dissolved in CDCl3, a relatively uncompetitive solvent with respect to hydrogen bonding, and 1H NMR spectra recorded (Scheme 2).
15 were dissolved in CDCl3, a relatively uncompetitive solvent with respect to hydrogen bonding, and 1H NMR spectra recorded (Scheme 2).
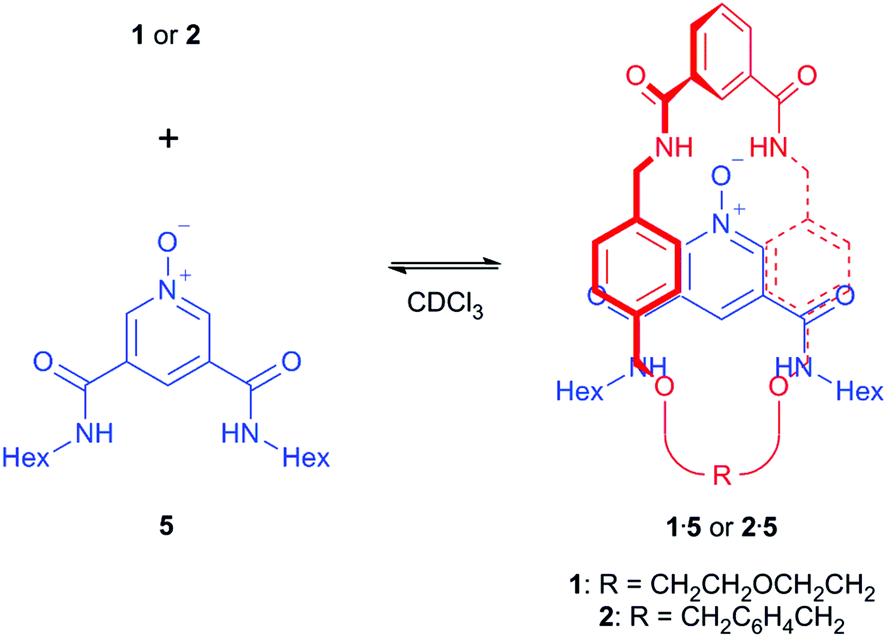 | ||
| Scheme 2 Attempted formation of pseudo-rotaxanes 1·5 and 2·5. | ||
Upon the addition of 5 to macrocycle 1, the aromatic resonances f and g split and moved upfield in the 1H NMR spectrum (Fig. 3). This is consistent with intercalation of the pyridine-N-oxide ring of 5 between the aromatic rings of macrocycle 1, confirming that pseudo-rotaxane 1·5 has formed.16 In contrast, only a very modest movement of the equivalent aromatic resonances of macrocycle 2 were observed in the 1H NMR spectrum upon addition of 5 (see ESI†), indicating that little (if any) pseudo-rotaxane formation occurred between 2 and 5.
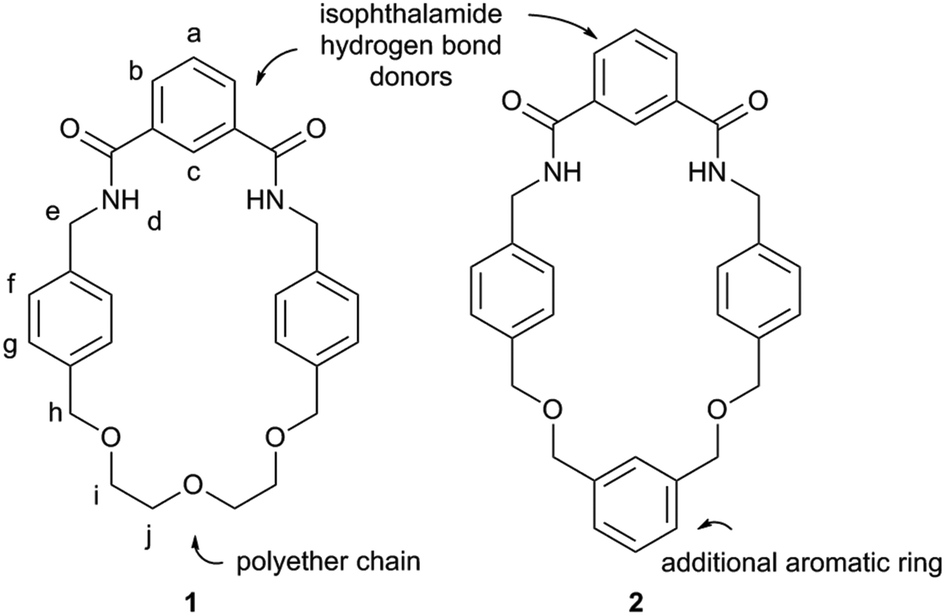 | ||
| Fig. 2 Structures of macrocycles 1 and 2. | ||
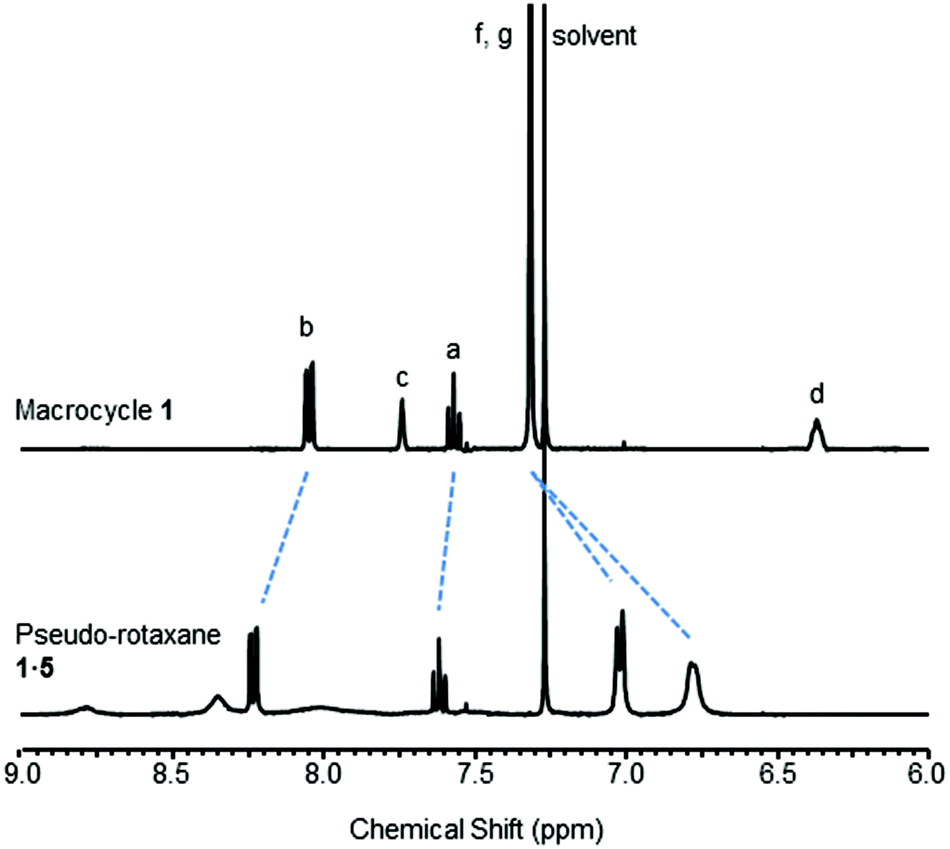 | ||
| Fig. 3 Partial 1H NMR spectra of macrocycle 1 and pseudo-rotaxane 1·5 (CDCl3, 5 mM, 400 MHz, 298 K). For atom labels see Fig. 2. | ||
Single crystals suitable for single crystal X-ray diffraction were grown by the slow diffusion of isopropyl ether into the deuterated chloroform solution containing 1 and 5. The solved structure revealed the interpenetrated structure of pseudo-rotaxane 1·5 (Fig. 4).
