
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elaboration of tetra-orthogonally-substituted aromatic scaffolds towards novel EGFR-kinase inhibitors†
Adam J.
Close
a,
Rhiannon N.
Jones
a,
Cory A.
Ocasio
a,
Paul
Kemmitt
b,
S. Mark
Roe
c and
John
Spencer
 *a
*a
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Falmer, BN1 9QJ, UK. E-mail: j.spencer@sussex.ac.uk
bAstraZeneca, Mereside Alderley Park, Macclesfield, SK10 4TG, UK
cDepartment of Biochemistry, School of Life Sciences, University of Sussex, Falmer, BN1 9QJ, UK
First published on 19th July 2016
Abstract
Nitration of three regioisomers of bromo-fluorobenzaldehyde proceeds regioselectively, notably with H2SO4/HNO3 at 0 °C. The thereby synthesized tetrasubstituted aromatics, endowed with orthogonal substituents, can be elaborated via Pd-catalysed coupling, reduction and reductive amination reactions. As a test-case, these compounds were converted into EGFR inhibitors related to Gefitinib, whose activity was rationalised by docking studies.
Introduction
Tetrasubstituted aromatics are commonplace in drug discovery yet regioselective routes towards these compounds, which often contain orthogonal groups, are rather scarce.1 Kinase inhibitors are a rapidly growing class of anticancer agents and many of these comprise such tetrasubstituted scaffolds, often built around an adenine-like quinazoline scaffold, a solubilising group (e.g. ethers, morpholine or piperazine groups) and a tri or tetra-substituted aromatic, hydrophobic group that imparts selectivity towards particular classes of kinase (Fig. 1).2–5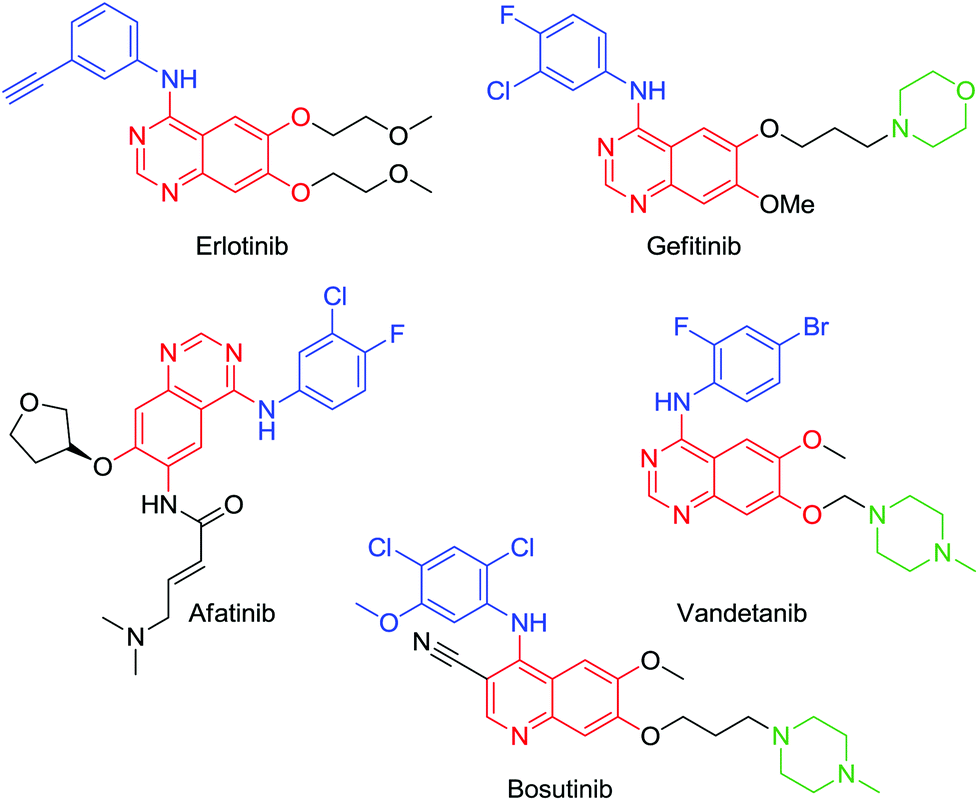 | ||
| Fig. 1 Kinase inhibitors, highlighting the aromatic hydrophobic (blue), the adenine mimic (red) and solubilising group (green). | ||
To underline their importance, the synthesis of the recently approved irreversible EGFR (epidermal growth factor receptor tyrosine kinase family; HER1) inhibitor Osimertinib, starts with a somewhat simple, yet non-trivial, tetrasubstituted precursor, which comprises four orthogonal substituents (Fig. 2).6
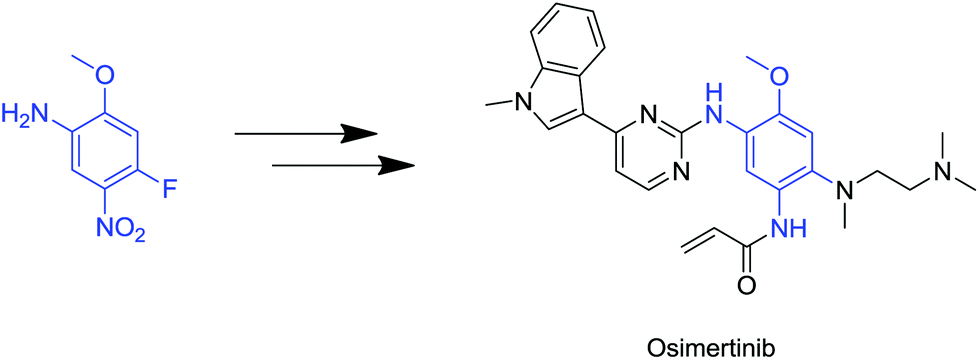 | ||
| Fig. 2 Demonstration of a tetrasubstituted aromatic used as a building block in anticancer molecules. | ||
We have recently reported procedures for forming poylsubstituted aromatics, mainly based on a MIDA boronate-substituted aryl scaffold.7,8 Our aim was to synthesise analogues of the type I and II (Fig. 3) as useful 1,2,3,4- and 1,2,4,5-substituted building blocks.
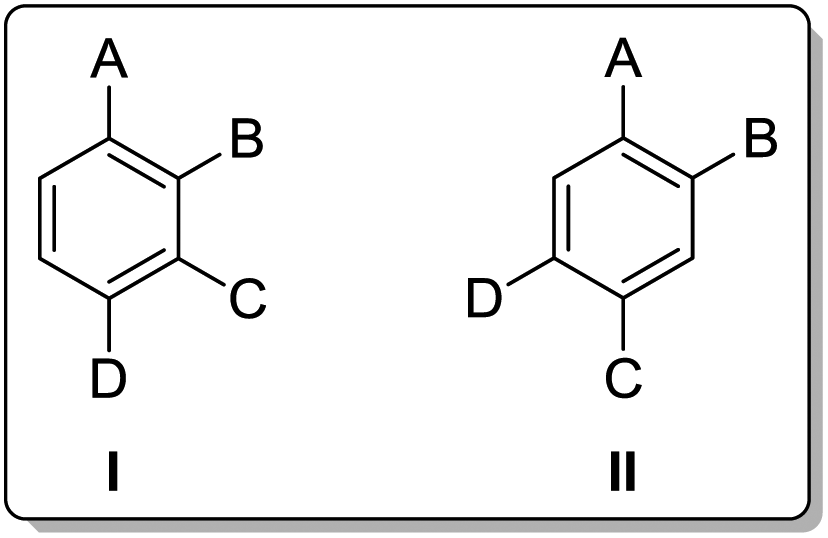 | ||
| Fig. 3 Tetrasubstituted aromatic building blocks. | ||
Here, we report our investigation of the synthesis of related scaffolds, which do not include a MIDAboronate aryl scaffold but are substituted with bromo, fluoro and formyl substituents. We show that these orthogonal groups can be modified in order to furnish useful drug-like fragments as well as tetrasubstituted units that can be incorporated into potential kinase inhibitors.
Results and discussion
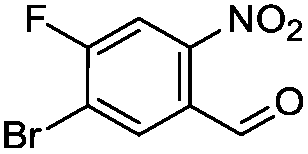
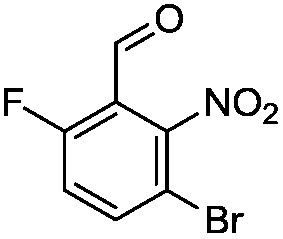
Brominations on trisubstituted aromatics are an effective means for synthesising tetrasubstitued aromatic units containing orthogonal (e.g. MIDA and bromide) groups. These electrophilic substitutions work with excellent regioselectivity when complementary directing groups are combined in the trisubstituted precursor. We now disclose our results on the related nitrations of trisubstituted (non-MIDA) aromatics and resulting functional group conversions thereafter affording novel tetrasubstituted frameworks. In order to maintain our ethos of elaborating orthogonally substituted aromatics we refrained from attempting the bromination of bromoaromatics.We attempted the nitration of three regioiomers of bromo-fluorobenzaldehyde (Table 1). Compounds 2a and 2b have been previously synthesised, described in patents, via nitration, using varying amounts of nitric acid in sulphuric acid.9,10 We required a generic, high yielding method for these three compounds and thus looked for alternative methods. Compound 1a was used to probe different nitration conditions due to the complementarity of the directing groups, which would be expected to give one regioisomer. Initial attempts included the use of potassium nitrate in sulphuric acid11 and Mn(acac)3 in DCM with nitric acid,12 neither of which gave good results, as analysed by crude 1H-NMR spectra.
