
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient and scalable synthesis of α,α-disubstituted β-amino amides†
Marianne Hagensen
Paulsen
a,
Magnus
Engqvist
b,
Dominik
Ausbacher
a,
Morten Bøhmer
Strøm
a and
Annette
Bayer
 *b
*b
aDepartment of Pharmacy, UiT – The Arctic University of Norway, NO-9037 Tromsø, Norway
bDepartment of Chemistry, UiT – The Arctic University of Norway, NO-9037 Tromsø, Norway. E-mail: annette.bayer@uit.no
First published on 12th July 2016
Abstract
A practical and efficient methodology for the preparation of 2-aminoethyl α,α-disubstituted β-amino amides in three steps from methyl cyanoacetate has been developed. The key step in the synthesis was the chemoselective reduction of the nitrile group in presence of an amide and aryl halide functionalities. Reduction with RANEY® Nickel catalyst, either with molecular hydrogen (8–10 bar) or under transfer hydrogenation conditions, necessitated in situ protection of the resulting amines with Boc2O, whereas aryl bromide containing nitriles could be chemoselectively reduced with ZnCl2/NaBH4 without debromination. The developed protocol involved only one chromatographic purification step and can be performed at gram scale.
Introduction
β-Amino acids are important building blocks that are found as components of natural products such as taxol1 and microcystin2 and in peptidomimetics3–5 to imitate the function of bioactive peptides. Due to the additional methylene group, proteases such as trypsin and α-chymotrypsin6,7 are unable to recognize β-amino acids. Thus, incorporation of β-amino acids into peptides is a known strategy to increase proteolytic stability.4,8–10 Peptidomimetics consisting of β-amino acids can also form unique secondary structures11,12 due to increased rotational possibilities that are useful for improving the biological activity of peptides.10,13Naturally occurring antimicrobial peptides (AMP) have attracted much attention due to their potential as a new class of antibiotics and anticancer agents.14 Both, antimicrobial and chemotherapeutic agents with a novel mechanism of action are strongly needed to fight the emergence of therapy resistant tumours and multi-drug resistant bacterial infections. Our group has reported a series of antimicrobial and cytotoxic α,α-disubstituted β-amino acid derivatives. These derivatives have an amphipathic structure consisting of two cationic groups and two lipophilic side-chains previously described as a pharmacophore model for small cationic AMPs.15–17 However, we have observed that the