
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biosynthesis-driven structure–activity relationship study of premonensin-derivatives†
A.
Ismail-Ali‡
a,
E. K.
Fansa‡
b,
N.
Pryk
a,
S.
Yahiaoui
c,
S.
Kushnir
a,
M.
Pflieger
 a,
A.
Wittinghofer
*b and
F.
Schulz
*a
a,
A.
Wittinghofer
*b and
F.
Schulz
*a
aFakultät für Chemie und Biochemie, Organische Chemie 1, Ruhr-Universität Bochum, Universitätsstr. 150, 44780 Bochum, Germany. E-mail: frank.schulz@rub.de
bMax-Planck-Institut für Molekulare Physiologie, Otto-Hahn-Str. 11, 44227 Dortmund, Germany. E-mail: alfred.wittinghofer@mpi-dortmund.mpg.de
cCentre d'Etudes et de Recherche sur le Médicament de Normandie UPRES EA 4258, Université de Caen Basse-Normandie, FR CNRS 3038 INC3 M, Bd Becquerel, 14032 Caen Cedex, France
First published on 19th July 2016
Abstract
The controlled derivatization of natural products is of great importance for their use in drug discovery. The ideally rapid generation of compound libraries for structure–activity relationship studies is of particular concern. We here use modified biosynthesis for the generation of such a library of reduced polyketides to interfere with the oncogenic KRas pathway. The polyketide is derivatized via side chain alteration, and variations in its redox pattern and in its backbone chain length through manipulation in the corresponding polyketide synthase. Structural and biophysical analyses revealed the nature of the interaction between the polyketides and KRas-interacting protein PDE6δ. Non-natural polyketides with low nanomolar affinity to PDE6δ were identified.
Introduction
Reduced polyketides form a well-known group of natural products with a multitude of bioactivities and high structural diversity. Their controlled derivatization is often limited to a few orthogonal functional groups available and thus is a main obstacle for their introduction into modern medicinal chemistry programmes.1 Engineered biosynthesis has emerged in recent years as a toolbox that complements total synthesis in pursuing this goal. Strategies in which the use of synthetic building blocks as supplements in fermentations reveal flexibility in the specificity of the biosynthetic enzymes and pave the way towards non-biological derivatives are of particular relevance.2 Structure–activity relationships (SAR) play a pivotal role in modern drug discovery. Low yields in engineered biosynthesis, are frequent and limit SAR to a few described cases.3 The knowledge-guided construction of focused compound libraries is important for efficient SAR studies using natural products. This requires different suitable approaches towards the modification of the biosynthetic machinery and, ideally, a preliminary understanding of the ligand–target interactions.Approaches to the derivatization of polyketides via artificial extender units have been reported in recent years, leading to derivatives of several different natural products. This was enabled by either site-directed mutagenesis or by the innate promiscuity of acyltransferase domains.4 In this context, we described the precursor-directed biosynthesis of the non-natural shunt product premonensin.5 In that study, the intrinsic substrate promiscuity of the monensin polyketide synthase (PKS) acyltransferase in module 5 was exploited by supplying small-scale cultures of S. cinnamonensis A495![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 with several malonic acid derivatives bearing different substituents at position 2. In the same study, premonensin B was shown to bind to the delta subunit of the human phosphodiesterase 6 (PDE6δ).5 The prenyl-binding protein PDE6δ mediates the shuttling of the oncoprotein KRas between membrane compartments, thereby, facilitating its plasma membrane localization and activity.7 Thus, small molecules inhibiting the interaction between KRas and PDE6δ have been considered as promising drug candidates targeting Ras.8 Based on these findings, we opted to investigate the interactions between premonensin and PDE6δ with the intention of identifying derivatives with an increased potency.