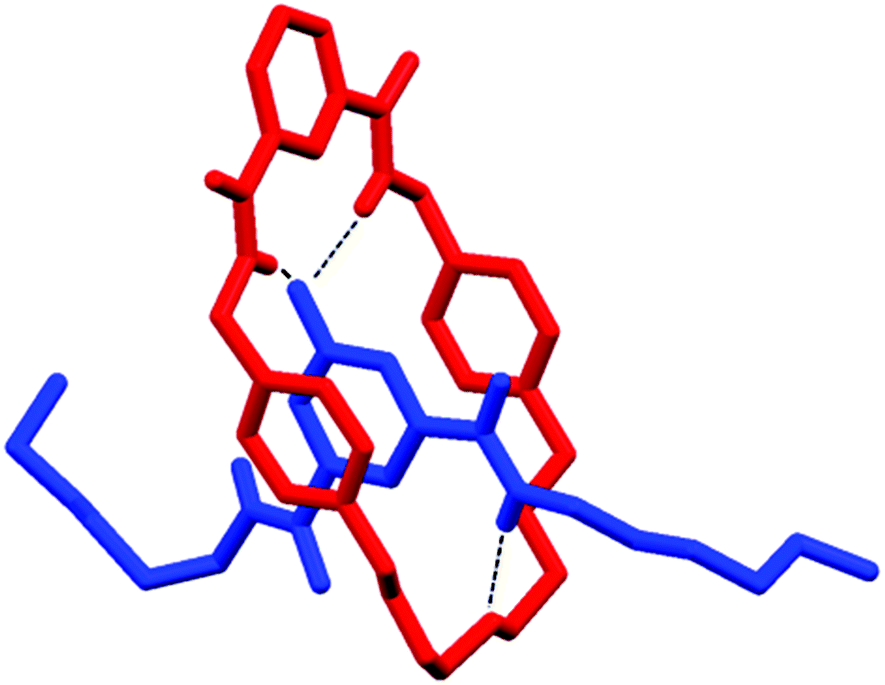 | ||
| Fig. 4 X-ray structure of pseudo-rotaxane 1·5. Hydrogen atoms (except N–Hs) and disorder in one of the hexyl chains are omitted for clarity. | ||
As expected there are hydrogen bonds between the isophthalamide N–Hs to the oxygen atom of the pyridine-N-oxide (N–H⋯O distances: 2.151 Å and 2.221 Å). In addition, there is a hydrogen bond interaction between the syn pyridine-N-oxide amide N–H to the central polyether oxygen (N–H⋯O distance: 2.129 Å). Based on this crystal structure, we tentatively attributed the failure of macrocycle 2 and thread 5 to form a pseudo-rotaxane in solution to the lack of an appropriately positioned atom in macrocycle 2 to form an equivalent hydrogen bond with the amide N–H of the pyridine-N-oxide bis-amide thread 5.
Computational study of pseudo-rotaxanes
To provide further insight into the experimental results of the pseudo-rotaxane studies, density-functional theory (DFT) calculations were undertaken on macrocycles 1 and 2, thread 5 and the possible pseudo-rotaxanes 1·5 and 2·5.17 The calculations were performed using the Gaussian 09 program,18 using the B3LYP functional19 together with the 6-31G* basis set for all atoms. Solvent effects (CHCl3) were modelled using the standard polarizable continuum model, with the default Gaussian 09 parameters for the solvent and cavity. Dispersion interactions were also approximated via Grimme's D3 dispersion correction.20 In each case, an optimised structure was obtained; vibrational frequencies were computed to ensure each structure was in fact a minimum on the potential energy surface, and to quantify the zero-point energy corrections.The isolated macrocycles were modelled first. In the case of macrocycle 1, the minimum energy structure in solution (see Fig. 5) was found to closely resemble that of the previously reported crystal structure,14 and the obtained structure would seem to favour the possibility of threading due to the potential for intermolecular hydrogen bonding and π–π stacking interactions. However, in the case of macrocycle 2, there were two distinct structures found; the first, based on the crystal structure, is the more stable, but the resultant structure (see Fig. 5) would not favour appropriate hydrogen bonding or π–π stacking interactions when threaded. The second, higher energy conformation (see ESI†), more closely resembles the minimized structure of macrocycle 1.
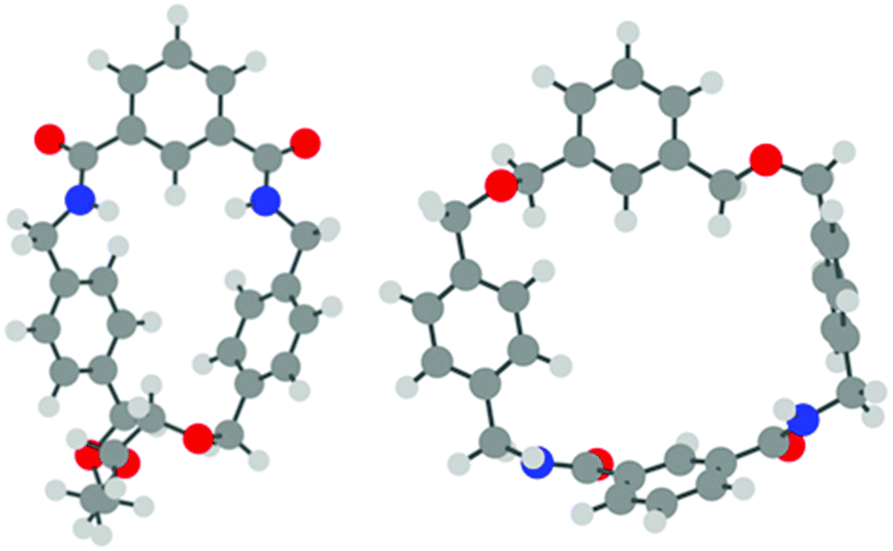 | ||
| Fig. 5 Minimum energy structures of macrocycles 1 (left) and 2 (right); each closely resembles the observed crystal structure of the respective macrocycle. | ||
A number of conformations of thread 5 were then considered. These included syn–syn, syn–anti and anti–anti conformations, additionally allowing for possible rearrangement in the terminal alkyl chains. The lowest energy conformation was found to be the syn–anti conformation, and the highest energy the anti–anti conformation, with the difference in energies between the most and least stable being less than 10 kJ mol−1.
After these initial calculations, the structural and energetic properties of the pseudo-rotaxanes 1·5 and 2·5 were considered. Three conformations (syn–syn, syn–anti and anti–anti) of thread 5 were examined, to determine which conformation was associated with the most stable pseudo-rotaxane; i.e., which of the three possible conformations of the thread is able to most strongly interact with the macrocycle unit.
