
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
General synthesis of P-stereogenic compounds: the menthyl phosphinate approach†
Olivier
Berger
and
Jean-Luc
Montchamp
*
Chemistry and Biochemistry, Box 298860. and Texas Christian University, Fort Worth, Texas 76129, USA. E-mail: j.montchamp@tcu.edu; Fax: +1 (817) 257 5851
First published on 13th July 2016
Abstract
Easily prepared menthyl phosphinates of high diastereoisomeric purity provide versatile intermediates for the synthesis of P-stereogenic compounds. Previous efforts starting about fifty years ago have been hampered by a lack of generality so the menthyl route has been nearly abandoned. Herein we provide a general solution to this long-standing problem and describe a general synthesis of menthyl H-phosphinate and disubstituted phosphinate esters. The method to prepare these versatile precursors relies on a simple and inexpensive process avoiding the use of phosphorus trichloride, Grignard reagents, and complicated cryogenic crystallizations. Established protocols can then be employed to synthesize P-stereogenic secondary and tertiary phosphine oxides and therefore P-stereogenic phosphine ligands.
Introduction
The synthesis of compounds featuring a chiral phosphorus atom (P-stereogenic compounds) has a long history dating as far back as the early twentieth century.1 However, it was not until the 1960's that major progress was made in the field. Most notably, this is also when the use of phosphinate esters of naturally occurring L-(−)-menthol first appeared in the classic work of Mislow2 and others.3 The methodology was used by Knowles and coworkers for the synthesis of the bisphosphine ligand diPAMP in 1975.4 Subsequent application of the ligand in the rhodium-catalyzed asymmetric hydrogenation of L-DOPA constitutes a milestone in asymmetric synthesis ultimately resulting in Knowles sharing the 2001 Nobel Prize in Chemistry with Noyori and Sharpless.Since these pioneering contributions, numerous methods have been devised to prepare P-stereogenic compounds, and many excellent reviews have appeared on the subject.5 Virtually all reactions available in the “asymmetric toolbox” have been employed for this purpose. Interestingly, menthyl phosphinates have largely fallen out of favor, in spite of their groundbreaking historical importance, and their well-established transformations into phosphine oxides (and ultimately phosphines). This situation is due to the fact that separating menthyl phosphinate diastereoisomers is often a very challenging task and each phosphinate requires different conditions.
The case of menthyl phenyl-H-phosphinate 1, undoubtedly the most widely employed of the menthyl phosphinates to date, is illustrative (Scheme 1). In 1968, Emmick and Letsinger prepared 1 in 47% yield (from PhPCl2, pyridine, menthol then NaHCO3) but reported that the diastereoisomers could not be separated.3a Shortly thereafter, Farnham, Murray, and Mislow reported the preparation of 1 in 90% de after crystallization at −78 °C, but the yield was not reported (Scheme 1a).2b In 1995, Hammer and coworkers separated the mixture using a chiral HPLC column, once again without a yield being reported.6 Han and coworkers then employed (RP)-1 in a series of publications,7 but the yield was never reported. Ultimately in 2015, they published a detailed experimental procedure for the improved synthesis of (RP)-1via low temperature hydrolysis of PhP(OMen)Cl, resulting in a 57% yield and >98% de after three recrystallizations at −30 °C (Scheme 1b).8 In 2011, Gatineau, Giordano, and Buono reported a detailed preparation of (RP)-1 using MenOPCl2 and PhMgCl, in 16% overall yield and 98% de after two recrystallizations at −20 °C (Scheme 1c).9 These authors also prepared o-tolyl and 1-naphthyl derivatives in comparable yields.9
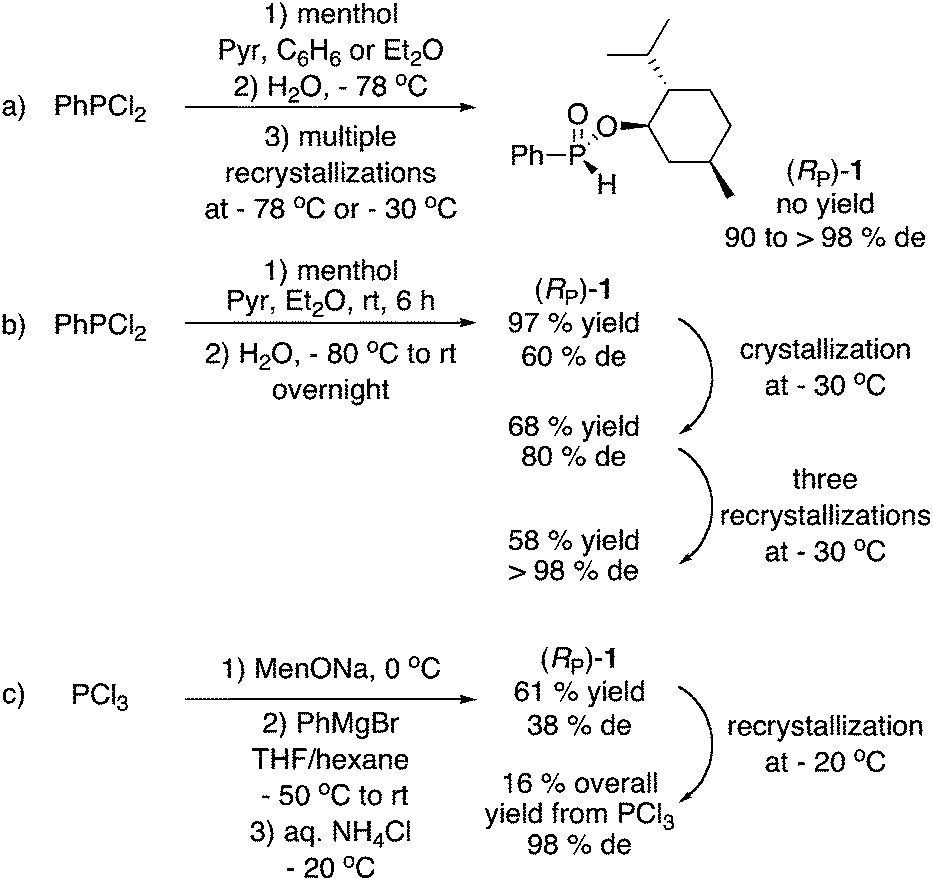 | ||
| Scheme 1 Literature preparations of menthyl phenyl-H-phosphinate (RP)-1. | ||
As can be seen in Scheme 1, the synthesis of (RP)-1 in good diasteromeric purity is challenging due to the fact that this compound is an oil at room temperature. Furthermore, all approaches employ P–Cl reagents, diethyl ether or THF as solvents, and cryogenic conditions during synthesis and/or purification. However, in spite of these practical issues, the reactions summarized in Scheme 1 have been conducted to prepare (RP)-1 in large quantity (ca. 50 g or 0.18 mol). Because the vast majority of P-stereogenic phosphine ligands contain a phenyl group, H-phosphinate 1 has been extensively employed as starting material, and numerous highly stereoselective/stereospecific reactions have been described.5c,7,9 Nonetheless, the ready availability of 1 remains questionable, let alone that of other menthyl H-phophinates.
We recently communicated what we believe to be a general solution to the problem of preparing a variety of menthyl phosphinates (including 1).10 Herein, the full account of this work is presented.
Results and discussion
Our methodology relies on two key discoveries: (1) the simple preparation of easily crystallized (hydroxymethyl)phosphinates,10 and (2) the stereoselective cleavage of the hydroxymethyl moiety using the Corey–Kim oxidation to unmask a P(O)H moiety, if desired.10,11 Of course, the hydroxymethyl methylene carbon can be preserved instead.Menthyl(hydroxymethyl)phosphinates
(Hydroxymethyl)-H-phosphinate 2 is the simplest member of this class of compounds.12 The preparation of 2 is straightforward, as shown in Scheme 2. Heating commercially available aqueous hypophosphorous acid with paraformaldehyde, followed by azeotropic distillation with menthol in toluene afforded 2 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers. In our initial report,10 the mixture was crystallized from ether at −18 °C in a standard freezer, however crystallization in a refrigerator at ∼0 °C gave similar results, thus recrystallization from Et2O at 0 °C gave (RP)-2 in 10% yield and >99% de. The absolute configuration was assigned via single X-ray crystallography.10 Whereas the overall yield of (RP)-2 is admittedly low, the process is very simple and does not require anhydrous conditions, base, organometallic reagent (Grignard or MenONa), nor cryogenic multiple recrystallizations. (Hydroxymethyl)-H-phosphinate (RP)-2 is also ideal for further elaboration of the remaining P(O)H functionality. The leftover mother liquor can also be exploited further to increase its value (vide infra). Replacing (−)-menthol with (+)-fenchyl alcohol and (−)-borneol gave the corresponding esters in 3% yield (92% de), and 17% yield (88% de), respectively (the absolute configuration at phosphorus was not established in those cases since menthol was superior and is one of the cheapest chiral auxiliaries).
:1 mixture of diastereoisomers. In our initial report,10 the mixture was crystallized from ether at −18 °C in a standard freezer, however crystallization in a refrigerator at ∼0 °C gave similar results, thus recrystallization from Et2O at 0 °C gave (RP)-2 in 10% yield and >99% de. The absolute configuration was assigned via single X-ray crystallography.10 Whereas the overall yield of (RP)-2 is admittedly low, the process is very simple and does not require anhydrous conditions, base, organometallic reagent (Grignard or MenONa), nor cryogenic multiple recrystallizations. (Hydroxymethyl)-H-phosphinate (RP)-2 is also ideal for further elaboration of the remaining P(O)H functionality. The leftover mother liquor can also be exploited further to increase its value (vide infra). Replacing (−)-menthol with (+)-fenchyl alcohol and (−)-borneol gave the corresponding esters in 3% yield (92% de), and 17% yield (88% de), respectively (the absolute configuration at phosphorus was not established in those cases since menthol was superior and is one of the cheapest chiral auxiliaries).
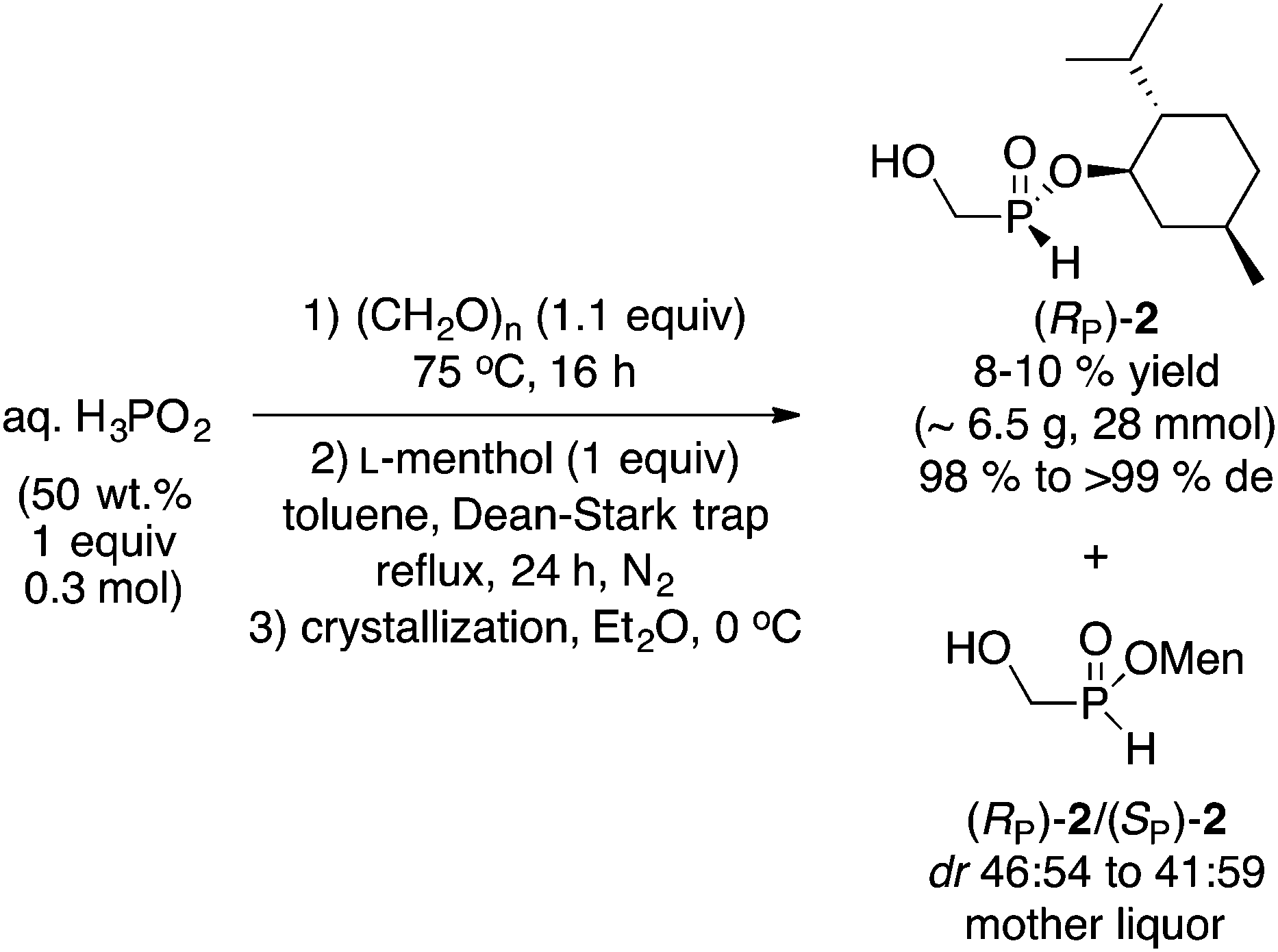 | ||
| Scheme 2 Synthesis of (hydroxymethyl)-H-phosphinate (RP)-2.10 | ||
A similar direct esterification approach is also successful with some H-phosphinic acids RPO2H2 (R = Ph, cinnamyl) as shown in Scheme 3.10 However, the steps must be reversed (menthol then paraformaldehyde) because only H-phosphinic acids can be esterified under Dean–Stark conditions. Menthyl(hydroxymethyl)phenylphosphinate (SP)-3 was obtained in 24% yield and 96% de after recrystallization at room temperature (similar results were obtained on a quarter scale).12 The absolute configuration of (SP)-3 was established by X-ray crystallography.10 Cinnamyl-H-phosphinic acid gave (RP)-4 in 32% yield and outstanding de.
