
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Truncated borrelidin analogues: synthesis by sequential cross metathesis/olefination for the southern fragment and biological evaluation†
Tülay
Gündemir-Durmaz
a,
Fabian
Schmid
a,
Yana
El Baz
a,
Annette
Häusser
a,
Carmen
Schneider
b,
Ursula
Bilitewski
b,
Guntram
Rauhut
c,
Delphine
Garnier
a,
Angelika
Baro
a and
Sabine
Laschat
*a
aInstitut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany. E-mail: sabine.laschat@oc.uni-stuttgart.de
bAG Compound Profiling and Screening, Helmholtz Zentrum für Infektionsforschung, Inhoffenstr. 7, D-38124 Braunschweig, Germany
cInstitut für Theoretische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
First published on 4th August 2016
Abstract
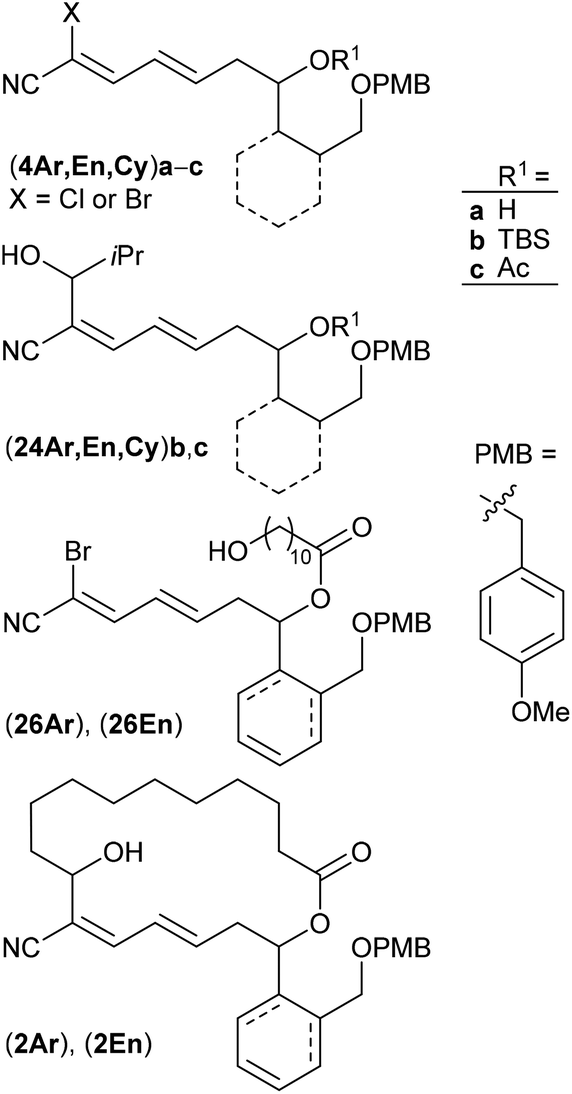
The construction of novel borrelidin analogues is reported in which the northern fragment is truncated to a simple hydroxyundecanecarboxylate and the original cyclopentanecarboxylic acid in the southern fragment is replaced with different six-membered rings. The required precursors were prepared by cross metathesis of the appropriate carbocycle-based homoallylic alcohol with crotonaldehyde followed by HWE olefination of the resulting enal with bromocyanophosphonate. The key aldehyde for intramolecular cross coupling was accessible by oxidation of the hydroxy group of the linked undecanecarboxylate unit. Grignard mediated macrocyclization finally yielded the borrelidin related products. The investigation is complemented by SAR studies and quantum-chemical calculations.
Introduction
Polyketide natural products consist of a huge variety of structurally diverse compounds originating from a modified fatty acid biosynthesis. They display a broad spectrum of biological activities relevant for medicinal chemistry and drug development.1 A prominent member of this family is (−)-borrelidin (1) (Scheme 1), which was isolated by Berger et al. in 1949 from Streptomyces rochei.2 Apart from its activity against borrelia, the major cause of lyme disease,2–4 a broad biological profile has been discovered. Borrelidin (1) exhibits antiviral activity,5 inhibits angiogenesis6 as well as several enzymes such as cyclin-dependent kinase Cdc28/Cln2 of Saccharomyces cerevisiae7 and threonyl tRNA synthetase.8 In addition, anti-malarial activity against chloroquine-resistant strains of Plasmodia falsiparum has been reported.9 The promising biological activities have stimulated several synthetic endeavours3 and up to now five total syntheses have been reported by the groups of Morken,10 Hanessian,11 Theodorakis,12 Omura13 and Minnaard14 complemented by synthetic studies.15 Hahn et al. synthesized complex intermediates in order to probe the dehydratase from borrelidin biosynthesis.16 By employing precursor-directed biosynthesis Wilkinson et al. prepared several borrelidin analogues with modified C-17 side chains and were able to separate anti-angiogenetic from cytotoxic activities.17,18 Omura's group reported that esterification of the free carboxylic acid with various triazolyl-containing alcohols led to improved anti-malarial activity and reduced cytotoxicity.9 With borrelidin B containing an aminomethyl group in place of the nitrile function the need for the CN group for threonyl tRNA synthetase inhibition was demonstrated.19 In a comparative cytological study loss of biological activity for borrelidin congeners with amide functions at C-12 and C-22 was observed.20 In all cases, however, the macrocycle of 1 was retained. These results motivated us to study borrelidin analogues with modified macrocyclic skeletons regarding their potential cytotoxic properties. Taking the concept of truncated natural products discussed by Gademann et al.21 and Maier22 and successfully demonstrated by Nakata23 for kendomycin analogues into account, we aimed to prepare the borrelidin related compounds 2 with different rings in the side chain at C-17 and the complex polyketide region truncated to a simple 11-hydroxyundecane chain (Scheme 1). The retrosynthesis is based on disconnection of 2 into the northern fragment 3 and the southern fragment 4, which can be traced back to known 11-bromoundecanoic acid and precursors 5. The latter requires different routes starting from known compounds.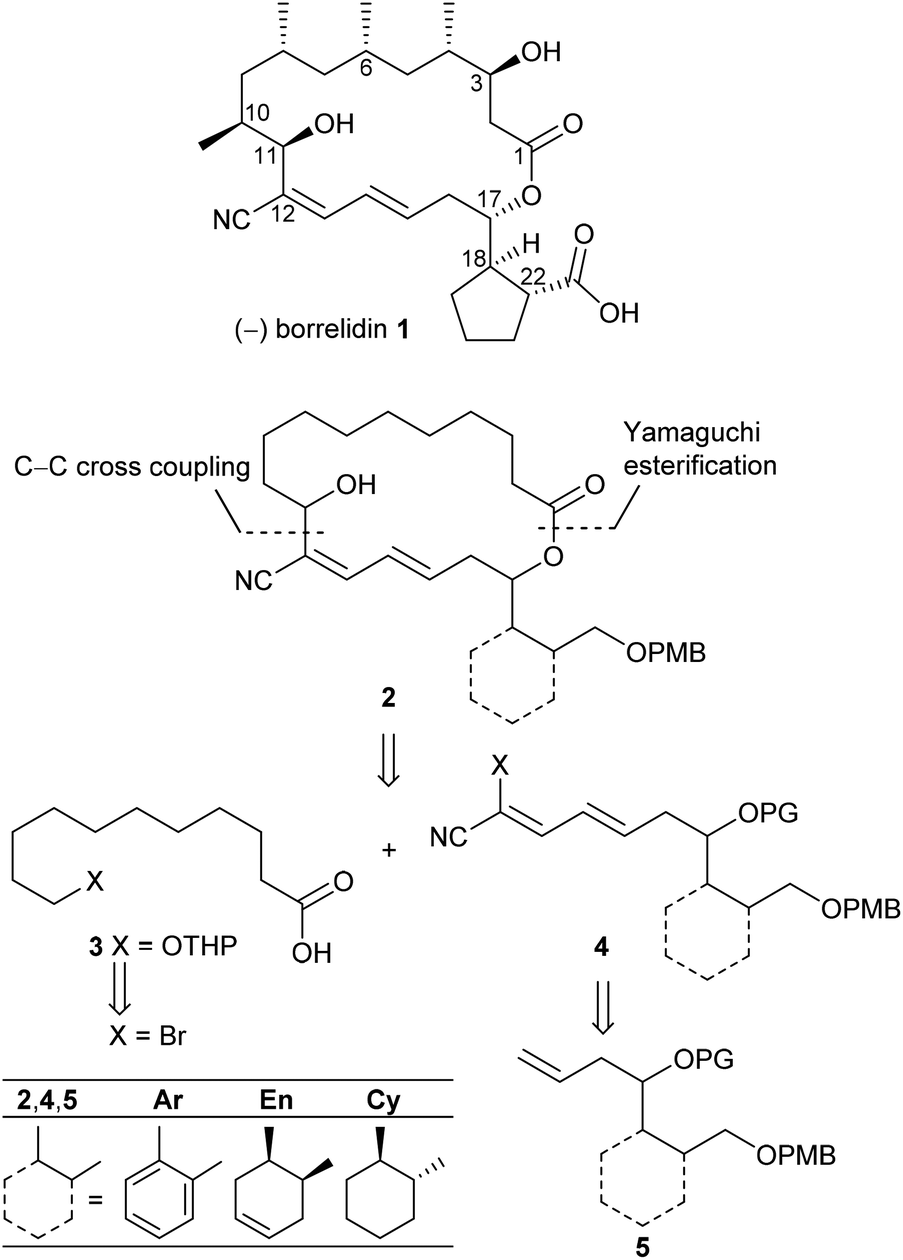 | ||
| Scheme 1 Retrosynthetic approach to borrelidin analogues 2 with different rings at C-17 and truncated northern fragment. | ||
As key steps for the connection of northern and southern fragments 3 and 4 an esterification and C–C coupling following Omura's strategy13 or, alternatively, Grignard coupling according to Iqbal's method24a were performed. To access cyanodienes 4 from precursors 5, the suitability of cross metathesis versus olefination or Knoevenagel-type condensation was studied based on seminal contributions by Iqbal.24b The results of the synthesis and biological investigation of southern fragments 4 and borrelidin analogues 2 are reported below.