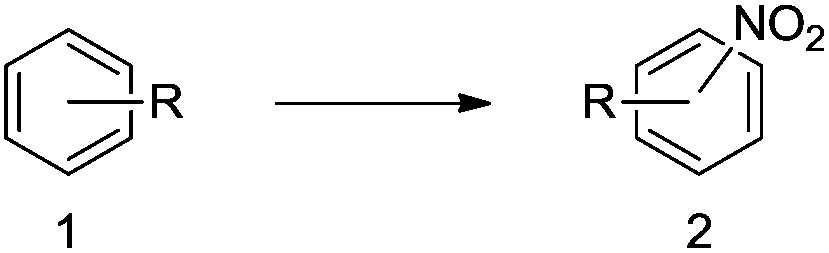
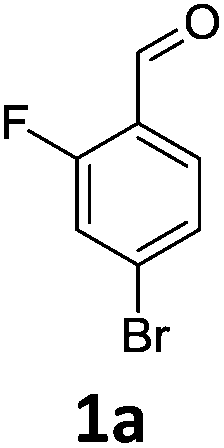
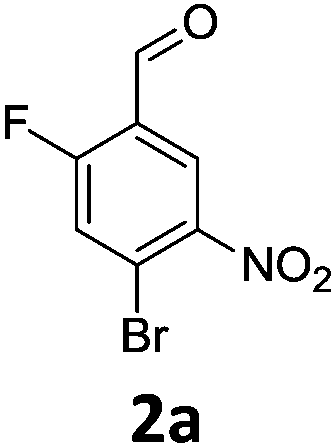
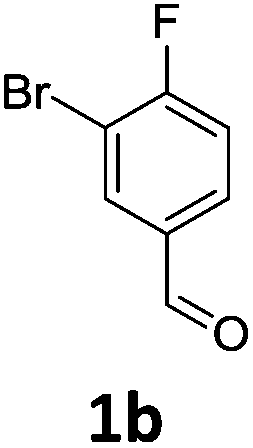
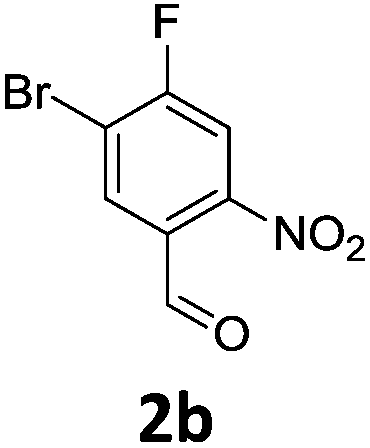
We were pleased that the NO2BF4 method showed comparable yields for the synthesis of 2a (entry 1, Table 1) to that of the nitric acid methods, adopted from the patents. For the synthesis of 2b and 2c, yields were slightly lower. In general a nitric/sulphuric acid mixture gave the best results and NO2BF4 proved to be ineffective in the attempted nitration of 1b and 1c.13
The regiochemistry of the products 2a–2c was confirmed by 1H and 13C NMR spectroscopy as well as by single crystal X-ray crystallography (Fig. 4).
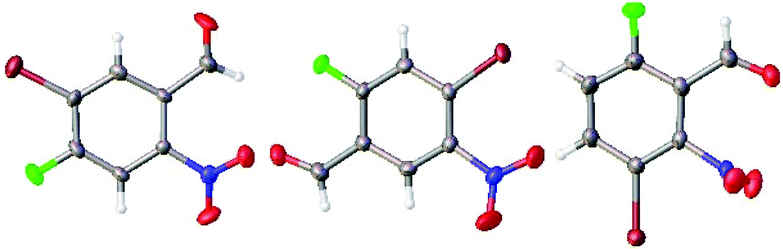 | ||
| Fig. 4 ORTEP diagrams showing crystal structures of 2b (left, CCDC 1485121), 2a (middle, CCDC 1485122) and 2c (right, CCDC 1485123). All are shown in Table 1. Red = oxygen blue = nitrogen, brown = bromine, green = fluorine, grey = carbon and white = hydrogen. | ||
Next, we sought a general method for reducing the nitro compounds 2 and we found that iron in acetic acid was an effective means for forming the anilines 3. Compound 3a has been synthesised previously using 10% platinum on carbon although we found it less effective than an alternative iron-mediated method (Scheme 1).14
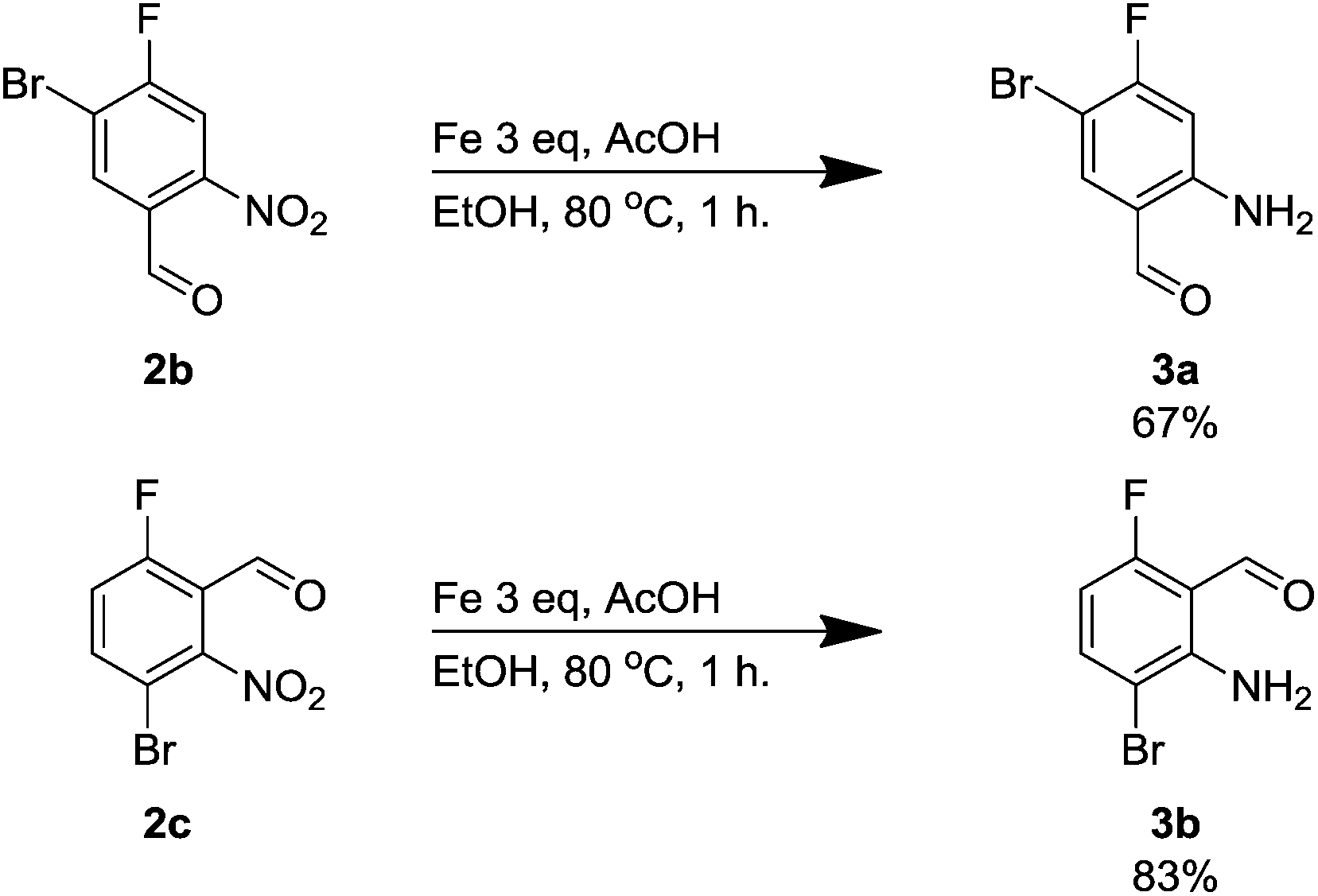 | ||
| Scheme 1 Iron-mediated reductions of compounds 2b and 2c. | ||
Unfortunately, a whole range of methods proved ineffective in the attempted reduction of the regioisomer 2a (Scheme 2). Nevertheless, by changing the reaction sequence (vide infra, Scheme 5) this issue can be circumvented and elaborated tetrasubstituted units can be synthesised.
