
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA short and modular approach towards 3,5-disubstituted indolizidine alkaloids†
Marco M.
Nebe
,
Sina
Zinn
and
Till
Opatz
*
Institute of Organic Chemistry, University of Mainz, Duesbergweg 10-14, D-55128 Mainz, Germany. E-mail: opatz@uni-mainz.de
First published on 28th June 2016
Abstract
3,5-Dialkyl indolizidines have been prepared in four linear steps from commercially available starting materials. The sequence involves two direct α-functionalization steps and a subsequent reductive amination and provides diastereoselective access to both C-3 epimers of the 5,9-trans-substituted indolizines. The naturally occurring indolizidines 195B and 223AB have been synthesized using this methodology.
Introduction
The indolizidine framework is part of a large number and variety of natural products which occur in microorganisms, plants, terrestrial animals, as well as marine creatures.1 Indolizidine alkaloids from terrestrial animals were essentially isolated from arthropods, i.e. ants and mites, as well as amphibians,2,3 the latter of which supposedly consume them via their arthropod prey (“dietary hypothesis”).4 An important class of these indolizidine alkaloids represented by at least 30 compounds, are the 3,5-disubstituted indolizidines, among which (+)-monomorine I (1, Fig. 1), a trail pheromone of the pharaoh ant, has received particular attention.2,5 Numerous synthetic approaches towards this class of alkaloids have been reported, most of which target a 5,9-cis-geometry, in which the residues at C-5 and C-9 occupy the same face of the molecule in a syn-fashion.6–14 This is certainly due to the more frequent occurrence of this relative configuration in nature. There are, however, also several examples of natural indolizidines featuring a 5,9-trans (anti-relation at C-5 and C-9) substitution (Fig. 1),2 and only a few synthetic methods selectively accessing this configuration have been reported so far.15–18 We herein report a four step modular access towards 3,5-disubstituted indolizidines, selectively producing a 5,9-trans substitution pattern. The developed route was applied to the synthesis of the naturally occurring 5,9-trans-dialkylindolizidines 195B and 223AB. In this way the (5E,9Z)- (2a and 3a) as well as the (5Z,9E)-isomers (2b and 3b) could be prepared.19 | ||
| Fig. 1 Absolute stereostructure of (+)-monomorine I (1) and relative stereostructures of 5,9-trans indolizidines 195B (2a and 2b) and 223AB (3a and 3b). | ||
Results and discussion
Our retrosynthetic approach for the synthesis of 3,5-disubstituted indolizidines (A) is shown in Scheme 1. The strategy involves the sequential double α-alkylation of an N-protected piperidine with an iodoalkane and an enone through the α-lithio-derivatives resulting in 2,6-trans-disubstitution (B), followed by an intramolecular reductive amination to form the bicyclic structure. This sequence would allow the formation of indolizidines bearing the desired substitution pattern, in which the substituents R1 and R2 are selected by the choice of iodoalkane C and the aldehyde D employed. | ||
| Scheme 1 Retrosynthetic approach for the synthesis of 3,5-dialkylindolizidines. | ||
The consecutive α-lithiation/alkylation to form 2,6-disubstituted piperidines B has originally been developed by Beak and Hoppe and has been extensively applied since.20–22 The reaction was later extended by Dieter et al. who introduced a transmetalation to copper(I) for the monoalkylation, allowing the utilization of softer electrophiles such as iodoalkanes as well as Michael-acceptors in the subsequent alkylation step (Scheme 2).23 Due to the inherent trans-selectivity of the second alkylation, the less common 2,6-trans-dialkylpiperidines and consequently the resulting 5,9-trans-indolizidines were expected to be the favored products of this sequence.20
 | ||
| Scheme 2 Lithiation/alkylation reactions developed by Beak and Dieter. | ||
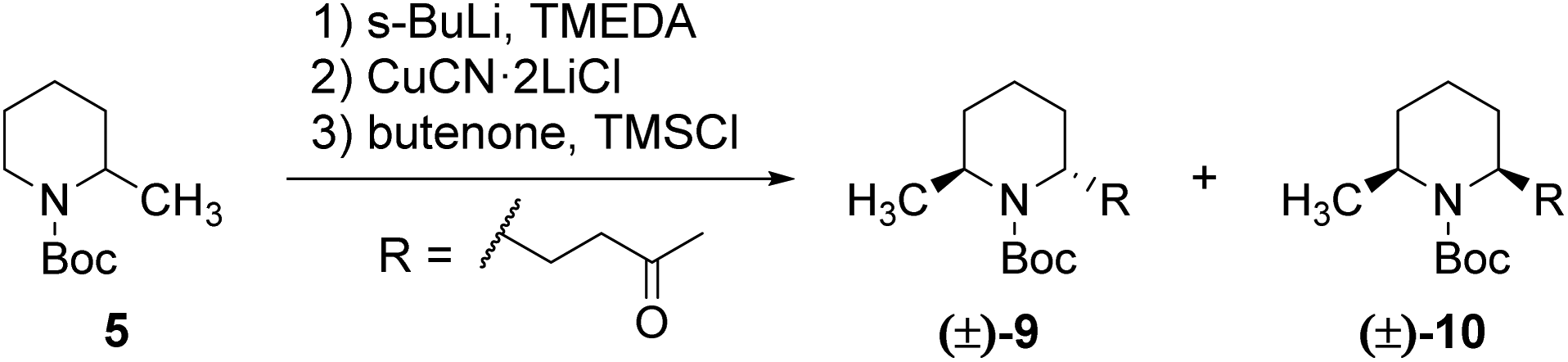
In search for a general protocol for the preparation of 3,5-dialkylindolizidines, we first focused on the model compound 3,5-dimethylindolizidine which should be accessible from commercially available 2-methylpiperidine and butenone (methyl vinyl ketone, MVK). The synthesis of the 2,6-trans-disubstituted piperidine 9 was achieved by a slightly modified protocol of Dieter et al., as displayed in Scheme 3.23 Deprotonation of the N-Boc-protected 2-methylpiperidine with s-BuLi in the presence of TMEDA leads to the formation of the intermediate complex 6, which undergoes Michael-addition to the α,β-unsaturated ketone after transmetalation to copper(I).