aromatic side-chains of the α,α-disubstituted β-amino acid derivatives are subjected to metabolism by liver microsomes, which can complicate drug development.18 Aromatic systems can be protected against such metabolic transformations by the introduction of electron withdrawing groups such as fluoro or bromo substituents. This spurred our interest in finding an efficient and scalable synthesis allowing for the preparation of halogenated analogues of α,α-disubstituted β-amino amides with the general structure shown in Fig. 1.
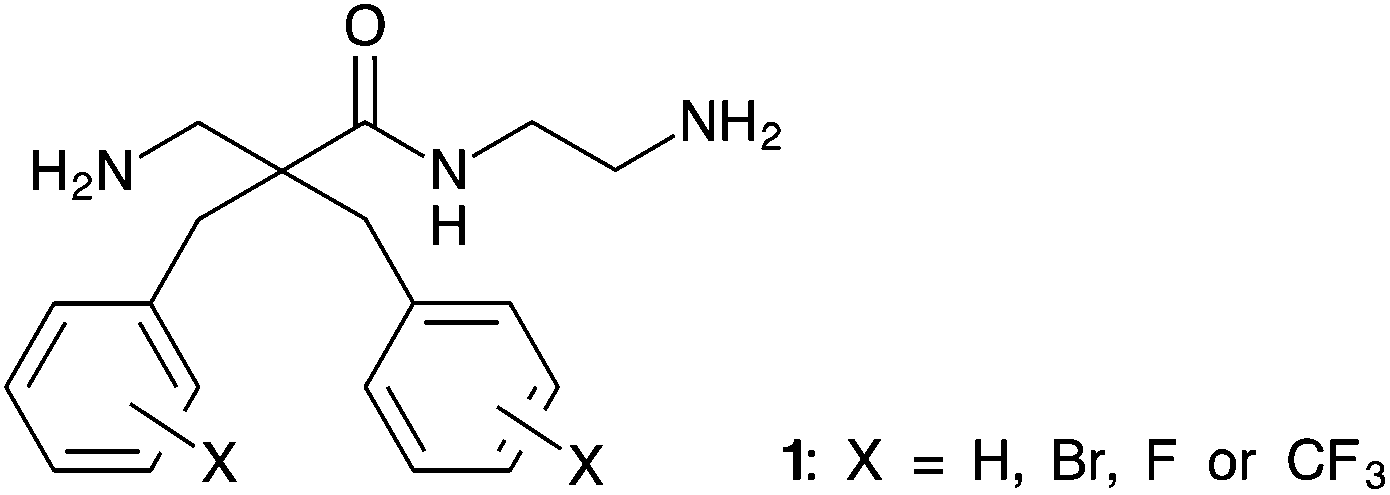 | ||
| Fig. 1 General structure of desired α,α-disubstituted β-amino acid derivatives containing two halogenated aromatic side chains and a 2-aminoethyl C-terminus. | ||
A number of methods for the preparation of α,α-disubstituted β-amino acids and derivatives have been reported (Scheme 1). Dialkylation of cyanoacetate alkyl ester followed by nitrile reduction and ester hydrolysis (A) was first described by Cronin and coworkers6 and has later been applied by us16,17 and others.19 Reddy et al. published an interesting approach based on palladium-catalysed directed C–H activation of cyanoacetamides followed by arylation (B1) and nitrile reduction with Pd/C (B2).20 Capone et al. reported the direct alkylation of protected β-alanine using KHMDS as base (C),21 while Lin et al. reported a related approach based on the alkylation and reduction of a β-alanine nickel(II) complex (D).22 Alkylated β-alanines were obtained after reduction of the nickel complexes by NaBH4 or Pd/C. Strategies based on the introduction of the amine on an alkylated precursor include the Mitsunobu reaction of 3-hydroxypropionate and phthalimide described by Ducry and coworkers (E),23 and Mannich or Vilsmeier–Haack type reactions between silyl enol ethers of 2,2-disubstituted acetic acids or esters and iminium salts (F and G) developed by the group of Karoyan24 and Bélanger,25 respectively. However, these reported protocols require either long reaction sequences, harsh reaction conditions or are not compatible with aryl bromides due to the use of transition metal catalysts, which are prone to lead to dehalogenation.
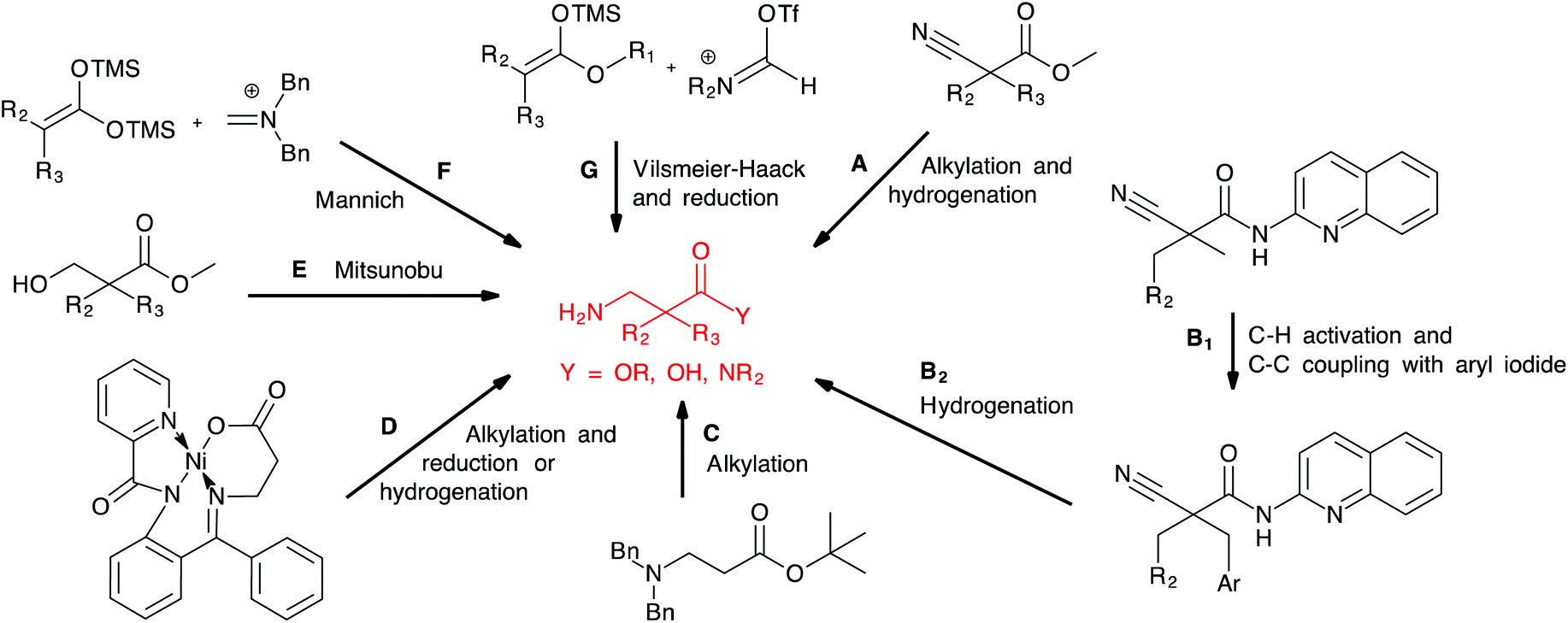 | ||
| Scheme 1 Reported approaches to α,α-disubstituted β-amino acids and derivatives. | ||
Here we present an efficient and easily scalable synthesis of 2-aminoethyl amides of α,α-disubstituted β-amino acids. Additionally, a bromide compatible variant was developed to allow for the preparation of aryl bromide substituted derivatives.