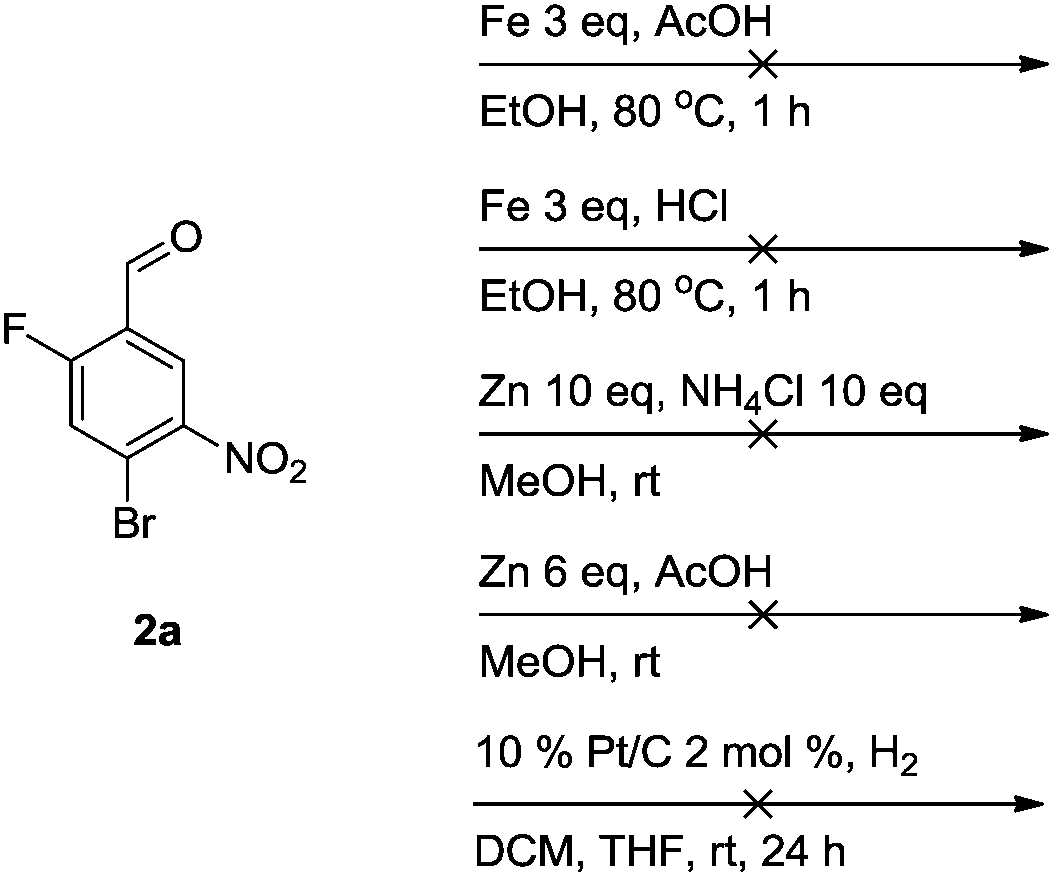 | ||
| Scheme 2 Nitro group reductions attempted on compound 2a. | ||
Alkyne groups are present in a number of kinase inhibitors such as Erlotinib (Fig. 1) and can also be used in e.g. indole forming reactions.15–17 We carried out Sonogashira couplings on 2a, 3a and 3b and obtained the silyl-protected alkynylbenzenes 4 in moderate to good yields (Scheme 3).
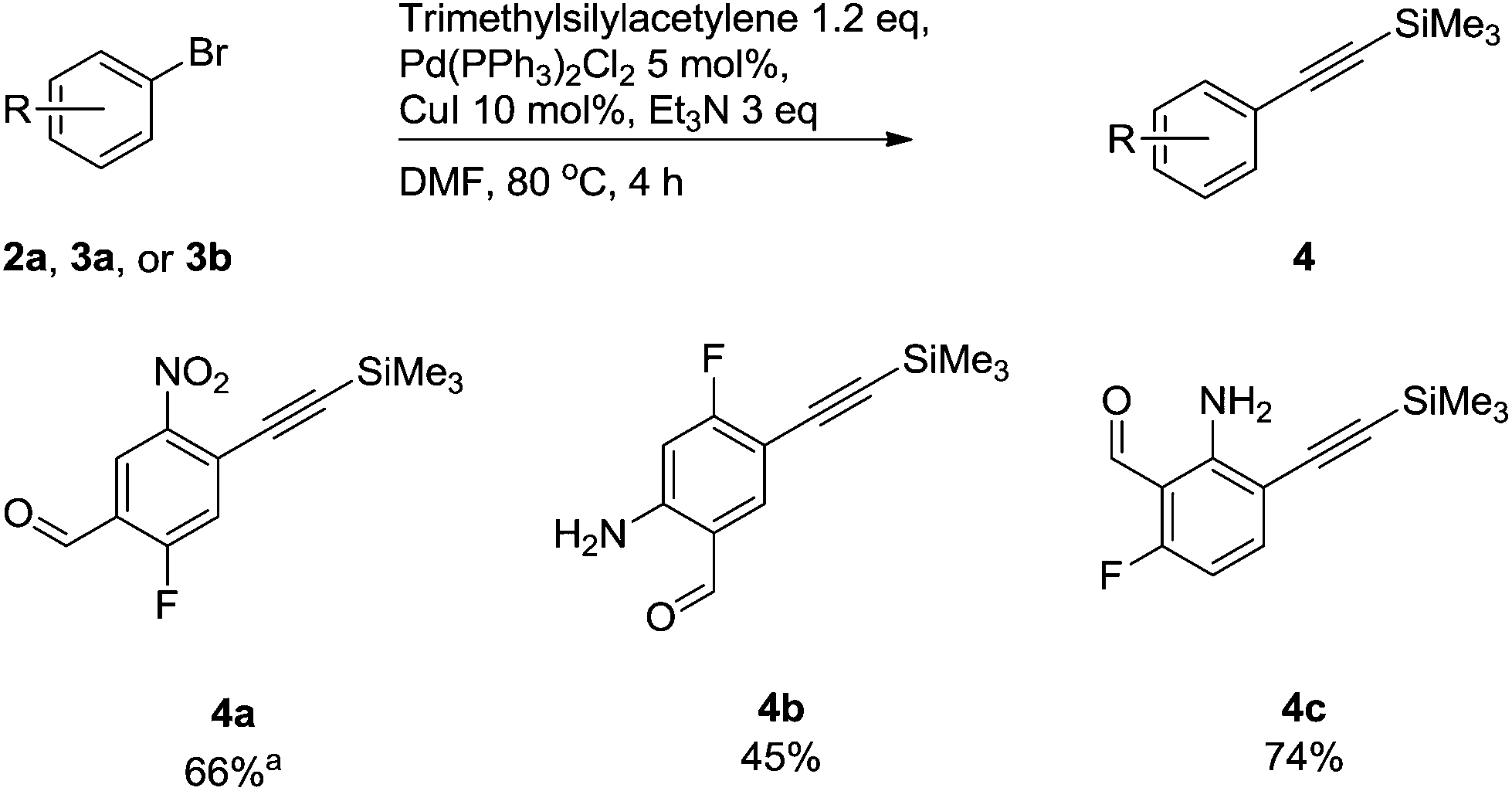 | ||
Scheme 3 Compounds synthesised via Sonogashira cross-coupling reactions of trimethylsilylacetylene. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction performed at rt for 1 hour. Reaction performed at rt for 1 hour. | ||
To show a broader synthetic scope, ethynyl MIDA phenylboronate derivatives were also included, although a slightly modified protocol was employed, since an excess of aryl halide was required as the halide could be removed with ease on silica gel (Scheme 4).18
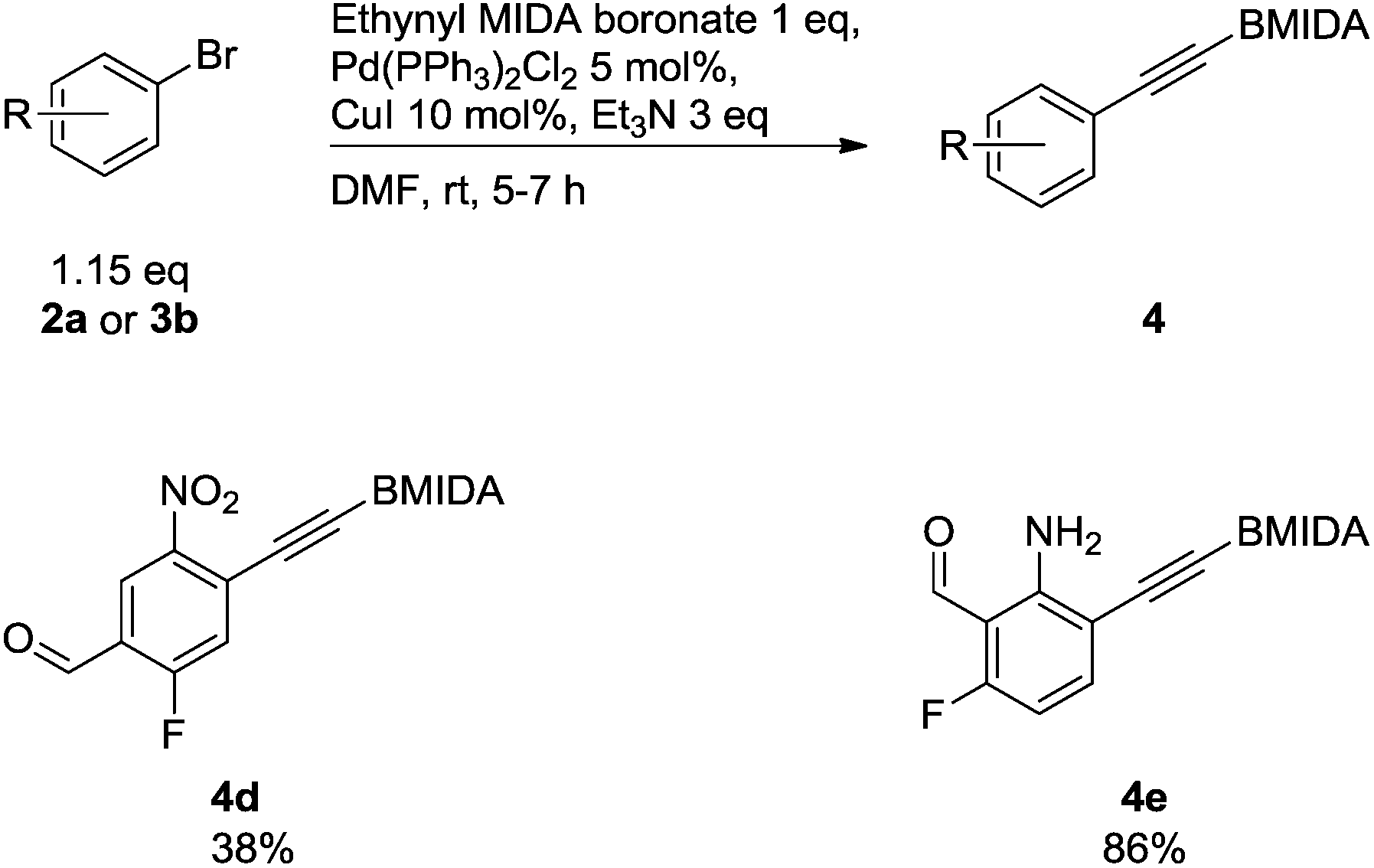 | ||
| Scheme 4 Compounds synthesised via Sonogashira cross-coupling reactions of ethynyl MIDAphenylboronates. | ||