For the geometry optimisations of pseudo-rotaxane 1·5, two sets of starting points were considered, to minimise bias and to maximise the chance of finding the global minimum on the potential energy surface. Firstly, structures based around the observed crystal structure of the pseudo-rotaxane, and secondly ‘free-hand’ structures drawn to maximise intermolecular interactions were considered. The structures based on the free-hand starting geometries were in each case found to give a final structure lower in energy (i.e., more stable) than the corresponding crystal-structure based starting points. However, the structures are qualitatively similar in all cases. Indeed, irrespective of the starting geometries, the stability of pseudo-rotaxane 1·5 was found to be greatest for the syn–syn thread conformation (depicted in Fig. 6), and least for the anti–anti conformation. A relatively small difference in stability between the syn–syn and syn–anti conformation was observed. Examination of the crystal structure data suggests that for pseudo-rotaxane 1·5, the syn–anti conformation is preferred; we attribute this preference to crystal packing effects in the solid state.
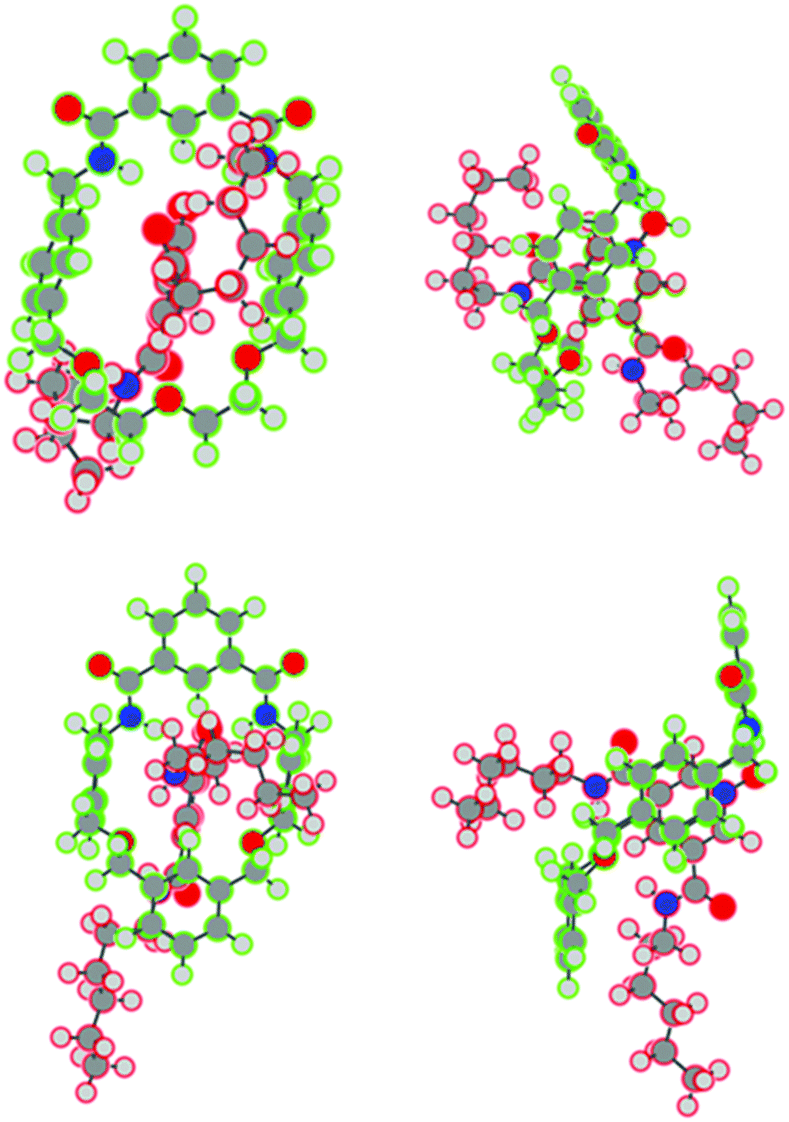 | ||
| Fig. 6 Optimised structures of pseudo-rotaxane 1·5 (top) and 2·5 (bottom) from DFT calculations. Two views of each pseudo-rotaxane are shown to highlight similarities of macrocycle-thread configurations and interactions in the two modelled pseudo-rotaxanes. | ||
There is good correlation between the O–Hc distance and the stability of the conformation (the shorter the hydrogen bond distance, the more stable the conformation) for pseudo-rotaxane 1·5 (Table 1). There are a range of average ring-ring distances in the range of 3.65 to 3.93 Å (the thread does not sit in the middle of the macrocycle), but these vary only slightly between the conformations, suggesting that the π–π interactions are similar irrespective of the conformation.
| 1 | 2 | |
|---|---|---|
| a Distances in Å. | ||
| syn–syn | 2.036 | 2.046 |
| syn–anti | 2.065 | 2.068 |
| anti–anti | 2.337 | 2.311 |
For the free-hand based starting geometries, the stability of pseudo-rotaxane 1·5 relative to individual 1 and 5 were then considered. To evaluate these quantities, the energy of the most stable found structures of the macrocycle and thread (irrespective of conformation) were subtracted from the energy of the pseudo-rotaxane with the thread in each of the three conformations. Here, the syn–syn conformation has the lowest energy. Relative to that lowest energy conformation, the syn–anti and anti–anti conformation was found to be 11 kJ mol−1 and 32 kJ mol−1 higher in energy respectively. Regardless of the choice of conformation of the thread, the pseudo-rotaxane was found to be stable relative to the individual macrocycle and thread. This suggests that although the strongest intermolecular interactions seem to occur in the syn–syn case (closely followed by the syn–anti case), there appears to be sufficient intermolecular interactions in each of the three cases to form a stable pseudo-rotaxane. A zero-point energy correction was also considered, and was found to have no significant impact on the size of the binding energies (slightly reducing the values, by less ten percent in each case). The zero-point corrected binding energy for the most stable conformation (syn–syn) was found to be around 224 kJ mol−1.
In the absence of a crystal structure, only ‘free-hand’ structures for pseudo-rotaxane 2·5, drawn to maximise intermolecular interactions as starting points for the geometry optimisations, were considered. The structures used were qualitatively similar to those obtained for pseudo-rotaxane 1·5. The results of the geometry optimisations were consistent with those of the other pseudo-rotaxane, with the syn–syn conformation being the most stable, closely followed by syn–anti, with the anti–anti conformation being the least stable. As for 1·5, there is correlation between the O–Hc distance and the stability of the conformation (see Table 1). The average ring–ring distances fall in the range 3.63–3.83 Å, which once again are similar to those observed for 1·5.
A similar examination of the relative stability of the pseudo-rotaxane 2·5 (with the three distinct thread conformations) was undertaken, quantifying the stability of the pseudo-rotaxane relative to the individual thread and macrocycle, and attempting to quantify the intermolecular interactions in the pseudo-rotaxane. Once again, the syn–syn conformation (depicted in Fig. 6) was the most stable relative to the individual thread and macrocycle, and the least stable was the anti–anti conformation. The relative behaviour of these two conformations was essentially the same as observed for pseudo-rotaxane 1·5, with the latter being 32 kJ mol−1 higher in energy. However, unlike for pseudo-rotaxane 1·5, the syn–anti conformation was similar in energy to the anti–anti conformation, rather than the syn–syn conformation, being 30 kJ mol−1 higher in energy than the syn–syn. Importantly, and perhaps counter-intuitively given that pseudo-rotaxane 2·5 is not experimentally observed, a similar binding energy for 2·5 to that of 1·5 was calculated. For 2·5, the zero-point corrected binding energy for the most stable conformation (syn–syn) was found to be around 213 kJ mol−1, only 10 kJ mol−1 smaller than in the case of 1·5.