6 with several malonic acid derivatives bearing different substituents at position 2. In the same study, premonensin B was shown to bind to the delta subunit of the human phosphodiesterase 6 (PDE6δ).5 The prenyl-binding protein PDE6δ mediates the shuttling of the oncoprotein KRas between membrane compartments, thereby, facilitating its plasma membrane localization and activity.7 Thus, small molecules inhibiting the interaction between KRas and PDE6δ have been considered as promising drug candidates targeting Ras.8 Based on these findings, we opted to investigate the interactions between premonensin and PDE6δ with the intention of identifying derivatives with an increased potency.
Results and discussion
We set up crystallization trials using different premonensin derivatives to gain structural insights into the mode of interaction between premonensin and PDE6δ. We were able to solve the structure of PDE6δ in complex with premonensin A at 2.4 Å (data collection and refinement statistics are summarized in Table S1†).Superimposition of the PDE6δ–premonensin A complex structure with the structure of the PDE6δ–Rheb complex (PDB code: 3T5G) (Fig. 1A; left) shows that premonensin A inserts into the hydrophobic cavity of PDE6δ, forming a similar hydrophobic interaction pattern as that mediated by the farnesyl moiety of Rheb (Fig. 1A; right). However, several additional interactions between premonensin A and PDE6δ can be identified. The ethyl group on C16 of premonensin A forms hydrophobic interactions with the side chains of residues W90 and I109 from PDE6δ (Fig. 1B; left). Moreover, the methyl ketone moiety of premonensin A mediates two H-bonds with the side chains of residues Q78 and R61 from PDE6δ (Fig. 1B; right).
 | ||
| Fig. 1 Crystal structure of premonensin A in complex with PDE6δ. (A) Superimposition of PDE6δ–premonensin A complex with the PDE6δ–Rheb complex (PDB code: 3T5G). Premonensin A (green) inserts into the hydrophobic pocket of PDE6δ (grey) and retains the hydrophobic contacts as mediated by the farnesyl moiety of Rheb (blue). (B) Selected additional contacts mediated by premonensin A with the side chains of PDE6δ. The ethyl group on C16 (arrow) forms hydrophobic interactions with W90 and I109 (left). The methyl ketone group forms two hydrogen bonds with Q78 and R61 (right). Note the pseudocyclic orientation of the region involving the methyl ketone. | ||
These additional contacts of premonensin A might lead to increasing affinity towards PDE6δ as compared to farnesylated Rheb (Kd 214 ± 10 nM). We decided to explore the SARs using an engineered biosynthesis approach to further scan the interaction in the binding pocket.
Initially, we opted for precursor-directed biosynthesis to replace the substituent at C16 with different alkyl-chains that would point towards W90 instead of the ethyl chain in Fig. 1. Based on our earlier investigations on the substrate flexibility of the monensin PKS, we supplied 1.8 L of SM16-cultures of S. cinnamonensis A495 with 10 mM of 2-propyl- and 2-butylmalonic acid-SNAC ester, respectively, and isolated 5.0 mg 16-propyl premonensin (1) and 3.8 mg 16-butyl premonensin (2) from the resulting fermentations. Identity of the compounds was confirmed by nuclear magnetic resonance (NMR) spectroscopy for the first time (Fig. 2 and ESI, chapter 7†).