A STAB (sodium triacetoxyborohydride)-mediated reductive amination of the formyl group in a selection of the compounds 2–4 was performed in order to form benzyl-substituted piperazine or morpholine derivatives (Table 2). Yields in general were good and these reactions proceeded selectively in the presence of an aniline substituent e.g. entries 2–5 (Table 2). Deprotected analogues of e.g.5d, may prove to be useful synthons for further derivatisation or as novel Ro3 (rule of three) fragments in drug discovery projects.19,20
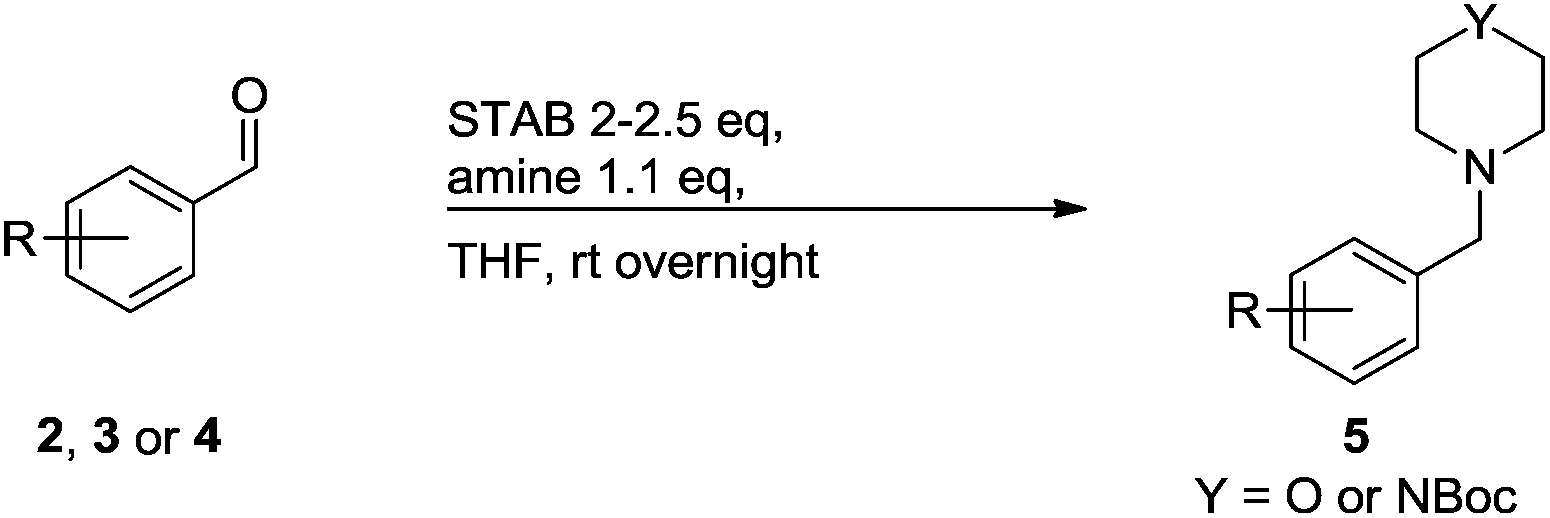
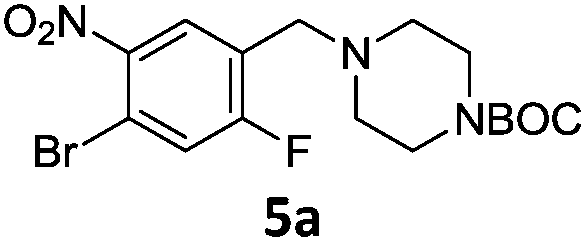
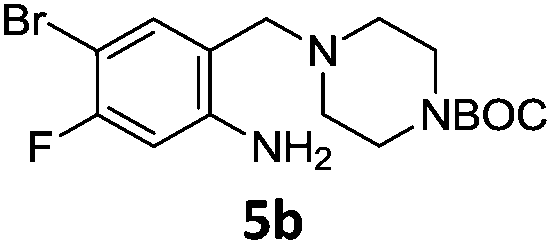
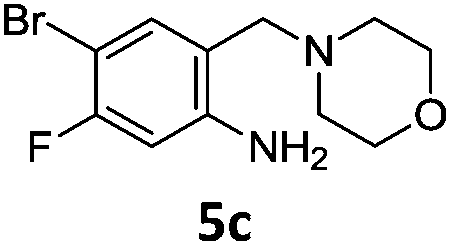
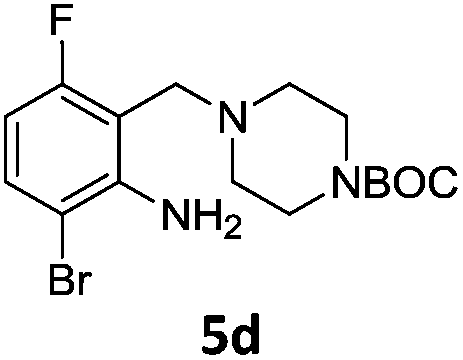
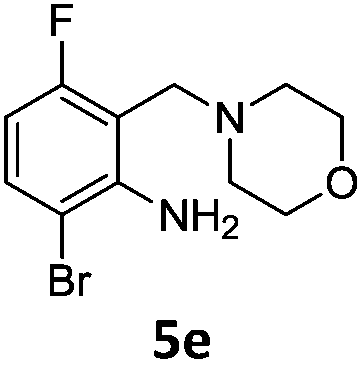
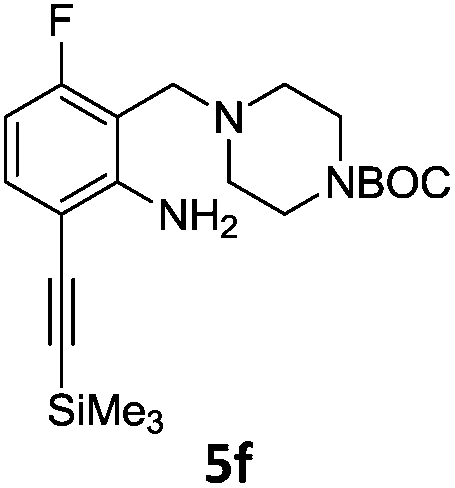
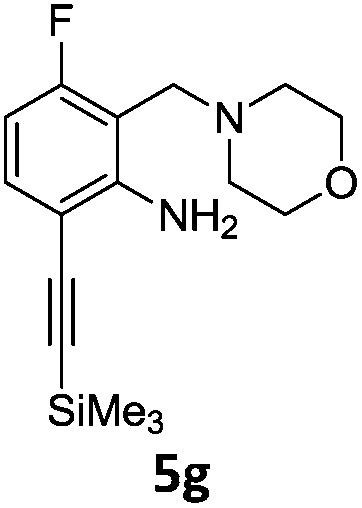
Nitro reduction of 5a was achieved, in moderate yield, using zinc in the presence of ammonium chloride, in order to maintain the acid-sensitive Boc protecting group. This represents an indirect route for obtaining the product 3c since it is a derivative of the recalcitrant nitro precursor 2a.
Having synthesized a range of tetrasubstituted anilines we wished to perform further modification of the aniline moiety. Firstly a Clauson-Kaas pyrrole synthesis was performed on the aniline 5d, which led to the pyrrole 6a (Scheme 6). Gratifyingly, the Boc-protecting group withstood the harsh microwave and acidic conditions.