To explain why pseudo-rotaxane 1·5 forms but 2·5 does not, requires a consideration of the minimized structures of macrocycles 1 and 2 derived from the crystal structures of these two species (which were found to be of lower energy than the minimized structures of 1 and 2 taken from the simulations of pseudo-rotaxanes 1·5 and 2·5). While the minimized structure of isolated macrocycle 1 is similar to that of the macrocycle in pseudo-rotaxane 1·5, isolated macrocycle 2 has a remarkably broad cavity – and clearly would require significant rearrangement to achieve the correct geometry to form the hydrogen bonds and π–π stacking of pseudo-rotaxane 2·5. We therefore propose that a high energy barrier exists that disfavours reorganization of macrocycle 2, to account for lack of pseudo-rotaxane formation in this case.
Rotaxane synthesis
Following the pseudo-rotaxane studies, attention turned to preparing fully interlocked rotaxanes by use of CuAAC conditions to stopper a bis-azide pyridine-N-oxide in the presence of an isophthalamide macrocycle (Scheme 3). The reactions attempted involved dissolving either macrocycle 1 or 2 in dichloromethane under an inert atmosphere, adding 1 equivalent of pyridine-N-oxide bis-azide 611 followed by 2 equivalents of alkyne stopper 7.21 Then 0.2 equivalents of Cu(CH3CN)4BF4 and TBTA were added, followed by 2.2 equivalents of N,N-diisopropylamine. The reactions were stirred overnight at room temperature, and then submitted to an aqueous work-up and purification by silica gel chromatography. Non-interlocked axle 10 was first prepared (by not using either 1 or 2) to verify these conditions would successfully react the components 6 and 7.22
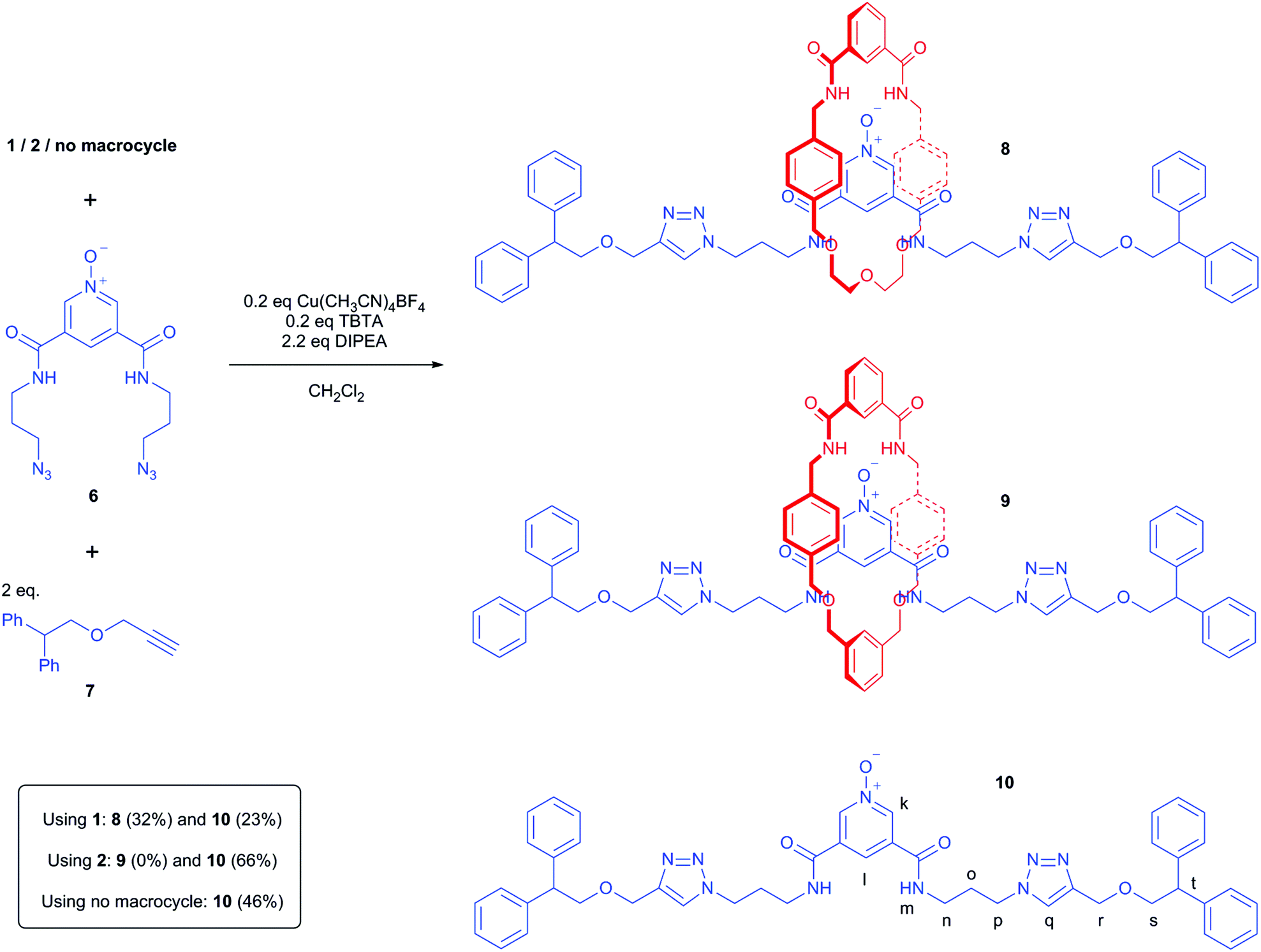 | ||
| Scheme 3 Attempted syntheses of rotaxanes 8 and 9 and non-interlocked axle 10. | ||
Using macrocycle 1, rotaxane 8 was isolated in a 32% yield. In contrast, using macrocycle 2, unsurprisingly no rotaxane 9 was observed in the crude 1H NMR spectrum, with only macrocycle 2 and non-interlocked axle 10 being isolated after column chromatography. These results were consistent with the pseudo-rotaxane studies using macrocycles 1 and 2 with pyridine-N-oxide thread 5.
The 1H NMR spectrum of rotaxane 8, along with that of non-interlocked macrocycle 1 and axle 10 for comparison, is shown in Fig. 7.23 The downfield shifts of protons c and d in the rotaxane compared to the non-interlocked macrocycle are indicative of hydrogen bonding to the oxygen atom of the pyridine-N-oxide of the axle component. The upfield shift and splitting of protons f and g are, as for pseudo-rotaxane 1·5, consistent with intercalation of the pyridine-N-oxide ring of the axle between the aromatic rings of the macrocycle.