 | ||
| Scheme 3 Synthesis of compound 9. (a) Boc2O, THF, 0 °C → r.t., 99%. | ||
Due to steric hindrance imposed by the tert-butyl group in the complex 6, the methyl group adopts an axial position, which leads to the above mentioned 2,6-trans configuration of the product. This was confirmed by a NOESY experiment which shows an intensive cross-peak between the protons of the methyl group at C-6 and the proton at C-2, which corresponds to a 1,3-diaxial arrangement of these substituents. The cis-isomer (10) shows no corresponding NOESY-contact as expected for an axial-equatorial arrangement of H-2 and CH3-6. We observed that the stereoselectivity is highly dependent on the temperature at which the transmetalation is carried out, as well as on the addition-sequence of MVK and TMSCl (see Table 1). While the transmetalation at −50 °C (entry 1), as described by Dieter et al. for the monoalkylation, leads to diastereomeric rations ranging from 2.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 6:1, a ratio of >95:5 was obtained, when all steps were carried out at −78 °C (entry 3). A ratio of 1.8:1 in favor of the cis-isomer was observed when the TMSCl was added 30 minutes after the addition of MVK (entry 2). This could be due to a configurational instability of the intermediate enolate 7 or to a low reactivity of the electrophile resulting in a configurational scrambling of the α-cuprated amine when no scavenger is present. Moreover, no reaction was observed, when the transmetalation-step was omitted (entry 4). It is noteworthy that this reaction exhibits a somewhat poor reproducibility and significant variations from one experiment to another can be observed in terms of yield and diastereomeric ratio. The same was reported by Dieter and coworkers for the corresponding reaction with unsubstituted N-Boc-piperidine. When the reaction parameters are thoroughly controlled, a satisfactory reproducibility can however be achieved and the dr is usually in excess of 95:5.
:1 to 6:1, a ratio of >95:5 was obtained, when all steps were carried out at −78 °C (entry 3). A ratio of 1.8:1 in favor of the cis-isomer was observed when the TMSCl was added 30 minutes after the addition of MVK (entry 2). This could be due to a configurational instability of the intermediate enolate 7 or to a low reactivity of the electrophile resulting in a configurational scrambling of the α-cuprated amine when no scavenger is present. Moreover, no reaction was observed, when the transmetalation-step was omitted (entry 4). It is noteworthy that this reaction exhibits a somewhat poor reproducibility and significant variations from one experiment to another can be observed in terms of yield and diastereomeric ratio. The same was reported by Dieter and coworkers for the corresponding reaction with unsubstituted N-Boc-piperidine. When the reaction parameters are thoroughly controlled, a satisfactory reproducibility can however be achieved and the dr is usually in excess of 95:5.
|
|
||||
|---|---|---|---|---|
| Entry | T t [°C] | t A (min) | Yield [%] | dr (9:10)b |
| a (1) s-BuLi (1.3 eq.), TMEDA (2.2 eq.), Et2O, −78 °C, 4 h, (2) CuCN·2LiCl (0.3 M in THF, 1 eq.), Tt, 1 h, (3) butenone (1 eq.), tA, TMSCl (5 eq.), −78 °C–r.t. b Determined by 1H-NMR-spectroscopy. c Isolated yield. d Reaction performed without the transmetalation step. | ||||
| 1 | −50 | 0 | 42c | 2.6:1–6:1 |
| 2 | −50 | 30 | 39b | 1:1.8 |
| 3 | −78 | 0 | 61c | >95:5 |
| 4d | — | 0 | no conv. | — |
The conversion of 9 to the corresponding indolizidine was carried out by acidic deprotection with subsequent reductive amination in situ (Scheme 4).
 | ||
| Scheme 4 Synthesis of 3,5-dimethyloctahydroindolizinium picrates 12a and 12b. | ||
Addition of acetyl chloride to an ethanolic solution of 9 leads to the formation of HCl, which efficiently cleaved the Boc-protecting group. Subsequent adjustment to pH 4–5 by addition of an acetate buffer facilitated the cyclization to the iminium ion 11, which could be monitored via LC-MS. The reduction to the tertiary amine was achieved by addition of sodium cyanoborohydride. Due to the high volatility of the tertiary amine, an isolation on a small scale was somewhat troublesome, hence crystallization as the picrate salt was performed. In this way, the indolizidine was isolated in 55% yield as a 1:1.2 mixture of the two C-3 epimers. The relative configuration could again be determined by NOESY-experiments which showed intensive cross-peaks between the proton at C-9 and the proton ((5E,9Z), 12a) or the methyl group ((5Z,9E), 12b) at C-3, respectively. Switching of the reducing agent to sodium triacetoxyborohydride surprisingly did not lead to the reduction of the intermediate iminium ion while upon treatment with hydrogen and Pd/C, only an incomplete conversion and a yield of 13% were obtained.
The optimized reaction sequence was then applied to the synthesis of (5E,9Z)- and (5Z,9E)-indolizidines 195B (2a, 2b) and 223AB (3a, 3b). The starting materials required for the desired substitution patterns of the indolizidines were prepared according to known procedures (Scheme 5). Boc-2-propylpiperidine (Boc-coniine, 15) was obtained in 58% yield similarly to 9 by a deprotonation/alkylation sequence of Boc-piperidine (14) with 1-iodopropane, based on a procedure described by Pizzuti et al. (Scheme 5).24 Hept-1-en-3-one (17) was prepared in two Steps via a Grignard addition with subsequent oxidation from valeraldehyde (16) and vinyl magnesium bromide.25,26 Deprotonation of the 2-substituted N-Boc-piperidines 5 and 15 with subsequent Michael-addition to hept-1-en-3-one (17) selectively furnished the respective 2,6-trans-disubstituted piperidines 18 and 19 in 49% and 48% yield (Scheme 6). The 2,6-trans-configuration could again be verified by NOESY. Reductive amination with sodium cyanoborohydride produced the two C-3 epimers of indolizidines 2 and 3 in 78% and 56% combined yield, respectively.
 | ||
| Scheme 5 Synthesis of Boc-coniine (15) and hept-1-en-3-one (17). (a) Boc2O, DCM, r.t., 94%. | ||
 | ||
| Scheme 6 Synthesis of (5E,9Z) and (5Z,9E) diastereomers of indolizidines 195B (2) and 223AB (3). | ||
As for the model compound, the isolation and purification of these alkaloids was somewhat challenging due to the still substantial volatility of these compounds, as well as their sensitivity towards aerial oxygen. When a purification by crystallization as described for picrate salts 12a and 12b was attempted, no precipitation of the respective salts could be accomplished upon the addition of picric acid. The same applied to acetic acid, trifluoroacetic acid, as well as HBr and HCl. Although chromatographic purification could be achieved on an NH2-functionalized silica gel, a successful separation of the two diastereomers was only achieved for indolizidines 2a and 2b. Chromatographic separation on silica gel or aluminum oxide as described in the literature was unsuccessful since no compound could be recovered from the column.27,28
The NMR-spectra of the isolated compounds were in agreement with the literature and the relative configuration could thus easily be assigned.16,27–29 As for compound 12, the (5E,9Z)-diastereomer was slightly favored in both cases over the (5Z,9E)-diastereomer with selectivities of 2.3:1 and 1.7:1, respectively.