Results and discussion
Synthesis of cyanodienes 4a–c
According to a procedure by Jay-Smith et al.25 1,2-benzenedimethanol 6 prepared from diethyl phthalate by LiAlH4 reduction26 was mono-protected with PMBCl to yield 7 in 88%. Subsequent Dess–Martin oxidation afforded the known aldehyde 8 in 95% yield (Scheme 2). While the Sakurai reaction of 8 with allyltrimethylsilane in the presence of various Lewis acids27 either resulted in no conversion at all or led to complete decomposition of the starting material, treatment of 8 with the corresponding Grignard reagent provided the desired alcohol (5Ar)a in 89% yield. TBS protection of (5Ar)a gave derivative (5Ar)b in 91% yield. As we aimed at a maximum flexibility with respect to the coupling of fragments 3 and 4 (Scheme 1), we also introduced the acetyl protecting group in (5Ar)a under standard acetylation conditions, which would allow a Yamaguchi macrolactonization as the intramolecular step. Precursor (5Ar)c served as a model acyl-protected substrate for the synthesis of the southern fragment.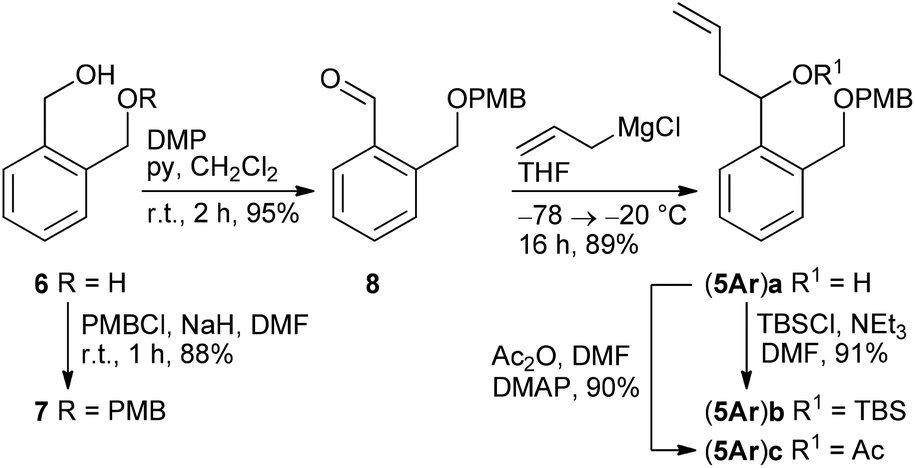 | ||
| Scheme 2 Synthesis of benzene-based homoallylic alcohols (5Ar)a–c. | ||
Cyclohexene-derived homoallylic alcohols (5En)a–c were prepared following a similar strategy (Scheme 3). Acidic hydrolysis of 4-cyclohexene-1,2-dicarboxylic anhydride 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 followed by LiAlH4 reduction and monoselective PMB-protection gave alcohol 10 in 76% overall yield (over three steps). After Dess–Martin oxidation, the resulting aldehyde 11 was subjected to a Grignard reaction to yield the homoallylic alcohol (5En)a in 95% as a diastereomeric mixture (dr 79:21), which was separated by HPLC for characterization. TBS-protection or acetylation of the diastereomeric alcohol (5En)a provided the target compounds (5En)b and (5En)c in 98% and 96% yield, respectively.
28 followed by LiAlH4 reduction and monoselective PMB-protection gave alcohol 10 in 76% overall yield (over three steps). After Dess–Martin oxidation, the resulting aldehyde 11 was subjected to a Grignard reaction to yield the homoallylic alcohol (5En)a in 95% as a diastereomeric mixture (dr 79:21), which was separated by HPLC for characterization. TBS-protection or acetylation of the diastereomeric alcohol (5En)a provided the target compounds (5En)b and (5En)c in 98% and 96% yield, respectively.
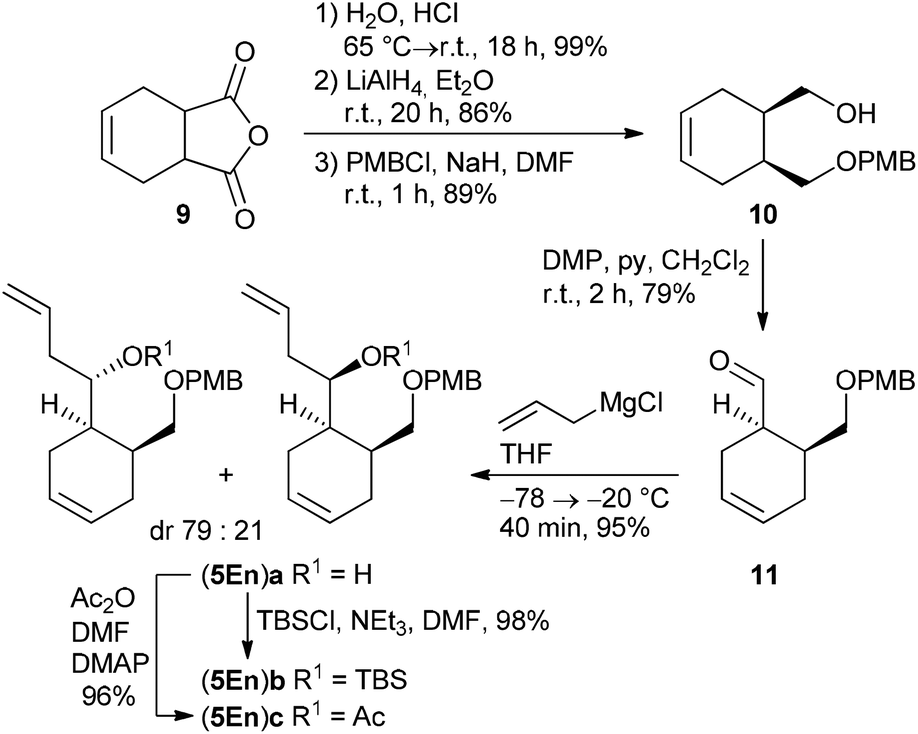 | ||
| Scheme 3 Synthesis of cyclohexene-based homoallylic alcohols (5En)a–c which were further used as diastereomeric mixtures. | ||
The cyclohexane-derived homoallylic alcohol (5Cy)a was prepared from diol 13, which was obtained from the known D-dimenthyl succinate 12 by a Yamamoto asymmetric carbocyclization16,29,30 followed by LiAlH4 reduction (Scheme 4).