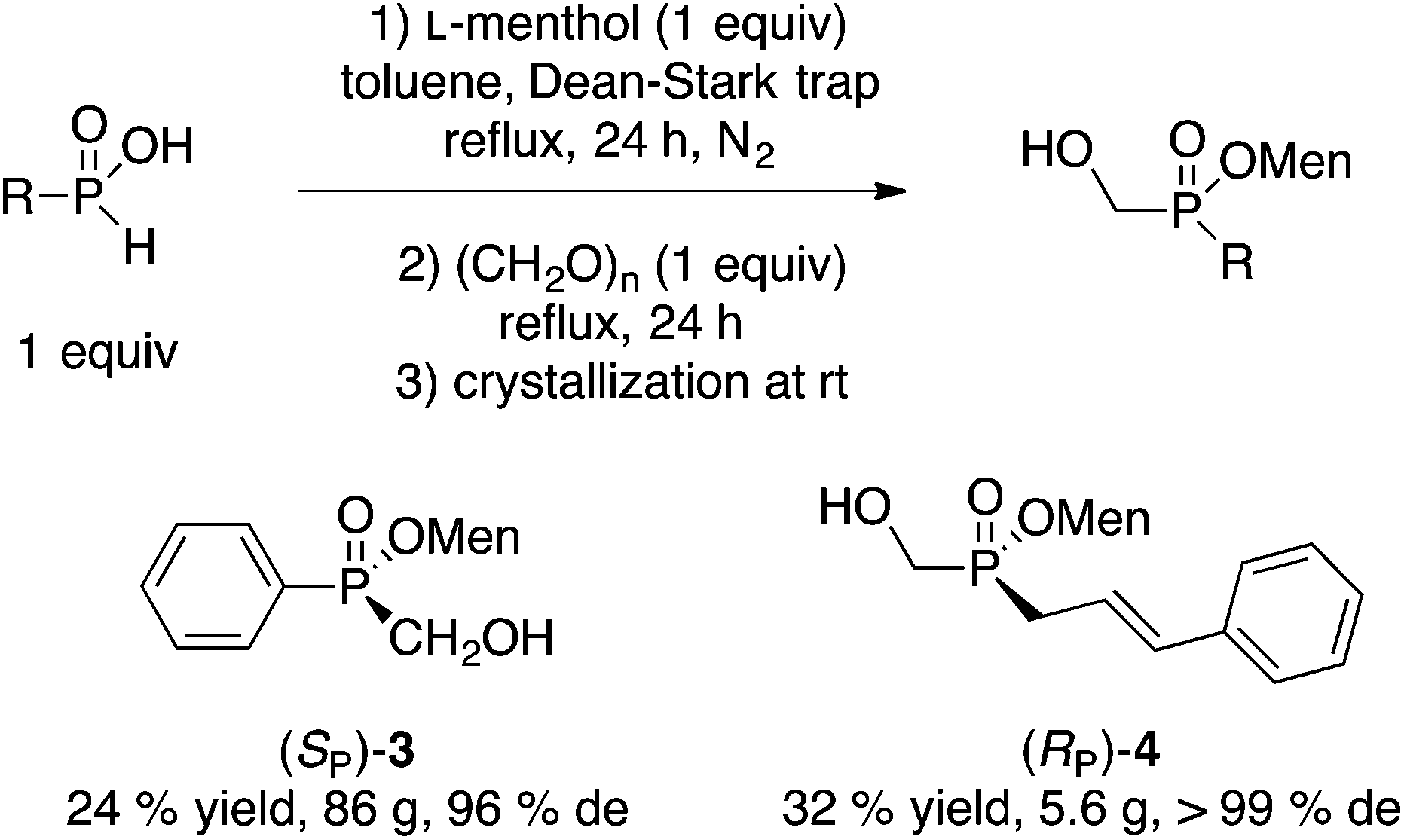 | ||
| Scheme 3 Direct reaction of H-phosphinic acids.10 | ||
In spite of the crystallinity typically displayed by (hydroxymethyl)phosphinates, the direct approach (Scheme 3) is not general and failed to provide an easily separable diastereoisomer with R = Me2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2, Ph3C, t-Bu, CH2OAc, CH2OBn, MeC(OEt)2 and Me2C(OH). Fortunately, (RP)-2 can be employed as a general starting material to access numerous disubstituted menthyl(hydroxymethyl)phosphinates through various reactions. Scheme 4 summarizes some palladium-catalyzed cross-coupling reactions.
CHCH2, Ph3C, t-Bu, CH2OAc, CH2OBn, MeC(OEt)2 and Me2C(OH). Fortunately, (RP)-2 can be employed as a general starting material to access numerous disubstituted menthyl(hydroxymethyl)phosphinates through various reactions. Scheme 4 summarizes some palladium-catalyzed cross-coupling reactions.
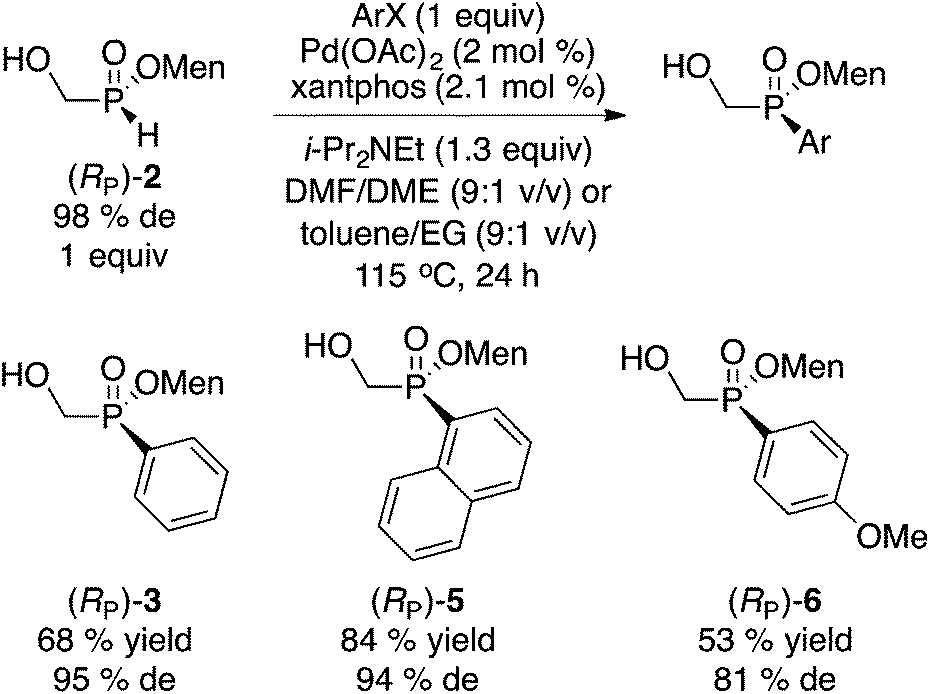 | ||
| Scheme 4 Palladium-catalyzed cross-couplings of (RP)-2. | ||
Interestingly, the cross-coupling of (RP)-2 with bromobenzene under conditions we previously developed13 (Scheme 4), resulted in the formation of (RP)-3 through expected retention of configuration, thereby delivering the opposite configuration at phosphorus when compared to that of the direct approach in Scheme 3. However, the cross-coupling reaction sometimes resulted in some erosion in the de as shown with p-anisyl (RP)-6. Nonetheless, the Pd-catalyzed coupling13 could be utilized to exploit the mother liquor leftover from the synthesis of (RP)-2. When a diastereomeric mixture of 2 ((SP)/(RP) 59:41) was reacted as in Scheme 4, room temperature crystallization of the crude product resulted in the formation of (SP)-3 in 23% yield (overall from H3PO2) and 97% de.10
We found that our palladium-catalyzed cross-coupling of allylic alcohols previously described with H-phosphinic acids14 is also successful on H-phosphinate esters.15 Thus, reaction of (RP)-2 with an equimolar amount of cinnamyl alcohol takes place smoothly to produce (RP)-4 in excellent yield and complete stereospecificity (eqn (1)).
Instead of the Pd-catalyzed cross-coupling, our Mn(II)-catalyzed/Mn(IV) promoted arylation16 can be employed. In this case the hydroxyl group in 2 must first be protected as the acetate. Acetylation of (RP)-2 (or of the diastereomeric mixture of 2) proceeds stereospecifically and quantitatively (Ac2O, Et3N, CH2Cl2, 0 °C to rt, 20 h) to deliver acetate 7.
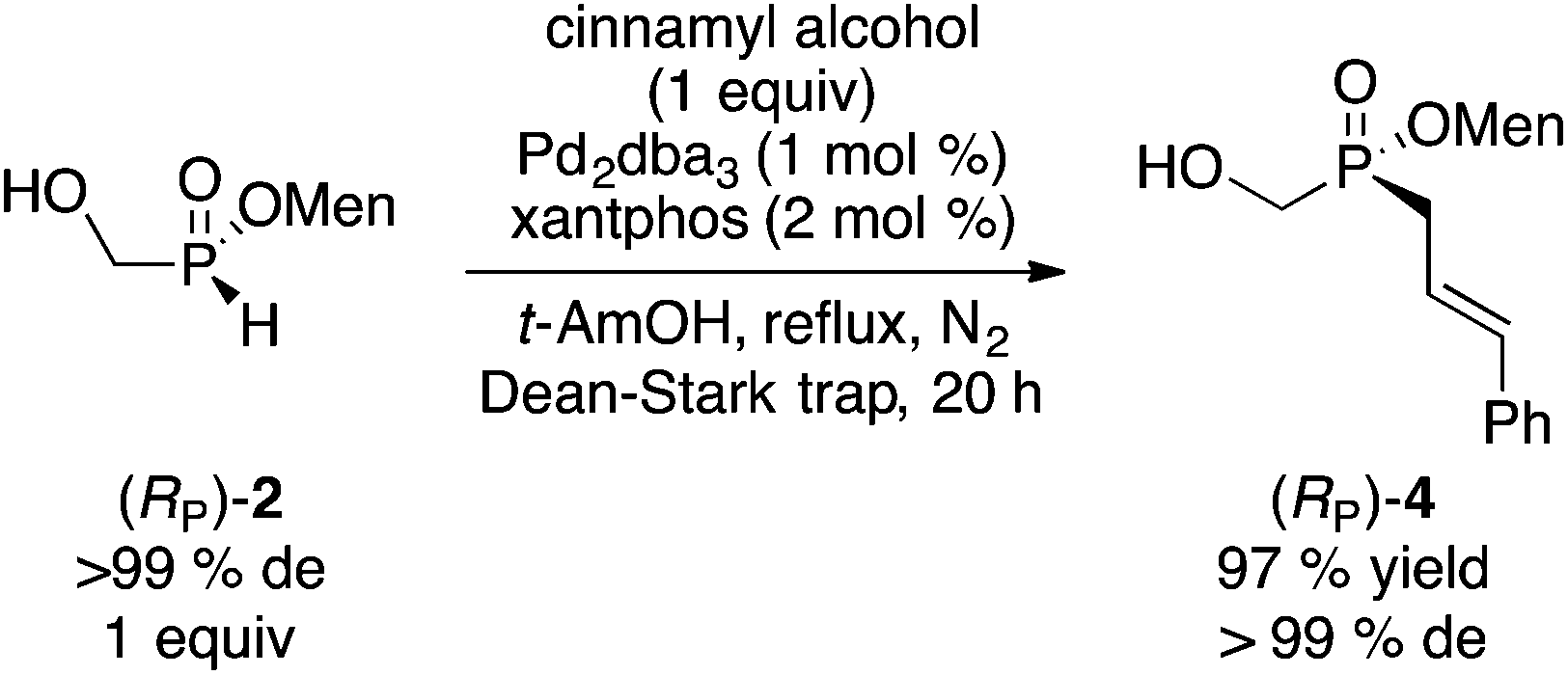 | (1) |
Acetate 7 was arylated as shown in Scheme 5. The reaction is stereospecific and proceeds with retention of configuration. Mesityl-substituted (RP)-8a was obtained in 79% yield without the need for a Grignard reagent. When 7 (obtained by acetylation of a mother liquor 2) was employed as starting material, compound 3a was obtained in 78% yield. Hydrolysis of 3a and crystallization at room temperature produces hydroxymethyl (SP)-3 in 16% overall yield (from the diastereomeric mixture of 7) and 95% de. Thus, although the yield is a little lower than in the Pd-catalyzed coupling we initially employed (23% yield, 97% de),10 the present sequence is simpler and about ten times less costly.
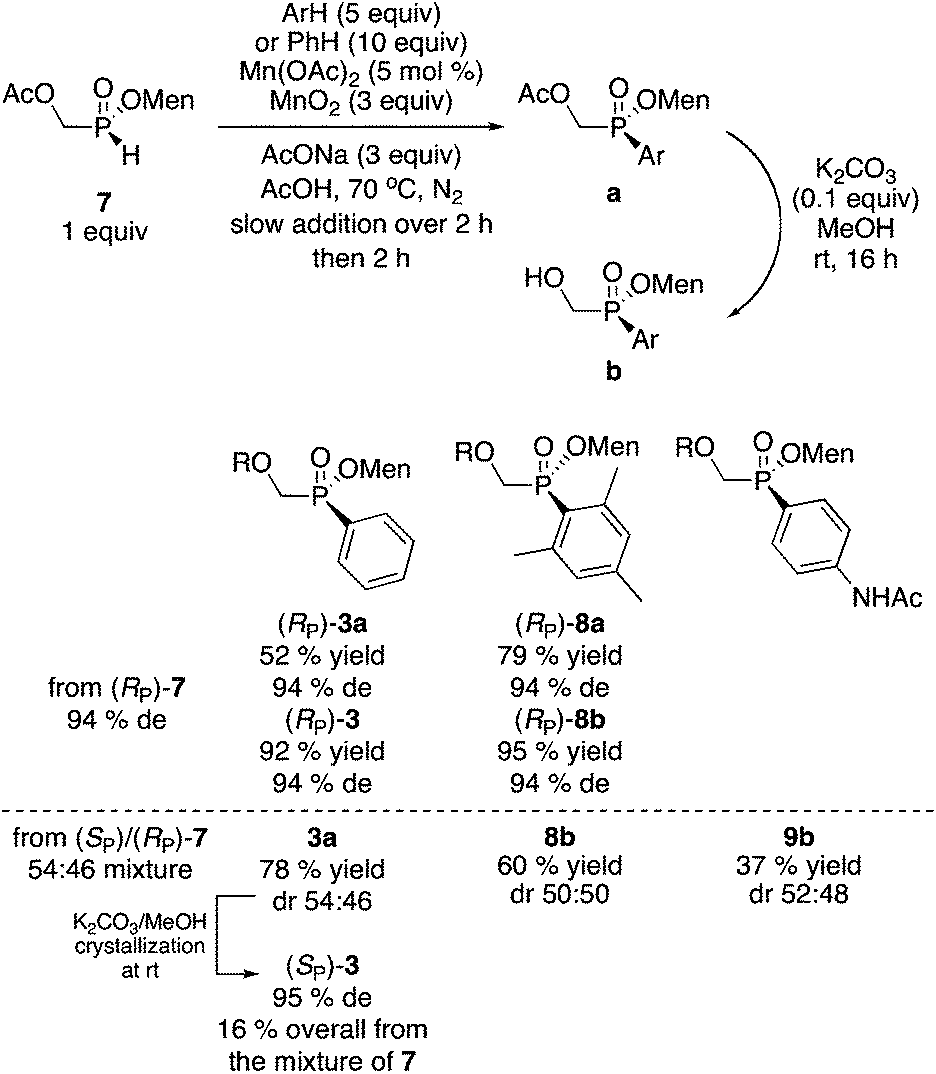 | ||
| Scheme 5 Manganese catalyzed/promoted arylation. | ||
Next, the alkylation of (RP)-2 was examined and the results are summarized in Scheme 6. Sila-Arbuzov alkylation17 was conducted by mild silylation with N,O-bis(trimethylsilyl)acetamide (BSA, 2 equiv.) followed by addition of the electrophile (MeI, allylBr, BnBr, TrBr, 2 equiv.). Overall, good yields and diastereoselectivities were observed.