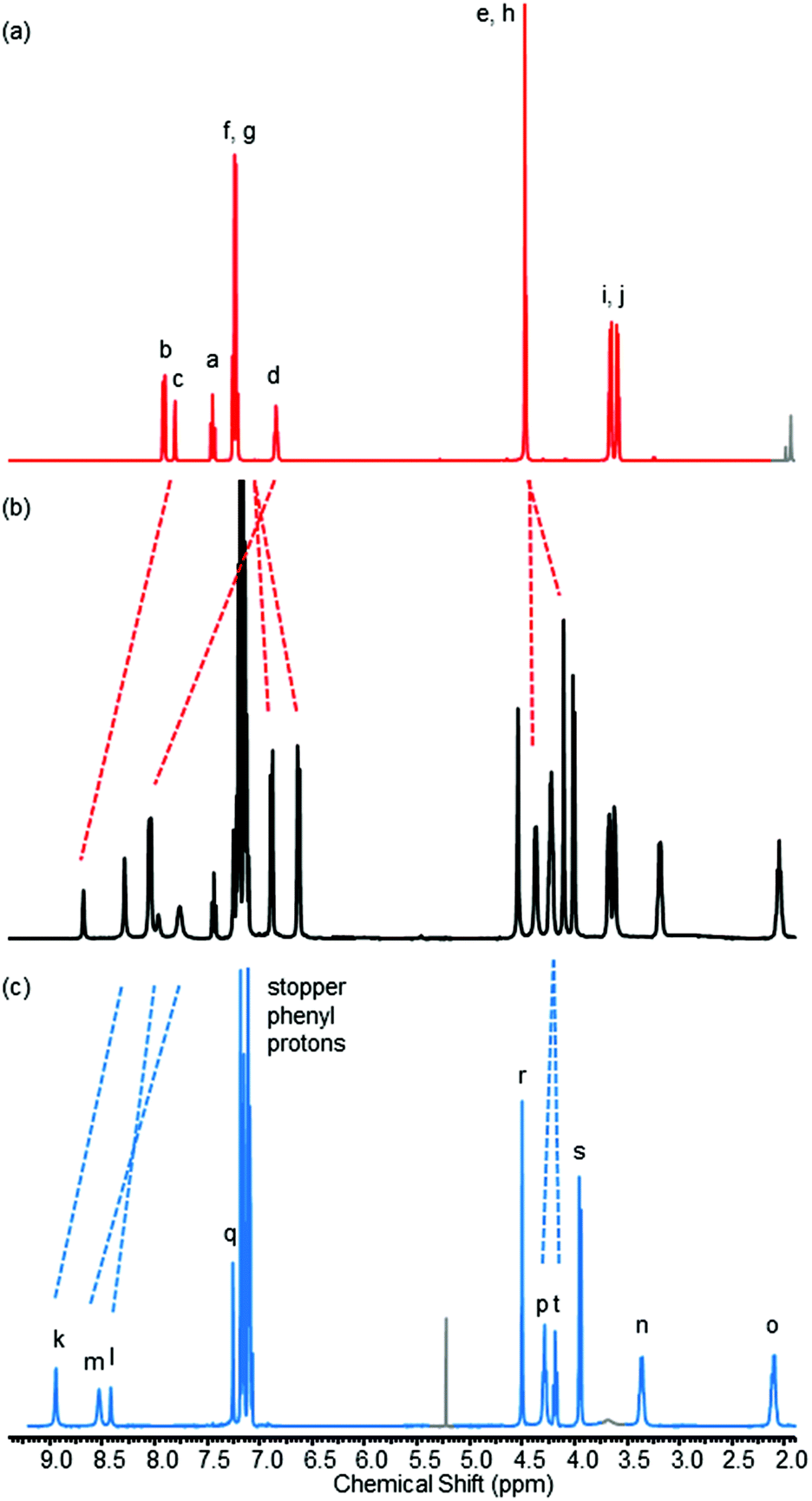 | ||
| Fig. 7 1H NMR spectra of (a) macrocycle 1 (b) rotaxane 8 and (c) axle 10 (CDCl3, 400 MHz, 298 K). For atom labels see Fig. 2 and Scheme 3. Trace solvent peaks are coloured grey. | ||
The interlocked nature of rotaxane 8 was further supported by the appearance of multiple through-space correlations being observed in the 1H–1H ROESY NMR spectrum between signals arising from protons in the macrocyclic and axle components (Fig. 8). Most clearly, the ortho-pyridinium proton k of the axle interacts with protons c, f and g (and possibly d) of the macrocyclic component. In addition, molecular ion peaks for the rotaxane are readily identifiable at m/z 1294.60 and 1316.59 corresponding to the H+ and Na+ adducts in the positive-ion electrospray MS (see ESI†).24
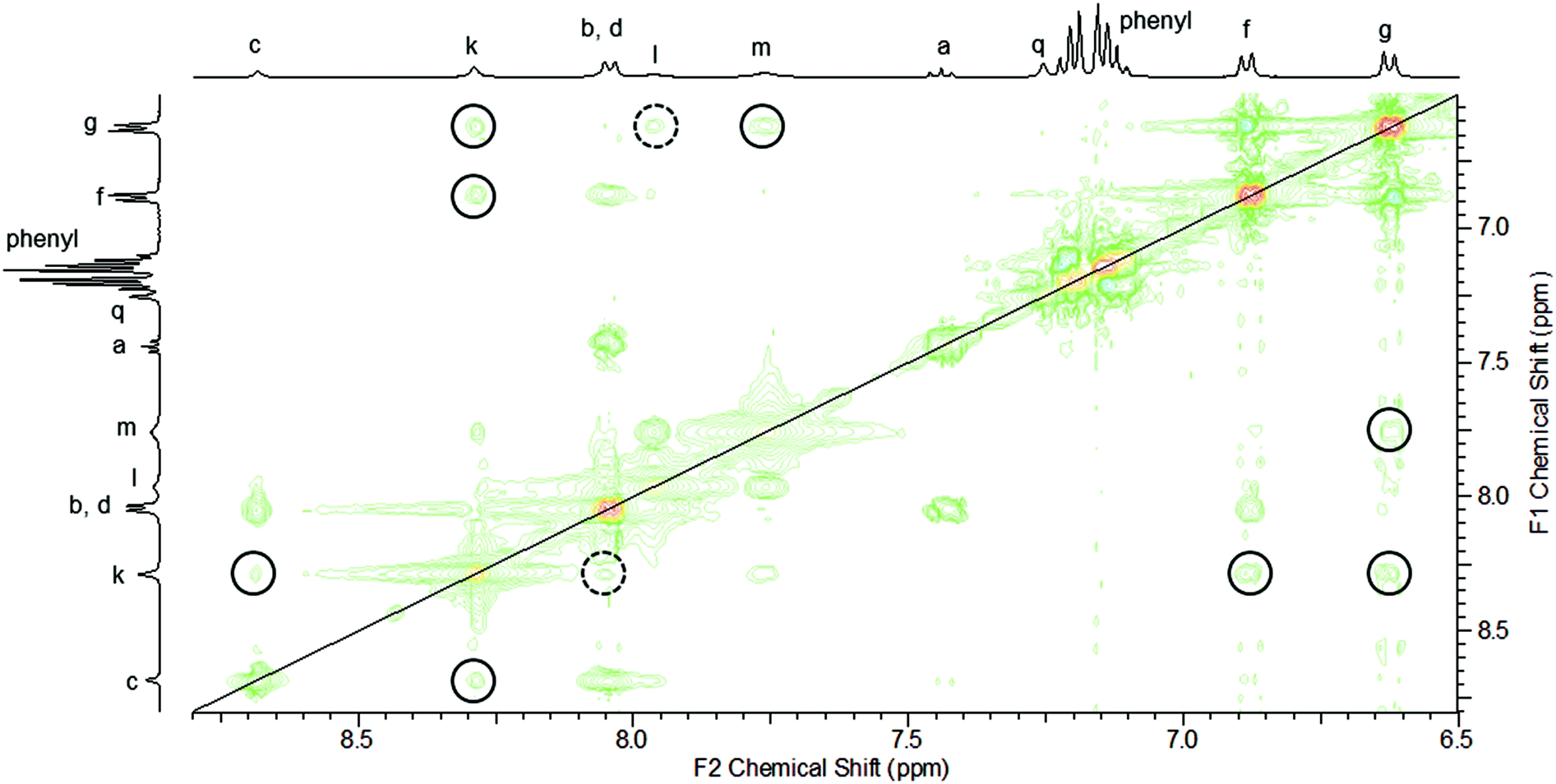 | ||
| Fig. 8 1H–1H ROESY NMR spectrum of rotaxane 8 (CDCl3, 400 MHz, 298 K). Inter-component through-space cross-peaks are circled. Cross-peaks that appear on only one side of the diagonal are enclosed by a dashed circle. | ||
Conclusions
The production in reasonable yield of a fully interlocked rotaxane comprising a 3,5-bisamide pyridine-N-oxide axle threaded through an isophthalamide macrocycle relying solely on hydrogen bond templation interactions is possible, without recourse to an additional ionic template. However, successful pseudo-rotaxane (and hence rotaxane) formation is dependent on the structure of the macrocycle, as rationalised in this study by a combination of NMR spectroscopy, X-ray crystallography and, importantly, computational modelling.The general architecture of rotaxane 8 presents numerous opportunities for incorporating functionality by use of appropriate precursors, for example on the axle and/or through the 5 position of the isophthalamide ring. The exploitation of this synthetic methodology, including the development of rotaxanes that can act as receptors for ionic and small molecular guests, is ongoing in our laboratories.