Conclusion
In summary, a highly modular synthetic protocol for the preparation of 3,5-dialkylindolizidines in only four linear steps from commercially available starting materials was developed. The nature of the two alkyl moieties is solely dependent on the alkyl iodide and aldehyde employed. The sequence involves two direct α-metalation steps of Boc-piperidine with a subsequent reductive amination. Due to the trans-selectivity of the second metalation, 5,9-trans-indolizidines were obtained selectively. To the best of our knowledge, no shorter access to this compound class bearing the indicated stereochemistry has been reported so far.Experimental section
General methods
All reactions including air or moisture sensitive compounds were carried out under an inert atmosphere of argon in flame- or oven dried glassware. sec-Butyllithium was purchased in solution (1.3 M in cyclohexane/n-hexane 92/8) from a commercial supplier and titrated against N-benzylbenzamide prior to use.30 All other reagents were reagent grade and used as purchased unless otherwise noted. The stated reaction temperatures refer to the temperature of the respective cooling bath. Melting points were determined in open capillary tubes using an electronic apparatus. NMR spectra were recorded on 300 MHz, 400 MHz or 600 MHz spectrometers equipped with a 5 mm BBFO probe head with z-gradient and ATM capability. Chemical shifts were referenced to the deuterated solvent (CDCl3, δ = 7.26 ppm and 77.16 ppm for 1H and 13C NMR, respectively) and reported in parts per million (ppm, δ) relative to tetramethylsilane (TMS, δ = 0.00 ppm).31 Coupling constants (J) are stated in Hz using the splitting abbreviations: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad. Standard pulse sequences were used for the 2D experiments. FT-Infrared spectra were recorded using a diamond ATR unit. High-resolution mass spectrometry was executed on a QToF-Instrument with a dual electrospray source and a suitable external calibrant. Thin-layer chromatography (TLC) was performed on 0.25 mm silica gel plates (60F254) visualizing with UV-light and developing with an anisaldehyde solution (4.1 mL of p-anisaldehyde, 1.7 mL AcOH and 5.6 mL H2SO4 in 450 mL EtOH) or potassium permanganate solution (2.0 g KMnO4 and 5.5 g NaCO3 in 250 mL H2O). Flash chromatography was carried out using the indicated solvent system on 35–70 μm silica gel or KP-NH silica gel from Biotage.General procedure for the preparation of CuCN·2LiCl solution
Lithium chloride (2.0 equiv.) was flame dried under vacuum at 140 °C for at least 4 h. Copper(I)-cyanide (1.0 equiv.) and THF (3.33 mL mol−1, producing a 0.3 M solution) were added under argon and the resulting mixture was stirred until all the solids had completely dissolved (usually about 1 h).General procedure (A) for the Michael-addition of 2 substituted tert-butylpiperidine-1-carboxylates to enones
The respective 2-substituted tert-butylpiperidine-1-carboxylate (1.0 equiv.) was dissolved in dry Et2O (3–5 mL mmol−1) under argon and TMEDA (2.3 equiv.) was added. The resulting solution was cooled to −78 °C and sec-butyllithium (1.2–1.4 equiv.) was added dropwise. The solution was stirred for 4 h at −78 °C, after which a 0.3 M solution of CuCN·2LiCl in THF (3.3 mL mmol−1, 1.0 equiv.) was added slowly. The resulting solution was stirred at −78 °C for 30 min. The respective enone (1.0 equiv.) and TMSCl (5.0 equiv.) were added dropwise and the resulting solution was warmed to ambient temperature overnight. To the resulting black solution was added TBAF (1 M in THF, 5.0 equiv.) at 0 °C and the mixture was stirred for 1 h. The mixture was filtered over a short plug of silica, eluting with hexanes/EtOAc (5:1), the solvents were removed under reduced pressure and the crude product was purified by silica gel column chromatography.
General procedure (B) for the synthesis of indolizidines via deblocking–reductive–amination sequence
The respective 2,6-disubstituted tert-butylpiperidine-1-carboxylate (1.0 equiv.) was dissolved in dry EtOH (ca. 5–15 mL mmol−1) under an inert nitrogen athmosphere. The solution was cooled to 0 °C and acetyl chloride was added portion-wise, generating an ethanolic HCl-solution. Stirring was continued overnight until LC-MS indicated full conversion of the starting material. A 1 M aqueous solution of NaOAc/HOAc (ca. 50 mL mmol−1) was added to adjust the pH to 4–5 and stirring was continued until LC-MS indicated complete formation of the Iminium-Ion (5–20 h). A portion of NaCNBH3 (3–4 equiv.) was added every 12 h, until LC-MS indicated complete reduction of the Iminium Ion. 2 M HCl(aq) (10 mL mmol−1) was added and the emerging HCN was removed by a constant stream of nitrogen (CAUTION!!!). The solution was washed with EtOAc (2 × 20 mL mmol−1) and the organic extracts were back-extracted with 2 M HCl(aq) (2 × 5 mL mmol−1). The combined aqueous phases were set to pH 10 by the addition of 2 M NaOH(aq) and extracted with CH2Cl2 (6 × 15 mL mmol−1). The combined organic extracts were dried over MgSO4 and the solvent was evaporated at a minimum pressure of 500 mbar.tert-Butyl 2-methylpiperidine-1-carboxylate (5)