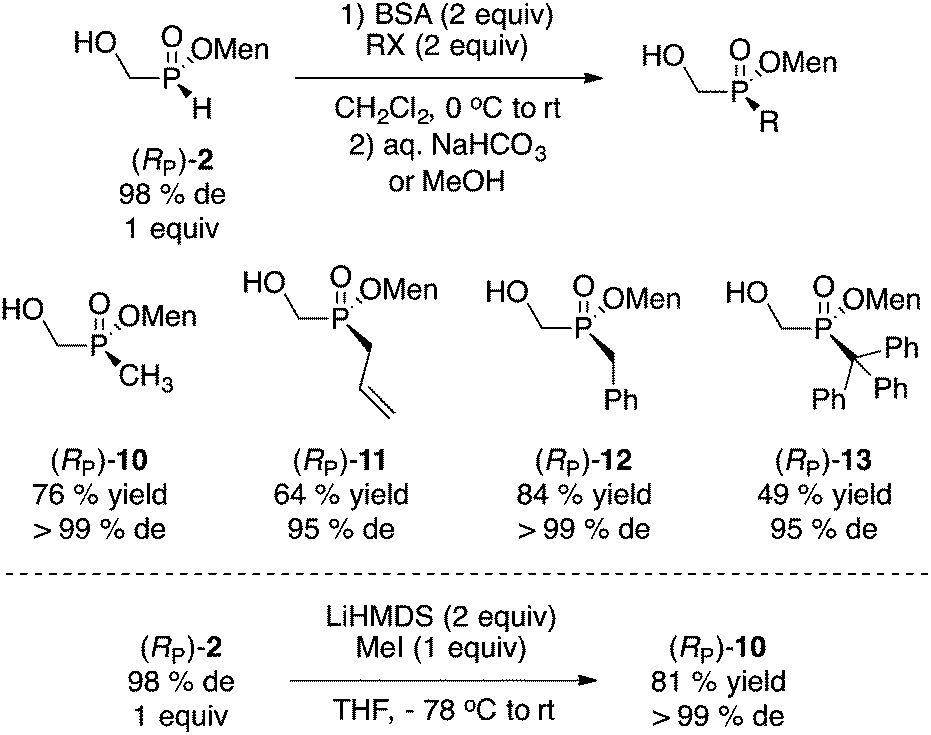 | ||
| Scheme 6 Alkylation of (RP)-2. | ||
Whereas the yield of trityl 13 is moderate, this is quite remarkable and potentially useful to ultimately prepare extremely bulky phosphine ligands.18 Early on, Mislow reported on the stereospecific (but condition-dependent) alkylation of (RP)-1 with sodium hydride and methyl iodide.2b With (RP)-2, alkylation under basic conditions (LiHMDS) gave a good result with methyl iodide as the electrophile (Scheme 6),10 but this was not investigated further considering the success and convenience of the sila-Arbuzov reaction.
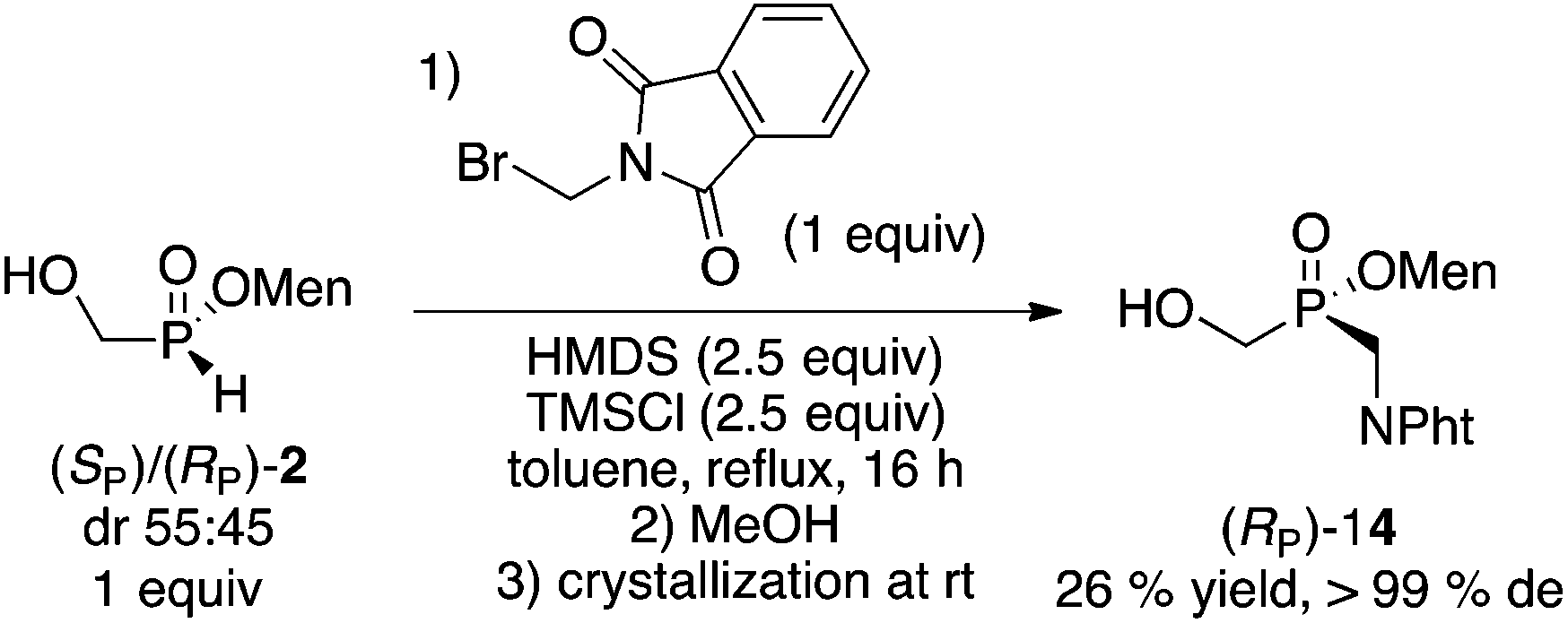
Finally, the reaction of unactivated (bromomethyl)phthalimide was examined using the more common silylation (HMDS/TMSCl)17 from the diastereoisomeric mixture (mother liquor) of 2. As shown in eqn (2), the overall yield was reasonable and the stereoselectivity excellent.
 | (2) |
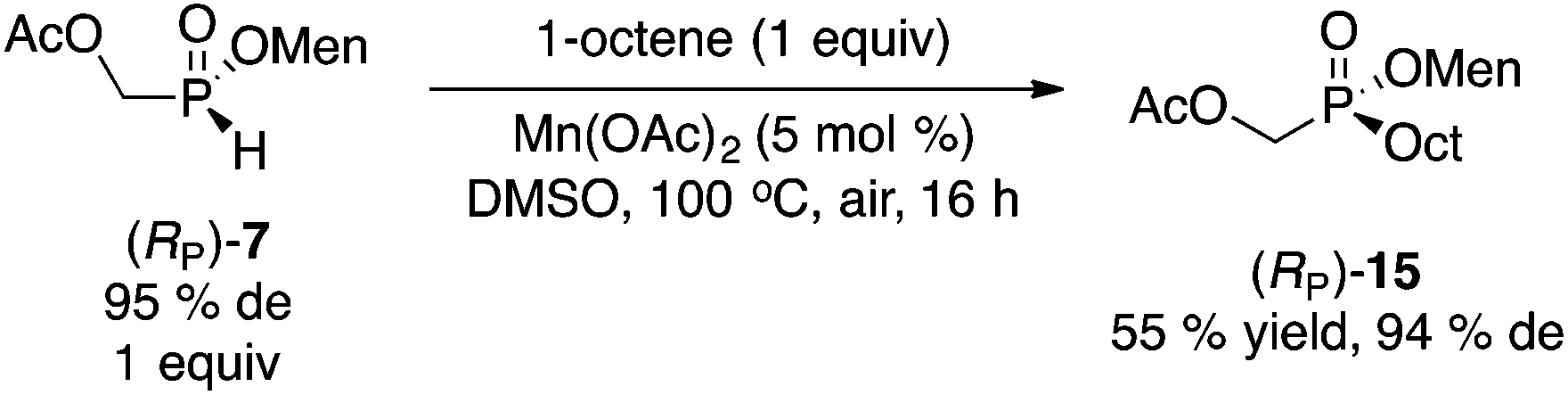 | (3) |
Next, we turned our attention to the free-radical reaction of 1-octene. Again, significant precedent exists with menthyl phenyl-H-phosphinate 1.19 However, aryl-H-phosphinates display special reactivities, because of their enhanced tautomerization properties.20 Whereas compounds 2 and 7 should also be activated because of the electron-withdrawing nature of the hydroxymethyl moiety (PhCOOH pKa = 4.18, HOCH2COOH pKa = 3.83, AcOCH2COOH pKa = 3.10),21,22 we chose conditions that should be generally applicable to any H-phosphinate ester. Therefore, our Mn(OAc)2-catalyzed reaction was examined (eqn (3)).23 As with the arylation chemistry (Scheme 5),16 protection of the hydroxyl as the acetate 7 was required. Product 15 was obtained in moderate yield but good diastereoselectivity.
Menthyl H-phosphinates
The mild oxidative cleavage of disubstituted (hydroxymethyl)phosphinates was investigated next. We showed previously that Corey–Kim oxidation (N-chlorosuccinimide/Me2S) proceeds in good yield to unmask a P(O)H group from the P(O)CH2OH precursor.10,11 Fortunately, this reaction is often stereospecific or at least highly stereoselective and proceeds in moderate to excellent yield. The results are shown in Scheme 7.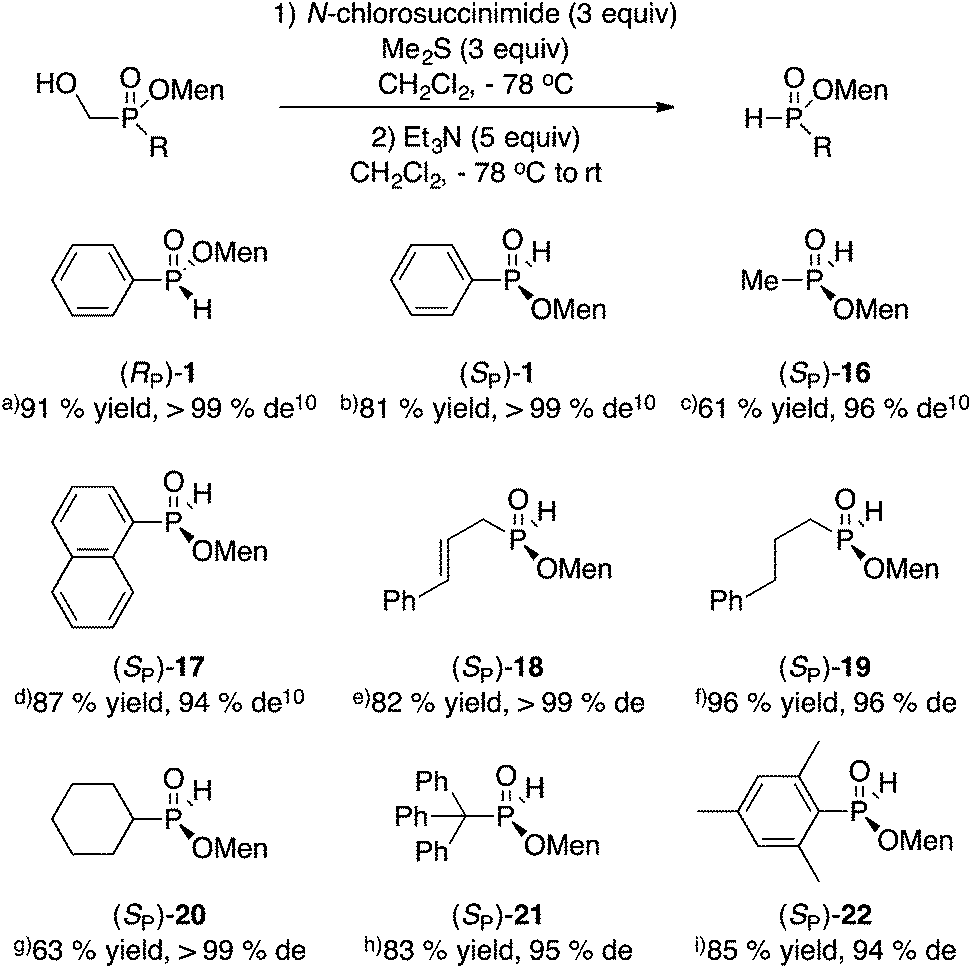 | ||
| Scheme 7 Synthesis of menthyl H-phosphinates. Configuration and de of the starting material: (a) (SP), >99%. (b) (RP), 95%. (c) (RP), >99%. (d) (RP), 94%. (e) (RP), >99%. (f) (RP), 98%. (g) (RP), >99%. (h) (RP), 95%. (i) (RP), 94%. | ||
Additionally, the oxidation can be conducted with odorless and recyclable dodecyl methyl sulfide.24 For example, the large scale (10 g, 32 mmol, >99% de) oxidation of (SP)-3 delivered (RP)-1 in 65% yield and >99% de. The long-chain sulfide was easily recovered quantitatively during purification and reused in other oxidations. An advantage of the Corey–Kim oxidation is that the sulfide can be used again directly without any intervening step, unlike oxidations involving the sulfoxide (Swern).