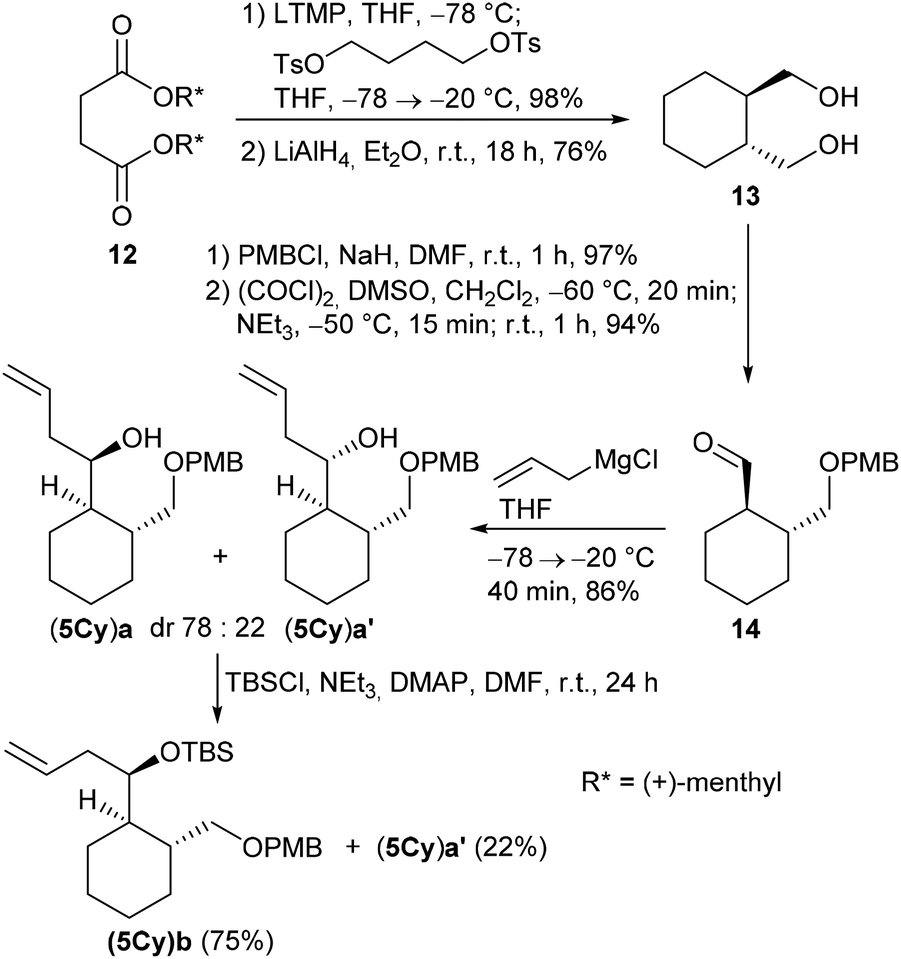 | ||
| Scheme 4 Synthesis of cyclohexane-based homoallylic alcohols (5Cy)a,b and (5Cy)a′. | ||
Mono-PMB protection of 13 and subsequent Swern oxidation afforded aldehyde 14 in 91% overall yield (over both steps). The Grignard reaction of 14 with allylmagnesium chloride led to an inseparable diastereomeric mixture of homoallylic alcohols (5Cy)a and (5Cy)a′ (dr 78:22) in 86%. However, when (5Cy)a and (5Cy)a′ were treated with TBSCl under our usual reaction conditions, only diastereomeric silylether (5Cy)b was isolated in 75% while the diastereomeric alcohol (5Cy)a′ remained unreacted and could be recovered in 22%. This diastereoselective kinetic resolution was rather unexpected, although some examples of diastereoselective Si–O couplings existed, e.g. employing silicon-stereogenic hydrosilanes and achiral Cu complexes.31,32 Presumably, the reactivity of the OH group in diastereomer (5Cy)a′ is diminished by hydrogen bonding between the OH group and the PMB group.
With homoallylic alcohols 5 in hand we investigated the cross metathesis with crotonaldehyde 15 (Table 1), which was reported to give better yields than acrolein when using the Grubbs II catalyst.33
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | 5 | R1 | Time (h) | 16 | Yieldb (%) | dr |
| a Reaction conditions: 5 (1.0 equiv.), 15 (1.0 equiv.), cat. (5 mol%). b Isolated yield. c Further addition of Grubbs II catalyst (5 mol%) after 18 h. | ||||||
| 1 | (5Ar)b | TBS | 20 | (16Ar)b | 35 | |
| 2 | (5Ar)c | Ac | 18 | (16Ar)c | 89 | |
| 3 | (5En)a | H | 19 | (16En)a | 88 | 79:21 |
| 4c | (5En)b | TBS | 20 | (16En)b | 92 | 83:17 |
| 5 | (5En)c | Ac | 18 | (16En)c | 84 | 73:27 |
| 6 | (5Cy)a′ | H | 18 | (16Cy)a′ | 79 | |
| 7c | (5Cy)b | TBS | 20 | (16Cy)b | 80 | |
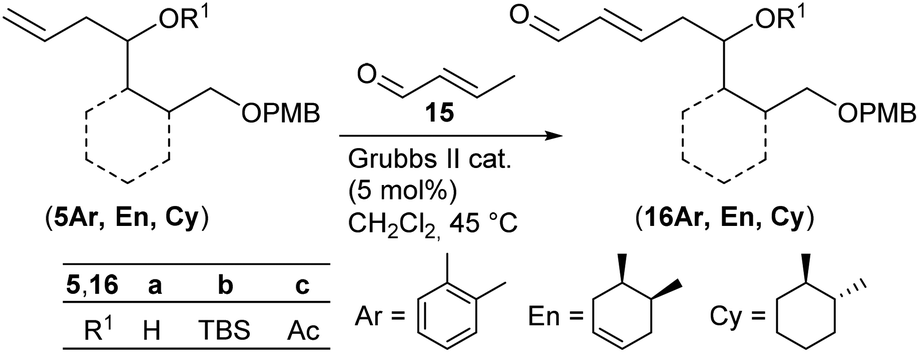
Treatment of the aromatic TBS-protected cross metathesis (CM) precursor (5Ar)b with crotonaldehyde 15 in the presence of the second generation Grubbs catalyst (Grubbs II) (5 mol%) in CH2Cl2 at 45 °C provided 35% of the desired product (16Ar)b (entry 1). Cross metathesis of cyclohexane (5Cy)b afforded 80% of enal (16Cy)b on further addition of the Grubbs II catalyst after 18 h of reaction time (entry 7). Under these conditions the corresponding cyclohexene derivative (5En)b gave 92% of the diastereomeric CM product (16En)b (dr 83:17 by 1H NMR of the CHO signal) (entry 4). As can be seen in Table 1, CM products (16En)a and (16Cy)a′ with free hydroxy groups were isolated in high yields of 88% and 79%, respectively (entries 3 and 6). This observation is in good agreement with the previous reports by Fuwa et al.34 and Lin and Davis35 suggesting that free hydroxy groups have a beneficial influence on cross metathesis reactions due to hydrogen-bonding of the OH group with the chlorine atom of the ruthenium carbene complex.36 Also the acetylated precursors (5Ar)c and (5En)c underwent cross metathesis under similar conditions providing the desired CM products (16Ar)c and (16En)c in comparable yields (entries 2 and 5).