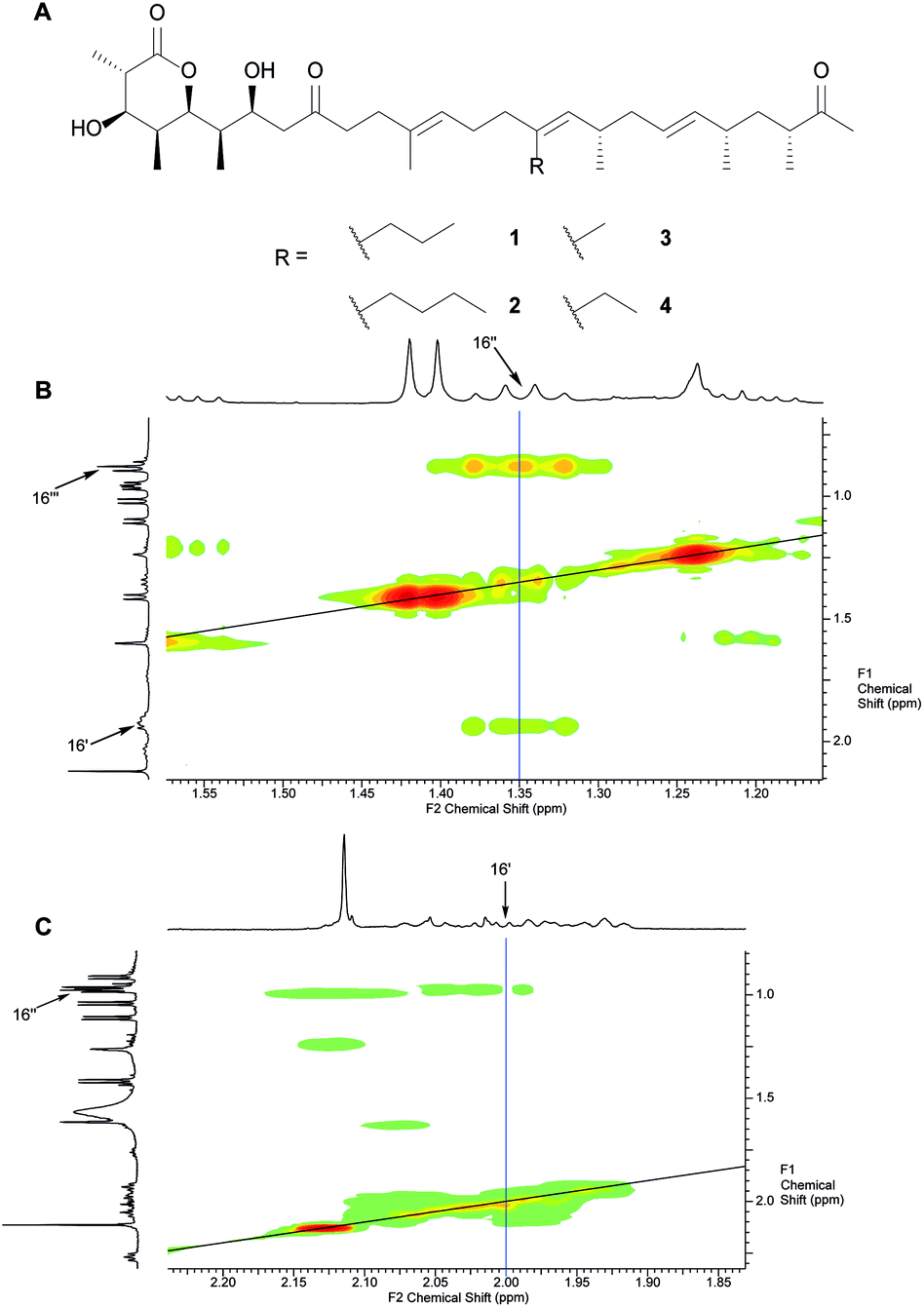 | ||
| Fig. 2 A. Premonensin derivatives obtained by precursor-directed biosynthesis. 16-Propyl premonensin (1) results from feeding 2-propylmalonic acid SNAC ester to S. cinnamonensis A495, 16-butyl premonensin (2) from an analogous experiment with 2-butylmalonic acid SNAC. 3 and 4 are premonensin B and A, resulting from a non-supplemented fermentation of S. cinnamonensis A495. B. H,H-COSY of 1, highlighting the 3J-coupling between protons at C16′′ with the neighbours at C16′′′ and C16′. C. H,H-COSY of 4, showing the 3J-coupling between protons at C16′ with its neighbours at C16′′, thereby, highlighting the key difference between the different compounds. See ESI (chapter 7)† for full assignments, including complete position numbering. | ||
Additionally, the pseudocyclic orientation of premonensin (Fig. 1B) is important for the formation of the directional H-bonds to Q78 and R61.