Experimental
General information
Commercially available solvents and chemicals were used without further purification unless stated. Dry solvents, NEt3 and DIPEA were purchased dry and stored under an inert atmosphere. Cu(CH3CN)4BF4 was stored in a desiccator over P4O10. Deionised water was used in all cases. Compounds 114 and 611 were prepared as previously reported. Details regarding the preparation of compounds 5 and 7 may be found in the ESI.†
Silica gel with a 60 Å particle size was used as the stationary phase for column chromatography. Analytical TLC was used to monitor the progress of column chromatography and analytical TLC plates were typically examined under short wavelength (λ = 254 nm) UV light. If required, ceric ammonium molybdate or potassium permanganate stains were used to develop the analytical TLC plates.
IR spectra were recorded on an Agilent Technologies Cary 630 FTIR spectrometer. NMR spectra were recorded on a Bruker Ultrashield 400 Plus spectrometer at 298 K, with the NMR data for 8 and 10 reported below assigned according to the atom labels to be found in Fig. 2 and Scheme 3. Mass spectra were recorded on a Thermofisher LTQ Orbitrap XL at the EPSRC UK National Mass Spectrometry Facility at Swansea University, UK, or a Shimadzu LCMS IT ToF instrument at Lancaster University, UK. Melting points were recorded on a Gallenkamp capillary melting point apparatus and are uncorrected.
Experimental procedures
:1 CH2Cl2/CH3OH) to yield the title compound, with (as identified by 1H NMR spectroscopy) one equivalent of CH2Cl2, as a white solid (177 mg, 38%). Rf = 0.27, 98:2 CH2Cl2/CH3OH. Mp multiple phase transitions between 208–220 °C. νmax/cm−1 (neat) 3330 (N–H), 2940 (C–H), 2860 (C–H), 2830 (C–H), 1640 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1530 (N–H), 1070 (C–O). δH(400 MHz; CDCl3) 7.94 (2H, d, 3J = 7.7 Hz, isophthalamide H4& H6), 7.81 (1H, s, isophthalamide H2), 7.48 (1H, t, 3J = 7.7 Hz, isophthalamide H5), 7.29–7.38 (12H, m, aromatic H), 6.74 (2H, br s, NH), 4.50–4.55 (12H, m, 3 × CH2). δC(100 MHz; CDCl3) 166.9 (CO), 138.4, 137.7, 137.2, 134.6, 130.9, 129.5, 128.5, 128.4, 128.2, 126.9, 126.6, 123.8 (12 Ar C environments), 71.9, 71.7 (2 × OCH2), 44.0 (NHCH2). m/z (ES) 541.1900 ([M + Cl]−, C32ClH30N2O4 requires 541.1900).
C), 1110 (C–O). δH(400 MHz; CDCl3) 7.64 (4H, d, 3J = 8.2 Hz, aromatic H), 7.47 (4H, d, 3J = 8.2 Hz, aromatic H), 7.31–7.40 (4H, m, aromatic H), 4.62 (4H, s, CH2), 4.60 (4H, s, CH2). δC(100 MHz; CDCl3) 143.7, 138.0, 132.2, 128.7, 127.7, 127.3, 127.0, 118.8, 111.3 (6 Ar C & the nitrile CN environments), 72.6, 71.2 (2 × CH2). m/z (ES) 386.1865 ([M + NH4]+, C24H24N3O2 requires 386.1863).
:2 to 92:8 CH2Cl2/CH3OH) to give the title compound as a foaming white solid (22 mg, 32%). Rf = 0.14, 98:2 CH2Cl2/CH3OH. Mp 150 °C (dec). νmax/cm−1 (neat) 3350 (N–H), 3060 (C–H), 2860 (C–H), 1650 (CO), 1540 (N–O). δH(400 MHz; CDCl3) 8.68 (1H, s, Hc), 8.29 (2H, s, Hk), 8.04–8.05 (5H, m, Hb & Hd), 7.96 (1H, s, Hl), 7.76 (2H, s, Hm), 7.44 (1H, t, 3J = 7.8 Hz, Ha), 7.10–7.25 (22H, m, aromatic H & Hq), 6.89 (4H, d, 3J = 7.8 Hz, Hf), 6.62 (4H, d, 3J = 7.8 Hz, Hg), 4.54 (4H, s, Hr), 4.37 (4H, s, He), 4.20–4.24 (6H, m, Hp & Ht), 4.10 (4H, s, Hh), 4.00 (4H, d, 3J = 7.2 Hz, Hs), 3.63–3.68 (8H, m, Hi & Hj), 3.18–3.20 (4H, m, Hn), 2.04–2.07 (4H, m, Ho). δC(100 MHz; CDCl3) 166.8, 162.5 (2 × CO), 145.0, 141.9, 140.0, 138.3, 135.4, 134.0, 132.3, 131.3, 129.1, 128.5, 128.4, 128.2, 126.5, 126.0, 124.9, 123.1 (17 pyridyl, aromatic & triazole C environments – including a coincident pair), 73.8, 70.9, 69.9, 64.4, 51.0, 47.8, 43.9, 37.2, 30.0 (10 × sp3 C – including a coincident pair). m/z (ES) 1294.6091 ([M + H]+, C75H80N11O10 requires 1294.6084); 1316.5940 ([M + Na]+, C75H79N11NaO10 requires 1316.5904).