Results and discussion
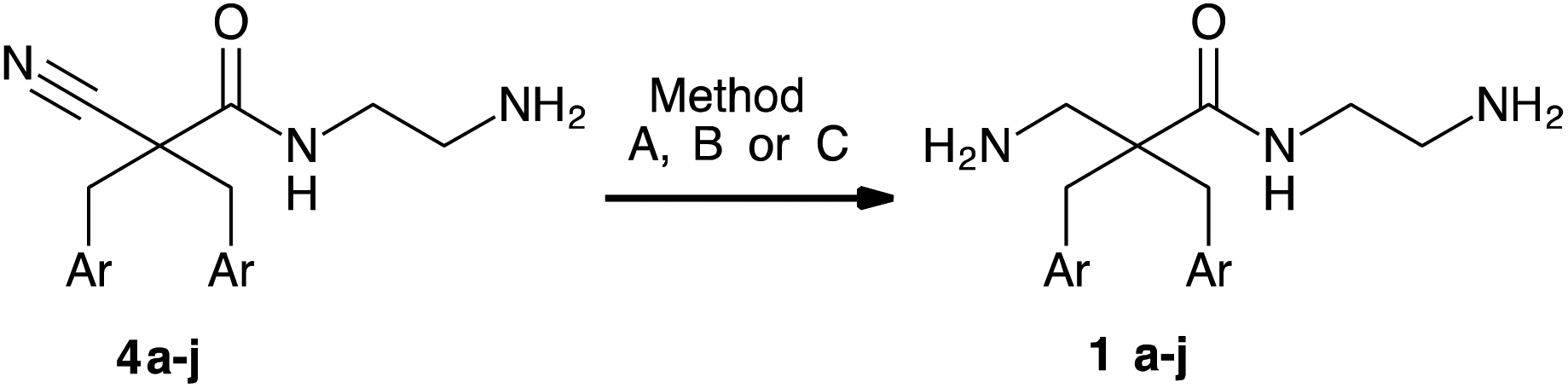
Our group has previously prepared analogues of the target compounds. In the reported synthesis16,17 (Scheme 2A), the methyl cyanoacetate was alkylated with a variety of alkyl and aryl halides. This was followed by reduction of the nitrile group with RANEY® nickel to provide the alkylated β-amino ester. After Boc-protection of the amine, the methyl ester was hydrolysed to the corresponding carboxylic acid. After activation of the acids with fluoro-N,N,N′,N′-tetramethylformamidinium hexafluorophosphate (TFFH), coupling with the desired amine provided the disubstituted α,α-disubstituted β-amino amides.16 Initial experiments revealed that this protocol consisted of several work-intensive steps and was not compatible with brominated substrates. Partial or complete debromination was observed during the nitrile reduction step with RANEY® nickel, leading to product mixtures that were difficult to separate or unwanted products. Consequently, we developed a more efficient synthesis as shown in Scheme 2B. Methyl cyanoacetate was dialkylated, followed by aminolysis and nitrile reduction. This strategy circumvented the amide coupling with TFFH and the protection/deprotection steps of the previous method and thereby significantly shortened the protocol. A key challenge was, however, to find a method for a chemoselective nitrile reduction.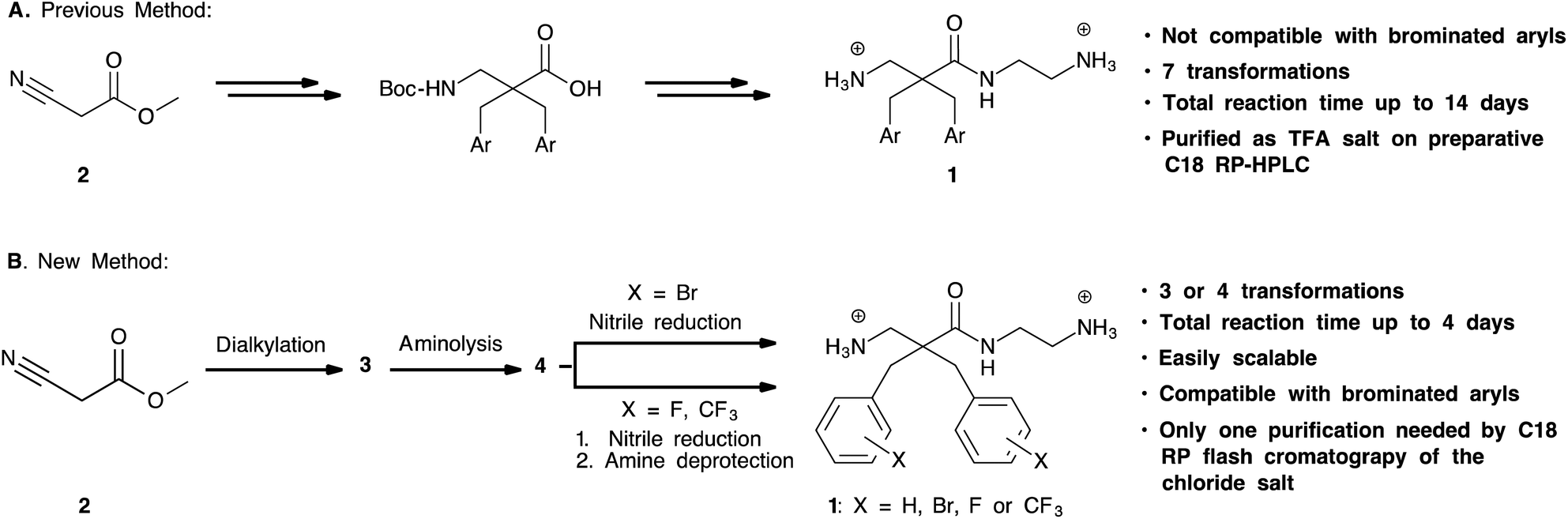 | ||
| Scheme 2 A. Previous reported method for synthesis of α,α-disubstituted β-amino amides; B. improved method described in this work. | ||
The dialkylation of cyanoacetate esters is well known.26–30 In the course of our work, several of the reported protocols were investigated. A modification of Oedigers method,30 the 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) mediated dialkylation of methyl cyanoacetate (2) in CH2Cl2, was found to be a fast and convenient procedure providing the dialkylated cyanoacetates (3a–j) in almost quantitative yields (>95% for all derivatives, except 3g) (Scheme 3). The reaction times were short and 10 min proved to be sufficient on a 1 mmol scale. The reaction was easily scaled up to a 50 mmol scale. The crude products were recrystallized in MeOH.
 | ||
| Scheme 3 Dialkylation of methyl cyanoacetate 2. | ||