The synthesis of cyanodiene fragments 4 was studied using first cross metathesis enals (16En)a,b (Scheme 5). Knoevenagel condensation of (16En)b with chloro- or bromoacetonitrile 17a,b37 to cyanodiene (4En)b, however, completely failed. An alternative strategy used a sequence of Wittig reaction/cross metathesis. While the Wittig olefination of enals (16En)a,b with methyltriphenylphosphonium bromide and nBuLi proceeded uneventfully to dienes (18En)a and (18En)b in 31% and 54% yield, respectively, the subsequent cross metathesis of (18En)b with substituted methacrylonitrile 1938 resulted in no conversion to cyanodiene (20En)b.
 | ||
| Scheme 5 Preliminary studies on olefination reactions of (16En)a,b to give fragments (4En)a,b (for details see the ESI†). | ||
Therefore, we decided to follow Omura's initial approach13 by utilizing electron-poor bromocyanophosphorane 2139 or cyanophosphonates 2240 and again enals (16En)a,b as benchmark substrates for the olefination. While the reaction of the unprotected derivative (16En)a with phosphorane 21 at room temperature in CH2Cl2 yielded the desired olefination product (4En)a in 38%, the corresponding TBS-protected (16En)b resulted in no conversion (Scheme 5). Afterwards neither (16Ar) nor (16Cy) enals reacted under these conditions (ESI†), and the Wittig reaction was abandoned.
Next, the HWE olefination of enals 16 with α-chloro- and α-bromocyanophosphonate 22a,b, respectively, was investigated. The results are summarized in Table 2. Deprotonation of α-chlorocyanophosphonate 22a with NaH in DMF at 0 °C followed by addition of substrate (16En)a with the free OH group or the TBS-protected analogue (16En)b at 0 °C provided the respective cyanodienes (4En)aCl and (4En)bCl in 57% and 53% yield (entries 4 and 8). In both cases, however, the reaction with the corresponding α-bromocyanophosphonate 22b under similar conditions did not lead to the olefination products (4En)aBr and (4En)bBr (entries 5 and 9). We surmised that α-bromocyanophosphonate 22b underwent nucleophilic displacement of the bromide by hydride rather than deprotonation of the acidic α-hydrogen.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entrya | Enal | R1 | 22 | Base | Solvent | Time (h) | Product | Yieldb (%) | drc |
| a Reaction conditions: 16 (1.0 equiv.), 22 (2.0 equiv.), LiCl (1.5 equiv.), DBU (1.5 equiv.); 16 (1.0 equiv.), NaH, 22 (1.25 equiv. each). b Isolated yield. c dr refers to stereocentre C-1 at the homoallylic ether position relative to the fixed cis or trans configuration in (4En) and (4Cy). d Temperature: −20 °C → r.t. e No conversion, starting materials were reisolated. f E/Z ratio at C-6. | |||||||||
| 1d | (16Ar)a | H | 22b | NaH | THF | 48 | (4Ar)aBr | — | — |
| 2 | (16Ar)c | Ac | 22a | DBU | MeCN | 1.5 | (4Ar)cCl | 11 | n.d. |
| 3 | (16Ar)c | Ac | 22b | DBU | MeCN | 1.5 | (4Ar)cBr | 65 | n.d. |
| 4 | (16En)a | H | 22a | NaH | DMF | 2.5 | (4En)aCl | 57 | 80:20 |
| 5 | (16En)a | H | 22b | NaH | DMF | 2.5 | (4En)aBr | —e | — |
| 6 | (16En)a | H | 22a | DBU | MeCN | 1 | (4En)aCl | 49 | 82:18 |
| 7 | (16En)a | H | 22b | DBU | MeCN | 1 | (4En)aBr | 49 | 80:20 |
| 8 | (16En)b | TBS | 22a | NaH | DMF | 3 | (4En)bCl | 53 | 76:24 |
| 9 | (16En)b | TBS | 22b | NaH | DMF | 2.5 | (4En)bBr | — | — |
| 10 | (16En)b | TBS | 22a | DBU | MeCN | 1 | (4En)bCl | 51 | 78:22 |
| 11 | (16En)b | TBS | 22b | DBU | MeCN | 1 | (4En)bBr | 48 | 78:22 |
| 12 | (16En)c | Ac | 22b | DBU | MeCN | 1.5 | (4En)cBr | 99 | 82:18 |
| 13 | (16Cy)a′ | H | 22a | NaH | DMF | 3 | (4Cy)a′Cl | — | — |
| 14 | (16Cy)a′ | H | 22a | DBU | MeCN | 1 | (4Cy)a′Cl | 43 | |
| 15 | (16Cy)a′ | H | 22b | DBU | MeCN | 1 | (4Cy)a′Br | 77 | |
| 16 | (16Cy)b | TBS | 22a | NaH | DMF | 2.5 | (4Cy)bCl | — | — |
| 17 | (16Cy)b | TBS | 22b | NaH | DMF | 2.5 | (4Cy)bBr | — | — |
| 18 | (16Cy)b | TBS | 22a | DBU | MeCN | 1 | (4Cy)bCl | 27 | 74:26f |
| 19 | (16Cy)b | TBS | 22b | DBU | MeCN | 1 | (4Cy)bBr | 76 | 85:15f |
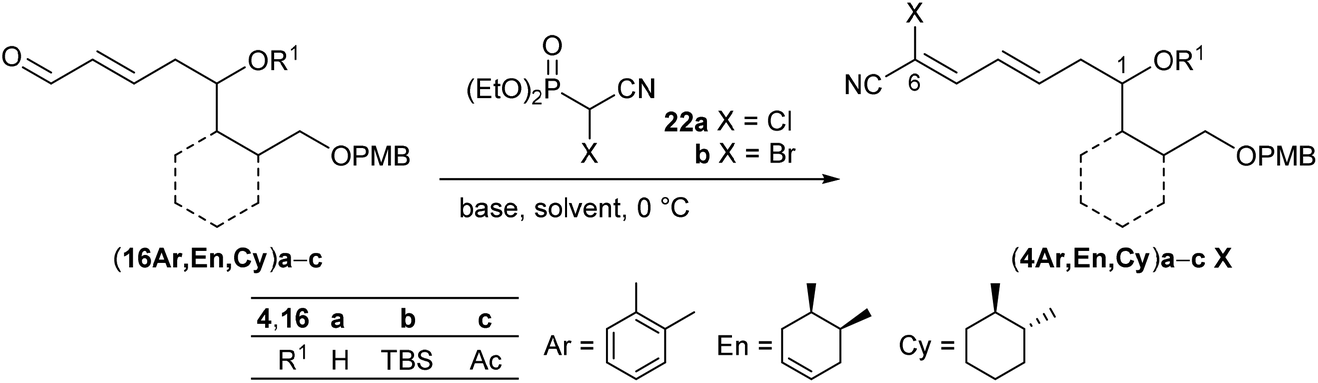
Following Omura's reaction conditions13 we finally succeeded in the preparation of both chloro- and bromocyanodienes 4. For this purpose, LiCl and subsequently DBU (1.5 equiv. each) were added to a solution of the respective substrate 16 and cyanophosphonate 22a or 22b in acetonitrile at 0 °C (Table 2). In this manner bromocyanodiene (4En)bBr could be isolated in 48% (entry 11). Similar yields were obtained for the unprotected enal (4En)a irrespective of the halide (entries 6 and 7). The HWE olefination of cyclohexane-based substrates (16Cy)a′,b resulted in good yields of bromocyanodienes (4Cy)a′Br (77%) and (4Cy)bBr (76%) (entries 15 and 19), while the yields of the corresponding chloro compounds (4Cy)a′Cl and (4Cy)bCl decreased to 43% and 27%, respectively (entries 14 and 18).
This difference in yields between chloro- and bromocyanodienes became even larger when the acetyl-protected enal (16Ar)c was olefinated to (4Ar)cCl (11%) and (4Ar)cBr (65%) (entries 2 and 3). A similar trend of increasing yield was observed for the olefination of acetylated (16En)c to (4En)cBr (99%) (entry 12).