:4 to 92:8 CH2Cl2/CH3OH) to give the title compound as a transparent colourless glass (22 mg, 47%). Rf = 0.17, 96:4 CH2Cl2/CH3OH. νmax/cm−1 (neat) 3270 (N–H), 3060 (C–H), 2860 (C–H), 1670 (CO), 1540 (N–O). δH(400 MHz; CDCl3) 8.94 (2H, s, Hk), 8.54 (2H, br s, Hm), 8.42 (1H, s, Hl), 7.26 (2H, s, Hq), 7.07–7.18 (20H, m, aromatic H), 4.50 (4H, s, Hr), 4.28 (4H, t, 3J = 6.4 Hz, Hp), 4.18 (4H, t, 3J = 7.3 Hz, Ht), 3.94 (4H, d, 3J = 7.3 Hz, Hs), 3.35–3.37 (4H, m, Hn), 2.10–2.13 (4H, m, Ho). δC(100 MHz; CDCl3) 162.2 (CO), 145.1, 141.9, 140.3, 133.7, 128.4, 128.2, 126.5, 123.2 (9 pyridyl, aromatic & triazole C environments – including a coincident pair), 73.8, 64.3, 50.9, 48.2, 37.5, 29.3 (6 × sp3 C). m/z (ES) 820.3939 ([M + H]+, C47H50N9O5 requires 820.3939).
O), 1530 (N–H), 1070 (C–O). δH(400 MHz; CDCl3) 7.94 (2H, d, 3J = 7.7 Hz, isophthalamide H4& H6), 7.81 (1H, s, isophthalamide H2), 7.48 (1H, t, 3J = 7.7 Hz, isophthalamide H5), 7.29–7.38 (12H, m, aromatic H), 6.74 (2H, br s, NH), 4.50–4.55 (12H, m, 3 × CH2). δC(100 MHz; CDCl3) 166.9 (CO), 138.4, 137.7, 137.2, 134.6, 130.9, 129.5, 128.5, 128.4, 128.2, 126.9, 126.6, 123.8 (12 Ar C environments), 71.9, 71.7 (2 × OCH2), 44.0 (NHCH2). m/z (ES) 541.1900 ([M + Cl]−, C32ClH30N2O4 requires 541.1900).
C), 1110 (C–O). δH(400 MHz; CDCl3) 7.64 (4H, d, 3J = 8.2 Hz, aromatic H), 7.47 (4H, d, 3J = 8.2 Hz, aromatic H), 7.31–7.40 (4H, m, aromatic H), 4.62 (4H, s, CH2), 4.60 (4H, s, CH2). δC(100 MHz; CDCl3) 143.7, 138.0, 132.2, 128.7, 127.7, 127.3, 127.0, 118.8, 111.3 (6 Ar C & the nitrile CN environments), 72.6, 71.2 (2 × CH2). m/z (ES) 386.1865 ([M + NH4]+, C24H24N3O2 requires 386.1863).
:2 to 92:8 CH2Cl2/CH3OH) to give the title compound as a foaming white solid (22 mg, 32%). Rf = 0.14, 98:2 CH2Cl2/CH3OH. Mp 150 °C (dec). νmax/cm−1 (neat) 3350 (N–H), 3060 (C–H), 2860 (C–H), 1650 (CO), 1540 (N–O). δH(400 MHz; CDCl3) 8.68 (1H, s, Hc), 8.29 (2H, s, Hk), 8.04–8.05 (5H, m, Hb & Hd), 7.96 (1H, s, Hl), 7.76 (2H, s, Hm), 7.44 (1H, t, 3J = 7.8 Hz, Ha), 7.10–7.25 (22H, m, aromatic H & Hq), 6.89 (4H, d, 3J = 7.8 Hz, Hf), 6.62 (4H, d, 3J = 7.8 Hz, Hg), 4.54 (4H, s, Hr), 4.37 (4H, s, He), 4.20–4.24 (6H, m, Hp & Ht), 4.10 (4H, s, Hh), 4.00 (4H, d, 3J = 7.2 Hz, Hs), 3.63–3.68 (8H, m, Hi & Hj), 3.18–3.20 (4H, m, Hn), 2.04–2.07 (4H, m, Ho). δC(100 MHz; CDCl3) 166.8, 162.5 (2 × CO), 145.0, 141.9, 140.0, 138.3, 135.4, 134.0, 132.3, 131.3, 129.1, 128.5, 128.4, 128.2, 126.5, 126.0, 124.9, 123.1 (17 pyridyl, aromatic & triazole C environments – including a coincident pair), 73.8, 70.9, 69.9, 64.4, 51.0, 47.8, 43.9, 37.2, 30.0 (10 × sp3 C – including a coincident pair). m/z (ES) 1294.6091 ([M + H]+, C75H80N11O10 requires 1294.6084); 1316.5940 ([M + Na]+, C75H79N11NaO10 requires 1316.5904).
:4 to 92:8 CH2Cl2/CH3OH) to give the title compound as a transparent colourless glass (22 mg, 47%). Rf = 0.17, 96:4 CH2Cl2/CH3OH. νmax/cm−1 (neat) 3270 (N–H), 3060 (C–H), 2860 (C–H), 1670 (CO), 1540 (N–O). δH(400 MHz; CDCl3) 8.94 (2H, s, Hk), 8.54 (2H, br s, Hm), 8.42 (1H, s, Hl), 7.26 (2H, s, Hq), 7.07–7.18 (20H, m, aromatic H), 4.50 (4H, s, Hr), 4.28 (4H, t, 3J = 6.4 Hz, Hp), 4.18 (4H, t, 3J = 7.3 Hz, Ht), 3.94 (4H, d, 3J = 7.3 Hz, Hs), 3.35–3.37 (4H, m, Hn), 2.10–2.13 (4H, m, Ho). δC(100 MHz; CDCl3) 162.2 (CO), 145.1, 141.9, 140.3, 133.7, 128.4, 128.2, 126.5, 123.2 (9 pyridyl, aromatic & triazole C environments – including a coincident pair), 73.8, 64.3, 50.9, 48.2, 37.5, 29.3 (6 × sp3 C). m/z (ES) 820.3939 ([M + H]+, C47H50N9O5 requires 820.3939).
Computational methodology
All calculations were performed using Gaussian 09 version E01;18 structures were visualised with Gaussview 5.09. Density-functional theory was employed to study the electronic and molecular structure of all of the systems, using the conventional Kohn–Sham approach. The B3LYP19 exchange–correlation functional was used (as defined in Gaussian 09, with VWN3 in the correlation functional definition, rather than the conventional VWN5 definition25), alongside the 6-31G* all-electron basis set on all atoms. The general use of larger basis sets was prohibitively expensive due to the size of the molecules (at least for the optimisations), and where tested did not change any of the conclusions. The default integration grid was employed for all calculations.For all calculations, solvent effects were modelled using the polarisable continuum model (PCM); the default Gaussian 09 cavity parameters were used based on the UFF radii, with chloroform as the continuum solvent.
Dispersion effects were approximated using the D3 approach of Grimme,20 with the parameters as recommended for the B3LYP functional.
Frequency calculations were undertaken to ensure that the stationary points found were in fact minima on the potential energy surfaces, and also to allow for zero-point energy corrections to the energetic data.
Acknowledgements
N. H. E. wishes to thank the Royal Society of Chemistry for a Research Grant that funded initial experiments on the work reported here. C. E. G. wishes to thank the Faculty of Science and Technology at Lancaster University for a PhD Studentship. We thank Dr Nathan Halcovitch (Lancaster University) for the X-ray structure determination of macrocycle 2 and pseudo-rotaxane 1·5. For the recording of mass spectrometry data we express our thanks to the EPSRC UK National Mass Spectrometry Facility (NMSF) at Swansea University, UK (samples of compounds 3, 4, 7 & 10), and Dr David Rochester at Lancaster University, UK (samples of compounds 2, 5 & 8). We thank Dr Geoff Akien (Lancaster University) for assistance in, and Dr Nick Rees (Oxford University) for advice about, the recording of the 1H–1H ROESY NMR spectrum of rotaxane 8.All underlying data for this paper are provided in the experimental section and the ESI.† Electronic copies of NMR spectra are available from DOI: 10.17635/lancaster/researchdata/90.