A common method for the formation of amide bonds is the peptide coupling of amines with activated carboxylic acids like acid chlorides or in situ activated carboxylic acids. A variety of attempts to couple in situ activated carboxylic acids, obtained after hydrolysis of 3, with N-Boc ethylene diamine in analogy to the previously reported method17 gave low yields and a significant amount of by-products thus requiring chromatographic purification. Therefore, aminolysis of the methyl esters was investigated as a more convenient and economic procedure (Table 1). The dialkylated methyl esters (3a–j) were dissolved in ethylene diamine as solvent and stirred until TLC indicated completion (0.5–24 h). Precipitation of the products by addition of water gave the corresponding 2-aminoethyl cyano amides 4a–j in good yields (72–98%) and purity (>95% as determined by analytical HPLC) (Table 1). The reaction was carried out up to a 50 mmol scale. Other protocols for assisted aminolysis31,32 were examined but did not provide significant improvement.

Initially, the chemoselective reductions of nitriles in presence of amides with RANEY® nickel were investigated for the cyano amides 4a–j. In general, the reductions were slow (1 bar, 45 °C, 5 days) and low yields were obtained (<30%). Not surprisingly, the reduction of the aryl bromide containing nitriles 4a and 4b resulted in substantial debromination. We hypothesise that the inefficient reduction with RANEY® nickel might relate to coordination by the amine functionalities and formation of byproducts. Therefore, reductions were performed in presence of Boc2O to affect simultaneous reduction and protection of the amine groups. By addition of Boc2O (4 eq.), reductions at 8–10 bar in MeOH (Table 2, Method A) gave full conversion within 18 h determined by MS and NMR of the crude. Deprotection (1.33 M HCl in dioxane) of the crude provided the target amino amides 1c–j (Table 2, entries 4–7, 9, 10 and 12). Yields were good (>70%) when purification by precipitation was possible (Table 2, entries 4, 5, 9 and 12). When chromatographic purification (C18 reverse phase flash chromatography) was necessary the yields dropped (25–52%) for most of the products (Table 2, entries 4, 6, 7, 9, 10 and 12). The reductions were routinely carried out at a 0.2–0.6 mmol scale. To test the scalability of method A, a reduction of 2.5 mmol of 4j (1 g) was carried out. The reaction at this scale did not result in the desired conversion within 18 h reaction time.