Our approach can deliver either absolute configuration at phosphorus, depending on the starting material that is chosen: (SP)-3 (Scheme 3) delivers (RP)-1, whereas (RP)-3 (Schemes 4 and 5) delivers (SP)-1. The preparation of (SP)-1via the literature procedures shown in Scheme 1 would require the use of D-(+)-menthol,7k which is about fifty times more expensive than L-(−)-menthol. If the ultimate goal is the preparation of a P-stereogenic tertiary phosphine, this does not matter since tertiary phosphine oxides can be reduced either with inversion or retention depending on the method employed.5d,w,25
On the other hand, the availability of either configuration of 1 using the same inexpensive L-(−)-menthol auxiliary could be an advantage for other applications, such as the synthesis of secondary phosphine oxide ligands.
Reactions preserving the methylene carbon in menthyl(hydroxymethyl)phosphinates
As mentioned earlier, one advantage of our hydroxymethyl route11 is that the carbon initially derived from formaldehyde can also be incorporated into P-stereogenic phosphinates. In the communication, we reported the Mitsunobu reaction of (SP)-3 with phthalimide (70% yield) and the Wittig rearrangement of allylated (SP)-3 in good yields and excellent stereoselectivity.Standard reactions of the alcohol moiety (Scheme 8) in (SP)-3 took place uneventfully. Elaboration of the products could also be conducted to prepare phosphinite–borane complex 27, thiophosphinates 28 and 29, and iodide 30 (Scheme 8).
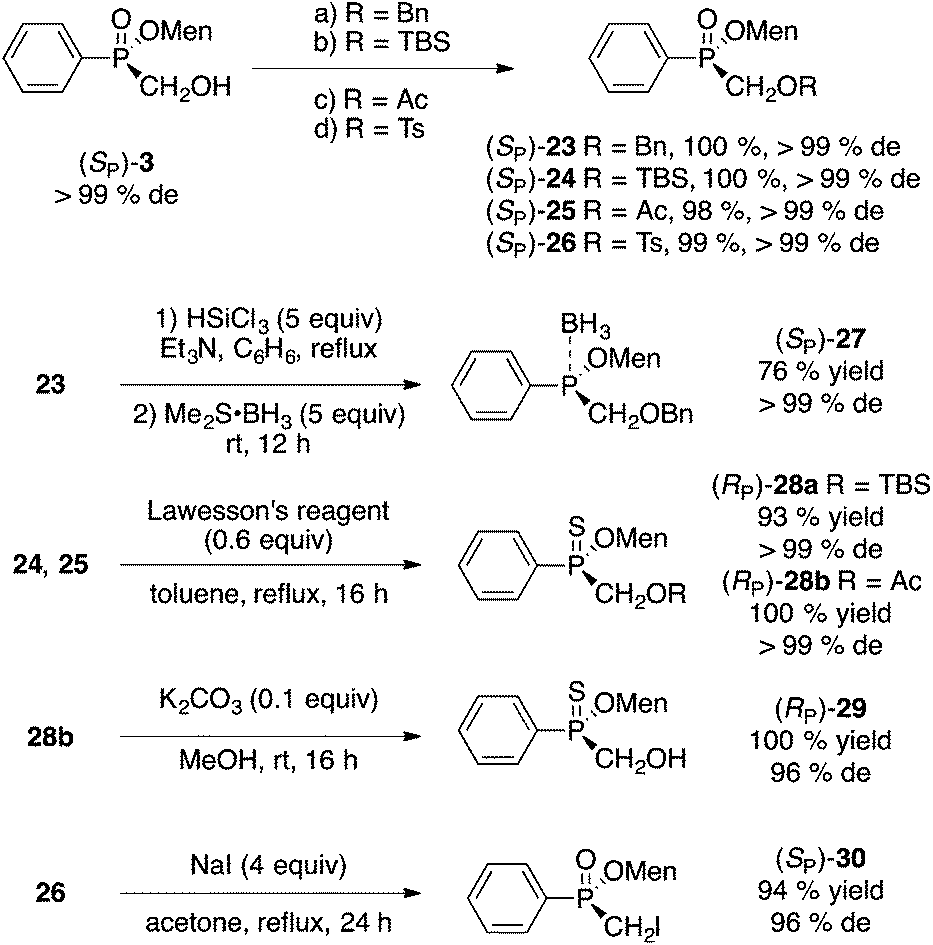 | ||
| Scheme 8 Modifications of (SP)-3. (a) NaH (1.5 equiv.), BnBr (1.2 equiv.), CH2Cl2, 0 °C to rt, 4 h. (b) t-BuMe2SiCl (1.5 equiv.), imidazole (2.7 equiv.), CH2Cl2, 0 °C to rt, 16 h. (c) Ac2O (1.2 equiv.), Et3N (1.25 equiv.), CH2Cl2, 0 °C to rt, 16 h. (d) TsCl (2 equiv.), i-Pr2NEt (2.5 equiv.), CH2Cl2, 0 °C to rt, 20 h. | ||
Magnesium–iodine exchange26 of 30 gave a Grignard reagent, which was dimerized to afford bisphosphinate 31 (eqn (4)) or reacted with chlorodiphenylphosphine to produce 1,1-bisphosphorus compound 32 in low yield (eqn (5)).
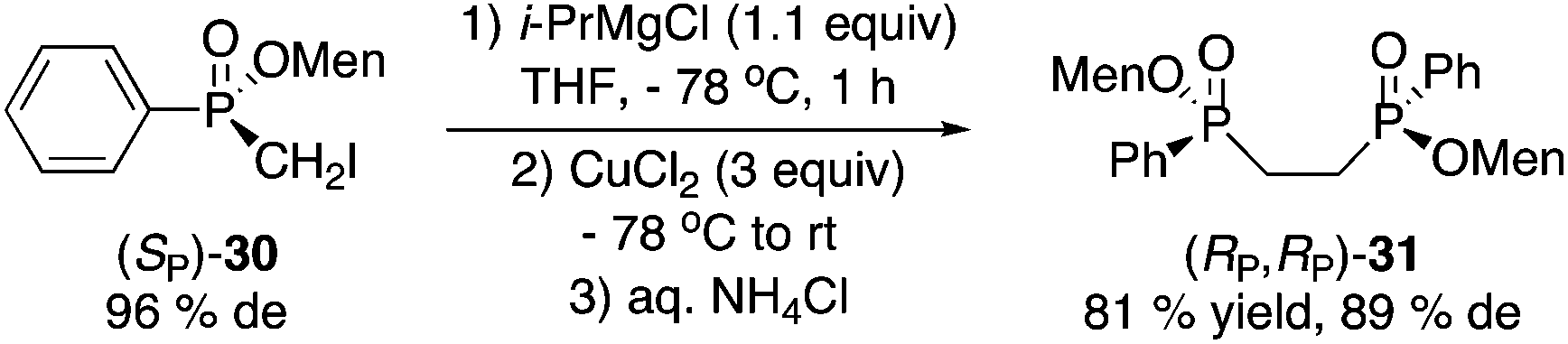 | (4) |
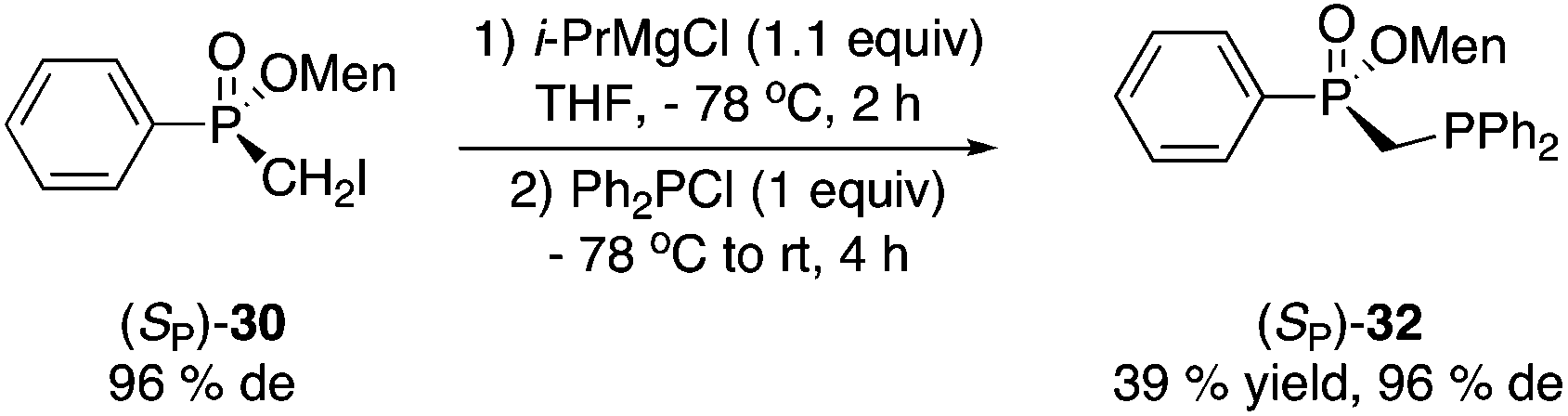 | (5) |
Miscellaneous reactions
Scheme 9 shows the formation of two phosphinate heterocycles. Hydrogenation of cinnamyl phosphinate (RP)-4 to 33, followed by oxidation11 gave (SP)-19 in excellent overall yield and good diastereoselectivity. Intramolecular Mn(II)/Mn(IV) arylation16,23 of (SP)-19 provided phosphinate 34 stereospecifically and in outstanding yield. On the other hand, annulation of (RP)-1 with diphenylacetylene under the same conditions delivered phosphinate 35 stereospecifically but in low yield (29%). It should be noted that this type of annulation has been described by Chen and Duan, with (RP)-1 (98% de) and diphenylacetylene but using Ag2O (2 equiv., DMF, 100 °C, 8 h) to produce 35 in a higher 40% yield, but with almost complete epimerization at the phosphorus center (26% de).27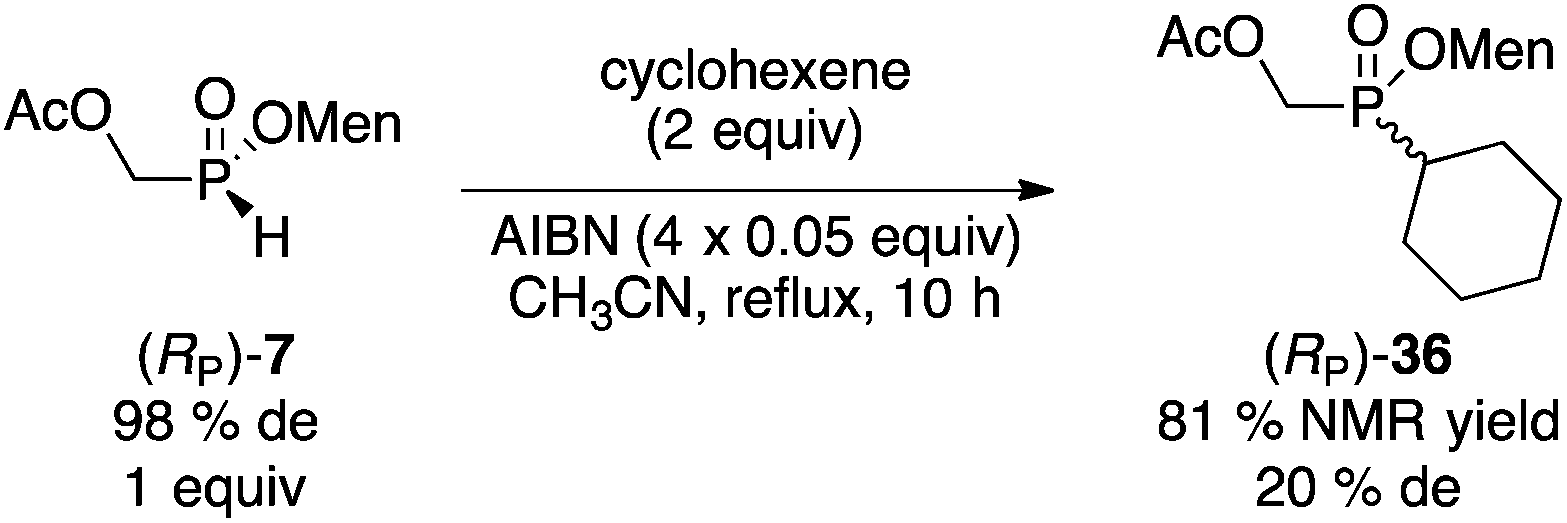 | (6) |
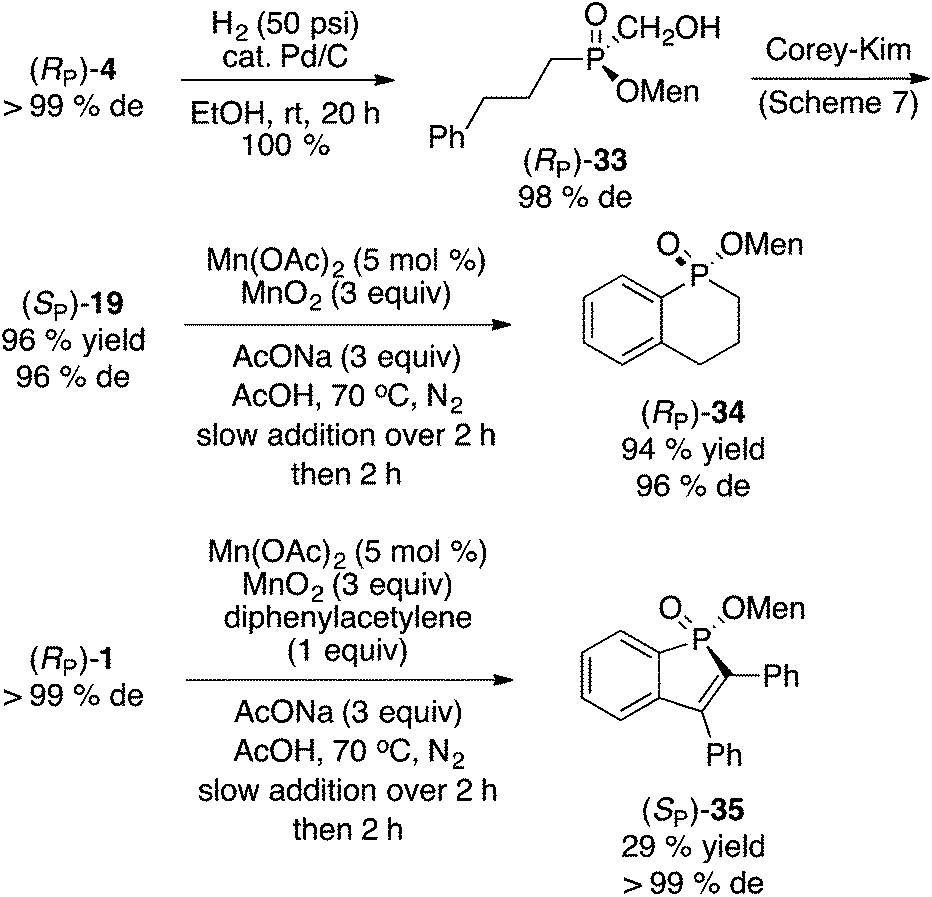 | ||
| Scheme 9 Formation of menthyl phosphinate heterocycles. | ||
Next, we investigated the preparation of menthyl cyclohexyl-H-phosphinate 20 since cyclohexyl-substituted phosphines are bulky and electron rich, and popular in catalysis. The AIBN-initiated free radical addition of (RP)-2 or (RP)-7 to cyclohexene was unsatisfactory. As shown in eqn (6), almost complete epimerization (20% de) took place and so 36 was not isolated. The observed NMR yield does however support the unusual reactivity of 7 over other less electron-deficient H-phosphinates.20 It is interesting to note that Han and coworkers reported the radical addition of (RP)-1 to be stereospecific.19
Initially, a circuitous route to the desired compound was devised starting from the mixture of diastereoisomers 7 (Scheme 10). Unfortunately, although intermediate 37 could be easily crystallized in acceptable diastereoselectivity, further erosion in diastereomeric excesses ultimately resulted in the production of 39 in low overall yield and poor 80% de.