Notes and references
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- For a review on functional rotaxanes, see: S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC.
- For some very recent examples of functional rotaxanes, see: (a) R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC; (b) M. Galli, J. E. M. Lewis and S. M. Goldup, Angew. Chem., Int. Ed., 2015, 54, 13545–13549 CrossRef CAS PubMed; (c) S. Guha, G. K. Shaw, T. M. Mitcham, R. R. Bouchard and B. D. Smith, Chem. Commun., 2016, 52, 120–123 RSC; (d) Y. Cakmak, S. Erbas-Cakmak and D. A. Leigh, J. Am. Chem. Soc., 2016, 138, 1749–1751 CrossRef CAS PubMed; (e) G. Yu, D. Wu, Y. Li, Z. Zhang, L. Shao, J. Zhou, Q. Hu, G. Tang and F. Huang, Chem. Sci., 2016, 7, 3017–3024 RSC.
- (a) C. Wu, P. R. Lecavalier, Y. X. Shen and H. W. Gibson, Chem. Mater., 1991, 3, 569 CrossRef CAS; (b) J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1992, 1131–1133 RSC; (c) L. Hogg, D. A. Leigh, P. J. Lusby, A. Morelli, S. Parsons and J. K. Y. Wong, Angew. Chem., Int. Ed., 2004, 43, 1218–1221 CAS; (d) A. M. Fuller, D. A. Leigh, P. J. Lusby, I. D. H. Oswald, S. Parsons and D. B. Walker, Angew. Chem., Int. Ed., 2004, 43, 3914–3918 CrossRef CAS PubMed; (e) S. M. Goldup, D. A. Leigh, P. J. Lusby, R. T. McBurney and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 6999–7003 CrossRef CAS PubMed; (f) V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed; (g) H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 CrossRef CAS PubMed; (h) A. Noor, S. C. Moratti and J. D. Crowley, Chem. Sci., 2014, 5, 4283–4290 RSC.
- (a) G. M. Hübner, J. Gläser, C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 383–386 CrossRef; (b) C. A. Schalley, G. Silva, C. F. Nising and P. Linnartz, Helv. Chim. Acta, 2002, 85, 1578–1596 CrossRef CAS; (c) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS PubMed; (d) E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289 CrossRef CAS PubMed; (e) L. M. Hancock and P. D. Beer, Chem. – Eur. J., 2009, 15, 42–44 CrossRef CAS PubMed; (f) M. J. Langton, L. C. Duckworth and P. D. Beer, Chem. Commun., 2013, 49, 8608–8610 RSC.
- (a) P. R. Ashton, M. Grognuz, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Tetrahedron Lett., 1991, 32, 6235–6238 CrossRef CAS; (b) R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS; (c) M. J. Gunter, N. Bampos, K. D. Johnstone and J. K. M. Sanders, New J. Chem., 2001, 25, 166–173 RSC.
- (a) F. G. Gatti, D. A. Leigh, S. A. Nepogodiev, A. M. Z. Slawin, S. J. Teat and J. K. Wong, J. Am. Chem. Soc., 2001, 123, 5983–5989 CrossRef CAS PubMed; (b) D. M. D'Souza, D. A. Leigh, L. Mottier, K. M. Mullen, F. Paolucci, S. J. Teat and S. Zhang, J. Am. Chem. Soc., 2010, 132, 9465–9470 CrossRef PubMed; (c) A. Altieri, V. Aucagne, R. Carrillo, G. J. Clarkson, D. M. D'Souza, J. A. Dunnett, D. A. Leigh and K. M. Mullen, Chem. Sci., 2011, 2, 1922–1928 RSC; (d) R. Ahmed, A. Altieri, D. M. D'Souza, D. A. Leigh, K. M. Mullen, M. Papmeyer, A. M. Z. Slawin, J. K. Y. Wong and J. D. Wollins, J. Am. Chem. Soc., 2011, 133, 12304–12310 CrossRef CAS PubMed; (e) A. Tron, P. J. Thornton, M. Rocher, H.-P. J. de Rouville, J.-P. Desvergne, B. Kauffmann, T. Buffeteau, D. Cavagnat, J. H. R. Tucker and N. D. McClenaghan, Org. Lett., 2014, 16, 1358–1361 CrossRef CAS PubMed.
- M. Chen, S. Han, L. Jiang, S. Zhou, F. Jiang, Z. Xu, J. Liang and S. Zhang, Chem. Commun., 2010, 46, 3932–3934 RSC.
- Additional Na+ or Ba2+ template: L. M. Hancock and P. D. Beer, Chem. Commun., 2011, 47, 6012–6014 RSC . Yields: (Na+) 50%, (Ba2+) 28%; (no cation) 10%.
- Additional Na+ template: R. C. Knighton and P. D. Beer, Chem. Commun., 2014, 50, 1540–1542 RSC . Yield: (Na+) 62%; (no cation) <10%.
- Additional Ln3+ template: F. Zapata, O. A. Blackburn, M. J. Langton, S. Faulkner and P. D. Beer, Chem. Commun., 2013, 49, 8157–8159 RSC . Yield: (Ln3+) 20%. In this system it was not possible to determine the yield in the absence of a cationic template, as the macrocycle would sequester the Cu cation being used in the CuAAC reaction being used to stopper the rotaxane.
- Additional Cl− template: J. M. Mercurio, F. Tyrrell, J. Cookson and P. D. Beer, Chem. Commun., 2013, 49, 10793–10795 RSC . Yields: (Cl−) 9%–17%; (no Cl−) 0%.
- L. M. Hancock, DPhil Thesis, University of Oxford, 2011.
- C. N. Marrs and N. H. Evans, Org. Biomol. Chem., 2015, 13, 11021–11025 CAS.
- See ESI† for synthetic details about thread 5.
- Due to peak broadening it is hard to assign with confidence peaks c and d in the 1H NMR of pseudo-rotaxane 1·3. It is expected that these peaks will have moved downfield due to hydrogen bonding to the oxygen of the pyridine-N-oxide.
- See ESI† for further images from the computational simulations and for coordinate data files.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (d) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1998, 37, 785–789 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- See ESI† for synthetic details about stopper 7.
- After work-up, analysis of the crude 1H NMR spectrum suggested a high conversion of 6 and 7 to axle 10, but after chromatographic purification, 10 was isolated in only a 46% yield. This is attributed to the polar nature of 10 meaning that material was lost on the polar silica stationary phase. Note that 10 was isolated in a 66% yield after column chromatography of the crude reaction mixture from the attempted synthesis of rotaxane 9.
- See ESI† for 13C NMR, IR and high resolution mass spectra of rotaxane 8.
- No peaks attributable to either macrocycle 1 or axle 10 were observed in the electrospray MS of rotaxane 8.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional notes on experimental procedures; copies of characterisation spectra of novel compounds; crystallographic data for macrocycle 2 and pseudo-rotaxane 1·5 and further computational modelling data. CCDC 1473491 and 1473492. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01435f |
| This journal is © The Royal Society of Chemistry 2016 |