|
|
||||
|---|---|---|---|---|
| Entry | Starting material | Methoda | Rxn. time (h) | Yield (%) |
| a Method A: i. RANEY® Ni, H2 (8–10 bar), Boc2O (approx. 4 eq.), MeOH, 45 °C ii. 1.33 M HCl in dioxane; Method B: i. RANEY® Ni, ammonium formate (4 eq.), Boc2O (4 eq.), EtOAc or MeOH, 45 °C ii. 1.33 M HCl in dioxane; Method C: ZnCl2/NaBH4. b Dehalogenated product. c Isolated yield after crystallisation. d Isolated yield after chromatographic purification. | ||||
| 1 | 4a | B | 1 | ndb |
| 2 | C | 1.5 | 75d | |
| 3 | 4b | C | 1.5 | 80d |
| 4 | 4c | A | 18 | 80c/25d |
| 5 | 4d | A | 18 | 90c |
| 6 | 4e | A | 18 | 40d |
| 7 | 4f | A | 18 | 50b |
| 8 | B | 48 | 30b | |
| 9 | 4g | A | 18 | 75c/50d |
| 10 | 4h | A | 18 | 52d |
| 11 | B | 48 | 62d | |
| 12 | 4i | A | 20 | 72c/35d |
| 13 | B | 24 | 27d | |
| 14 | 4j | B | 48 | 80d |
Additionally, reduction of the nitrile group with a modified protocol at 1 bar using transfer hydrogenation conditions (Method B) with ammonium formate as hydrogen source was investigated (Table 2, entries 8, 11, 13 and 14). For substances 4h and 4j good yields (62% of 4h and 80% of 4j) were obtained within 48 h (entries 11 and 14), while other substrates (4f and 4i) showed low conversion (<30%) (entries 8 and 13). The reduction of 4j under transfer hydrogenation conditions (method B) was performed at a 2.6 mmol scale with good results (80% yield).
An important aim of this study was the preparation of halogen-containing analogues of 1. As expected, the modified RANEY® nickel reduction did not prevent the unwanted debromination. In the reduction of 4a by RANEY® nickel with ammonium formate at 1 bar in presence of Boc2O (Method B), substantial debromination was observed already after 1 h reaction time (Table 2, entry 1). Caddick and coworkers described selective reductions of nitriles in presence of either amides or aryl bromides employing catalytic nickel(II) chloride with excess sodium borohydride.33 In our case, this protocol resulted in dehalogenation. Dehalogenation also occurred with CoCl2/NaBH4.19,34,35 Finally, we achieved the desired chemoselectivity with a system of ZnCl2/NaBH4 as reducing agent.36
Reduction of nitriles 4a and 4b with ZnCl2/NaBH4 gave the corresponding amines 1a in 75% and 1b in 80% yield after decomplexation of any amine–boron complexes by boiling in 6 M aqueous HCl and chromatographic purification (C18 RP flash chromatography). Interestingly, no significant hydrolysis of the amides 1a and 1b was observed during the acidic treatment, however prolonged acidic treatment resulted in decarboxylative loss of the amide functionality.
Conclusions
In summary, we have describe a convenient and robust approach to α,α-dialkylated β-amino amides comprising of a dialkylation with DBU, followed by aminolysis and chemoselective reduction of the nitrile group. A nitrile reduction method that tolerates the presence of bromoaryl substituents was developed. Only one chromatographic purification was needed to yield >95% pure compounds in a three step procedure with an overall yield of 21–84%.Experimental section
Materials and methods
All reagents and solvents were purchased from commercial sources and used as supplied unless otherwise stated. Anhydrous THF was prepared by storage over 4 Å molecular sieves. RANEY® nickel was bought from Sigma Aldrich (CAS no. 7440-02-0, 2800, slurry, in H2O, active catalyst). The hydrogenations with RANEY® nickel at higher pressure (8–10 bar) were carried out on a Parr Instrument, Series 4590 Micro Stirred reactor, 50 mL, attached to a Parr 4843 Modular Controller. Reactions were monitored by thin-layer chromatography (TLC) with Merck pre-coated silica gel plates (60 F254). Visualization was accomplished with either UV light or by immersion in potassium permanganate or phosphomolybdic acid (PMA) followed by light heating with a heating gun. Purification of reactions was carried out by chromatography using a RP C18 column preloaded on a Samplet® cartridge belonging to a Biotage SP-1. Analytical HPLC was carried out on a Waters 2695 Separations Module equipped with an XBridge™ C18 5 μm, 4.6 mm × 250 mm column and analysed at wavelengths 214 and 254 nm with a Waters 996 PDA detector spanning from wavelengths 210 to 310 nm. The compounds were eluted with water and acetonitrile, both containing 0.1% TFA. The gradient started at 10% acetonitrile, 3 minutes isocratic gradient, followed by a linear increase to 90% acetonitrile over 17 minutes, followed by an 8 min acetonitrile wash and then 10 min equilibration to the starting gradient. The flow rate was 1 ml min−1. NMR spectra were obtained on a 400 MHz Bruker Avance III HD equipped with a 5 mm SmartProbe BB/1H (BB = 19F, 31P-15N). Data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), coupling constant (J, Hz) and integration. Chemical shifts (δ) are reported in ppm relative to the residual solvent peak (CDCl3: δH 7.26 and δC 77.16; Methanol-d4: δH 3.31 and δC 49.00). Positive ion electrospray ionization mass spectrometry was conducted on a Thermo electron LTQ Orbitrap XL spectrometer.General procedure for dialkylation of methyl cyanoacetate 2
Methyl cyanoacetate 2 was dissolved in CH2Cl2 (approx. 0.1 M, prefiltered through K2CO3), cooled to 0 °C, added DBU (2 eq.) and stirred for 2 min. The alkyl halide (2 eq.) was added in small portions to avoid increase in temperature. The reaction was left to stir at RT until completion. The reaction was monitored on TLC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 EtOAc/toluene). After completion, the reaction was quenched with water and extracted with EtOAc. The organic phase was washed with water (3 times) and brine, dried over Na2SO4, filtered and evaporated to dryness. To remove residual EtOAc, chloroform was added and re-evaporated. If necessary, MeOH was added to precipitate the crude. The resulting solid was recrystallized in MeOH.