Di-tert-butyl dicarbonate (10.68 g, 48.93 mmol, 1.0 equiv.) was dissolved in dry THF (50 mL) under argon at 0 °C. 2-Methylpiperidine (5.90 mL, 49.97 mmol, 1.0 equiv.) was added dropwise. The mixture was stirred at 0 °C for 30 min and then at ambient temperature overnight. The mixture was washed with sat. aq. NaHCO3 (50 mL) and the aqueous phase extracted with Et2O (3 × 50 mL). The combined organic extracts were washed with sat. aq. NaHCO3, then brine (50 mL), dried over MgSO4 and the solvent was evaporated under reduced pressure to yield 5 (9.69 g, 48.62 mmol, 99%) as a colorless oil. Rf = 0.23 (SiO2, hexanes/EtOAc 20:1); IR (ATR) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 2934, 1687, 1406, 1364, 1337, 1274, 1172, 1141, 1075, 770; 1H NMR (300 MHz, CDCl3) δ (ppm) = 4.33–4.24 (m, 1H, 2-H), 3.83 (d, br, J = 13.1 Hz, 1H, CH2A-6), 2.72 (td, J = 13.1, 2.8 Hz, 1H, CH2B-6), 1.63–1.19 (m, 6H, 3-H, 4-H, 5-H), 1.37 (s, 9H, C(CH3)3), 1.04 (d, J = 7.0 Hz, 3H, CH3-1′); 13C NMR (75.5 MHz, CDCl3) δ (ppm) = 155.2 (C
(cm−1) = 2934, 1687, 1406, 1364, 1337, 1274, 1172, 1141, 1075, 770; 1H NMR (300 MHz, CDCl3) δ (ppm) = 4.33–4.24 (m, 1H, 2-H), 3.83 (d, br, J = 13.1 Hz, 1H, CH2A-6), 2.72 (td, J = 13.1, 2.8 Hz, 1H, CH2B-6), 1.63–1.19 (m, 6H, 3-H, 4-H, 5-H), 1.37 (s, 9H, C(CH3)3), 1.04 (d, J = 7.0 Hz, 3H, CH3-1′); 13C NMR (75.5 MHz, CDCl3) δ (ppm) = 155.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 79.1 (C(CH3)3), 46.2 (C-2), 38.8 (C-6), 30.2 (C-3), 28.7 (3C, C(CH3)3), 25.8 (C-5), 18.8 (C-4), 15.8 (CH3). The analytical data are in accordance with the literature.32
O), 79.1 (C(CH3)3), 46.2 (C-2), 38.8 (C-6), 30.2 (C-3), 28.7 (3C, C(CH3)3), 25.8 (C-5), 18.8 (C-4), 15.8 (CH3). The analytical data are in accordance with the literature.32
tert-Butyl piperidine-1-carboxylate (14)
Di-tert-butyl dicarbonate (3.70 g, 16.95 mmol, 1.0 equiv.) was dissolved in CH2Cl2 (50 mL) and piperidine (1.85 mL, 18.68 mmol, 1.1 equiv.) was added dropwise. The solution was stirred overnight and the solvent was removed under reduced pressure. Purification by bulb-to-bulb distillation (7 mbar, 150 °C) yielded 14 (2.96 g, 15.99 mmol, 94%) as a colorless oil. Rf = 0.67 (SiO2, cyclohexane/EtOAc 5:1); IR (ATR) (cm−1) = 2976, 2934, 2858, 1690, 1411, 1365, 1268, 1170, 1147; 1H NMR (300 MHz, CDCl3) δ (ppm) = 3.36–3.32 (m, 4H, 2,6-H), 1.60–1.45 (m, 6H, 3,5-H; 4-H), 1.44 (s, 9H, CH3); 13C NMR (75.5 MHz, CDCl3) δ (ppm) = 155.1 (CO), 79.2 (C(CH3)3), 44.9 (br, C-2,6), 28.6 (3C, CH3), 25.9 (C-3,5), 24.6 (C-4). The analytical data are in accordance with the literature.32
Hept-1-en-3-one (17)
A solution of vinylmagnesium bromide (0.7 M, 50 mL, 35.00 mmol, 1.2 equiv.) was added to a solution of pentanal (3.20 mL, 29.74 mmol, 1.0 equiv.) in dry THF (50 mL) under argon at −20 °C. After complete addition, the cooling bath was removed and the mixture was allowed to warm to ambient temperature. After TLC indicated full consumption of the starting material (2 h), 1 M aq. HCl (60 mL) was added and stirring was continued for 10 min. The organic layer was separated and the aqueous phase was extracted with Et2O (3 × 60 mL). The combined organic extracts were washed with brine, dried over MgSO4 and the solvents removed under reduced pressure (40 °C, 100 mbar). Purification by bulb-to-bulb distillation (10–20 mbar, 90–100 °C) yielded hept-1-en-3-ol (2.28 g, 19.97 mmol, 67%) as a colorless liquid. Rf = 0.21 (SiO2, hexanes/EtOAc 5:1); IR (ATR) (cm−1) = 3417, 2957, 2933, 2872, 2864, 1720, 1466, 1379, 1146, 962; 1H NMR (300 MHz, CDCl3) δ (ppm) = 5.86 (ddd, J = 17.1, 10.4, 6.3 Hz, 1H, 2-H), 5.20 (d pseudo-t, Jd = 17.1, Jpseudo-t ≈ 1 Hz, 1H, CH2A-1), 5.09 (d pseudo-t, Jd = 10.4, Jpseudo-t ≈ 1 Hz, 1H, CH2B-1), 4.08 (pseudo-q pseudo-t, Jpseudo-q ≈ 6 Hz, Jpseudo-t ≈ 1 Hz, 1H, 3-H), 1.66 (s br, 1H, OH), 1.57–1.48 (m, 2H, 4-H), 1.42–1.24 (m, 4H, 5-H, 6-H), 0.91 (m, 3H, 7-H); 13C NMR (75.5 MHz, CDCl3) δ (ppm) = 141.5 (C-2), 114.7 (C-1), 73.4 (C-3), 36.9 (C-4), 27.6, 22.8 (C-5, C-6), 14.2 (C-7). The analytical data are in accordance with the literature.26 Hept-1-en-3-ol (2.51 g, 21.98 mmol, 1.0 equiv.) was dissolved in CH2Cl2 (50 mL) and a freshly ground mixture of KMnO4 (2.0 g, 12.66 mmol, 0.58 equiv.) and freshly prepared MnO233 (6.0 g, 69.01 mmol, 3.1 equiv.) was added. The resulting suspension was stirred at ambient temperature for 48 h. After filtration over Celite with CH2Cl2, the solvent was evaporated under reduced pressure (40 °C, 200 mbar) to yield 17 (1.73 g, 15.42 mmol, 72%) as a colorless liquid. Rf = 0.51 (SiO2, hexanes/EtOAc 5:1); IR (ATR) (cm−1) = 2958, 2929, 2872, 1727, 1465, 1380, 1368, 1286, 1126, 1074; 1H NMR (400 MHz, CDCl3) δ (ppm) = 6.35 (dd, J = 17.7, 10.5 Hz, 1H, 2-H), 6.21 (dd, J = 17.7, 1.3 Hz, 1H, CH2A-1), 5.81 (dd, J = 10.5, 1.3 Hz, 1H, CH2B-1), 2.59 (t, J = 7.4 Hz, 2H, 4-H), 1.64–1.56 (m, 2H, 5-H), 1.38–1.29 (m, 2H, 6-H), 0.91 (t, J = 7.3 Hz, 3H, 7-H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) = 201.3 (C-3), 136.7 (C-1), 128.0 (C-2), 39.5 (C-4), 26.3, 22.5 (C-5, C-6), 14.0 (C-7). The analytical data are in accordance with the literature.34
tert-Butyl 2-propylpiperidine-1-carboxylate (15)
tert-Butyl piperidine-1-carboxylate (14, 1.03 g, 5.53 mmol, 1.0 equiv.) was dissolved in dry Et2O (40 mL) under argon and TMEDA (2.0 mL, 13.25 mmol, 2.4 equiv.) was added. The resulting solution was cooled to −78 °C and sec-butyllithium (13.25 mmol, 2.4 equiv.) was added dropwise. The solution was stirred for 4 h at −78 °C, after which a 0.3 M solution of CuCN·2LiCl in THF (44.1 mL, 13.23 mmol, 2.4 equiv.) was added slowly. The resulting solution was allowed to warm to −50 °C and stirred at this temperature for 1 h. It was again cooled to −78 °C and 1-iodopropane (1.3 mL, 13.31 mmol, 2.4 equiv.) was added dropwise and the resulting solution was warmed to ambient temperature overnight. To the resulting black solution was added sat. NH4Cl(aq) (50 mL) and the mixture was stirred for 1 h. The aqueous phase was extracted with EtOAc (3 × 60 mL). The combined organic extracts were washed with water (60 mL) and brine (60 mL), dried over MgSO4 and the solvent was evaporated under reduced pressure. Purification by column chromatography (SiO2, hexanes/EtOAc 30:1, 1% NEt3) yielded 15 (0.73 g, 3.21 mmol, 58%) as a colorless oil. Rf = 0.41 (SiO2, cyclohexane/EtOAc 5:1); IR (ATR) (cm−1) = 2955, 2931, 2865, 1687, 1414, 1364, 1244, 1170, 1144, 767; 1H NMR (300 MHz, CDCl3) δ (ppm) = 4.20 (s br, 1H, 2-H), 3.95 (d, br, J = 12.4 Hz, 1H, CH2A-6), 2.74 (m, 1H, CH2B-6), 1.71–1.16 (m, 10H, 3-H, 4-H, 5-H, 1′-H, 2′-H), 1.44 (s, 9H, C(CH3)3), 0.91 (d, J = 7.3 Hz, 3H, 3′-H); 13C NMR (75.5 MHz, CDCl3) δ (ppm) = 154.8 (CO), 78.8 (C(CH3)3), 50.2 (C-2), 38.7 (C-6), 31.7 (C-1′), 28.5 (4C, C-3, C(CH3)3), 25.7 (C-5), 19.5 (C-2′), 19.0 (C-4), 14.1 (C-3′). The analytical data are in accordance with the literature.35
tert-Butyl trans-2-methyl-6-(3-oxobutyl)piperidine-1-carboxylate (9)
The title compound was prepared according to general procedure A using tert-butyl 2-methylpiperidine-1-carboxylate (1.06 g, 5.32 mmol, 1.0 equiv.), Et2O (20 mL), TMEDA (1.8 mL, 11.93 mmol, 2.2 equiv.), sec-butyllithium (1.3 M, 5.4 mL, 7.02 mmol, 1.3 equiv.), CuCN·2LiCl (0.3 min dry THF, 18 mL, 5.4 mmol, 1.0 equiv.), TMSCl (3.4 mL, 26.60 mmol, 5.0 equiv.) and butenone (0.46 mL, 5.45 mmol, 1.0 equiv.). Purification by column chromatography (SiO2, hexanes/EtOAc 30:1) yielded 9 (0.88 g, 3.27 mmol, 61%) as a colorless oil. Rf = 0.17 (SiO2, cyclohexane/EtOAc 5:1); IR (ATR) (cm−1) = 2967, 2931, 2872, 1717, 1684, 1454, 1392, 1364, 1324, 1175; 1H NMR, COSY, NOESY (600 MHz, CDCl3) δ (ppm) = 3.90–3.86 (m, 1H, H-2), 3.85–3.80 (m, 1H, H-6), 2.47 (t, J = 7.7 Hz, 2H, H-2′′), 2.14 (s, 3H, H-4′′), 1.95–1.89 (m, 1H, CH2A-1′′), 1.88–1.78 (m, 2H, CH2A-3, CH2A-5), 1.77–1.70 (m, 1H, CH2B-1′′), 1.66–1.56 (m, 3H, CH2-4, CH2B-5), 1.54–1.48 (m, 1H, CH2B-3), 1.45 (s, 9H, C(CH3)3), 1.22 (d, J = 6.7 Hz, 3H, CH3-1′); 13C NMR, HSQC, HMBC (75.5 MHz, CDCl3) δ (ppm) = 208.9 (C-3′′), 155.7 (NCO2), 79.2 (C(CH3)3), 51.1 (C-6), 47.5 (C-2), 41.3 (C-2′′), 30.2 (C-4′′), 28.7 (C(CH3)3), 28.6 (C-1′′), 27.1 (C-3), 24.6 (C-5), 20.8 (C-1′), 14.2 (C-4); HRMS (ESI) m/z 292.1877 ([M + Na]+, calcd for C15H27NO3Na 292.1889).
tert-Butyl trans-2-methyl-6-(3-oxoheptyl)piperidine-1-carboxylate (18)
The title compound was prepared according to general procedure A using tert-butyl 2-methylpiperidine-1-carboxylate (0.97 g, 4.87 mmol, 1.0 equiv.), Et2O (20 mL), TMEDA (1.68 mL, 11.13 mmol, 2.3 equiv.), sec-butyllithium (1.3 M, 5.22 mL, 6.79 mmol, 1.4 equiv.), CuCN·2LiCl (0.3 min dry THF, 16.3 mL, 4.89 mmol, 1.0 equiv.), TMSCl (3.1 mL, 24.25 mmol, 5.0 equiv.) and hept-1-en-3-one (17, 0.65 mL, 4.87 mmol, 1.0 equiv.). Purification by column chromatography (SiO2, hexanes/EtOAc 15:1) yielded 18 (0.75 g, 2.40 mmol, 49%) as a slight yellow oil. Rf = 0.34 (SiO2, cyclohexane/EtOAc 5:1); IR (ATR) (cm−1) = 2957, 2933, 2872, 1684, 1456, 1392, 1364, 1324, 1174, 1121, 772; 1H NMR, COSY, NOESY (600 MHz, CDCl3) δ (ppm) = 3.90–3.86 (m, 1H, H-2), 3.84–3.80 (m, 1H, H-6), 2.43 (t, J = 7.7 Hz, 2H, H-2′′), 2.40 (t, J = 7.5 Hz, 2H, H-4′′), 1.95–1.89 (m, 1H, CH2A-1′′), 1.88–1.78 (m, 2H, CH2A-3, CH2A-5), 1.77–1.70 (m, 1H, CH2B-1′′), 1.66–1.48 (m, 6H, CH2B-3, CH2-4, CH2B-5, CH2-5′′), 1.45 (s, 9H, C(CH3)3), 1.33–1.26 (m, 2H, CH2-6′′), 1.22 (d, J = 6.7 Hz, 3H, CH3-1′); 0.89 (t, J = 7.4 Hz, 3H, CH3-7′′); 13C NMR, HSQC, HMBC (75.5 MHz, CDCl3) δ (ppm) = 211.3 (C-3′′), 155.7 (NCO2), 79.2 (C(CH3)3), 51.3 (C-6), 47.5 (C-2), 42.8 (C-4′′), 40.4 (C-2′′), 28.7 (C(CH3)3), 28.6 (C-1′′), 27.1 (C-3), 26.2 (C-5′′), 24.6 (C-5), 22.5 (C-6′′), 20.8 (C-1′), 14.3 (C-4), 14.0 (C-7′′); HRMS (ESI) m/z 334.2354 ([M + Na]+, calcd for C18H33NO3Na 334.2358).
tert-Butyl trans-2-(3-oxoheptyl)-6-propylpiperidine-1-carboxylate (19)
The title compound was prepared according to general procedure A using tert-butyl 2-propylpiperidine-1-carboxylate (0.43 g, 1.88 mmol, 1.0 equiv.), Et2O (10 mL), TMEDA (0.66 mL, 4.37 mmol, 2.3 equiv.), sec-butyllithium (1.3 M, 2.05 mL, 2.67 mmol, 1.4 equiv.), CuCN·2LiCl (0.3 min dry THF, 6.3 mL, 1.89 mmol, 1.0 equiv.), TMSCl (1.2 mL, 9.39 mmol, 5.0 equiv.) and hept-1-en-3-one (17, 0.25 mL, 1.87 mmol, 1.0 equiv.). Purification by column chromatography (SiO2, hexanes/EtOAc 15:1) yielded 19 (0.30 g, 0.89 mmol, 48%) as a slight yellow oil. Rf = 0.33 (SiO2, cyclohexane/EtOAc 5:1); IR (ATR) (cm−1) = 2957, 2933, 2872, 1713, 1682, 1455, 1391, 1364, 1170, 773; 1H NMR, COSY, NOESY (600 MHz, CDCl3) δ (ppm) = 3.71–3.66 (m, 2H, H-2, H-6), 2.45–2.38 (m, 4H, H-2′, H-4′), 2.00–1.94 (m, 1H, CH2A-1′), 1.77–1.51 (m, 10H, CH2-3, CH2-4, CH2-5, CH2B-1′, CH2A-1′′), 1.47–1.39 (m, 1H, CH2B-1′′), 1.45 (s, 9H, C(CH3)3), 1.33–1.26 (m, 4H, CH2-6′, CH2-2′′), 0.91 (t, J = 7.5 Hz, 3H, CH3-3′′); 0.89 (t, J = 7.5 Hz, 3H, CH3-7′); 13C NMR, HSQC, HMBC (75.5 MHz, CDCl3) δ (ppm) = 211.4 (C-3′′), 155.9 (NCO2), 79.2 (C(CH3)3), 52.3 (C-6), 51.7 (C-2), 42.8 (C-4′), 40.3 (C-2′), 36.2 (C-1′′), 28.7 (C(CH3)3), 28.2 (C-1′), 26.1 (C-5′), 25.7, 24.8 (C-3, C-5), 22.5 (C-6′), 20.4 (C-2′′), 15.6 (C-4), 14.2 (C-3′′), 14.0 (C-7′); HRMS (ESI) m/z 362.2664 ([M + Na]+, calcd for C20H37NO3Na 362.2671).
5,9-trans-3,5-Dimethyloctahydroindolizinium 2,4,6-trinitrophenolate (12)
The title compound was prepared according to general procedure B using tert-butyl trans-2-methyl-6-(3-oxobutyl)piperidine-1-carboxylate (9, 174 mg, 0.646 mmol, 1.0 equiv.), EtOH (4.5 mL), AcCl (2 × 0.4 mL), 1 M NaOAc/HOAc-buffer (12.5 mL), NaCNBH3 (1 × 145 mg, 2.31 mmol, 3.6 eq.). The product was isolated as its picrate-salt, by dissolving in EtOH (4 mL) and adding a saturated solution of picric acid in EtOH (4 mL). The resulting solution was heated to reflux briefly and cooled until full precipitation. The salt was filtered and washed with EtOH to yield 12 (136 mg, 0.356 mmol, 55%) as a yellow solid. The product was isolated as a 1:1.2 mixture (1H NMR) of C-3 epimers. Mp 200–201 °C dec. (EtOH); Rf = 0.26 (SiO2, CHCl3/MeOH); IR (ATR) (cm−1) = 3025, 2954, 2918, 2849, 1631, 1564, 1365, 1316, 1297, 1269; 1H NMR, COSY, NOESY, TOCSY (600 MHz, CDCl3) δ (ppm) = Major Isomer (5E,9Z, 12a): 9.49 (br, s, 1H, NH), 8.86 (s, 2H, Ar–H), 4.20–4.14 (m, 1H, H-5), 3.28–3.20 (m, 1H, H-3), 3.19–3.11 (m, 1H, H-9), 2.39–1.94 (m, 5H, H-1, Ha-2, Ha-6, Ha-8), 1.93–1.79 (m, 2H, Hb-2, Hb-8), 1.79–1.57 (m, 3H, Hb-6, H-7), 1.36 (d, J = 6.5 Hz, 3H, C3-CH3), 1.29 (d, J = 7.1 Hz, 3H, C5–CH3); Minor Isomer (5Z,9E, 12b): 9.49 (br, s, 1H, NH), 8.86 (s, 2H, Ar–H), 4.20–4.14 (m, 1H, H-9), 3.83–3.73 (m, 1H, H-3), 3.40–3.33 (m, 1H, H-5), 2.39–1.94 (m, 3H, Ha-1, Ha-2, Ha-8), 1.93–1.79 (m, 3H, Hb-1, Hb-2, Ha-6), 1.79–1.57 (m, 4H, Hb-6, H-7, Hb-8), 1.52 (d, J = 6.7 Hz, 3H, C5–CH3), 1.49 (d, J = 6.7 Hz, 3H, C3–CH3); 13C NMR, HSQC, HMBC, HSQC-TOCSY (75.5, 150.9 MHz, CDCl3) δ (ppm) = Major Isomer (5E,9Z, 12a): 161.7 (CAr-1), 141.9 (CAr-2,6), 128.0 (CAr-4), 126.5 (CAr-3,5), 60.1 (C-9), 59.0 (C-3), 50.9 (C-5), 28.7 (C-6), 28.5 (C-8), 27.9 (C-2), 26.2 (C-1), 17.5 (C-7), 14.6 (C3–CH3), 10.0 (C5–CH3); Minor Isomer (5Z,9E, 12b): 161.7 (CAr-1), 141.9 (CAr-2,6), 128.0 (CAr-4), 126.5 (CAr-3,5), 59.8 (C-3), 59.2 (C-9), 53.3 (C-5), 29.3 (C-2), 27.2 (C-6), 26.8 (C-1), 25.3 (C-8), 18.1 (C3–CH3), 17.4 (C5–CH3), 16.9 (C-7); HRMS (ESI) m/z 154.1595 ([M + H]+, calcd for C10H20N 154.1596).
5,9-trans-3-Butyl-5-methyloctahydroindolizine (2)
The title compound was prepared according to general procedure B using tert-butyl trans-2-methyl-6-(3-oxoheptyl)-piperidine-1-carboxylate (18, 84 mg, 0.270 mmol, 1.0 equiv.), EtOH (5 mL), AcCl (3 × 0.3 mL), 1 M NaOAc/HOAc-buffer (25 mL), NaCNBH3 (2 × 63 mg, 1.00 mmol, 3.7 eq.). Purification by column chromatography (KP-NH, n-pentane/Et2O 100:1) yielded 2a (5E,9Z, 28 mg, 0.143 mmol, 53%) and 2b (5Z,9E, 12 mg, 0.061 mmol, 23%) as colorless oils. Rfa = 0.42 (KP-NH, n-pentane/Et2O 10:1), Rfb = 0.25 (KP-NH, n-pentane/Et2O 10:1); IR (ATR) (cm−1) = 2953, 2921, 2852, 1646, 1465, 1377, 1299, 1242; 1H NMR, COSY, NOESY, TOCSY (400, 600 MHz, CDCl3) δ (ppm) = Major Isomer (5E,9Z, 2a): 3.39–3.34 (m, 1H, H-5), 2.45–2.38 (m, 2H, H-3, H-9), 1.79–1.65 (m, 5H, Ha-1, Ha-2, Ha-6, Ha-8, Ha-1′), 1.55–1.50 (m, 2H, Hb-6, Ha-7), 1.49–1.41 (m, 1H, Hb-7), 1.37–1.08 (m, 8H, Hb-1, Hb-2, Hb-8, Hb-1′, H-2′, H-3′), 0.91–0.86 (m, 6H, 4′-CH3, C5–CH3); Minor Isomer (5Z,9E, 2b): 3.31–3.28 (m, 1H, H-9), 2.95–2.90 (m, 1H, H-3), 2.88–2.84 (m, 1H, H-5), 2.04–1.98 (m, 1H, Ha-2), 1.79–1.65 (m, 2H, Ha-1, Ha-6), 1.63–1.52 (m, 2H, Ha-7, Ha-1′), 1.50–1.41 (m, 2H, Hb-1, Hb-7), 1.40–1.20 (m, 9H, Hb-2, Hb-6, H-8, Hb-1′, H-2′, H-3′), 1.17 (d, J = 6.7 Hz, 3H, C5–CH3); 0.89 (t, J = 7.0 Hz, 3H, H-4′); 13C NMR, HSQC, HMBC, H2BC, HSQC-TOCSY (150.9 MHz, CDCl3) δ (ppm) = Major Isomer (5E,9Z, 2a): 59.3 (C-3), 55.5 (C-9), 47.5 (C-5), 32.6 (C-8), 32.5 (C-1′), 31.7 (C-6), 29.4 (C-2), 29.0 (C-2′), 28.3 (C-1), 23.3 (C-3′), 19.5 (C-7), 14.3 (C-4′), 7.7 (C5-CH3); Minor Isomer (5Z,9E, 2b): 59.8 (C-3), 55.4 (C-9), 48.7 (C-5), 36.4 (C-1′), 29.3 (C-2′), 29.1 (C-2), 28.7 (C-1), 27.1 C-8), 27.0 (C-6), 23.2 (C-3′), 20.7 (C5-CH3), 19.2 (C-7), 14.3 (C-4′); HRMS (ESI) m/z 196.2058 ([M + H]+, calcd for C13H26N 196.2065). The analytical data are in accordance with the literature.28
5,9-trans-3-Butyl-5-propyloctahydroindolizine (3)
The title compound was prepared according to general procedure B using tert-butyl trans-2-(3-oxoheptyl)-6-propylpiperidine-1-carboxylate (19, 108 mg, 0.318 mmol, 1.0 equiv.), EtOH (5 mL), AcCl (3 × 0.3 mL), 1 M NaOAc/HOAc-buffer (25 mL), NaCNBH3 (2 × 63 mg, 1.00 mmol, 3.7 eq.). Purification by column chromatography (KP-NH, n-pentane/Et2O 100:1) yielded 3 (40 mg, 0.179 mmol, 56%) as a colorless oil. The product was isolated as a 1:1.7 mixture (determined by 1H NMR) of C-3 epimers. Rfa = 0.58 (KP-NH, n-pentane/Et2O 10:1), Rfb = 0.44 (KP-NH, n-pentane/Et2O 10:1); IR (ATR) (cm−1) = 2955, 2922, 2852, 1647, 1464, 1377, 1299, 1243; 1H NMR, COSY, NOESY, TOCSY (600 MHz, CDCl3) δ (ppm) = Major Isomer (5E,9Z, 3a): 3.07–3.04 (m, 1H, H-5), 2.57–2.52 (m, 1H, H-3), 2.43–2.38 (m, 1H, H-9), 1.80–1.73 (m, 2H, Ha-2, Ha-1′), 1.71–1.62 (m, 4H, Ha-1, Ha-6, Ha-8, Ha-1′′), 1.59–1.49 (m, 2H, Hb-6, Ha-7), 1.48–1.39 (m, 1H, Hb-7), 1.38–1.08 (m, 11H, Hb-1, Hb-2, Hb-8, Hb-1′, Hb-1′′, H-2′, H-2′′, H-3′), 0.93–0.87 (m, 6H, 4′-CH3, 3′′-CH3); Minor Isomer (5Z,9E, 3b): 3.25–3.22 (m, 1H, H-9), 2.94–2.89 (m, 1H, H-3), 2.72–2.68 (m, 1H, H-5), 2.02–1.95 (m, 1H, Ha-2), 1.80–1.73 (m, 1H, Ha-1), 1.71–1.62 (m, 1H, Ha-6), 1.59–1.49 (m, 3H, Ha-7, Ha-1′, Ha-1′′), 1.48–1.39 (m, 2H, Hb-7, Hb-1′′), 1.38–1.08 (m, 12H, Hb-1, Hb-2, Hb-6, H-8, Hb-1′, H-2′, H-2′′, H-3′), 0.93–0.87 (m, 6H, 4′-CH3, 3′′-CH3); 13C NMR, HSQC, HMBC, H2BC, HSQC-TOCSY (100.6, 150.9 MHz, CDCl3) δ (ppm) = Major Isomer (5E,9Z, 3a): 58.6 (C-5), 56.3 (C-9), 52.6 (C-5), 32.7 (C-1′), 32.6 (C-8), 29.6 (C-1), 28.9 (C-2′), 28.5 (C-2), 27.9 (C-6), 23.3 (C-3′), 23.0 (C-1′′), 21.0 (C-2′′), 19.5 (C-7), 14.7 (C-3′′), 14.3 (C-4′); Minor Isomer (5Z,9E, 3b): 58.6 (C-5), 55.1 (C-9), 52.4 (C-5), 36.1 (C-1′), 35.6 (C-1′′), 29.2 (C-1), 28.9 (C-2′), 28.8 (C-2), 27.4 (C-8), 23.3 (C-3′), 23.0 (C-6), 20.5 (C-2′′), 19.5 (C-7), 14.4, 14.3 (C-3′′, C-4′); HRMS (ESI) m/z 224.2375 ([M + H]+, calcd for C15H30N 224.2378). The analytical data are in accordance with the literature.16
Acknowledgements
This work was supported by the Carl Zeiss foundation. The authors thank Dr J. C. Liermann and Lars Andernach (both Mainz) for NMR spectroscopy and Dr N. Hanold (Mainz) for mass spectrometry.Notes and references
- J. P. Michael, in The Alkaloids: Chemistry and Biology, ed. H.-J. Knölker, Academic Press, 2016, vol. 75, pp. 1–498 Search PubMed.