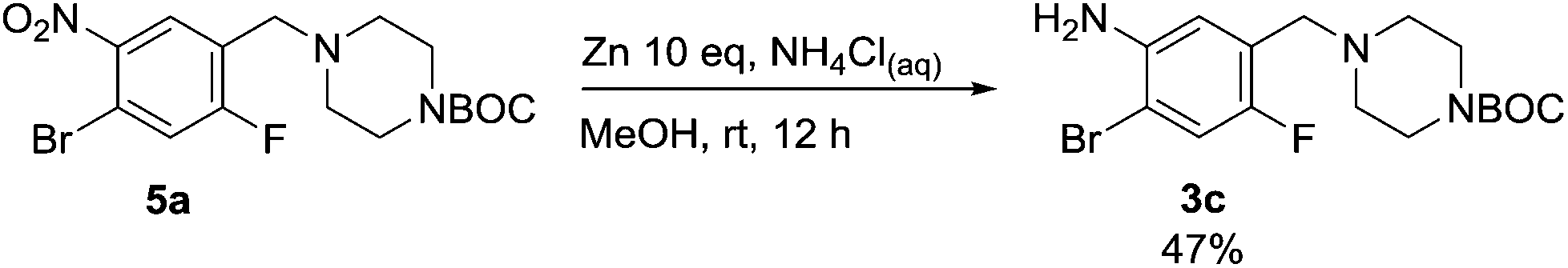 | ||
| Scheme 5 Zinc-mediated nitro reductions of compound 5a. | ||
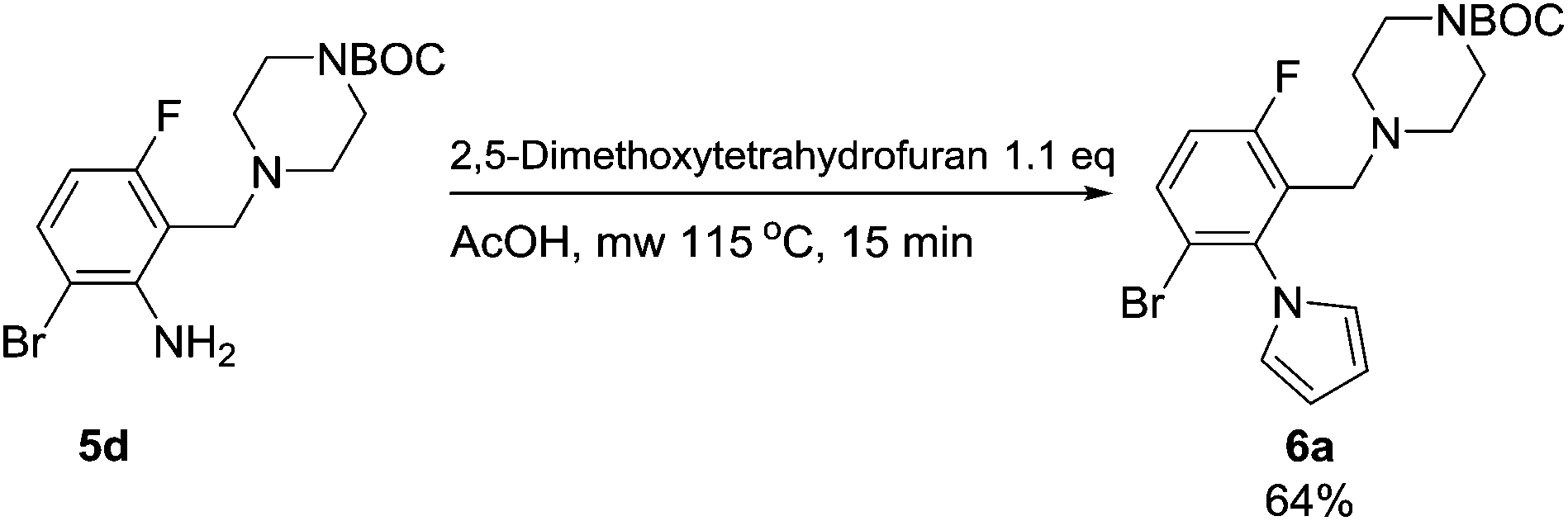 | ||
| Scheme 6 Clauson-Kaas pyrrole synthesis under microwave conditions on compound 5d. | ||
As a proof of principle, based on structural similarity to the EGFR inhibitor Gefitinib and with the emergence of tetra-substituted scaffolds leading to the development of anticancer therapeutics (e.g. Osimertinib), we rationalised that compounds 7–9 are likely to target the receptor tyrosine kinase, EGFR. In order to assess the biological activity of our panel of tetra-substituted small-molecules, we carried out a dual-pass biological evaluation including in vitro and cellular-based assays. Compound potency against EGFR (wild-type, Exon 20) was established using a homogeneous time-resolved fluorescence (HTRF) kinase assay, which measures the extent of internal tyrosine phosphorylations. The hERG-CHO cell line over-expresses the human Ether-à-go-go Related Gene (hERG), which is a gene (KCNH2) that encodes a K+ channel (Kv11.1) and is a red light toxicity alert in many drug discovery programmes (Table 3).
| Compounds synthesised from 2b | Compounds synthesised from 2c | Compounds synthesised from 2a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||||
| Compound | EGFR IC50a |
Mean hERG inhibitionb (%) | Dock scorec | Compound | EGFR IC50a |
Mean hERG inhibitionb (%) | Dock scorec | Compound | EGFR IC50a |
Mean hERG inhibitionb (%) | Dock scorec |
| a EGFR Ex20 WT HTRF CR GMean IC50 (μM). b hERG Hu CHO IF EPhs SS GD mean, at 10 μM. c Using Schrodinger Glide. | |||||||||||
|
0.0184 | 61.1 | −7.849 |
|
0.935 | 44.5 | −8.895 |
|
>21.3 | 33.7 | −5.986 |
|
0.377 | 96.5 |
|
21.5 | 21.5 |
|
27.1 | 46.4 | |||
|
0.546 | 55.6 |
|
>30 | 43.9 | ||||||
|
>30 | 19.5 | |||||||||
As a cautionary note, we did not embark on a drug discovery programme; for example, the biological evaluation (vide infra) was performed a long time after the synthesis of the compounds, preventing for example, any new syntheses and fine tuning of the products to address pharmacokinetics or off-target issues or to further investigate SAR (structure activity relationships).
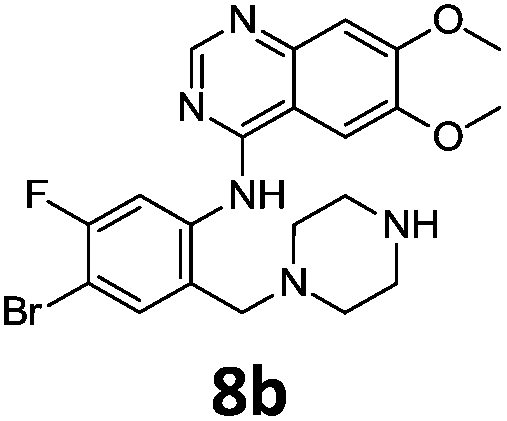
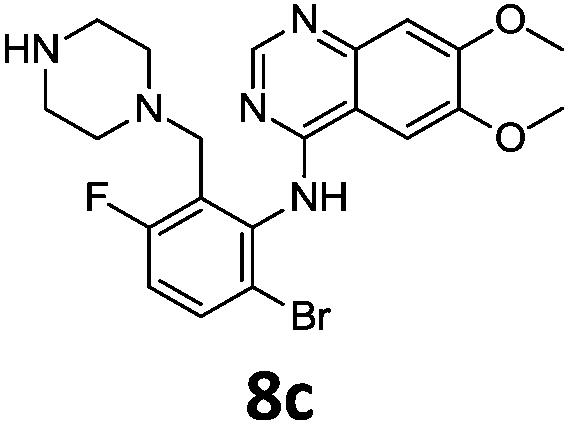
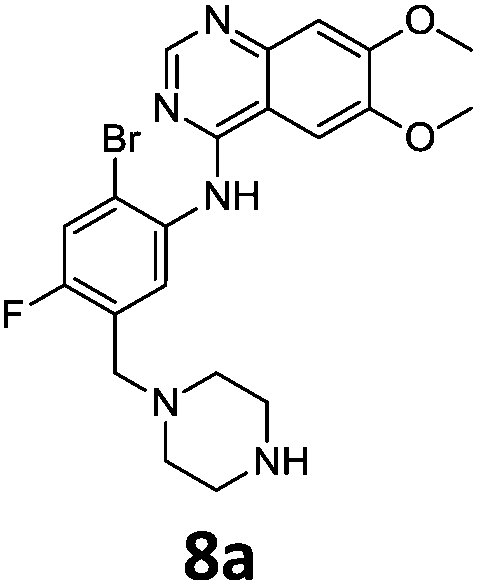
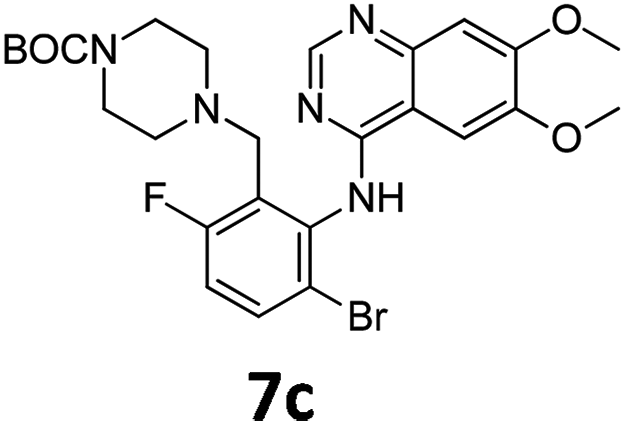
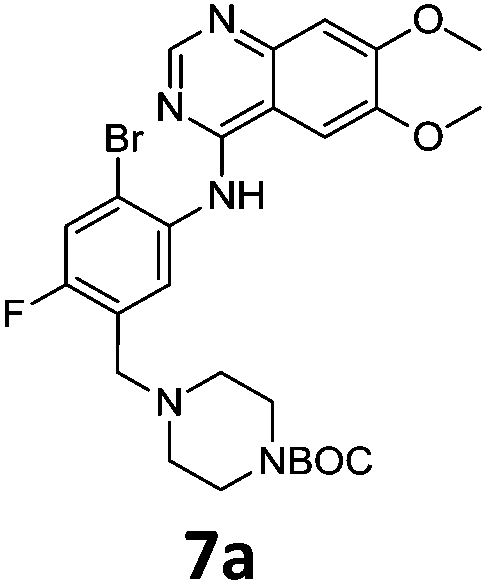
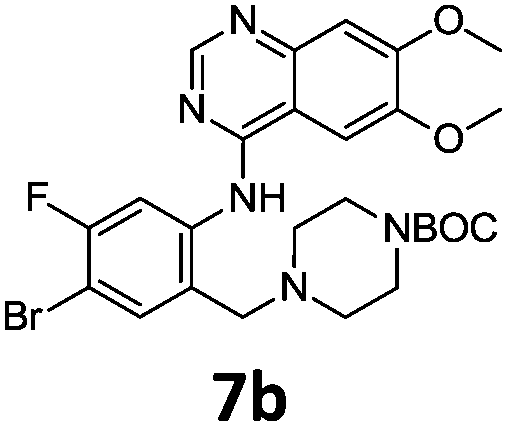
Commercially available 4-chloro-6,7-dimethoxyquinazoline was reacted with anilines 3c, 5b, and 5d to afford 7a–7c in moderate to good yields (Scheme 7). We found that the use of sodium bis(trimethylsilyl)amide (NaHDMS) gave the product without deprotection of the Boc piperazine, unlike commonly used acid catalysed protocols.21 Boc-deprotection was achieved by the addition of hydrochloric acid in 1,4-dioxane (Scheme 8) and purification was achieved by simply using solid phase extraction (strong cation exchange, SCX, column), giving 8a–8c.