:4 EtOAc/toluene). After completion, the reaction was quenched with water and extracted with EtOAc. The organic phase was washed with water (3 times) and brine, dried over Na2SO4, filtered and evaporated to dryness. To remove residual EtOAc, chloroform was added and re-evaporated. If necessary, MeOH was added to precipitate the crude. The resulting solid was recrystallized in MeOH.
General procedure for aminolysis of 3a–j
The procedure was performed under N2. The alkylated methylcyanoacetate (3a–j) was added ethylenediamine and stirred at room temperature until completion. The reaction was monitored on TLC. After completion, the reaction mixture was cooled on ice and water was added to the reaction mixture until precipitation occurred. The product was filtered off, washed carefully with water and dried under vacuum.General procedure for reduction with RANEY® Nickel at 8 bar (Method A)
One spoon of RANEY® Nickel (approx. 5 g) was transferred to the 50 mL Parr hydrogenation bomb, washed with 3 × 15 mL MeOH before addition of substrate (0.2–2.5 mmol) and Boc2O (approx. 4 eq.). The reaction was stirred overnight (18 h) at 45 °C at 8–10 bar. After completion, the bomb was cooled to room temperature, the reaction mixture was purged and the catalyst was filtered off through a pad of sand and celite under N2 and the filtrate evaporated to dryness. The crude was dissolved in dioxane and added 4 M HCl/dioxane and H2O and heated to 60 °C for 2 h. The reaction mixture was evaporated to dryness and purified on C18 RP flash chromatography (acetonitrile/water) or dissolved in EtOAc and precipitated by addition of Et2O.Warning: Reactions at larger scale (12 mmol) are not recommended in the 50 mL Parr bomb. Delayed, uncontrolled gas evolution after the release of hydrogen pressure was observed.
General procedure for reduction with RANEY® Nickel at 1 bar (Method B)
One spoon of RANEY® Nickel (approx. 5 g) was transferred to a round bottom flask, washed with 3 × 15 mL MeOH and 3 × 15 mL EtOAc before addition of substrate dissolved in EtOAc. The reaction was stirred for 48 h (unless otherwise stated) at 45 °C with a H2 containing balloon attached. The reaction mixture was cooled to room temperature before the catalyst was filtered off through a pad of sand and celite under N2, washed with brine, dried with Na2SO4 and evaporated to dryness. The product was purified by C18 RP flash chromatography (acetonitrile/water).General procedure for reduction with ZnCl2/NaBH4 (Method C)
The reducing agent was prepared by stirring ZnCl2 (1 eq., 1.15 g) and NaBH4 (2 eq., 0.68 g) in dry THF (40 mL) overnight. The substrate (1 eq.) was dissolved in the reducing agent (2 eq.) and refluxed for 1.5 h. The reaction mixture was allowed to cool down to RT, quenched with 0.1 mL water followed by 1 mL 6 M aqueous HCl. The reaction mixture was refluxed for 10 min until the boron-compound-complex had dissociated (followed by MS). The reaction mixture evaporated to dryness, redissolved in MeOH and purified by C18 RP flash chromatography (acetonitrile/water).Method A: 4f (0.24 mmol, 0.11 g), Boc2O (0.95 mmol, 0.20 g) and 1 spoon of RANEY® Nickel in MeOH gave the di-Boc-protected intermediate. The intermediate was added 4 mL dioxane, 0.5 mL H2O and 2 mL 4 M HCl/dioxane to yield the crude, which was purified by C18 RP flash chromatography to yield the title compound as clear crystals (0.053 g, 50%).
Method B: 4f (0.50 mmol, 0.22 g), Boc2O (6 eq.), ammonium formate (4 eq.) and 1 spoon of RANEY® Nickel in EtOAc gave the di-Boc-protected intermediate. The intermediate was added 4 mL dioxane, 0.5 mL H2O and 2 mL 4 M HCl/dioxane to yield the crude, which was purified by C18 RP flash chromatography to yield the title compound (0.066 g, 30%).
1H NMR (400 MHz, Methanol-d4): δ 7.77 (dd, J = 7.9, 1.4, 2H), 7.64 (td, J = 7.7, 1.4, 2H), 7.51 (t, J = 7.6, 2H), 7.40 (d, J = 7.8, 2H), 3.54 (t, J = 6.9, 2H), 3.44 (dAB, J = 15.7, 2H), 3.37 (dAB, J = 16.0, 2H), 3.03 (s, 2H). 13C NMR (101 MHz, Methanol-d4): δ 177.3, 134.8, 133.4, 132.7, 130.8 (q, J = 29.4), 129.1, 128.1 (q, J = 5.7), 125.8 (q, J = 274.7), 45.2, 39.9, 38.7, 37.7. HRMS (ESI) m/z: [M + H]+ Calculated for C21H24F6N3O 448.1824; Found: 448.1804.
Method A: 4h (0.52 mmol, 0.30 g), Boc2O (2.74 mmol, 0.60 g) and 1 spoon of RANEY® Nickel in MeOH gave the di-Boc-protected intermediate. The intermediate was added 4 mL dioxane, 0.5 mL H2O and 2 mL 4 M HCl/dioxane to yield the crude, which was purified by C18 RP flash chromatography to yield the title compound as clear crystals (0.067 g, 23%).
Method B: 4h (0.59 mmol, 0.35 g), Boc2O (6 eq.), ammonium formate (4 eq.) and 1 spoon of RANEY® Nickel in EtOAc gave the di-Boc-protected intermediate. The intermediate was added 4 mL dioxane, 0.5 mL H2O and 2 mL 4 M HCl/dioxane to yield the crude (0.34 g, 97%).
1H NMR (400 MHz, Methanol-d4): δ 7.94 (s, 6H), 3.58–3.47 (m, 4H), 3.29 (dAB, J = 14.4, 2H), 3.15 (t, J = 6.2, 2H), 3.10 (s, 2H).
13C NMR (101 MHz, Methanol-d4): δ 175.2, 139.7, 132.9 (q, J = 33.2), 132.3–131.9 (m), 124.8 (q, J = 273.0), 122.5 (p, J = 3.9), 50.6, 43.4, 40.6, 40.1, 38.5. HRMS (ESI) m/z: [M + H]+ Calculated for C23H22F12N3O 584.1572; Found: 584.1567.
Method A: 4i (0.606 mmol, 0.254 g), Boc2O (2.42 mmol, 0.53 g) and 1 spoon of RANEY® nickel in MeOH gave the di-Boc-protected intermediate. The intermediate was added 4 mL dioxane, 0.5 mL H2O and 2 mL 4 M HCl/dioxane to yield the crude, which was purified by C18 RP flash chromatography to yield the title compound as clear crystals (0.092 g, 35%).
Method B: 4i (1.27 mmol, 0.53 g), Boc2O (2 eq.), ammonium formate (4 eq.) and 1 spoon of RANEY® Nickel in EtOAc (24 h) gave the di-Boc-protected intermediate. The intermediate was added 4 mL dioxane, 0.5 mL H2O and 2 mL 4 M HCl/dioxane to yield the crude, which was purified by C18 RP flash chromatography to yield the title compound (0.14 g, 27%).
1H NMR (400 MHz, Methanol-d4): δ 7.39 (d, J = 8.3, 4H), 7.17 (d, J = 8.3, 4H), 3.51 (t, J = 6.4, 2H), 3.22 (A dAB, J = 14.2, 2H), 3.11 (t, J = 6.4, 2H), 3.00 (s, 2H), 2.94 (dAB, J = 14.2, 2H), 1.31 (s, 18H).13C NMR (101 MHz, Methanol-d4): δ 177.3, 151.6, 133.4, 131.2, 126.6, 50.5, 43.6, 40.7, 40.6, 38.6, 35.3, 31.7. HRMS (ESI) m/z: [M + H]+ Calculated for C27H42N3O 424.3327; Found: 424.3320.
Acknowledgements
This work was financially supported by NRC grant (no: 214493/F20 – “fellesløftet”) and a UiT – The Arctic University of Norway grant (no: A23259).Notes and references
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325 CrossRef CAS PubMed.
- D. P. Botes, A. A. Tuinman, P. L. Wessels, C. C. Viljoen, H. Kruger, D. H. Williams, S. Santikarn, R. J. Smith and S. J. Hammond, J. Chem. Soc., Perkin Trans. 1, 1984, 2311 RSC.
- T. Godballe, L. L. Nilsson, P. D. Petersen and H. Jenssen, Chem. Biol. Drug Des., 2011, 77, 107 CAS.
- C. Adessi and C. Soto, Curr. Med. Chem., 2002, 9, 963 CrossRef CAS PubMed.
- B. B. Jayendra, C. Tiffany, B. Libero and P. H. Rickey, Curr. Top. Med. Chem., 2013, 13, 3205 CrossRef.
- J. R. Cronin, G. U. Yuen and S. Pizzarello, Anal. Biochem., 1982, 124, 139 CrossRef CAS PubMed.