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed.
- R. A. Saporito, R. A. Norton, N. R. Andriamaharavo, H. M. Garraffo and T. F. Spande, J. Chem. Ecol., 2011, 37, 213–218 CrossRef CAS PubMed.
- R. A. Saporito, T. F. Spande, H. Martin Garraffo and M. A. Donnelly, Heterocycles, 2009, 79, 277–297 CrossRef CAS.
- S. Leclercq, J. C. Braekman, D. Daloze and J. M. Pasteels, in Fortschritte der Chemie organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Falk, G. W. Kirby and R. E. Moore, Springer Vienna, Vienna, 2000, vol. 79, pp. 115–229 Search PubMed.
- Y. Koriyama, A. Nozawa, R. Hayakawa and M. Shimizu, Tetrahedron, 2002, 58, 9621–9628 CrossRef CAS.
- A. B. Smith and D.-S. Kim, J. Org. Chem., 2006, 71, 2547–2557 CrossRef CAS PubMed.
- Z. Sun, S. Yu, Z. Ding and D. Ma, J. Am. Chem. Soc., 2007, 129, 9300–9301 CrossRef CAS PubMed.
- S. R. Angle and M. Kim, J. Org. Chem., 2007, 72, 8791–8796 CrossRef CAS PubMed.
- H. Liu, D. Su, G. Cheng, J. Xu, X. Wang and Y. Hu, Org. Biomol. Chem., 2010, 8, 1899–1904 CAS.
- M. Gartner, R. Weihofen and G. Helmchen, Chem. – Eur. J., 2011, 17, 7605–7622 CrossRef PubMed.
- F. Abels, C. Lindemann, E. Koch and C. Schneider, Org. Lett., 2012, 14, 5972–5975 CrossRef CAS PubMed.
- F. Abels, C. Lindemann and C. Schneider, Chem. – Eur. J., 2014, 20, 1964–1979 CrossRef CAS PubMed.
- N. N. Rao, B. B. Parida and J. K. Cha, Org. Lett., 2014, 16, 6208–6211 CrossRef CAS PubMed.
- R. V. Stevens and A. W. M. Lee, J. Chem. Soc., Chem. Commun., 1982, 103–104 RSC.
- D. J. Hart and Y. M. Tsai, J. Org. Chem., 1982, 47, 4403–4409 CrossRef CAS.
- E. Conchon, Y. Gelas-Mialhe and R. Remuson, Tetrahedron: Asymmetry, 2006, 17, 1253–1257 CrossRef CAS.
- G. Barbe, G. Pelletier and A. B. Charette, Org. Lett., 2009, 11, 3398–3401 CrossRef CAS PubMed.
- The expressions (5E,9Z) and (5Z,9E) refer to the stereochemical terminology introduced by Daly, et al. (ref. 2) and describes the configuration at C-5 and C-9 relative to the configuration at C-3. In this case (5E,9Z) refers to a 3,5-trans- and 3,9-cis-geometry respectively. Likewise (5Z,9E) refers to a 3,5-cis- and 3,9-trans-geometry.
- P. Beak, A. Basu, D. J. Gallagher, Y. S. Park and S. Thayumanavan, Acc. Chem. Res., 1996, 29, 552–560 CrossRef CAS.
- D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282–2316 CrossRef CAS.
- E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092–10142 CrossRef CAS PubMed.
- R. K. Dieter, C. M. Topping and L. E. Nice, J. Org. Chem., 2001, 66, 2302–2311 CrossRef CAS PubMed.
- M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Org. Biomol. Chem., 2008, 6, 3464–3466 CAS.
- A. Shaabani, P. Mirzaei, S. Naderi and D. G. Lee, Tetrahedron, 2004, 60, 11415–11420 CrossRef CAS.
- F. Felluga, C. Forzato, F. Ghelfi, P. Nitti, G. Pitacco, U. M. Pagnoni and F. Roncaglia, Tetrahedron: Asymmetry, 2007, 18, 527–536 CrossRef CAS.
- A. Brandi, F. Cordero and C. Querci, J. Org. Chem., 1989, 54, 1748–1750 CrossRef CAS.
- N. Yamazaki and C. Kibayashi, J. Am. Chem. Soc., 1989, 111, 1396–1408 CrossRef CAS.
- D. R. Artis, I.-S. Cho, S. Jaime-Figueroa and J. M. Muchowski, J. Org. Chem., 1994, 59, 2456–2466 CrossRef CAS.
- A. F. Burchat, J. M. Chong and N. Nielsen, J. Organomet. Chem., 1997, 542, 281–283 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- D. Stead, G. Carbone, P. O'Brien, K. R. Campos, I. Coldham and A. Sanderson, J. Am. Chem. Soc., 2010, 132, 7260–7261 CrossRef CAS PubMed.
- J. Attenburrow, A. F. B. Cameron, J. H. Chapman, R. M. Evans, B. A. Hems, A. B. A. Jansen and T. Walker, J. Chem. Soc., 1952, 1094–1111 RSC.
- M. Vavrecka and M. Hesse, Helv. Chim. Acta, 1991, 74, 438–444 CrossRef CAS.
- D. Gagnon, S. Lauzon, C. Godbout and C. Spino, Org. Lett., 2005, 7, 4769–4771 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob01308b |
| This journal is © The Royal Society of Chemistry 2016 |