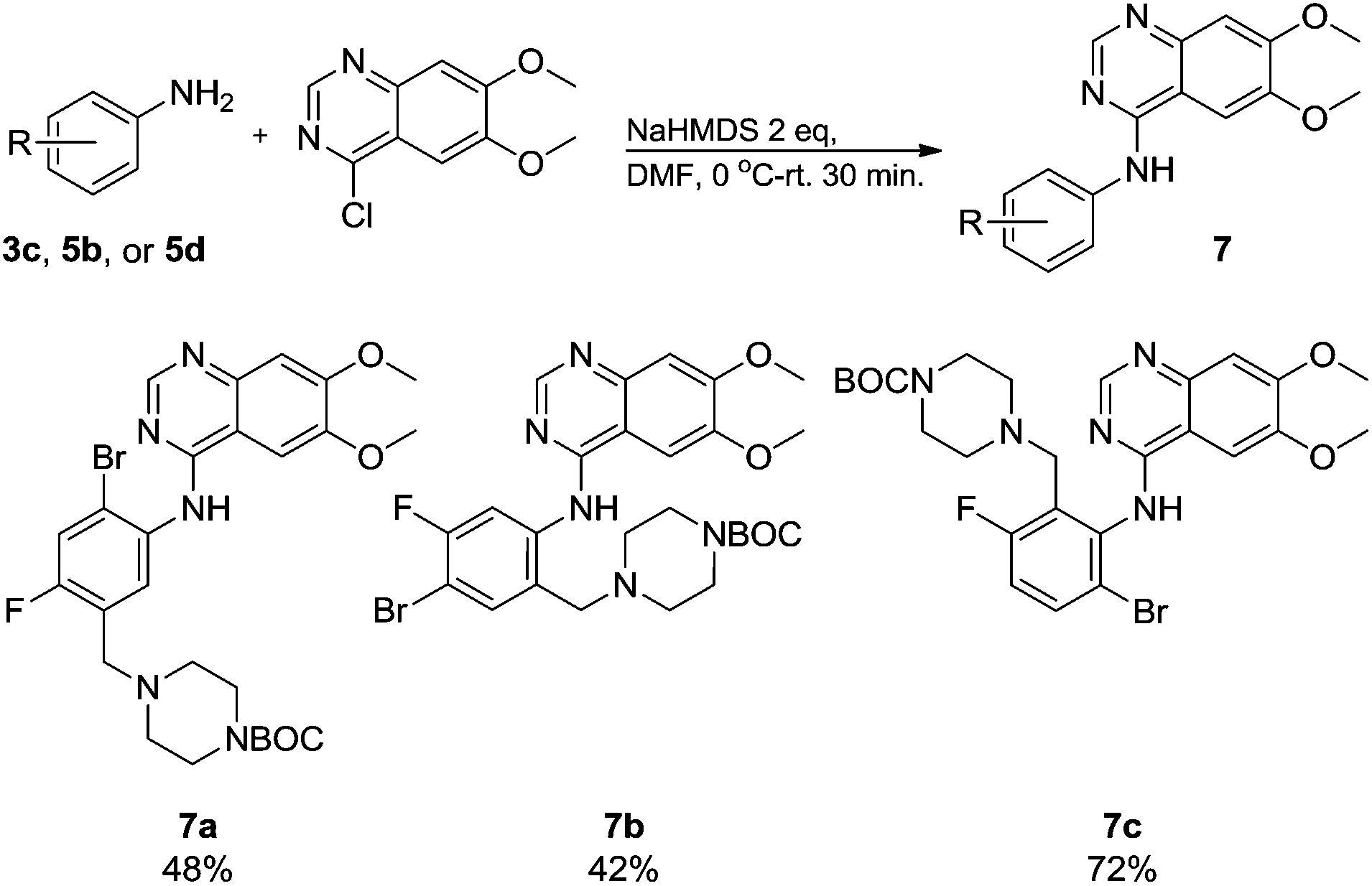 | ||
| Scheme 7 Linking of the quinazoline unit. | ||
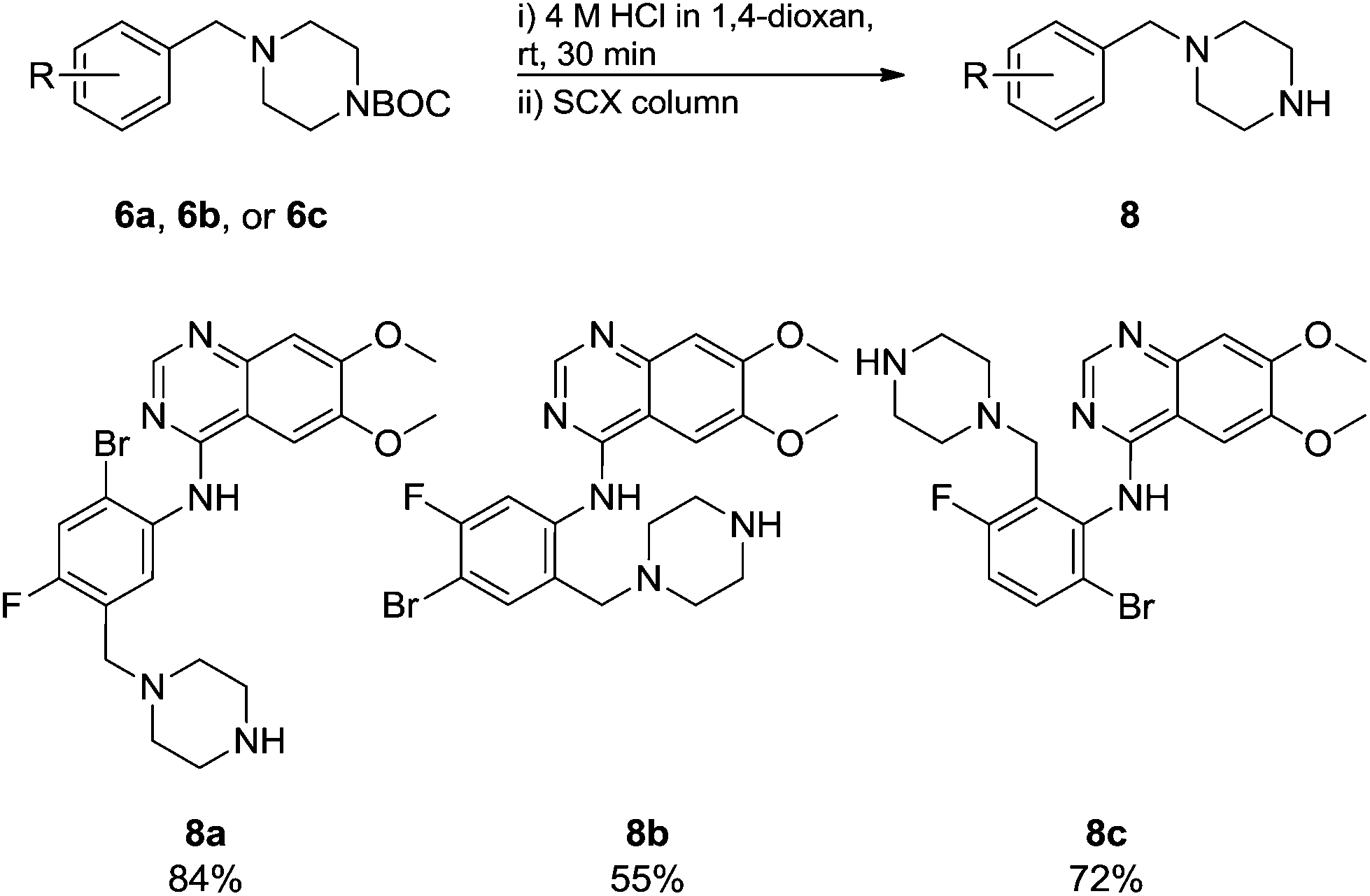 | ||
| Scheme 8 Boc cleavage. | ||
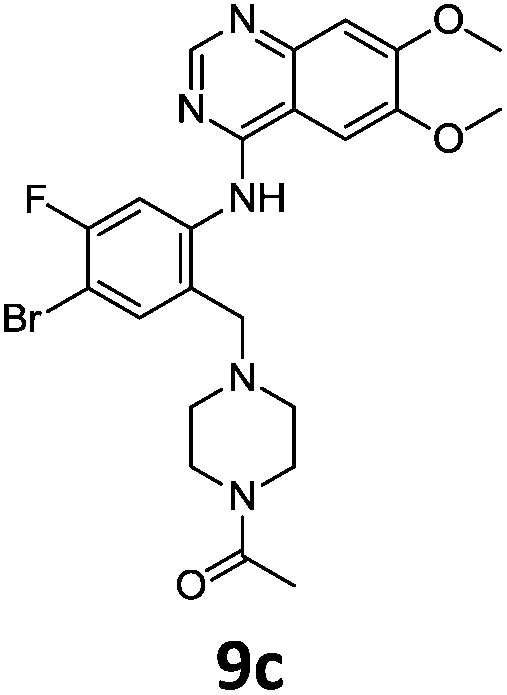
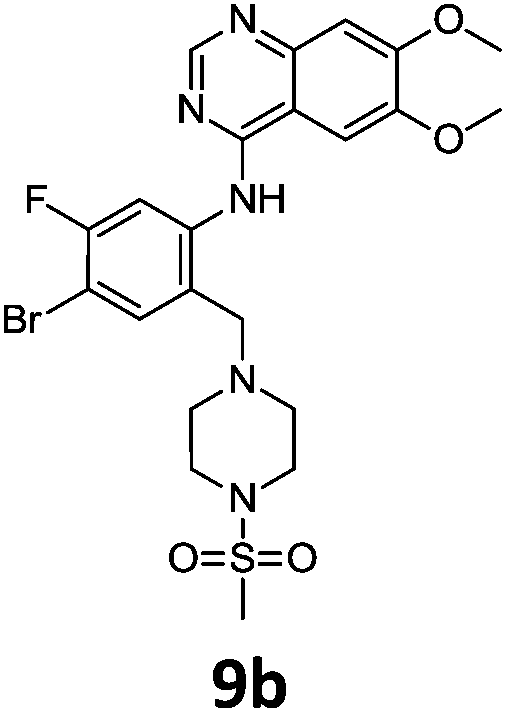
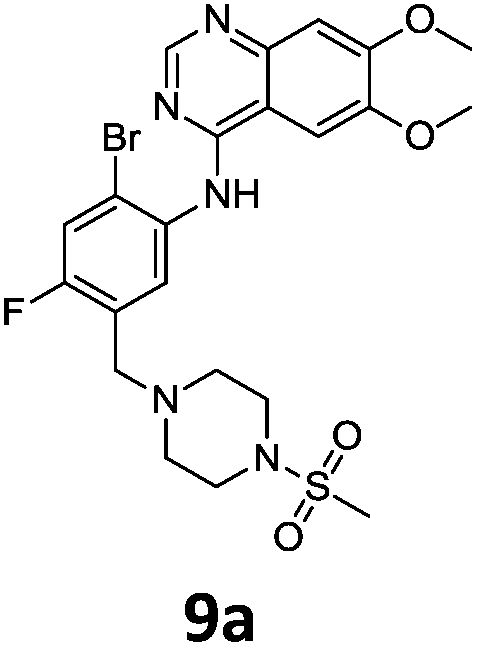
These newly synthesised secondary amines could be easily functionalised to sulphonamides or amides in high yields particularity in the case of 9b and 9c (Scheme 9).
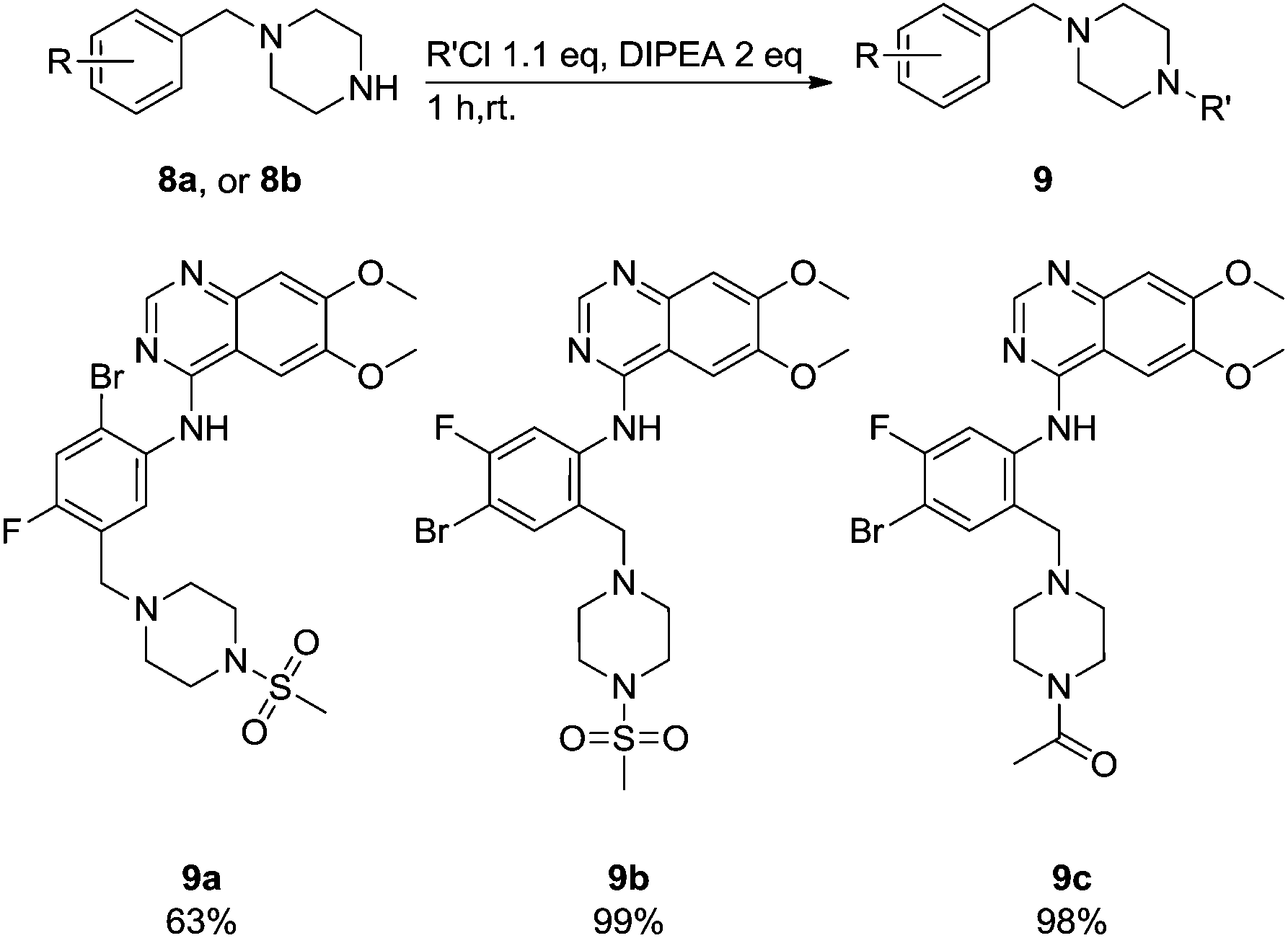 | ||
| Scheme 9 Functionalization of the piperazine unit. | ||
Compounds 7–9 were tested for inhibition of wild-type EGFR (Table 3). These results show that all of the compounds containing a BOC protecting groups were inactive except for 7c, which had a high IC50 (21.5 μM). None of the compounds with regiochemistry resulting from the 2a series i.e.8a, 7a and 9a showed any appreciable activity. The most active compound was 8b (entry 8) which gave an IC50 of 18.4 nM. All of the compounds in the 2b series gave good results i.e. all IC50's were less than 1 μM, with the exception of the BOC protected compound 7b, which showed no activity. Unfortunately the percentage hERG inhibition increased as the activity of the compounds improved, as exemplified by 8b (Table 3). There are ways in which to design out hERG inhibitions e.g. by reducing lipophilicity, reducing the pKa of the nitrogen atoms, increasing steric hindrance around the nitrogen atoms or decreasing the number of hydrogen bond acceptors, but, as mentioned above, this was not addressed.22
The biological data suggest that the solubilising group piperazine ortho to the aniline moiety (linking to the quinazoline unit) gave compounds of higher activity i.e. compounds in the 2b and 2c series. However, the series where the aniline and the piperazine groups are mutually meta, i.e. the 2a series, leads to a loss of activity. To look in to the relationship between the meta and ortho compounds we modelled the binding mode of the compounds 8a–8c and produced docking poses (Table 3/Fig. 5).