We, hence, chose a compound from our premonensin library reported previously to probe this interaction. In premonensin-ER20 (Fig. 3A, 5 and 6),9 the pseudocyclic orientation of the premonensin chain to allow for the interaction between the methyl ketone and R61 and Q78 would be disfavoured in comparison to 1–4, as rotation around the π-bond between C22 and C23 is hindered (highlighted in red in Fig. 3A). The antibiotically active premonensin-ER20 is the result of an inactivation of the enoylreductase domain in module 2 of the monensin PKS in S. cinnamonensis through point mutations in its NADPH-binding Rossmann fold.
 | ||
| Fig. 3 A. Premonensin ER20-A (5) and -B (6). B. Truncated premonensin A (7) and B (8). C. H-NMR of the vinylic region of 7 (top) and 3 (bottom) showing the missing vinylic proton. See ESI (chapter 7)† for full assignments. | ||
The long, hydrophobic chain of premonensin may be important for two aspects. Firstly, it can serve as a scaffold for positioning the polar head groups as well as the side-chain at C16 in proper orientations. Secondly, it can contribute to the affinity by hydrophobic interactions with the equally long and hydrophobic-binding pocket of PDE6δ. To probe these interactions, we isolated the novel premonensin derivatives that were produced by a spontaneous variant of S. cinnamonensis A495. The latter appeared on several independent occasions during experiments on the genetic modification of the strain. The NMR analysis of the fermentation products demonstrates the production of shortened derivatives of premonensin (7 and 8, see Fig. 3B). Following the colinearity principle of polyketide biosynthesis, these products must arise from a bimodular skipping event of modules 7 and 8 in the monensin PKS. This pathway towards compounds 7 and 8 is further corroborated by the isolation of a shunt product after module 6 (see ESI, Fig. 21–28†), which indicates a slow transfer between module 6 and the subsequent extension module, in this case, module 9. To the best of our knowledge, this is the first bimodular skipping event reported for type I PKS. The underlying biosynthetic mechanism remains elusive to date.
We used fluorescently labelled farnesylated and carboxy methylated Rheb peptide and performed competitive fluorescence polarization measurements, as previously described, to measure the interaction of derivatives with PDE6δ.8 The results showed that premonensin derivatives bind to PDE6δ with affinities in the low nanomolar or submicromolar range (Fig. 4).