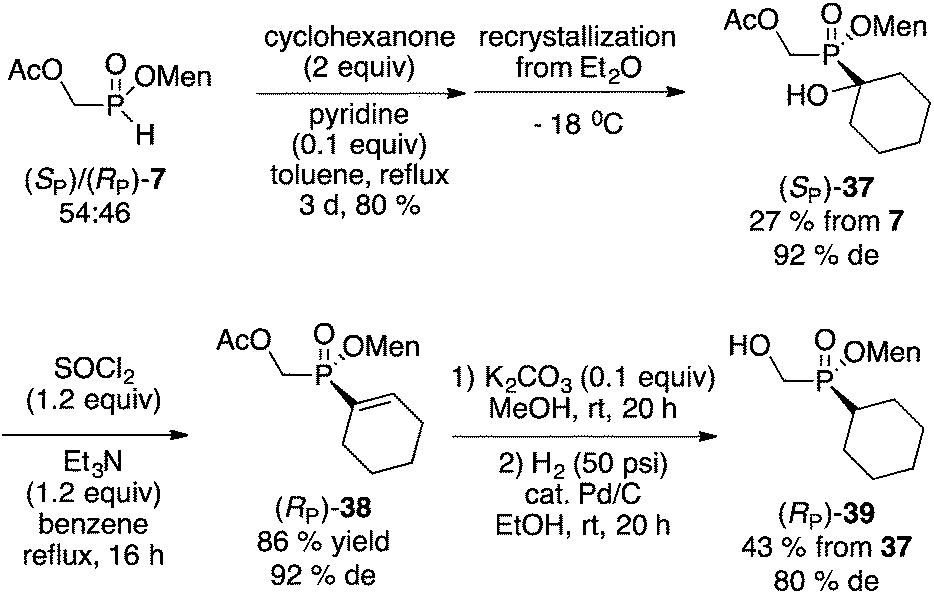 | ||
| Scheme 10 Alternate route to menthyl cyclohexyl-(hydroxymethyl)-phosphinate 39. | ||
Instead of separating the diastereoisomers early in the synthesis on cyclohexanone-adduct 37 (Scheme 10), postponing this key step to the final product gave a much better outcome (Scheme 11). Thus, the mixture of 39 obtained from the mixture of 7 was easily crystallized to furnish (RP)-39 in excellent diastereomeric purity and acceptable overall yield.
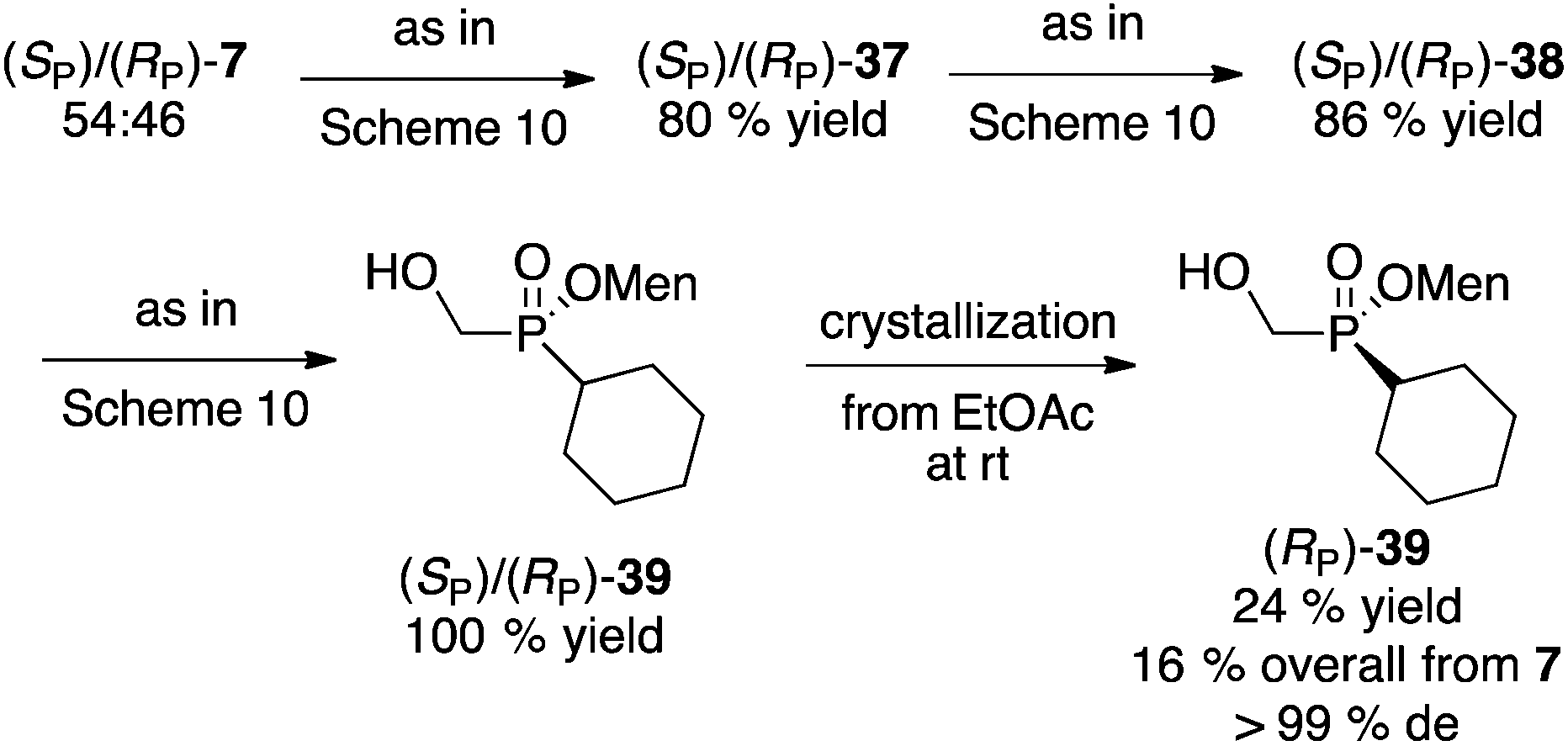 | ||
| Scheme 11 Synthesis of (RP)-39via late separation of the diastereoisomers. | ||
Alternatively, the Mn(OAc)2-catalyzed addition of H-phosphinates could be employed (Scheme 12).23 Again, the mixture of acetate diastereoisomers 7 (from the acetylation of the mother liquor) was employed as the starting material as a way to further increase the value of the process shown in Scheme 2. Addition to cyclohexene proceeded in moderate yield (50%) to deliver 36. Acetate hydrolysis under standard conditions produced 39 as a mixture of diastereoisomers, but crystallization then gave 39 in reasonable yield and excellent diastereoselectivity. In contrast, the Mn-catalyzed reaction23 of (RP)-7 (96% de) with cyclohexene produced (RP)-39 in 90% de but only in 40% isolated yield.
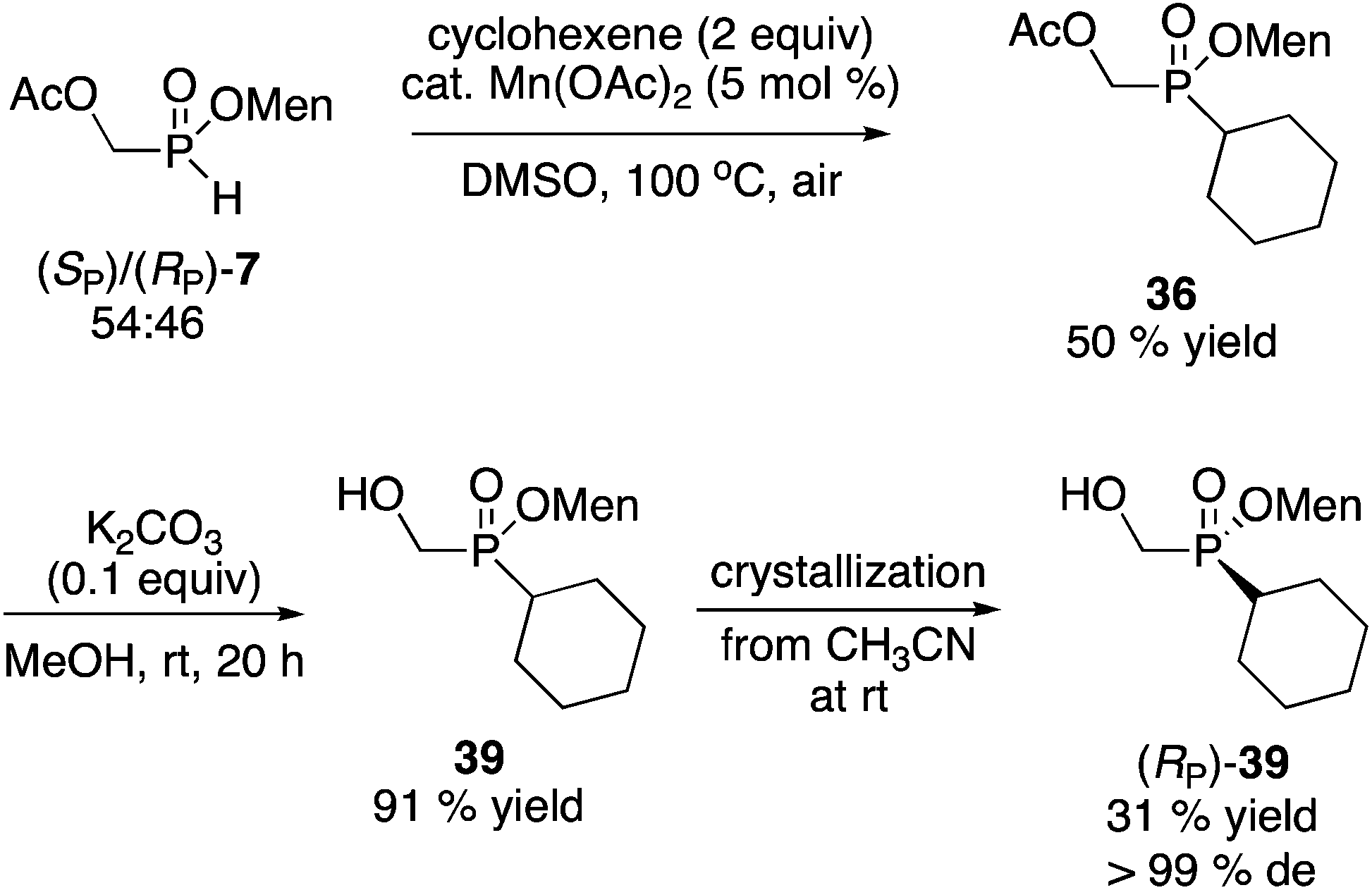 | ||
| Scheme 12 Alternative route to (SP)-39. | ||
Although it was not tried, in the final analysis the best route might be the AIBN-initiated addition of the mixture of diastereoisomers 7 to cyclohexene, followed by hydrolysis and crystallization as in Schemes 11 and 12. No matter the route, compound 39 can then be converted into menthyl cyclohexyl-H-phosphinate 20 as shown on Scheme 7.
The synthesis of other building blocks was investigated. Yokomatsu and coworkers investigated the preparation of P-stereogenic building blocks as precursors of phosphinyl dipeptide isosteres.28 Their enzymatic resolution of the key ethyl 1,1-diethoxyethyl(hydroxymethyl)phosphinate (MeC(OEt)2P(O)(OEt)CH2OH) is important because in principle, the product could be elaborated into a variety of other organophosphorus compounds. However, the stereospecificity of the acetal cleavage was not established, and the resolution requires a 3.5:1 w/w ratio between enzyme (lipase AK) and substrate.28 Nonetheless, the product could be obtained in 40% yield and 99% ee, with isoprenyl acetate as the reagent.28 We decided to see if the menthyl phosphinate approach could be employed starting from the mixture of 2 or 7. Protection of 7 using triethyl orthoacetate proceeded as we reported29 to afford 40, and subsequent hydrolysis gave 41 (Scheme 13). Unfortunately, neither 40 nor 41 could be crystallized to a pure diastereoisomer. Ciba–Geigy reagent 4230 could be transesterified (Ti(Oi-Pr)4, menthol) to furnish menthyl(1,1-diethoxyethyl)-H-phosphinate 43, but the diastereoisomers could not be separated by crystallization either.
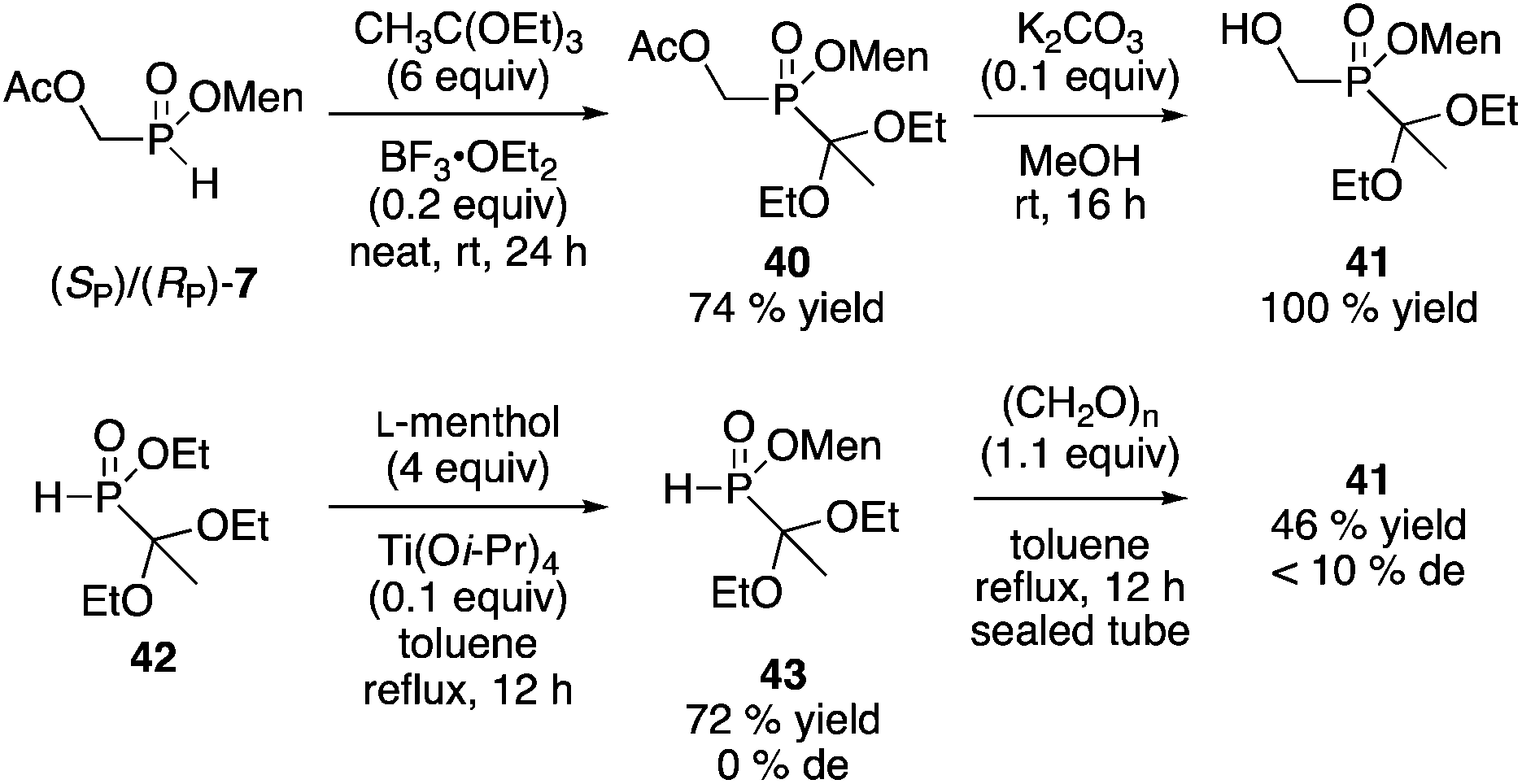 | ||
| Scheme 13 Attempted preparation of acetal-protected building blocks. | ||
Interestingly, the transesterification of H-phosphinate esters with menthol has not been described in the literature, though we have not yet investigated the generality of this process. Besides, one of our goals is to avoid the need to find crystallization conditions for numerous compounds, and instead to proceed from a handful of building blocks, especially 2 and 7, as demonstrated in the present manuscript.
Finally, the separation of differentially-substituted bis(hydroxymethyl)phosphinate derivatives was briefly examined (Scheme 14). Sila-Arbuzov alkylation17 of 2 gave benzyl ether 44. Alternatively, reaction of 7 with paraformaldehyde afforded acetate 45. Neither product could be enriched by crystallization. However, reaction of 45 with 3,5-dinitrobenzoyl chloride gave the corresponding diester, which was easily crystallized from toluene at room temperature, to afford 46 as a single diastereoisomer (the absolute configuration at phosphorus has not been established) in 28% overall yield from the mother liquor 2.
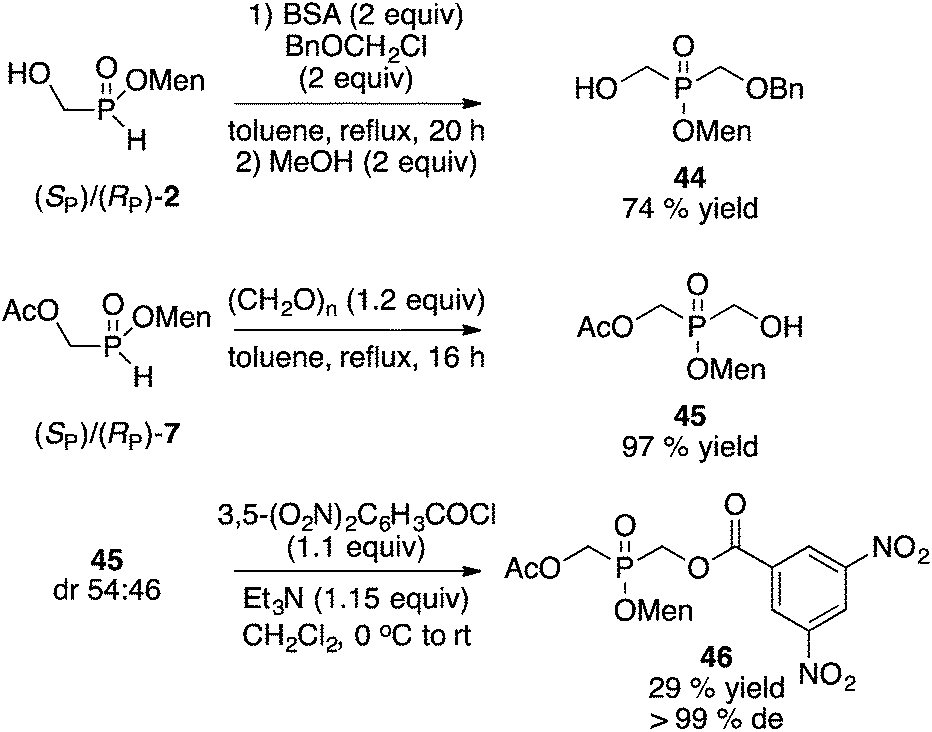 | ||
| Scheme 14 Differentially-substituted bis(hydroxymethyl)phosphinates. | ||
As mentioned in the introduction, numerous reactions have been described in the literature using (RP)-1,5c,7,31 and few other menthyl H-phosphinates5c,9,32 but to a much lesser extent. In our communication,10 we also reported various reactions of (RP)-1, such as alkylation and cross-coupling. For example, menthyl methylphenylphosphinate (Knowles’ intermediate in the synthesis of di-PAMP)4a was prepared through the sila-Arbuzov reaction as for (RP)-2 in Scheme 6. Thus, (RP)-1 (95% de) gave (RP)-menthyl methylphenylphosphinate in 90% yield and 94% de.
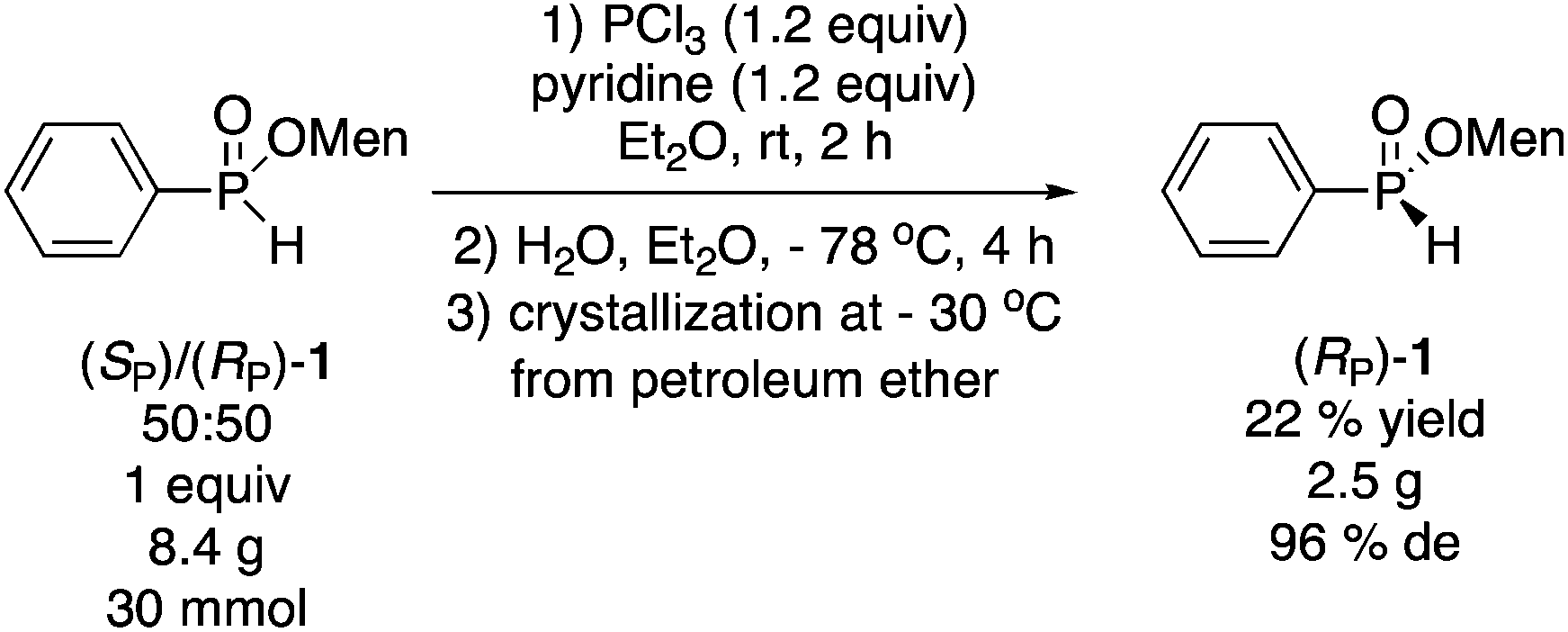
Having found that the hydrolysis of PhP(OMen)Cl can result in significant diastereoselectivity (see Scheme 1b), Han and coworkers reported the enrichment of (RP)/(SP)-1 mixtures through chlorination with PCl3 and subsequent hydrolysis.8 Thus, a ca. 50:50 mixture of diastereoisomers 1 could be converted into an 88:12 mixture. In our case, the starting 50:50 mixture is an intermediate in the preparation of (SP)-3 (Scheme 3), and surprisingly, this Dean–Stark synthesis of 1 (0% de) has not been reported previously, even though it can be conducted very easily on a large scale. Reaction of the mixture (8.4 g, 30 mmol) with PCl3 and low temperature hydrolysis as described by Han, gave a 65% yield of a 82:18 diastereomeric mixture (64% de), which was crystallized from petroleum ether at −30 °C to afford (RP)-1 in 22% overall isolated yield and 96% de (eqn (7)).
 | (7) |
Menthyl H-phosphinates of high diastereroisomeric purities are very useful intermediates because they can be converted into disubstituted compounds as described here and elsewhere.33 Disubstituted menthyl phosphinates have been transformed into tertiary phosphine oxides through the reaction with organometallics (inversion)2e,4a,34 or through lithiation with lithium di-tert-butylbiphenylide (LDBB) followed by alkylation (retention).35 Alternatively, Han showed that chlorination of menthyl-H-phosphinate esters with CuCl2 or CCl4 followed by substitution with Grignard reagents or other nucleophiles, takes place in excellent yields and stereoselectivities (inversion).36
Perhaps more importantly, the direct conversion of menthyl H-phosphinates into secondary phosphine oxides is well-established in the literature.7k,9,37 The use of H-phosphinates has been limited mainly by the availability of the starting materials in good optical purity, a problem the present work has now largely solved (Scheme 7). Giordano and coworkers demonstrated the versatility of P-stereogenic adamantyl H-phosphinates RP(O)(OAd)H as “universal precursors of P-stereogenic compounds”.38
Borane complexes derived from menthyl phosphinates are another type of compounds whose popularity has been increasing.39 Pietrusiewicz and Buono have devised methods to synthesize secondary phosphine–borane complexes from menthyl H-phosphinates.39a,b Early on, Imamoto prepared phosphinite–borane complexes39c–f from a different route (PhPCl2, RMgX, MenOH/Pyr, and separation by preparative thin layer chromatography) and showed that these products react stereospecifically (substitution with MeLi). Compound 27 (Scheme 8) is one example of a phosphinite–borane complex we synthesized in good yield and stereospecifically, but the scope of this reaction has not yet been examined. On the other hand, the direct borane reduction of disubstituted phosphinates possessing an unprotected hydroxyl group has been studied.40
Conclusions
In spite of its historical importance, the use of P-stereogenic menthyl phosphinate esters has been hampered by their limited availability. Significant efforts have been devoted to the synthesis of H-phosphinates because of their synthetic flexibility: (1) displacement to secondary phosphine oxides, (2) functionalization to disubstituted menthyl phosphinates. Nonetheless, except perhaps for menthyl phenyl-H-phosphinate 1, other P-stereogenic menthyl H-phosphinates have been elusive until now. Our methodology delivers versatile menthyl(hydroxymethyl)-H-phosphinate (RP)-2 in multigram quantities, through a very simple process that employs inexpensive reactants (aq. H3PO2, paraformaldehyde, and L-(−)-menthol) and straightforward crystallization. The reagent can be converted into numerous menthyl phosphinates through reactions like sila-Arbuzov alkylation, palladium-catalyzed cross-coupling, and manganese-catalyzed/promoted radical hydrophosphinylation/arylation. Furthermore, the mother liquor containing the mixture of diastereoisomers 2 can also be exploited to synthesize a variety of other valuable menthyl(hydroxymethyl)phosphinates through similar reactions. Corey–Kim oxidation of the hydroxymethyl intermediates affords the corresponding menthyl H-phosphinates in good yields and excellent stereoselectivities. Alternatively, the hydroxymethyl carbon can be incorporated into the products. The overall strategy avoids the need for chlorophosphines, Grignard reagents, strict anhydrous conditions, chiral HPLC separation, enzymatic resolution, cumbersome crystallization below −20 °C, or expensive auxiliaries like ephedrine.The present method offers practical and general synthesis of P-stereogenic menthyl phosphinates (including H-phosphinates), which have been established as important intermediates for the preparation of various P-stereogenic phosphine ligands. Thus, our approach constitutes a solution to the more than fifty year-old menthyl phosphinate problem of the asymmetric synthesis of P-stereogenic organophosphorus compounds.
Experimental section
(Rp)-Menthyl(hydroxymethyl)-H-phosphinate 2
Paraformaldehyde (9.91 g, 330 mmol, 1.1 equiv.) and hypophosphorous acid (39.6 g, 300 mmol, 1 equiv., 50% in water) were introduced in a round bottom flask and the reaction mixture was stirred for 24 h at 75 °C. The reaction mixture was cooled down to rt and the residue was diluted in toluene (300 mL). L-Menthol (46.9 g, 300 mmol, 1 equiv.) was added and the reaction mixture was stirred for 24 h at reflux under N2 in a flask equipped with a Dean–Stark trap. The solvent was then removed under vacuum and the residue obtained was dissolved in a mixture of diethyl ether/hexane (50 mL/200 mL) and the flask was placed in the refrigerator for 4 h (2 °C). The solid obtained was filtered and solubilized in diethyl ether (200 mL) and placed in the fridge (2 °C) for 3 h to afford the product as white needles (6.54 g, 10%, >99% de). Mp = 101–102 °C; 31P NMR (162 MHz, CDCl3): δ = 34.9 (dm, J = 542 Hz); 1H NMR (400 MHz, CDCl3): δ = 7.16 (dm, J = 542 Hz, 1H), 4.04–4.23 (m, 2H), 3.82–4.00 (m, 2H), 2.14–2.24 (m, 1H), 1.98–2.11 (m, 1H), 2.04 (dquint., J = 2.4 and 7.0 Hz, 1H), 1.62–1.73 (m, 2H), 1.34–1.52 (m, 2H), 1.24 (q, J = 12.0 Hz, 1H), 0.93 (d, J = 6.7 Hz, 6H), 0.76–1.10 (m, 2H), 0.80 (d, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ = 79.3 (d, JPOC = 8.3 Hz), 59.7 (d, JPC = 111 Hz), 48.5 (d, JPOCC = 5.5 Hz), 43.3, 33.8, 31.5, 25.6, 22.9, 21.8, 20.8, 15.7; HRMS (ESI+) m/z calcd for C11H23O3P ([M + Na]+) 257.1385, found 257.1423; [α]22D = −61.4° (chloroform).(Sp)-Menthyl(hydroxymethyl)phenylphosphinate 3
To a solution of phenylphosphinic acid (42.6 g, 300 mmol, 1 equiv.) in toluene (300 mL) was added L-menthol (46.9 g, 300 mmol, 1 equiv.). The reaction mixture was then stirred at reflux for 24 h under N2 in a flask equipped with a Dean–Stark trap. After cooling down the reaction to rt, paraformaldehyde (9.01 g, 300 mmol, 1 equiv.) was added and the reaction mixture was stirred at reflux for 24 h under N2. The solvent was then removed under vacuum and the crude obtained was recrystallized at rt in diethyl ether (200 mL) to afford the product as colorless crystals (24.2 g, 26%, 97% de). Mp = 138–139 °C; 31P NMR (162 MHz, CDCl3): δ = 37.2 (s); 1H NMR (400 MHz, CDCl3): δ = 7.77–7.87 (m, 2H), 7.52–7.60 (m, 1H), 7.42–7.51 (m, 2H), 4.29–4.43 (m, 2H), 3.93–4.10 (m, 2H), 2.26 (dquint., J = 2.6 and 7.0 Hz, 1H), 1.80–1.91 (m, 1H), 1.57–1.73 (m, 2H), 1.26–1.47 (m, 2H), 0.96 (d, J = 7.1 Hz, 3H), 0.74–1.13 (m, 3H), 0.89 (d, J = 7.0 Hz, 3H), 0.78 (d, J = 6.4 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ = 132.3 (d, JPCCCC = 2.8 Hz), 131.7 (d, JPCCC = 9.9 Hz, 2C), 130.6 (d, JPC = 123 Hz), 128.3 (d, JPCC = 12.1 Hz, 2C), 77.1 (d, JPOC = 8.3 Hz), 60.2 (d, JPC = 117 Hz), 48.7 (d, JPOCC = 6.1 Hz), 43.2, 34.0, 31.4, 25.5, 22.8, 21.9, 21.1, 15.7; HRMS (EI+) m/z calcd for C16H28O3P ([M + H]+) 311.1776, found 311.1766; [α]22D = −46.7° (chloroform).Representative procedure for the Corey–Kim oxidation. (Rp)-Menthyl phenyl-H-phosphinate 1
To a solution of N-chlorosuccinimide (4.0 g, 30 mmol, 3 equiv.) in dichloromethane (150 mL) at −78 °C and under N2 was added dropwise a solution of dimethyl sulfide (2.2 mL, 30 mmol, 3 equiv.) in dichloromethane (10 mL). After 30 minutes at −78 °C, a solution of (Sp)-menthyl(hydroxymethyl)phenylphosphinate 3 (3.1 g, 10 mmol, 1 equiv.) in dichloromethane (30 mL) was added over 20 minutes. After 1 h at −78 °C, triethylamine (7 mL, 50 mmol, 5 equiv.) was added over 15 minutes and the reaction was stirred for 30 minutes at −78 °C. After warming up to rt, water was added and the two layers were separated. The aqueous layer was extracted with dichloromethane twice. The combined organic layer was dried over MgSO4, filtered and concentrated under vacuum. The crude obtained was purified by column chromatography (hexane/ethyl acetate 9:1 to 8:2) to afford the product as a colorless oil (2.58 g, 92%, >99% de). 31P NMR (162 MHz, CDCl3): δ = 24.7 (dm, J = 553 Hz); 1H NMR (400 MHz, CDCl3): δ = 7.73–7.84 (m, 2H), 7.66 (d, J = 553 Hz, 1H), 7.46–7.64 (m, 3H), 4.22–4.36 (m, 1H), 2.14–2.27 (m, 2H), 1.62–1.75 (m, 2H), 1.38–1.54 (m, 2H), 1.24 (q, J = 11.2 Hz, 1H), 0.78–1.13 (m, 2H), 0.96 (d, J = 7.0 Hz, 3H), 0.90 (d, J = 6.4 Hz, 3H), 0.86 (d, J = 7.0 Hz, 3H); [α]23D = −35.5° (chloroform, literature with 90% de: −21.0° in benzene).
Author contributions
All authors have given approval to the final version of the manuscript.Competing interest
The authors declare no competing financial interest.Acknowledgements
This material is based in part upon work supported by the National Science Foundation under grant CHE-1262254.References
- (a) A. Grabulosa, in P-Stereogenic Ligands in Enantioselective Catalysis, RSC Publishing, Cambridge, UK, 1st edn, 2011, pp. 1–501 Search PubMed; (b) H. B. Kagan, in Asymmetric Synthesis, ed. J. D. Morrison, Academic Press, New York, 1985, vol. 5, pp. 1–39 Search PubMed; (c) D. Valentine Jr., in Asymmetric Synthesis, ed. J. D. Morrison and J. W. Scott, Academic Press, New York, 1984, vol. 4, pp. 263–312 Search PubMed.
- (a) J. Donohue, N. Mandel, W. B. Farnham, R. K. Murray Jr., K. Mislow and H. P. Benschop, J. Am. Chem. Soc., 1971, 93, 3792–3793 CrossRef; (b) W. B. Farnham, R. K. Murray Jr. and K. Mislow, J. Am. Chem. Soc., 1970, 92, 5809–5810 CrossRef CAS; (c) R. A. Lewis and K. Mislow, J. Am. Chem. Soc., 1969, 91, 7009–7012 CrossRef CAS; (d) O. Korpiun, R. A. Lewis, J. Chickos and K. Mislow, J. Am. Chem. Soc., 1968, 90, 4842–4846 CrossRef CAS; (e) O. Korpiun and K. Mislow, J. Am. Chem. Soc., 1967, 89, 4784–4786 CrossRef CAS.
- (a) T. L. Emmick and R. L. Letsinger, J. Am. Chem. Soc., 1968, 90, 3459–3465 CrossRef CAS; (b) L. P. Reiff and H. S. Aaron, J. Am. Chem. Soc., 1970, 92, 5275–5276 CrossRef CAS; (c) A. Nudelman and D. J. Cram, J. Am. Chem. Soc., 1971, 36, 335–337 Search PubMed; (d) A. Nudelman and D. J. Cram, J. Org. Chem., 1968, 90, 3869–3870 CAS.
- (a) B. D. Vineyard, W. S. Knowles, M. J. Sabacky, G. L. Bachman and D. J. Weinkauff, J. Am. Chem. Soc., 1977, 99, 5946–5952 CrossRef CAS; (b) W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567–2568 CrossRef CAS.
- Reviews on P-stereogenic compounds: (a) P. Bagi, V. Uji, M. Czugler, E. Fogassy and G. Keglevich, Dalton Trans., 2016, 45, 1823–1842 RSC; (b) O. I. Kolodiazhnyi, Top. Curr. Chem., 2015, 360, 161–236 CrossRef CAS PubMed; (c) T. Chen and L.-B. Han, Synlett, 2015, 1153–1163 CAS; (d) D. Hérault, D. H. Nguyen, D. Nuel and G. Buono, Chem. Soc. Rev., 2015, 44, 2508–2528 RSC; (e) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2012, 23, 1–46 CrossRef CAS; (f) T. M. Shaikh, C.-M. Weng and F.-E. Hong, Coord. Chem. Rev., 2012, 256, 771–803 CrossRef CAS; (g) T. Nemoto and Y. Hamada, Tetrahedron, 2011, 67, 667–687 CrossRef CAS; (h) N. Oka and T. Wada, Chem. Soc. Rev., 2011, 40, 5829–5843 RSC; (i) J. S. Harvey and V. Gouverneur, Chem. Commun., 2010, 46, 7477–7485 RSC; (j) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25–90 CrossRef CAS; (k) D. S. Glueck, Synlett, 2007, 2627–2634 CrossRef CAS; (l) R. Engel and J. I. Rizzo, Curr. Org. Chem., 2006, 10, 2393–2405 CrossRef CAS; (m) M. J. Johansson and N. C. Kann, Mini-Rev. Org. Chem., 2004, 1, 233–247 CrossRef CAS; (n) P.-H. Leung, Acc. Chem. Res., 2004, 37, 169–177 CrossRef CAS PubMed; (o) K. V. L. Crépy and T. Imamoto, Top. Curr. Chem., 2003, 229, 1–40 CrossRef; (p) L. A. Wozniak and A. Okruszek, Chem. Soc. Rev., 2003, 32, 158–169 RSC; (q) P. Guga, A. Okruszek and W. J. Stec, Top. Curr. Chem., 2002, 200, 169–200 CrossRef; (r) F. Maienza, F. Spindler, M. Thommen, B. Pugin and A. Mezzeti, Chimia, 2001, 55, 694–698 CAS; (s) Y. Yamanoi and T. Imamoto, Rev. Heteroat. Chem., 1999, 20, 227–248 CAS; (t) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 1998, 9, 1279–1332 CrossRef CAS; (u) M. Ohff, J. Holz, M. Quirmbach and A. Börner, Synthesis, 1998, 1391–1415 CrossRef CAS; (v) A. Marinetti, V. Kruger and F.-X. Buzin, Coord. Chem. Rev., 1998, 178–180, 755–770 CrossRef CAS; (w) K. M. Pietrusiewicz and M. Zablocka, Chem. Rev., 1994, 94, 1375–1411 CrossRef CAS; (x) Z. J. Lesnikowski, Bioorg. Chem., 1993, 21, 127–155 CrossRef CAS; (y) H. B. Kagan and M. Sasaki, Optically active phosphines: preparation, uses and chiroptical properties, Patai's The Chem. Organophosphorus Compd., 1990, 1, 51–102 CAS.
- M. de Fatima Fernandez, C. P. Vlaar, H. Fan, Y.-H. Liu, F. R. Fronczek and R. P. Hammer, J. Org. Chem., 1995, 60, 7390–7391 CrossRef.
- (a) Y. Zhao, X. Chen, T. Chen, Y. Zhou, S.-F. Yin and L.-B. Han, J. Org. Chem., 2015, 80, 62–69 CrossRef CAS PubMed; (b) J. Yang, T. Chen, Y. Zhou, S.-F. Yin and L.-B. Han, Organometallics, 2015, 34, 5095–5098 CrossRef CAS; (c) H. Zhang, Y.-M. Sun, L. Yao, S.-Y. Ji, C.-Q. Zhao and L.-B. Han, Chem. – Asian J., 2014, 9, 1329–1333 CrossRef CAS PubMed; (d) B. Xiong, M. Li, Y. Liu, Y. Zhou, C. Zhao, M. Goto, S.-F. Yin and L.-B. Han, Adv. Synth. Catal., 2014, 356, 781–794 CrossRef CAS; (e) X.-B. Wang, M. Goto and L.-B. Han, Chem. – Eur. J., 2014, 20, 3631–3635 CrossRef CAS PubMed; (f) Y.-M. Sun, Z.-Y. Xu, L.-J. Liu, F.-J. Meng, H. Zhang, B.-C. Fu, L.-J. Sun, M.-J. Niu, S.-W. Cong, C.-Q. Zhao and L.-B. Han, Tetrahedron: Asymmetry, 2014, 25, 1520–1526 CrossRef CAS; (g) Y.-M. Sun, N. Xin, Z.-Y. Xu, L.-J. Liu, F.-J. Meng, H. Zhang, B.-C. Fu, Q.-J. Liang, H.-X. Zheng, L.-J. Sun, C.-Q. Zhao and L.-B. Han, Org. Biomol. Chem., 2014, 12, 9457–9465 RSC; (h) T. Chen, Y. Zhou, C. Guo and L.-B. Han, Chem. Lett., 2013, 13, 1065–1067 CrossRef; (i) B. Xiong, R. Shen, M. Goto, S.-F. Yin and L.-B. Han, Chem. – Eur. J., 2012, 18, 16902–16910 CrossRef CAS PubMed; (j) G. Wang, R. Shen, Q. Xu, M. Goto, Y. Zhao and L.-B. Han, J. Org. Chem., 2010, 75, 3890–3892 CrossRef CAS PubMed; (k) Q. Xu, C.-Q. Zhao and L.-B. Han, J. Am. Chem. Soc., 2008, 130, 12648–12655 CrossRef CAS PubMed; (l) L.-B. Han, C.-Q. Zhao, S.-Y. Onozawa, M. Goto and M. Tanaka, J. Am. Chem. Soc., 2002, 124, 3842–3843 CrossRef CAS PubMed; (m) See also ref. 5c.
- W. M. Wang, L.-J. Liu, C.-Q. Zhao and L.-B. Han, Eur. J. Org. Chem., 2015, 2342–2345 CrossRef CAS.
- D. Gatineau, L. Giordano and G. Buono, J. Am. Chem. Soc., 2011, 133, 10728–10731 CrossRef CAS PubMed.
- O. Berger and J.-L. Montchamp, Angew. Chem., Int. Ed., 2013, 52, 11377–11380 CrossRef CAS PubMed.
- O. Berger, L. Gavara and J.-L. Montchamp, Org. Lett., 2012, 14, 3404–3407 CrossRef CAS PubMed.
- J.-L. Montchamp and O. Berger, The Synthesis of P-Chiral Compounds, US pat, 8877957B2, 2014 Search PubMed. Compounds (RP)-2 and (RP)-3 are commercially available from Strem Chemicals: #15-2915 and #15-2928, respectively.
- (a) O. Berger, C. Petit, E. L. Deal and J.-L. Montchamp, Adv. Synth. Catal., 2013, 355, 1361–1373 CrossRef CAS; (b) E. L. Deal, C. Petit and J.-L. Montchamp, Org. Lett., 2011, 13, 3270–3273 CrossRef CAS PubMed.
- (a) K. Bravo-Altamirano, I. Abrunhosa-Thomas and J.-L. Montchamp, J. Org. Chem., 2008, 73, 2292–2301 CrossRef CAS PubMed; (b) K. Bravo-Altamirano and J.-L. Montchamp, Org. Lett., 2006, 8, 4169–4171 CrossRef CAS PubMed; (c) K. Bravo-Altamirano and J.-L. Montchamp, Org. Synth., 2008, 85, 96–105 CrossRef CAS.
- A. Fers-Lidou, O. Berger and J.-L. Montchamp, manuscript in preparation.
- O. Berger and J.-L. Montchamp, Chem. – Eur. J., 2014, 20, 12385–12388 CrossRef CAS PubMed.
- Selected examples: (a) J. K. Thottathil, C. A. Przybyla and J. L. Moniot, Tetrahedron Lett., 1984, 25, 4737–4740 CrossRef CAS; (b) M. V. Livantsov, A. A. Prishchenko and I. F. Lutsenko, Russ. J. Gen. Chem., 1987, 928–935 Search PubMed; (c) J. Rudovsky, J. Kotek, P. Hermann, I. Lukes, V. Mainero and S. Aime, Org. Biomol. Chem., 2005, 112–117 RSC; (d) M. Matziari, D. Georgiadis, V. Dive and A. Yiotakis, Org. Lett., 2001, 3, 659–662 CrossRef CAS PubMed; (e) D. M. Bartley and J. K. Coward, J. Org. Chem., 2005, 70, 6757–6774 CrossRef CAS PubMed; (f) G. Bianchini, M. Aschi, G. Cavicchio, M. Crucianelli, S. Preziuso, C. Gallina, A. Nastari, E. Gavuzzo and F. Mazza, Bioorg. Med. Chem., 2005, 13, 4740–4749 CrossRef CAS PubMed; (g) N. G. Anderson, B. M. Ciaramella, A. F. Feldman, D. A. Lust, J. L. Moniot, L. Moran, R. E. Polomski and S. S. Y. Wang, Org. Process Res. Dev., 1997, 1, 211–216 CrossRef CAS; (h) C. Fougère, E. Guénin, J. Hardouin and M. Lecouvey, Eur. J. Org. Chem., 2009, 6048–6054 CrossRef.
- (a) H. H. Hatt, J. Chem. Soc., 1933, 776–786 RSC; (b) V. Plack, J. R. Goerlich, H. Thönnessesn, P. G. Jones and R. Schmutsler, Z. Anorg. Allg. Chem., 1995, 621, 1082–1092 CrossRef; (c) V. Plack, J. R. Goerlich, H. Thönnessesn, P. G. Jones and R. Schmutsler, Z. Anorg. Allg. Chem., 1999, 625, 1278–1286 CrossRef CAS; (d) V. Plack, J. R. Goerlich and R. Schmutsler, Z. Anorg. Allg. Chem., 1998, 624, 1940–1942 CrossRef CAS; (e) A. F. Gushwa, Y. Belabassi, J.-L. Montchamp and A. F. Richards, J. Chem. Crystallogr., 2009, 39, 337–347 CrossRef CAS; (f) C. Dobrota, A. Duraud, M. Toffano and J.-C. Fiaud, Eur. J. Org. Chem., 2008, 2439–2445 CrossRef CAS.
- (a) T. Hirai and L.-B. Han, Org. Lett., 2007, 9, 53–55 CrossRef CAS PubMed; (b) L.-B. Han and C.-Q. Zhao, J. Org. Chem., 2005, 70, 10121–10123 CrossRef CAS PubMed.
- B. J. Janesko, H. C. Fisher, M. J. Bridle and J.-L. Montchamp, J. Org. Chem., 2015, 80, 10025–10032 CrossRef CAS PubMed.
- J. Layole, A. Lattes, B. Battie, H. Zamarlik and J. Carles, J. Chromatogr., 1973, 76, 441–454 CrossRef CAS.
- As expected, the R3B-initiated radical addition of 2 and 7 to alkenes proceeds at room temperature: J. Gatignol, J.-L. Montchamp, unpublished results.
- H. C. Fisher, O. Berger, F. Gelat and J.-L. Montchamp, Adv. Synth. Catal., 2014, 356, 1199–1204 CrossRef CAS.
- K. Nishide, S.-I. Ohsugi, M. Fudesaka, S. Kodama and M. Node, Tetrahedron Lett., 2002, 43, 5177–5179 CrossRef CAS.
- (a) C. Petit, E. Poli, A. Favre-Réguillon, L. Khrouz, S. Denis-Quanquin, L. Bonneviot, G. Mignani and M. Lemaire, ACS Catal., 2013, 3, 1431–1438 CrossRef CAS; (b) K. V. Rajendran and D. C. Gilheany, Chem. Commun., 2012, 48, 817–819 RSC; (c) E. H. Krenske, J. Org. Chem., 2012, 77, 3969–3977 CrossRef CAS PubMed; (d) E. H. Krenske, J. Org. Chem., 2012, 77, 1–4 CrossRef CAS PubMed; (e) T. Imamoto, S.-I. Kikuchi, T. Miura and W. Wada, Org. Lett., 2001, 3, 87–90 CrossRef CAS PubMed; (f) K. Naumann, G. Zon and K. Mislow, J. Am. Chem. Soc., 1969, 91, 2788–2789 CrossRef CAS.
- P. Coutrot, M. Youssefi-Tabrizi and C. Grison, J. Organomet. Chem., 1986, 316, 13–18 CrossRef CAS.
- Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754–16757 CrossRef CAS PubMed.
- T. Yamagishi, J.-I. Mori, T. Haruki and T. Yokomatsu, Tetrahedron: Asymmetry, 2011, 22, 1358–1363 CrossRef CAS.
- L. Coudray and J.-L. Montchamp, Eur. J. Org. Chem., 2009, 4646–4654 CrossRef CAS PubMed.
- (a) J. G. Dingwall, J. Ehrenfreund, R. G. Hall and J. Jack, Phosphorus, Sulfur Relat. Elem., 1987, 30, 571–574 CrossRef CAS; (b) P. P. McCleery and B. Tuck, J. Chem. Soc., Perkin Trans. 1, 1989, 1319–1329 RSC; (c) J. G. Dingwall, J. Ehrenfreund and R. G. Hall, Tetrahedron, 1989, 45, 3787–3808 CrossRef CAS; (d) E. K. Baylis, Tetrahedron Lett., 1995, 36, 9385–9388 CrossRef CAS; (e) E. K. Baylis, Tetrahedron Lett., 1995, 36, 9389–9392 CrossRef CAS; (f) W. Froestl, S. J. Mickel, R. G. Hall, G. von Sprecher, P. J. Diel, D. Strub, P. A. Baumann, F. Brugger, C. Gentsch, J. Jaekel, H.-R. Olpe, G. Rihs, A. Vassout, P. C. Waldmeier and H. Bittiger, J. Med. Chem., 1995, 38, 3297–3312 CrossRef CAS PubMed; (g) W. Froestl, S. J. Mickel, G. von Sprecher, P. J. Diel, R. G. Hall, L. Maier, D. Strub, V. Melillo, P. A. Baumann, R. Bernasconi, C. Gentsch, K. Hauser, J. Jaekel, G. Karlsson, K. Klebs, L. Maitre, C. Marescaux, M. F. Pozza, M. Schmutz, M. W. Steinmann, H. van Riezen, A. Vassout, C. Mondadori, H.-R. Olpe, P. C. Waldmeier and H. Bittiger, J. Med. Chem., 1995, 38, 3313–3331 CrossRef CAS PubMed; (h) S. N. L. Bennett and R. G. Hall, J. Chem. Soc., Dalton Trans., 1995, 1145–1151 RSC.
- (a) Z.-Y. Zhou, H. Zhang, L. Yao, J.-H. Wen, S.-Z. Nie and C.-Q. Zhao, Chirality, 2016, 28, 132–135 CrossRef CAS PubMed; (b) Y.-N. Ma and S.-D. Yang, Chem. – Eur. J., 2015, 21, 6673–6677 CrossRef CAS PubMed; (c) A. Kinbara, M. Ito, T. Abe and T. Yamagishi, Tetrahedron, 2015, 71, 7614–7619 CrossRef CAS; (d) G. Kumuraswamy and R. Raju, Adv. Synth. Catal., 2014, 356, 2591–2598 CrossRef; (e) A.-X. Zhou, L.-L. Mao, G.-W. Wang and S.-D. Yang, Chem. Commun., 2014, 50, 8529–8532 RSC; (f) L.-L. Mao, A.-X. Zhou, N. Liu and S.-D. Yang, Synlett, 2014, 2727–2732 CAS; (g) K. D. Reichl, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2013, 135, 9354–9357 CrossRef CAS PubMed; (h) L. Liu, Y. Wang, Z. Zeng, P. Xu, Y. Gao, Y. Yin and Y. Zhao, Adv. Synth. Catal., 2013, 355, 659–666 CrossRef CAS; (i) N. Oka, K. Ito, F. Tomita and K. Ando, Chem. Lett., 2012, 41, 1630–1632 CrossRef CAS; (j) X. Zhang, H. Liu, X. Hu, G. Tang, J. Zhu and Y. Zhao, Org. Lett., 2011, 13, 3478–3481 CrossRef CAS PubMed.
- R. Bodalski and J. Koszuk, Phosphorus, Sulfur, Silicon Relat. Elem., 1989, 44, 99–102 CrossRef CAS.
- A. J. Bloomfield and S. B. Herzon, Org. Lett., 2012, 14, 4370–4373 CrossRef CAS PubMed.
- Y.-N. Ma and S.-D. Yang, Chem. – Eur. J., 2015, 21, 6673–6677 CrossRef CAS PubMed.
- Y. Koide, A. Sakamoto and T. Imamoto, Tetrahedron Lett., 1991, 32, 3375–3376 CrossRef CAS.
- (a) Y. Zhou, G. Wang, Y. Saga, R. Shen, M. Goto, Y. Zhao and L.-B. Han, J. Org. Chem., 2010, 75, 7924–7927 CrossRef CAS PubMed; (b) B. Xiong, Y. Zhou, C. Zhao, M. Goto, S.-F. Yin and L.-B. Han, Tetrahedron, 2013, 69, 9373–9380 CrossRef CAS; (c) G. Wang, R. Shen, Q. Xu, M. Goto, Y. Zhao and L.-B. Han, J. Org. Chem., 2010, 75, 3890–3892 CrossRef CAS PubMed; (d) Y. Zhou, J. Yang, T. Chen, S.-F. Yin, D. Han and L.-B. Han, Bull. Chem. Soc. Jpn., 2014, 87, 400–402 CrossRef CAS.
- (a) A. Leyris, J. Bigeault, D. Nuel, L. Giordano and G. Buono, Tetrahedron Lett., 2007, 48, 5247–5250 CrossRef CAS; (b) R. Bigler and A. Mezzetti, Org. Process Res. Dev., 2016, 20, 253–261 CrossRef CAS.
- D. Gatineau, D. H. Nguyen, D. Hérault, N. Vanthuyne, J. Leclaire, L. Giordano and G. Buono, J. Org. Chem., 2015, 80, 4132–4141 CrossRef CAS PubMed.
- (a) M. Stankevič and K. M. Pietrusiewicz, J. Org. Chem., 2007, 72, 816–822 CrossRef PubMed; (b) D. Moraleda, D. Gatineau, D. Martin, L. Giordano and G. Buono, Chem. Commun., 2008, 3031–3033 RSC; (c) T. Imamoto, T. Yoshizawa, K. Hirose, Y. Wada, H. Masuda, K. Yamaguchi and H. Seki, Heteroat. Chem., 1995, 6, 99–104 CrossRef CAS; (d) T. Imamoto, M. Matsuo, T. Nonomura, K. Kishikawa and M. Yanagawa, Heteroat. Chem., 1993, 4, 475–486 CrossRef CAS; (e) T. Oshiki and T. Imamoto, J. Am. Chem. Soc., 1992, 114, 3975–3977 CrossRef CAS; (f) T. Imamoto, T. Oshiki, T. Onozawa, M. Matsuo, T. Hikosaka and M. Yanagawa, Heteroat. Chem., 1992, 3, 563–575 CrossRef CAS; (g) P. Kielbasinski, M. Albrycht, R. Zurawinski and M. Mikolajczyk, J. Mol. Catal., B: Enzym., 2006, 39, 45–49 CrossRef CAS.
- (a) M. Kwiatkowska, G. Krasinski, M. Cypryk, T. Cierpal and P. Kielbasinski, Tetrahedron: Asymmetry, 2011, 22, 1581–1590 CrossRef CAS; (b) S. Lemouzy, D. H. Nguyen, D. Gatineau, L. Giordano, D. Hérault and G. Buono, Pure Appl. Chem., 2016, 88, 333–339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectral data, 1H-, 13C-, 31P-NMR, and HRMS (PDF). See DOI: 10.1039/c6ob01413e |
| This journal is © The Royal Society of Chemistry 2016 |