- J. D. Sadowsky, J. K. Murray, Y. Tomita and S. H. Gellman, ChemBioChem, 2007, 8, 903 CrossRef CAS PubMed.
- F. Kudo, A. Miyanaga and T. Eguchi, Nat. Prod. Rep., 2014, 31, 1056 RSC.
- V. Tørfoss, D. Ausbacher, C. d. A. Cavalcanti-Jacobsen, T. Hansen, B.-O. Brandsdal, M. Havelkova and M. B. Strøm, J. Pept. Sci., 2012, 18, 170 CrossRef PubMed.
- D. Seebach, M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913 CrossRef CAS.
- D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015 RSC.
- K. Gademann, T. Hintermann and J. V. Schreiber, Curr. Med. Chem., 1999, 6, 905 CAS.
- T. Beke, C. Somlai and A. Perczel, J. Comput. Chem., 2006, 27, 20 CrossRef CAS PubMed.
- D. W. Hoskin and A. Ramamoorthy, Biochim. Biophys. Acta, 2008, 1778, 357 CrossRef CAS PubMed.
- M. B. Strøm, B. E. Haug, M. L. Skar, W. Stensen, T. Stiberg and J. S. Svendsen, J. Med. Chem., 2003, 46, 1567 CrossRef PubMed.
- T. Hansen, T. Alst, M. Havelkova and M. B. Strøm, J. Med. Chem., 2010, 53, 595 CrossRef CAS PubMed.
- T. Hansen, D. Ausbacher, G. E. Flaten, M. Havelkova and M. B. Strøm, J. Med. Chem., 2011, 54, 858 CrossRef CAS PubMed.
- T. Hansen, M. K. Moe, T. Anderssen and M. B. Strøm, Eur. J. Drug Metab. Pharmacokinet., 2012, 37, 191 CrossRef CAS PubMed.
- A. Gaucher, Y. Zuliani, D. Cabaret, M. Wakselman and J. P. Mazaleyrat, Tetrahedron: Asymmetry, 2001, 12, 2571 CrossRef CAS.
- M. D. Reddy and E. B. Watkins, J. Org. Chem., 2015, 80, 11447 CrossRef CAS PubMed.
- S. Capone, P. Walde, D. Seebach, T. Ishikawa and R. Caputo, Chem. Biodiversity, 2008, 5, 16 CAS.
- D. Lin, G. Deng, J. Wang, X. Ding, H. Jiang and H. Liu, J. Org. Chem., 2010, 75, 1717–1722 CrossRef CAS PubMed.
- L. Ducry, S. Reinelt, P. Seiler, F. Diederich, D. R. Bolin, R. M. Campbell and G. L. Olson, Helv. Chim. Acta, 1999, 82, 2432 CrossRef CAS.
- R. Moumné, B. Denise, A. Parlier, S. Lavielle, H. Rudler and P. Karoyan, Tetrahedron Lett., 2007, 48, 8277 CrossRef.
- A. Romanens and G. Bélanger, Org. Lett., 2015, 17, 322 CrossRef CAS PubMed.
- F. Zhou, G.-J. Cheng, W. Yang, Y. Long, S. Zhang, Y.-D. Wu, X. Zhang and Q. Cai, Angew. Chem., Int. Ed., 2014, 53, 9555 CrossRef CAS PubMed.
- Y.-C. Ko and J.-L. Zhu, Synthesis, 2007, 3659 CAS.
- E. Doni, B. Mondal, S. O'Sullivan, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2013, 135, 10934 CrossRef CAS PubMed.
- J. C. Hessler, J. Am. Chem. Soc., 1913, 35, 990 CrossRef CAS.
- H. Oediger and F. Möller, Liebigs Ann., 1976, 348 CrossRef CAS.
- E. C. d. Lima, C. C. d. Souza, R. d. O. Soares, B. G. Vaz, M. N. Eberlin, A. G. Dias and P. R. R. Costa, J. Braz. Chem. Soc., 2011, 22, 2186 CrossRef.
- X. Yang and V. B. Birman, Org. Lett., 2009, 11, 1499 CrossRef CAS PubMed.
- S. Caddick, D. B. Judd, A. K. d. K. Lewis, M. T. Reich and M. R. V. Williams, Tetrahedron, 2003, 59, 5417 CrossRef CAS.
- J. Seyden-Penne, Reductions by the Alumino- and Borohydrides in Organic Synthesis, Wiley-VCH, USA, 2nd edn, 1997 Search PubMed.
- K. Nagata, D. Sano, Y. Shimizu, M. Miyazaki, T. Kanemitsu and T. Itoh, Tetrahedron: Asymmetry, 2009, 20, 2530 CrossRef CAS.
- S. Narasimhan and R. Balakumar, Aldrichimica Acta, 1998, 31, 19 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/c6ob01219a |
| This journal is © The Royal Society of Chemistry 2016 |