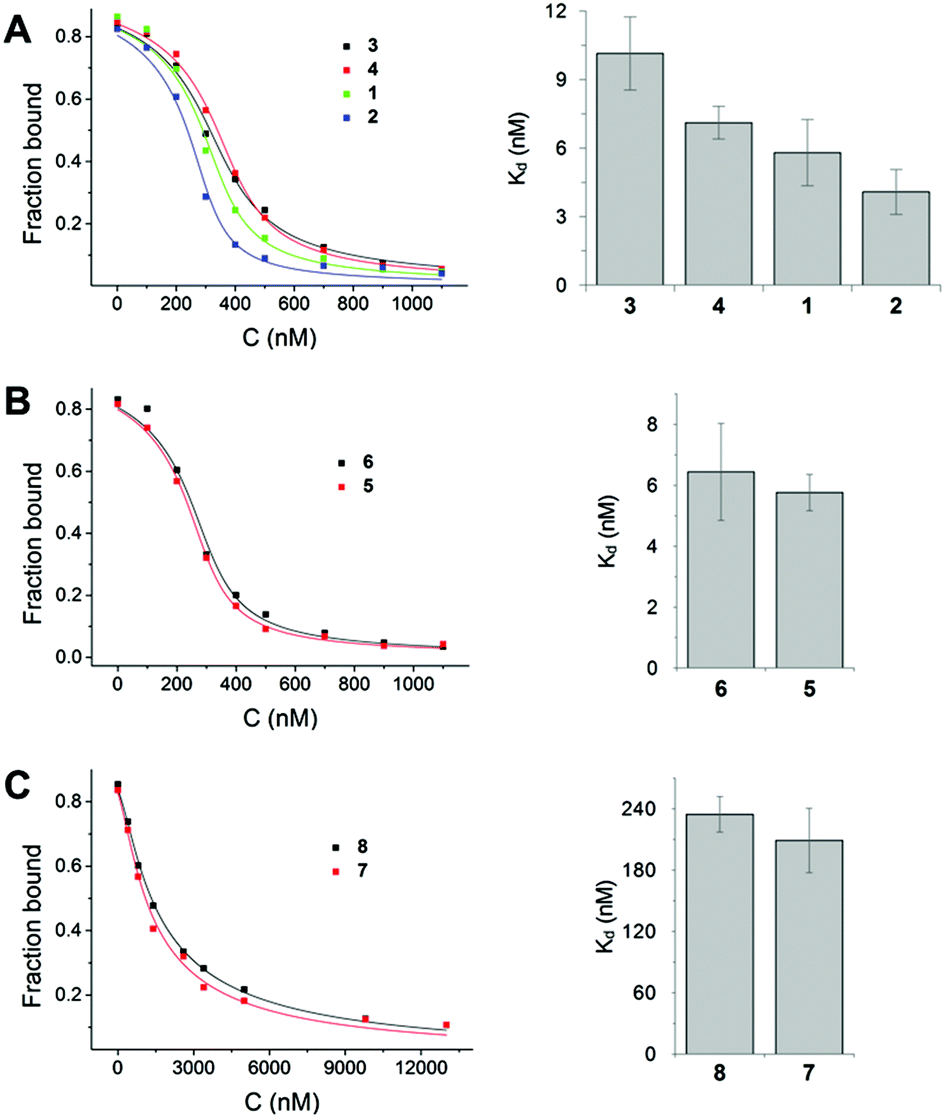 | ||
| Fig. 4 Affinity determination of premonensin derivatives to PDE6δ. Competitive fluorescence polarization measurements by titration of increasing concentrations of premonensins into a preformed complex of 25 nM FITC-labelled Rheb peptide and 350 nM PDE6δ. Titration data were fitted with a competition model and show displacement of the ligand from PDE6δ by the polyketides. | ||
Based on the affinity values and the chemical structure, we classified the eight premonensin derivatives into three binding modes. Compounds 1, 2, 3 and 4 bind to PDE6δ with affinities of 5.8, 4.0, 10.1 and 7.1 nM, respectively, as shown in Fig. 4A. These four derivatives differ only in the length of the alkyl side chains at C16 and can, thus, maintain the same interaction mode with PDE6δ. The increase in the affinity from compound 4 (premonensin A) to compound 2 (16-butylpremonensin) could be explained by the increase of hydrophobic interactions between the alkyl chains on C16 and the interacting residues W90 and I109 of PDE6δ.
Compounds 5 and 6 also bind to PDE6δ with low nanomolar affinities of 5.7 nM and 6.4 nM, respectively (Fig. 4B). However, the interaction of these two derivatives is expected to be different from compounds 1, 2, 3 and 4, as rotation around the double bond between C22 and C23 is prevented, thus, perturbing the orientation of the methyl ketone moiety towards Q78 and R61 in PDE6δ, as seen in the crystal structure with 4.
The shorter chain length of compounds 7 and 8, compared to the other premonensin derivatives, resulted in an approximately 50-fold lower affinity towards PDE6δ, with Kd values of 208.9 nM and 234.5 nM, respectively (Fig. 4C). This could be explained by the fewer hydrophobic interactions within the hydrophobic cavity of PDE6δ or the lack of an ideal conformation that would facilitate further non-covalent interactions.
Compounds 3, 4, 7 and 8 were subjected to proliferation assays using the KRAS-dependent cell line RPMI-8226 (multiple myeloma). Compounds 1 and 2 were found to be poorly soluble, while compounds 5 and 6 yielded inconclusive results due to partial decomposition under assay conditions.10 Compounds 3 and 4 showed an IC50 value of 11.2 μM and 8.2 μM, respectively. The truncated derivatives 7 and 8 showed low activity (IC50 > 18 μM) (see ESI, Table 2,† for further details).