Synthesis of borrelidin analogues 2
In order to optimize the reaction conditions for the envisioned cross coupling of fragments 3 and 4 we first studied the reaction of bromocyanodienes (4)Br with isobutyraldehyde 23 as a model substrate for fragment 3 (Table 3).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Diene | Method | Product | Yieldb (%) | drc | 25 | Yieldb (%) |
| a Reaction conditions: 4 (1.0 equiv.), 23 (1.0 equiv.), SmI2 (29 equiv.), DMPU (19 equiv.); iPrMgBr (1.1 equiv.). b Isolated yield. c Determined by integration in 1H NMR spectra. | |||||||
| 1 | (4Ar)cBr | A | (24Ar)c | 18 | 66:34 |
— | — |
| 2 | (4Ar)cBr | B | (24Ar)c | 28 | 52:48 |
(25Ar)c | 41 |
| 3 | (4En)bBr | B | (24En)b | 37 | 83:17 |
— | — |
| 4 | (4En)cBr | A | (24En)c | 8 | 67:33 |
— | — |
| 5 | (4Cy)bBr | B | (24Cy)b | 48 | 89:11 |
(25Cy)b | 28 |

According to Omura's conditions,13b the aryl-substituted acetoxycyanodiene (4Ar)cBr was treated with aldehyde 23 and a large excess of SmI2 (29 equiv.) in the presence of DMPU instead of HMPA in THF (method A). The secondary alcohol (24Ar)c could be isolated, albeit in a low yield of 18% (dr 66:34) (entry 1). In contrast, treatment of (4Ar)cBr with iPrMgBr in THF according to Iqbal's method,24 followed by addition of 23 (method B) slightly increased the yield to 28% (dr 52:48), but alcohol (24Ar)c was accompanied by a byproduct (25Ar)c being isolated in 41% as an E/Z mixture (11:89) (entry 2). Its presence indicated that the initial attack of the isopropyl Grignard to fragment (4Ar)cBr and subsequent halogen metal exchange had indeed taken place, but subsequent addition of the in situ formed Grignard seemed to be too slow and thus, hydrolysis of the cyanodiene Grignard species occurred during workup giving byproduct (25Ar)c. Under both Grignard and SmI2 conditions, cyanodienes (4En)bBr and (4En)cBr reacted to alcohols (24En)b and (24En)c which were isolated in low yields of 37% and 8%, respectively (entries 3 and 4). The Grignard addition of the cyclohexyl-substituted fragment (4Cy)bBr gave alcohol (24Cy)b in 48% yield (dr 89:11) together with 28% of the dehalogenated byproduct (25Cy)b (entry 5). The results in Table 3 revealed that intermolecular cross coupling could be achieved under both SmI2 and Grignard mediated conditions, however, with less satisfactory yields for the SmI2 method. We thus anticipated a poorer performance of SmI2 in the intramolecular cyclization compared to the Grignard reaction.
Keeping the obtained results in mind we continued with the synthesis of the macrocyclic borrelidin analogues 2 as outlined in Scheme 6. Homoallylic alcohols (5Ar)a and (5En)a were esterified under Yamaguchi conditions41 with the northern fragment 3 prepared in 99% yield by tetrahydropyranylation38b of 11-hydroxyundecanoic acid42 to yield the corresponding carboxylates (5Ar)d and (5En)d in 86% and 91%, respectively. Cross metathesis with crotonaldehyde 15 in the presence of the Grubbs II catalyst under the optimized conditions provided enals (16Ar)d and (16En)d in 81% and 54% yield, respectively. The HWE reaction between the latter and bromocyanophosphonate 22b gave the olefination products (4Ar)dBr and (4En)dBr in 59% and 42%, respectively. The decreased yields might be caused by a lower solubility of the less polar precursors (16Ar)d and (16En)d in acetonitrile compared to the model compound (16Ar)c having a simple acetate protecting group. THP deprotection with PPTS in EtOH proceeded uneventfully to afford alcohols (26Ar) and (26En) in 84% and 93% yield, which were subsequently oxidized with TPAP under Ley conditions43 to yield aldehydes (27Ar) and (27En) in 88% and 78%, respectively, the key intermediates of the intramolecular cross coupling. The intermolecular cross coupling conditions (Table 3) were transferred to the intramolecular coupling reaction.
 | ||
Scheme 6 Synthesis of borrelidin analogues (2Ar) and (2En). For attempts to assign the stereochemistry of the terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in derivative (4En)dBr see the ESI.† C double bond in derivative (4En)dBr see the ESI.† | ||
The SmI2 mediated Reformatsky-type macrocyclization of (27Ar) under high dilution according to Omura (method A) failed to give the desired macrocycle (2Ar). Only the open-chain cyanodiene (25Ar)e (R1 = HO(CH2)10CO–, see also Table 3 and ESI†), resulting from SmI2 induced reduction of the aldehyde moiety to the primary alcohol and simultaneous reductive debromination, was isolated in 27%. In contrast, the cross coupling of (27Ar) with iPrMgBr in THF (method B) by the Grignard reaction succeeded. The amount of iPrMgBr, however, was increased to 2 equivalents and the reaction time for halogen metal exchange prolonged to 1 h. After stirring at room temperature for 36 h followed by aqueous workup, the desired borrelidin analogue (2Ar) was isolated in 11% yield. In the case of cyclohexenyl-derived compound (27En), only small amounts of the target macrocycle (2En) were detected after 36 h reaction time under similar conditions. However, reaction time extension to 9 days resulted in the macrocyclic target compound (2En) in 11% yield.
We assume that the northern fragment 3 might be responsible for the moderate yields of the Grignard induced macrocyclization and the complete failure of SmI2 promoted intramolecular reactions of aldehydes 27. In comparison, Omura obtained 60% of the desired macrocycle with the 1,3,5,7-tetramethylated northern fragment in the case of the parent borrelidin synthesis.13 These results might arise from the different conformations of the non-branched and the methyl-branched fragments. In order to investigate this aspect in detail, we have performed quantum-chemical calculations based on density functional theory (B3LYP/cc-pVDZ) for (27Ar). Out of a large number of conformers, we isolated 27A and 27B as shown in Fig. 1.
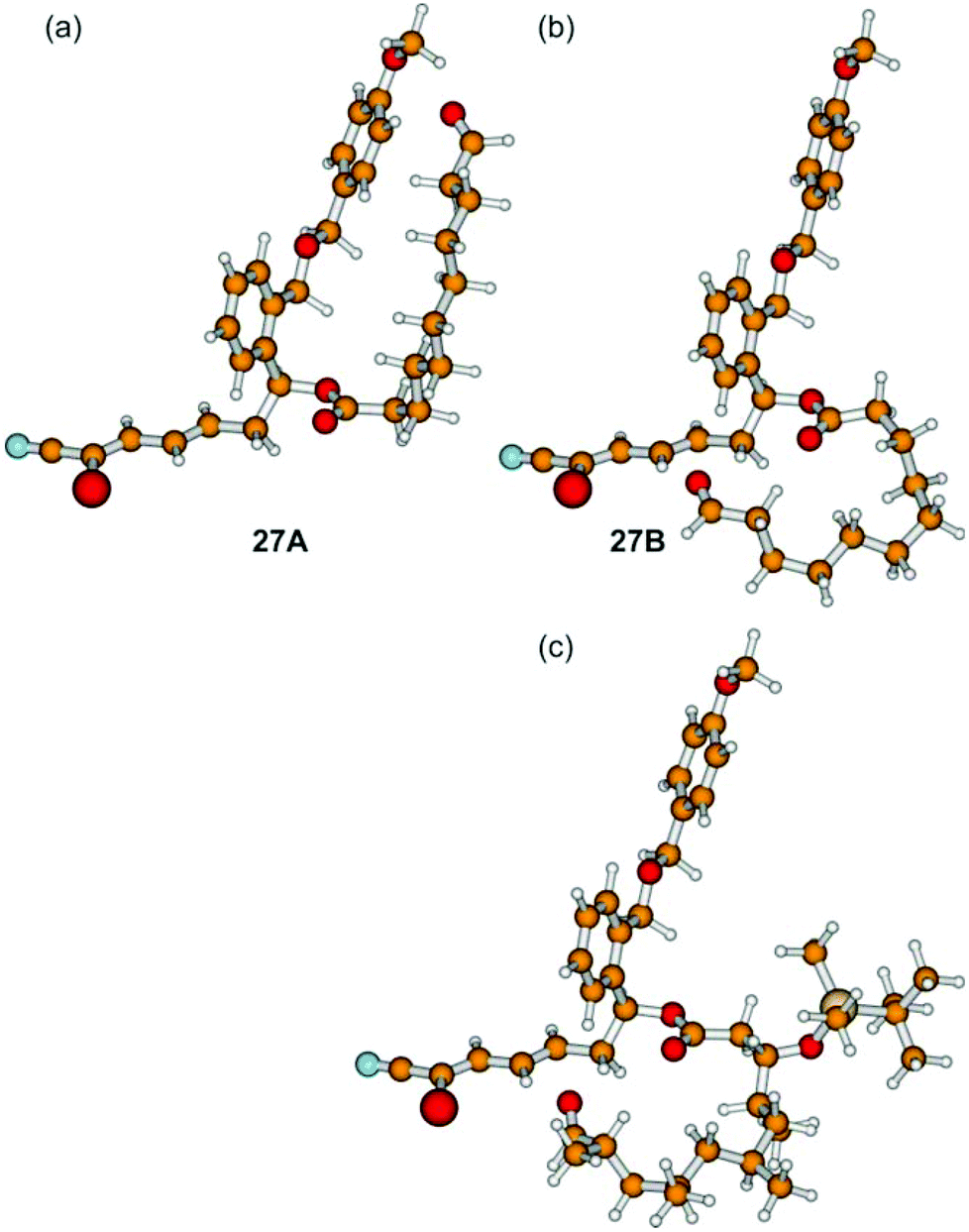 | ||
| Fig. 1 Different conformations of (27Ar) (a, b). Conformer 27A is about 2.6 kcal mol−1 lower in energy than 27B (B3LYP/cc-pVDZ) (for further conformers see the ESI†). For comparison the methyl-branched borrelidin analogue of 27B is depicted (c). | ||
After correction for the zero-point vibrational energy, conformer 27A was found to be 2.6 kcal mol−1 lower in energy than conformer 27B, which constitutes a possible precursor to the cyclization reaction. As slight distortions in the 27B structure led to conformer 27A, the barrier between these two conformers must be very low. The opposite was found for the methyl-branched system: distortions of the analogue of conformer 27B did not lead to the analogue of conformer 27A, but the system was always trapped in the local minimum of the structure of the 27B analogue. As a consequence, it is the occurrence of a multitude of conformations (including those with a linear northern fragment), which are lower in energy than the precursor 27B shown in Fig. 1, and which are connected by low barriers, being responsible for low yields and/or long reaction times.
Biological studies of cyanodienes and borrelidin analogues 2
The ability of a set of cyanodiene derivatives 4, 24, 26, and borrelidin analogues (2Ar) and (2En) to inhibit the proliferation of the L-929 mouse fibroblast cell line was examined using the WST-1 cytotoxicity assay according to the protocol given by the manufacturer.44 The results are summarized in Table 4.|
|
|||||
|---|---|---|---|---|---|
| Entrya | Compound | IC50 (μM) | Entrya | Compound | IC50 (μM) |
| a IC50 values were calculated by fitting the concentration dependence of the signals from the WST-1 cytotoxicity assay44 with the 4-parameter equation and are given as mean ± S.D. of four replicates. | |||||
| 1 | Borrelidin 1 | 0.49 ± 0.19 | 11 | (4En)cBr | 27.1 ± 1.5 |
| 2 | (2Ar) | >50 | 12 | (4Cy)a′Cl | 12.9 ± 2.7 |
| 3 | (2En) | >50 | 13 | (4Cy)aBr | 16.9 ± 3.4 |
| 4 | (4Ar)aBr | 25.0 ± 0.7 | 14 | (4Cy)bCl | >50 |
| 5 | (4Ar)cCl | 37.6 ± 10.8 | 15 | (4Cy)bBr | 37.8 ± 5.1 |
| 6 | (4Ar)cBr | >50 | 16 | (24Ar)c | — |
| 7 | (4En)aCl | 23.3 ± 7.9 | 17 | (24En)b | 16.8 ± 4.7 |
| 8 | (4En)aBr | 20.5 ± 9.6 | 18 | (24Cy)b | 14.5 ± 4.4 |
| 9 | (4En)bCl | — | 19 | (26Ar) | 22.3 ± 9.8 |
| 10 | (4En)bBr | — | 20 | (26En) | 16.4 ± 3.9 |
As can be seen from the data in Table 4, the studied derivatives 4, 24, and 26 generally revealed only moderate cytotoxicity against the L-929 mouse fibroblast cell line with IC50 values in the range of approximately 13–38 μM (entries 4, 5, 7, 8, 11–13, 15 and 17–20), which are approximately 2 orders of magnitude higher than the IC50 value of borrelidin itself (entry 1).
The cyclohexane-based fragment (4Cy)a′Cl with the free hydroxy group was the most active one in this series (IC50 12.9 ± 2.7 μM). The presence of 11-hydroxyundecanecarboxylate in compound (26Ar) seems to influence the cytotoxic activity when compared with acetyl-protected (4Ar)cBr (entries 6 and 19), as only (26Ar) displayed potency against the cell line (IC50 22.3 ± 9.8 μM). This effect was less pronounced for the corresponding cyclohexene-based derivatives (4En)cBr and (26En) (entries 11 and 20). Also the activity of cyanodienes 24 appeared to depend on the O-protecting group. While acetyl-protected (24Ar)c was inactive, TBS-ethers (24En)b and (24Cy)b revealed IC50 values between 15 and 17 μM, respectively (entries 16–18). Both borrelidin analogues (2Ar) and (2En) were inactive (entries 2 and 3) showing the relevance of the residue at C-17 for the biological activity of intact borrelidin. However, when only the southern fragment is present, the cytotoxicities seem to be only slightly affected by the kind of the six-membered ring system.
It should be noted that Sugawara et al. reported a significant decrease of cytotoxicity in the human diploid embryonic cell line MRC-5 when carboxylic acid at C-22 in borrelidin (1) was replaced by a methylester.9a Furthermore, the cytotoxic activity was almost lost upon acetylation of the 3-OH and 11-OH groups of 1. Although comparison of these data should be handled with great care and taking into consideration that the mode of action of these analogues and the natural product might be different due to significant structural differences, the results in Table 4 suggest that an unbiased halogenocyanodiene moiety apparently favors cytotoxicity, whereas the macrocyclic ring deteriorates the activity (e.g. compare entries 2, 4 and 19). The increase of biological activity of borrelidin (1) by one order of magnitude upon hydrogenation of the C12–C15 diene moiety9a indicates that the contribution of the cyanodiene unit to the biological mode of action as well as the role of the northern fragment and the interplay between the northern and the southern fragment need to be further studied.
Conclusion
We have prepared a dedicated library of cyanodienes employing a sequence of cross metathesis and HWE olefination as key steps while alternative approaches met with little success. Cyanodienes were designed as structural analogues of the southern fragment of borrelidin (1). Attempts to cyclize cyanodienes (27Ar) and (27En) to the corresponding macrocyclic borrelidin analogues (2Ar) and (2En) by using Omura's SmI2 method were not successful. However, this key transformation could be achieved by Grignard cross coupling providing the borrelidin analogues (2Ar) with an aryl side chain at C17 and the corresponding analogue (2En) with a cyclohexenyl side chain at C17 in 6 steps starting from fragments (5Ar)a and (5En)a. Quantum-chemical calculations were performed for (27Ar) to interpret the macrocyclization. The calculation suggests that the northern fragment, i.e. the 11-hydroxyundecanecarboxylate, makes the macrocyclization entropically less favorable due to a multitude of flexible conformations which are connected by low barriers. The opposite was found for the highly methyl-branched fragment in the natural product 1. For the latter a helical conformation has already been reported11,15a which seems to facilitate the macrocyclization. SAR studies employing the L-929 mouse fibroblast cell line indicate a higher cytotoxicity for unbiased cyanodienes 4, 24, and 26 than for the truncated borrelidin derivatives 2. Future work must demonstrate whether more rigid northern fragments are beneficial with regard to both macrocycle formation and biological activity.Acknowledgements
Generous financial support by the Dr Eugen Ebert Stiftung (fellowship for T. G.-D.), the Ministerium für Wissenschaft, Forschung und Kunst des Landes Baden-Württemberg, the Fonds der Chemischen Industrie and the GDCh (travel grant for T. G.-D.) is gratefully acknowledged. The authors gratefully acknowledge the technical assistance of Yasmin Wenzel (AG COPS, HZI) and the support of Dr Aruna Raja (Dept. CBIO, HZI) with the biological profiling of the compounds.Notes and references
- Reviews: (a) S. M. Dalby and I. Paterson, Curr. Opin. Drug Discovery Dev., 2010, 13, 777–794 CAS; (b) C. Hertweck, Angew. Chem., 2009, 121, 4782–4811 ( Angew. Chem., Int. Ed. , 2009 , 48 , 4688–4716 ) CrossRef.
- J. Berger, L. M. Jampolsky and M. W. Goldberg, Arch. Biochem., 1949, 22, 476–478 CAS.
- Review: T. Nagamitsu, Y. Harigaya and S. Omura, Proc. Jpn. Acad., Ser. B, 2005, 81, 244–256 CrossRef CAS.
- (a) T. C. Schüz and H. Zähner, FEMS Microbiol. Lett., 1993, 114, 41–45 CrossRef; (b) H. Maehr and R. H. Evans, J. Antibiot., 1987, 40, 1455–1456 CrossRef CAS PubMed; (c) S. K. Singh, S. Gurusiddaiah and J. W. Whalen, Antimicrob. Agents Chemother., 1985, 27, 239–245 CrossRef CAS PubMed; (d) M. Lumb, P. E. Macey, J. Spyvee, J. M. Whitmarsh and R. D. Wright, Nature, 1965, 206, 263–265 CrossRef CAS PubMed.
- L. Dickinson, A. J. Griffiths, C. G. Mason and R. F. N. Mills, Nature, 1965, 206, 265–268 CrossRef CAS PubMed.
- (a) T. Kawamura, D. Liu, M. J. Towle, R. Kageyama, N. Tsukahara, T. Wakabayashi and B. A. Littlefield, J. Antibiot., 2003, 56, 709–715 CrossRef CAS PubMed; (b) T. Wakabayashi, R. Kageyama, N. Naruse, N. Tsukahara, Y. Funahashi, K. Kitoh and Y. Watanabe, J. Antibiot., 1997, 50, 671–676 CrossRef CAS PubMed.
- E. Tsuchiya, M. Yukawa, T. Miyakawa, K. Kimura and H. Takahashi, J. Antibiot., 2001, 54, 84–90 CrossRef CAS PubMed.
- W. Paetz and G. Nass, Eur. J. Biochem., 1973, 35, 331–337 CrossRef CAS PubMed.
- (a) A. Sugawara, T. Tanaka, T. Hirose, A. Ishiyama, M. Iwatsuki, Y. Takahashi, K. Otoguro, S. Omura and T. Sunazuka, Bioorg. Med. Chem. Lett., 2013, 23, 2302–2305 CrossRef CAS PubMed; (b) K. Otoguro, H. Ui, A. Ishiyama, M. Kobayashi, H. Togashi, Y. Takahashi, R. Masuma, H. Tanaka, H. Tomoda, H. Yamada and S. Omura, J. Antibiot., 2003, 56, 727–729 CrossRef CAS PubMed; (c) K. Otoguro, A. Ishiyama, H. Ui, M. Kobayashi, C. Manabe, G. Yan, Y. Takahashi, H. Tanaka, H. Yamada and S. Omura, J. Antibiot., 2002, 55, 832–834 CrossRef CAS PubMed; (d) K. Otoguro, A. Kohana, C. Manabe, A. Ishiyama, H. Ui, K. Shiomi, H. Yamada and S. Omura, J. Antibiot., 2001, 54, 658–663 CrossRef CAS PubMed.
- M. O. Duffey, A. LeTiran and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 1458–1459 CrossRef CAS PubMed.
- (a) S. Hanessian, S. Giroux and V. Mascitti, Synthesis, 2006, 1057–1076 CrossRef CAS; (b) S. Hanessian, N. Chahal and S. Giroux, J. Org. Chem., 2006, 71, 7403–7411 CrossRef CAS PubMed; (c) S. Hanessian, Y. Yang, S. Giroux, V. Mascitti, J. Ma and F. Raeppel, J. Am. Chem. Soc., 2003, 125, 13784–13792 CrossRef CAS PubMed.
- (a) B. G. Vong, S. H. Kim, S. Abraham and E. A. Theodorakis, Angew. Chem., 2004, 116, 4037–4041 ( Angew. Chem., Int. Ed. , 2004 , 43 , 3947–3951 ) CrossRef; (b) B. G. Vong, S. Abraham, A. X. Xiang and E. A. Theodorakis, Org. Lett., 2003, 5, 1617–1620 CrossRef CAS PubMed.
- (a) T. Nagamitsu, D. Takano, K. Marumoto, T. Fukuda, K. Furuya, K. Otoguro, K. Takeda, I. Kuwajima, Y. Harigaya and S. Omura, J. Org. Chem., 2007, 72, 2744–2756 CrossRef CAS PubMed; (b) T. Nagamitsu, D. Takano, T. Fukuda, K. Otoguro, I. Kuwajima, Y. Harigaya and S. Omura, Org. Lett., 2004, 6, 1865–1867 CrossRef CAS PubMed.
- (a) A. V. R. Madduri and A. J. Minnaard, Chem. – Eur. J., 2010, 16, 11726–11731 CrossRef CAS PubMed; (b) F. López, S. R. Harutyunyan, A. Meetsma, A. J. Minnaard and B. L. Feringa, Angew. Chem., 2005, 117, 2812–2816 ( Angew. Chem., Int. Ed. , 2005 , 44 , 2752–2756 ) CrossRef; (c) R. Des Mazery, M. Pullez, F. López, S. R. Harutyunyan, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 9966–9967 CrossRef CAS PubMed.
- (a) M. Theurer, Y. El Baz, K. Koschorreck, V. B. Urlacher, G. Rauhut, A. Baro and S. Laschat, Eur. J. Org. Chem., 2011, 4241–4249 CrossRef CAS; (b) J. S. Yadav, P. Bezawada and V. Chenna, Tetrahedron Lett., 2009, 50, 3772–3775 CrossRef CAS; (c) C. Herber and B. Breit, Chem. – Eur. J., 2006, 12, 6684–6691 CrossRef CAS PubMed; (d) T. Novak, Z. Tan, B. Liang and E.-I. Negishi, J. Am. Chem. Soc., 2005, 127, 2838–2839 CrossRef CAS PubMed; (e) N. Haddad, M. Grishko and A. Brik, Tetrahedron Lett., 1997, 38, 6075–6078 CrossRef CAS; (f) N. Haddad, A. Brik and M. Grishko, Tetrahedron Lett., 1997, 38, 6079–6082 CrossRef CAS.
- F. Hahn, N. Kandziora, S. Friedrich and P. F. Leadlay, Beilstein J. Org. Chem., 2014, 10, 634–640 CrossRef PubMed.
- B. Wilkinson, M. A. Gregory, S. J. Moss, I. Carletti, R. M. Sheridan, A. Kaja, M. Ward, C. Olano, C. Mendez, J. A. Salas, P. F. Leadlay, R. vanGinckel and M.-Q. Zhang, Bioorg. Med. Chem. Lett., 2006, 16, 5814–5817 CrossRef CAS PubMed.
- (a) J. Woolard, W. Vousden, S. J. Moss, A. Krishnakumar, M. V. R. Gammons, D. G. Nowak, N. Dixon, J. Micklefield, A. Spannhoff, M. T. Bedford, M. A. Gregory, C. J. Martin, P. F. Leadlay, M. Q. Zhang, S. J. Harper, D. O. Bates and B. Wilkinson, Chem. Sci., 2011, 2, 273–278 RSC; (b) S. J. Moss, I. Carletti, C. Olano, R. M. Sheridan, M. Ward, V. Math, M. Nur-E-Alam, A. F. Braña, M. Q. Zhang, P. F. Leadlay, C. Méndez, J. A. Salas and B. Wilkinson, Chem. Commun., 2006, 2341–2343 RSC; (c) C. Olano, B. Wilkinson, C. Sánchez, S. J. Moss, R. Sheridan, V. Math, A. J. Weston, A. F. Braña, C. J. Martin, M. Oliynyk, C. Méndez, P. F. Leadlay and J. A. Salas, Chem. Biol., 2004, 11, 87–97 CAS; (d) C. Olano, B. Wilkinson, S. J. Moss, A. F. Braña, C. Méndez, P. F. Leadlay and J. A. Salas, Chem. Commun., 2003, 2780–2782 RSC.
- C. J. Schulze, W. M. Bray, F. Loganzo, M.-H. Lam, T. Szal, A. Villalobos, F. E. Koehn and R. G. Linington, J. Nat. Prod., 2014, 77, 2570–2574 CrossRef CAS PubMed.
- A. Sidhu, J. R. Miller, A. Tripathi, D. M. Garshott, A. L. Brownell, D. J. Chiego, C. Arevang, Q. Zeng, L. C. Jackson, S. A. Bechler, M. U. Callaghan, G. H. Yoo, S. Sethi, H.-S. Lin, J. H. Callaghan, G. Tamayo-Castillo, D. H. Sherman, R. J. Kaufman and A. M. Fribley, ACS Med. Chem. Lett., 2015, 6, 1122–1127 CrossRef CAS PubMed.
- (a) E. A. Crane and K. Gademann, Angew. Chem., Int. Ed., 2016, 55, 3882–3902 CrossRef CAS PubMed; (b) J.-Y. Wach and K. Gademann, Synlett, 2012, 163–170 CAS.
- M. E. Maier, Org. Biomol. Chem., 2015, 13, 5302–5343 CAS.
- K. Tanaka, H. Matsuyama, M. Watanabe, Y. Fujimori, K. Ishibashi, T. Ozawa, T. Sato, Y. Saikawa and M. Nakata, J. Org. Chem., 2014, 79, 9922–9947 CrossRef CAS PubMed.
- (a) C. V. Krishna, V. R. Bhonde, A. Devendar, S. Maitra, K. Mukkanti and J. Iqbal, Tetrahedron Lett., 2008, 49, 2013–2017 CrossRef CAS; (b) C. V. Krishna, S. Maitra, R. V. Dev, K. Mukkanti and J. Iqbal, Tetrahedron Lett., 2006, 47, 6103–6106 CrossRef CAS.
- M. Jay-Smith, D. P. Furkert, J. Sperry and M. A. Brimble, Synlett, 2011, 1395–1398 CAS.
- Y. Ueno, Y. Watanabe, A. Shibata, T. Yoshikawa, T. Takano, M. Kohara and Y. Kitade, Bioorg. Med. Chem., 2009, 17, 1974–1981 CrossRef CAS PubMed.
- (a) A. Marx and H. Yamamoto, Angew. Chem., 2000, 112, 182–184 ( Angew. Chem., Int. Ed. , 2000 , 39 , 178–181 ) CrossRef; (b) A. Hosomi and H. Sakurai, Tetrahedron Lett., 1976, 16, 1295–1298 CrossRef; (c) A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1976, 5, 941–942 CrossRef.
- J. de Mier-Vinué, A. M. Montaña, V. Moreno and M. J. Prieto, Z. Anorg. Allg. Chem., 2005, 631, 2054–2061 CrossRef.
- (a) T. P. Hilditch, J. Chem. Soc. Trans., 1909, 95, 1570–1577 RSC; (b) A. E. Martin, T. M. Ford and J. E. Bulkowski, J. Org. Chem., 1982, 47, 412–415 CrossRef CAS; (c) M. Ouchi, Y. Inoue, Y. Liu, S. Nagamune, S. Nakamura, K. Wada and T. Hakushi, Bull. Chem. Soc. Jpn., 1990, 63, 1260–1262 CrossRef CAS.
- (a) A. Misumi, K. Iwanaga, K. Furuta and H. Yamamoto, J. Am. Chem. Soc., 1985, 107, 3343–3345 CrossRef CAS; (b) O. Fujimura, F. J. de la Mata and R. H. Grubbs, Organometallics, 1996, 15, 1865–1871 CrossRef CAS.
- A. Weickgenannt, J. Mohr and M. Oestreich, Tetrahedron, 2012, 68, 3468–3479 CrossRef CAS.
- R. D. Crouch, Synth. Commun., 2013, 43, 2265–2279 CrossRef CAS.
- Y. Zhang, C. C. Arpin, A. J. Cullen, M. J. Mitton-Fry and T. Sammakia, J. Org. Chem., 2011, 76, 7641–7653 CrossRef CAS PubMed.
- (a) H. Fuwa, H. Yamaguchi and M. Sasaki, Org. Lett., 2010, 12, 1848–1851 CrossRef CAS PubMed; (b) H. Fuwa, A. Saito and M. Sasaki, Angew. Chem., 2010, 122, 3105–3108 ( Angew. Chem., Int. Ed. , 2010 , 49 , 3041–3044 ) CrossRef.
- Review: Y. A. Lin and B. G. Davis, Beilstein J. Org. Chem., 2010, 6, 1219–1228 CrossRef CAS PubMed.
- A. H. Hoveyda, P. J. Lombardi, R. V. O'Brien and A. R. Zhurgralin, J. Am. Chem. Soc., 2009, 131, 8378–8379 CrossRef CAS PubMed.
- D. Prajapati, K. C. Lekhok, J. S. Sandhu and A. C. Ghosh, J. Chem. Soc., Perkin Trans. 1, 1996, 959–960 RSC.
- (a) R. Csuk, U. Höring and M. Schaade, Tetrahedron, 1996, 52, 9759–9776 CrossRef CAS; (b) N. Ravindranath, C. Ramesh and B. Das, Synlett, 2001, 1777–1778 CrossRef CAS.
- (a) R. A. Aitken, N. A. Al-Awadi, O. M. E. El-Dusouqui, D. M. M. Farrell and A. Kumar, Int. J. Chem. Kinet., 2006, 38, 496–502 CrossRef CAS; (b) H. J. Bestmann and M. Schmidt, Angew. Chem., 1987, 99, 64–65 ( Angew. Chem., Int. Ed. Engl. , 1987 , 26 , 79–81 ) CrossRef CAS.
- B. Iorga, L. Ricard and P. Savignac, J. Chem. Soc., Perkin Trans. 1, 2000, 3311–3316 RSC.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef CAS.
- D. Y. Duveau, P. M. Arce, R. A. Schoenfeld, N. Raghav, G. A. Cortopassi and S. M. Hecht, Bioorg. Med. Chem., 2010, 18, 6429–6441 CrossRef CAS PubMed.
- S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 639–666 CrossRef.
- M. Ishiyama, M. Shiga, K. Sasamoto, M. Mizoguchi and P. He, Chem. Pharm. Bull., 1993, 41, 1118–1122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures and syntheses of the described derivatives, preliminary studies on olefination reactions, NMR studies on the stereochemistry of the terminal double bond in bromocyanodiene (4En)dBr, quantum-chemical calculation of (27Ar) as well as 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c6ob01358a |
| This journal is © The Royal Society of Chemistry 2016 |