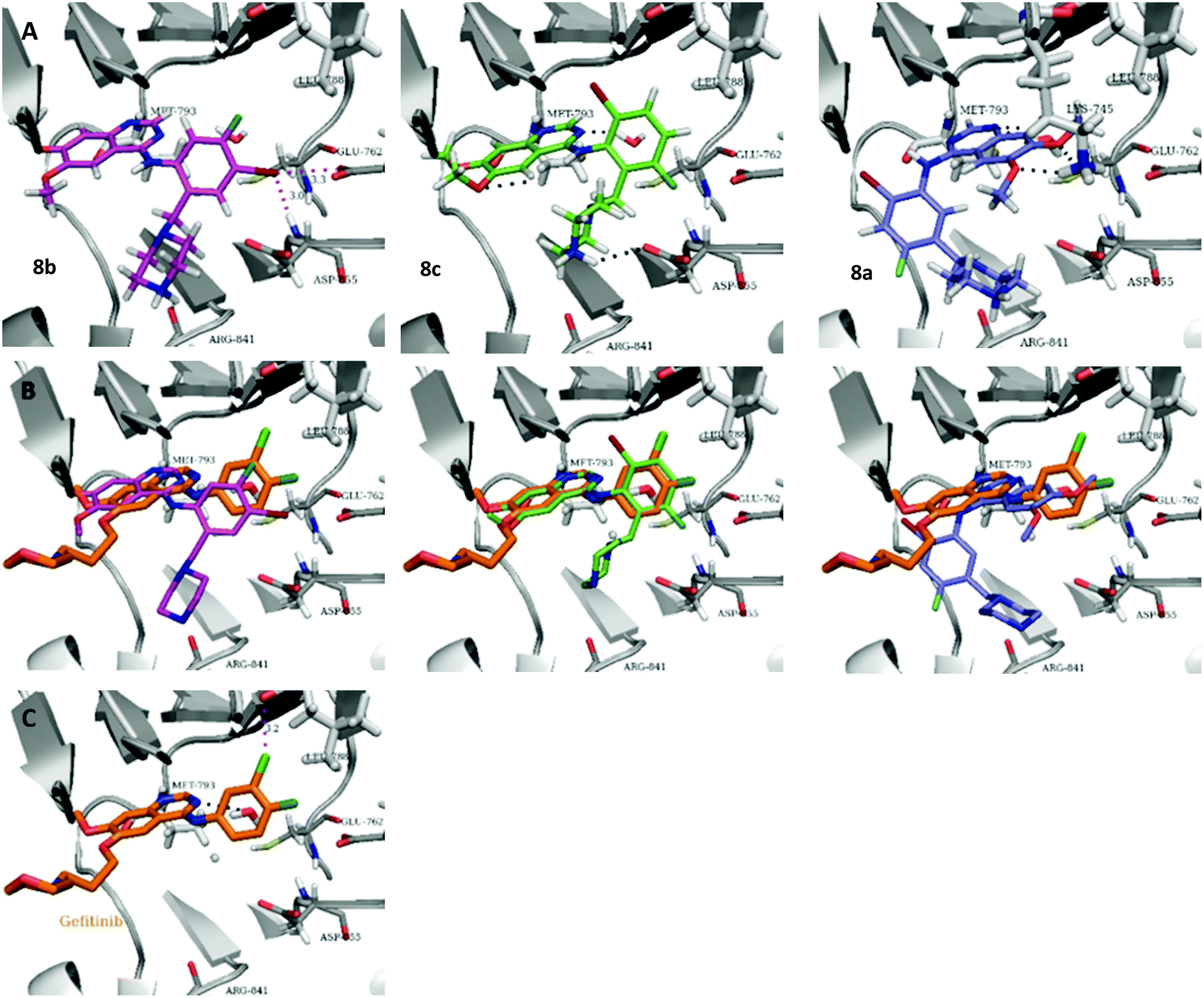 | ||
| Fig. 5 Docked 8a–c and comparison to Gefitinib, using Schrodinger Glide. (A) Docking poses of 8a–c. (B) Comparison of docked 8a–c with cocrystalized Gefitinib. (C) Cocrystal structure of gefitinib in EGFR (PDB code: 2ITY). | ||
To explore the binding mode of 8a–c in EGFR we performed docking studies using the structure of the Gefitinib-EGFR complex.23 We found that 8c was able to bind in a way similar to Gefitinib forming archetypal hydrogen bonds between the backbone N–H of Met793 and a structural water from the nitrogen atoms of the quinazoline core (Fig. 5). An additional hydrogen bond was formed from the protonated piperazine and the carbonyl of Asp855. The halogenated phenyl group was accommodated in the hydrophobic pocket however, halogen substitution in 8c was not aligned so as to form the halogen bonding exhibited in Gefitinib between the Cl⋯C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Leu788.
O of Leu788.
Compound 8b, with halogen substitution meta- and para- to the aniline group, was docked similarly with the quinazoline core and phenyl ring slightly shifting to form Br⋯H–N (Asp855) and Br⋯OC (Glu762) halogen bonds. The key interaction between Met793 and the quinazoline nitrogen was formed but the bond with the structural water was not conserved. An additional interaction between Arg841 and the protonated piperazine was formed.
Compound 8a, with the piperazine meta- to the aniline cannot be docked in a mode similar to gefitinib and forces an alternative binding mode whereby the methoxy-groups of the quinazoline are forced into the hydrophobic pocket forming hydrogen bonds between the oxygen of the MeO– and the H–N of Lys745. This suggests, and is in agreement, that 8a will be inactive or show a much lower potency towards EGFR.
Conclusions
A library of tetrasubstituted aromatics has been synthesized starting with robust nitration chemistry. The library has been elaborated into a series of useful potential intermediates for drug discovery and final drug-like entities as exemplified by the formation of a range of EGFR inhibitors that display low nM inhibition.Acknowledgements
AstraZeneca (ACJ) and the University of Sussex (AJC, RNJ) are thanked for PhD studentship funding. Worldwide Cancer Research (grant no. 14-1002) partly funded this work through the provision of Schrodinger Glide software. The EPSRC UK National Mass Spectrometry Facility at Swansea University is thanked for assistance.Notes and references
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 CAS.
- J. C. M. Uitdehaag, F. Verkaar, H. Alwan, J. De Man, R. C. Buijsman and G. J. R. Zaman, Br. J. Pharmacol., 2012, 166, 858–876 CrossRef CAS PubMed.
- L. L. Remsing Rix, U. Rix, J. Colinge, O. Hantschel, K. L. Bennett, T. Stranzl, A. Müller, C. Baumgartner, P. Valent, M. Augustin, J. H. Till and G. Superti-Furga, Leukemia, 2009, 23, 477–485 CrossRef CAS PubMed.
- P. Martin, S. Oliver, S. J. Kennedy, E. Partridge, M. Hutchison, D. Clarke and P. Giles, Clin. Ther., 2012, 34, 221–237 CrossRef CAS PubMed.
- P. Wu, T. E. Nielsen and M. H. Clausen, Drug Discovery Today, 2015, 21, 5–10 CrossRef PubMed.
- M. R. V. Finlay, M. Anderton, S. Ashton, P. Ballard, P. A. Bethel, M. R. Box, R. H. Bradbury, S. J. Brown, S. Butterworth, A. Campbell, C. Chorley, N. Colclough, D. A. E. Cross, G. S. Currie, M. Grist, L. Hassall, G. B. Hill, D. James, M. James, P. Kemmitt, T. Klinowska, G. Lamont, S. G. Lamont, N. Martin, H. L. Mcfarland, M. J. Mellor, J. P. Orme, D. Perkins, P. Perkins, G. Richmond, P. Smith, R. A. Ward, M. J. Waring, D. Whittaker, S. Wells and G. L. Wrigley, J. Med. Chem., 2014, 57, 8249–8267 CrossRef CAS PubMed.
- A. J. Close, P. Kemmitt, M. K. Emmerson and J. Spencer, Tetrahedron, 2014, 70, 9125–9131 CrossRef CAS.
- A. J. Close, P. Kemmitt, S. M. Roe and J. Spencer, Org. Biomol. Chem., 2016, 14, 6751–6756 CAS.
- M. R. Barbachyn, P. J. Dobrowolski, A. R. Hurd, D. J. Mcnamara, J. R. Palmer, A. G. Romero, J. C. Ruble, D. A. Sherry, L. M. Thomasco, P. L. Toogood, G. L. Bundy, G. E. Martin and D. L. Romero, WO Pat., 031195 A1, 2004 Search PubMed.
- E. Baxter, WO Pat., 097401 A1, 2009 Search PubMed.
- L. M. Gaster, F. E. Blaney, S. Davies, D. M. Duckworth, P. Ham, S. Jenkins, A. J. Jennings, G. F. Joiner, F. D. King, K. R. Mulholland, P. A. Wyman, J. J. Hagan, J. Hatcher, B. J. Jones, D. N. Middlemiss, G. W. Price, G. Riley, C. Roberts, C. Routledge, J. Selkirk and P. D. Slade, J. Med. Chem., 1998, 41, 1218–1235 CrossRef CAS PubMed.
- U. Yadav, H. Mande and P. Ghalsasi, J. Chem. Educ., 2012, 89, 268–270 CrossRef CAS.
- Abdulla, S. Amina and Y. Kumar, Synth. Commun., 2011, 41, 2946–2951 CrossRef.
- N. A. Paras, J. Brown, Y. Cheng, S. Hitchcock, T. Judd, P. Lopez, A. E. Minatti, T. Nixey, T. Powers, C. M. Tegley, Q. Xue, B. Yang and W. Zhong, WO Pat., 090911 A1, 2011 Search PubMed.
- J. M. W. Chan, G. W. Amarante and F. D. Toste, Tetrahedron, 2011, 67, 4306–4312 CrossRef CAS PubMed.
- A. Arcadi, G. Bianchi and F. Marinelli, Synthesis, 2004, 610–618 CAS.
- D. R. Adams, M. A. J. Duncton, J. R. A. Roffey and J. Spencer, Tetrahedron Lett., 2002, 43, 7581–7583 CrossRef CAS.
- J. R. Struble, S. J. Lee and M. D. Burke, Tetrahedron, 2010, 66, 4710–4718 CrossRef CAS.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876–877 CrossRef PubMed.
- R. A. E. Carr, M. Congreve, C. W. Murray and D. C. Rees, Drug Discovery Today, 2005, 10, 987–992 CrossRef CAS PubMed.
- L. Francois, A. Hennequin, K. M. Foote and K. H. Gibson, WO Pat., 004732 A1, 2004 Search PubMed.
- A. M. Aronov, J. Med. Chem., 2006, 49, 6917–6921 CrossRef CAS PubMed.
- C. H. Yun, T. J. Boggon, Y. Li, M. S. Woo, H. Greulich, M. Meyerson and M. J. Eck, Cancer Cell, 2007, 11, 217–227 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1485121–1485123. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01394e |
| This journal is © The Royal Society of Chemistry 2016 |