Conclusions
In conclusion, we demonstrate here the structure activity relationship between PDE6δ and premonensin derivatives afforded by different approaches to engineered biosynthesis. Additionally, we report on the first described bimodular skipping event involving a type I PKS. A crystal structure showed premonensin A binding to the hydrophobic cavity of PDE6δ and Kd measurements demonstrated nanomolar affinities for the derivatives. Thus, these polyketides, originating from engineered biosynthesis, are shown to perturb the interaction between PDE6δ and farnesylated Ras proteins, rendering them potential inhibitors of the oncogenic KRas pathway.Experimental
Crystallography
Stock solutions of premonensin derivatives (10 mM) were mixed with PDE6δ (500 μM) at a 1:1 molar ratio in the crystallization buffer (30 mM Tris-HCL, pH 7.5, 150 mM NaCl and 3 mM DTE). The crystallization hits of premonensin A in complex with PDE6δ appeared in the Core II suite from Qiagen (0.16 M ammonium sulphate, 0.08 M sodium acetate anhydrous pH 4.6, 20% PEG 4000 and 20% glycerol) at 20 °C. The diffraction dataset was collected at the X10SA beam-line of the Suisse Light Source, Villigen. Data processing was performed using the XDS programme. PDE6δ from the PDE6δ–Rheb complex structure (PDB code: 3T5G) was used as a model. The structure was solved by molecular replacement and, finally, refined by several rounds using Molrep and REFMAC5 programmes from CCP4 (suite).
Competitive fluorescence polarization
Fluorescence polarization measurements were carried out using a Fluoromax-4 spectrophotometer (HORIBA Jobin Yvon, Munich, Germany) in the crystallization buffer at 20 °C. Excitation and emission wavelengths of 495 nm and 520 nm, respectively, were used for the fluorescein-labelled Rheb peptide. Stock solutions of premonensin derivatives (10 mM) were prepared as described.5 Affinities were determined by titrating increasing amounts of premonensin derivatives into a preformed complex of FITC-Rheb peptide with PDE6δ. Data analysis was performed with OriginPro 9.0 with a competition model derived from the law of mass action as described.8Acknowledgements
This work was supported by the Cluster of Excellence RESOLV (EXC 1069) and by the Deutsche Forschungsgemeinschaft (DFG). A. Ismail-Ali is a fellow of the International Max-Planck Research School of Chemical and Molecular Biology. N. Pryk acknowledges a predoctoral stipend from the Jürgen Manchot-Foundation. Axel Choidas (Lead Discovery Center GmbH) is thanked for the cellular assays. The authors thank Herbert Waldmann for helpful discussions.Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed; G. M. Cragg, P. G. Grothaus and D. J. Newman, J. Nat. Prod., 2014, 77, 703–723 CrossRef PubMed.
- S. Friedrich and F. Hahn, Tetrahedron, 2015, 71, 1473–1508 CrossRef CAS; A. Kirschning and F. Hahn, Angew. Chem., Int. Ed., 2012, 51, 4012–4022 CrossRef PubMed.
- E. Schax, J.-G. Walter, H. Märzhäuser, F. Stahl, T. Scheper, D. A. Agard, S. Eichner, A. Kirschning and C. Zeilinger, J. Biotechnol., 2014, 180, 1–9 CrossRef CAS PubMed; M. A. Gregory, A. L. Kaja, S. G. Kendrew, N. J. Coates, T. Warneck, M. Nur-e-Alam, R. E. Lill, L. S. Sheehan, L. Chudley, S. J. Moss, R. M. Sheridan, M. Quimpere, M.-Q. Zhang, C. J. Martin and B. Wilkinson, Chem. Sci., 2013, 4, 1046–1052 RSC; E. M. Novoa, N. Camacho, A. Tor, B. Wilkinson, S. Moss, P. Marín-García, I. G. Azcárate, J. M. Bautista, A. C. Mirando, C. S. Francklyn, S. Varon, M. Royo, A. Cortés and L. Ribas de Pouplana, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5508–E5517 CrossRef PubMed.
- U. Sundermann, K. Bravo-Rodriguez, S. Klopries, S. Kushnir, H. Gomez, E. Sanchez-Garcia and F. Schulz, ACS Chem. Biol., 2013, 8, 443–450 CrossRef CAS PubMed; M. C. Walker, B. W. Thuronyi, L. K. Charkoudian, B. Lowry, C. Khosla and M. C. Chang, Science, 2013, 341, 1089–1094 CrossRef PubMed; I. Koryakina, J. McArthur, S. Randall, M. M. Draelos, E. M. Musiol, D. C. Muddiman, T. Weber and G. J. Williams, ACS Chem. Biol., 2012, 8, 200–208 CrossRef PubMed; K. Bravo-Rodriguez, S. Klopries, K. Koopmans, U. Sundermann, S. Yahiaoui, J. Arens, S. Kushnir, F. Schulz and E. Sanchez-Garcia, Chem. Biol., 2015, 22, 1425–1430 CrossRef PubMed.
- K. Bravo-Rodriguez, A. F. Ismail-Ali, S. Klopries, S. Kushnir, S. Ismail, E. K. Fansa, A. Wittinghofer, F. Schulz and E. Sanchez-Garcia, ChemBioChem, 2014, 15, 1991–1997 CrossRef CAS PubMed.
- M. Oliynyk, C. B. W. Stark, A. Bhatt, M. A. Jones, Z. A. Hughes-Thomas, C. Wilkinson, Z. Oliynyk, Y. Demydchuk, J. Staunton and P. F. Leadlay, Mol. Microbiol., 2003, 49, 1179–1190 CrossRef CAS PubMed; A. Bhatt, C. B. W. Stark, B. M. Harvey, A. R. Gallimore, Y. A. Demydchuk, J. B. Spencer, J. Staunton and P. F. Leadlay, Angew. Chem., Int. Ed., 2005, 44, 7075–7078 CrossRef PubMed.
- M. Schmick, N. Vartak, B. Papke, M. Kovacevic, D. C. Truxius, L. Rossmannek and P. I. H. Bastiaens, Cell, 2014, 157, 459–471 CrossRef CAS PubMed.
- G. Zimmermann, B. Papke, S. Ismail, N. Vartak, A. Chandra, M. Hoffmann, S. A. Hahn, G. Triola, A. Wittinghofer, P. I. H. Bastiaens and H. Waldmann, Nature, 2013, 497, 638–642 CrossRef CAS PubMed; G. Zimmermann, C. Schultz-Fademrecht, P. Küchler, S. Murarka, S. Ismail, G. Triola, P. Nussbaumer, A. Wittinghofer and H. Waldmann, J. Med. Chem., 2014, 57, 5435–5448 CrossRef PubMed; B. Papke, S. Murarka, H. A. Vogel, P. Martín-Gago, M. Kovacevic, D. C. Truxius, E. K. Fansa, S. Ismail, G. Zimmermann, K. Heinelt, C. Schultz-Fademrecht, A. Al Saabi, M. Baumann, P. Nussbaumer, A. Wittinghofer, H. Waldmann and P. I. H. Bastiaens, Nat. Commun., 2016, 7, 11360 CrossRef PubMed.
- S. Kushnir, U. Sundermann, S. Yahiaoui, A. Brockmeyer, P. Janning and F. Schulz, Angew. Chem., Int. Ed., 2012, 51, 10664–10669 CrossRef CAS PubMed.
- C. Scholl, S. Fröhling, I. F. Dunn, A. C. Schinzel, D. A. Barbie, S. Y. Kim, S. J. Silver, P. Tamayo, R. C. Wadlow, S. Ramaswamy, K. Döhner, L. Bullinger, P. Sandy, J. S. Boehm, D. E. Root, T. Jacks, W. C. Hahn and D. G. Gilliland, Cell, 2009, 137, 821–834 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C6OB01201A |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |