
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoronic acid/Brønsted acid co-catalyst systems for the synthesis of 2H-chromenes from phenols and α,β-unsaturated carbonyls†
Victoria
Dimakos
,
Tishaan
Singh
and
Mark S.
Taylor
*
Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada. E-mail: mtaylor@chem.utoronto.ca
First published on 17th June 2016
Abstract
Protocols for the synthesis of substituted 2H-chromenes from α,β-unsaturated carbonyls and phenols are described. Optimal combinations of arylboronic acids and Brønsted acids have been identified, such that both can be employed in catalytic quantities to accelerate these condensations. The method has been used to synthesize a variety of substituted 2H-chromenes, as well as photochromic naphthopyrans. The use of pentafluorophenylboronic acid and diphenylphosphinic acid enabled an expansion of the electrophile scope to include α,β-unsaturated ketones. Hall's ‘phase-switching’ of boronic acids has been exploited to achieve the separation of the two co-catalysts from unpurified reaction mixtures by a simple liquid–liquid extraction.
Introduction
The 2H-chromene ring system is a core structure of numerous biologically active natural products,1 and its annelated congeners (naphthopyrans and related structures) display useful photochromic properties.2 Several methods exist for the synthesis of 2H-chromenes, enabling access to a variety of substitution patterns.3 Transition metal-catalyzed transformations that have been exploited for 2H-chromene synthesis include alkyne hydroarylation,4 allylic substitution5 and olefin metathesis.6 Approaches based on Brønsted or Lewis acid-catalyzed condensations7–9 or cyclizations10 have also been reported.Boronic acid-promoted condensations of phenols with α,β-unsaturated carbonyl compounds provide access to 2H-chromenes from readily available starting materials (Scheme 1). This mode of reactivity was initially reported by scientists at Merck,11 and was further developed into a general method for 2H-chromene synthesis by Snieckus and co-workers.12 The likely reaction pathway involves formation of a benzodioxaborinine intermediate by Nagata alkylation,13 followed by fragmentation to an ortho-quinone methide and electrocyclic ring closure. Over the years, boronic acid-mediated condensations of this type have been applied to the synthesis of several classes of benzopyran-based natural products.14–16
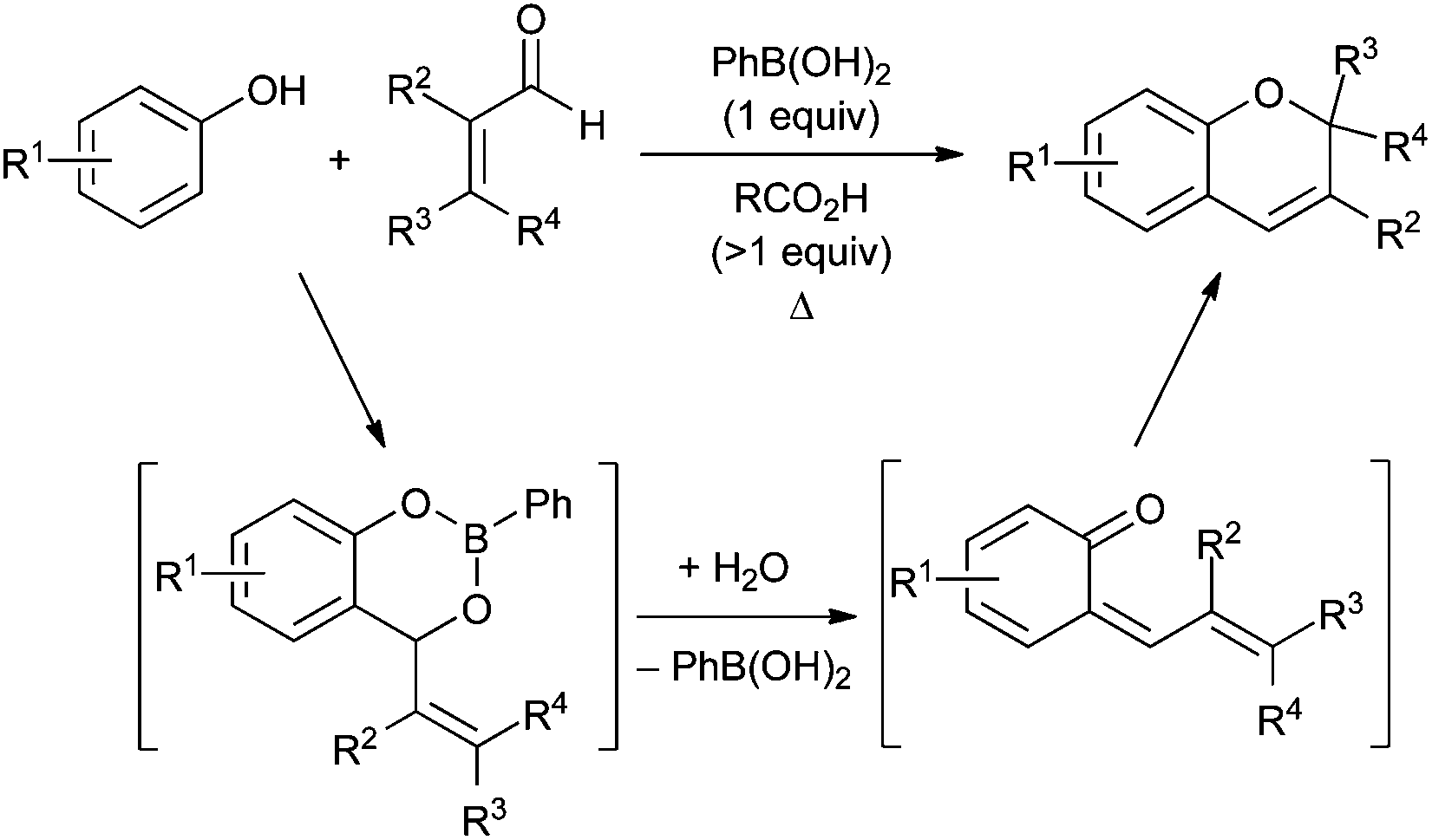 | ||
| Scheme 1 Phenylboronic acid-mediated condensations of phenols and α,β-unsaturated aldehydes. A proposed pathway is shown below the reaction equation. | ||
The protocol for boronic acid-promoted 2H-chromene synthesis has changed relatively little since Dufresne's initial report more than two decades ago. This described the use of 1.6 equivalents of phenylboronic acid (PhB(OH)2) and 30 mol% of propionic acid relative to phenol, with removal of water by azeotropic reflux in toluene using a Dean–Stark trap.11 Snieckus and co-workers reported similar conditions, employing 1 equivalent of PhB(OH)2 and an excess (>85 equiv.) of acetic acid, also in toluene with a Dean–Stark apparatus.12 A survey of Brønsted acids (including carboxylic acids, sulfonic acids and H2SO4) was carried out in 2007, revealing that propionic acid (110 equiv.) provided best results for the coupling of umbelliferone and 3-methyl-2-butenal (1 equiv. of PhB(OH)2 in toluene at reflux). Each of these studies points towards the combination of stoichiometric phenylboronic acid and a carboxylic acid promoter.
Although the thermolysis of the benzodioxaborinine intermediate provides a pathway for catalyst turnover, examples of protocols that employ substoichiometric quantities of arylboronic acid are scarce. Wilson and co-workers showed that 25 mol% of PhB(OH)2 was able to promote the condensation of phloroglucinol with 3-methyl-2-butenal, yielding a mixture of bis- and tris-annulated products (39% and 55% yields, respectively, for a total of >9 turnovers).17 As part of the synthesis of a series of pyranocarbazole alkaloids, Knölker's group found that condensations of citral with a hydroxycarbazole could be achieved in 75% yield using 20 mol% of PhB(OH)2 in toluene at reflux.15 Another relevant study by McCubbin showed that pentafluorophenylboronic acid (10 mol%) was able to catalyse condensations of 2-naphthol with propargylic alcohols, generating substituted naphthopyran products.18 These same conditions were used to effect Friedel–Crafts propargylations of other electron-rich aromatics, and it is not clear whether there is a direct mechanistic connection to the above-mentioned phenol–enal annulations. In any case, it appeared to us that evaluating a range of organoboron acid and Brønsted acid catalysts might be an opportunity to improve the efficiency or expand the scope of such condensations, especially given the impressive progress that has been achieved in the development of ‘designer’ boronic acids for use in catalysis in recent years.19,20
Here, we describe a systematic evaluation of organoboron acid/Brønsted acid combinations for the synthesis of 2H-chromenes from phenols and α,β-unsaturated carbonyl compounds. By variation of the two promoters, we have developed efficient and operationally simple protocols in which both are employed in catalytic quantities. These conditions have been applied to a range of reaction partners, including α,β-unsaturated ketones as well as aldehydes.
Results and discussion
3-Methoxyphenol (2a) and enone 3a were chosen as the substrates for evaluation of organoboron acid and Brønsted acid co-catalysts (Table 1). Previous reports of boronic acid-promoted 2H-chromene synthesis describe couplings of α,β-unsaturated aldehydes, but not ketones, and so we anticipated that screening against this substrate combination might be useful for identifying particularly active catalysts.|
|
|||
|---|---|---|---|
| Entry | Organoboron acid | Brønsted acid HX | Yielda |
| a Determined by 1H NMR spectroscopy of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. Reactions were carried out under inert atmosphere. | |||
| 1 | Ph2BOH | — | <5% |
| 2 | 1a | — | 5% |
| 3 | Ph2BOH | PhCO2H | 15% |
| 4 | 1a | PhCO2H | 30% |
| 5 | PhB(OH)2 | PhCO2H | 20% |
| 6 | 1b | PhCO2H | 30% |
| 7 | 1c | PhCO2H | 40% |
| 8 | 1d | PhCO2H | 45% |
| 9 | 1e | PhCO2H | 45% |
| 10 | PhB(OH)2 | CH3CH2CO2H | 10% |
| 11 | PhB(OH)2 | Ph2PO2H | 60% |
| 12 | 1c | CH3CH2CO2H | 60% |
| 13 | 1c | Ph2PO2H | 70% |
| 14 | 1c | CSA | <5% |
| 15 | 1e | CH3CH2CO2H | 15% |
| 16 | 1e | Ph2PO2H | 70% |
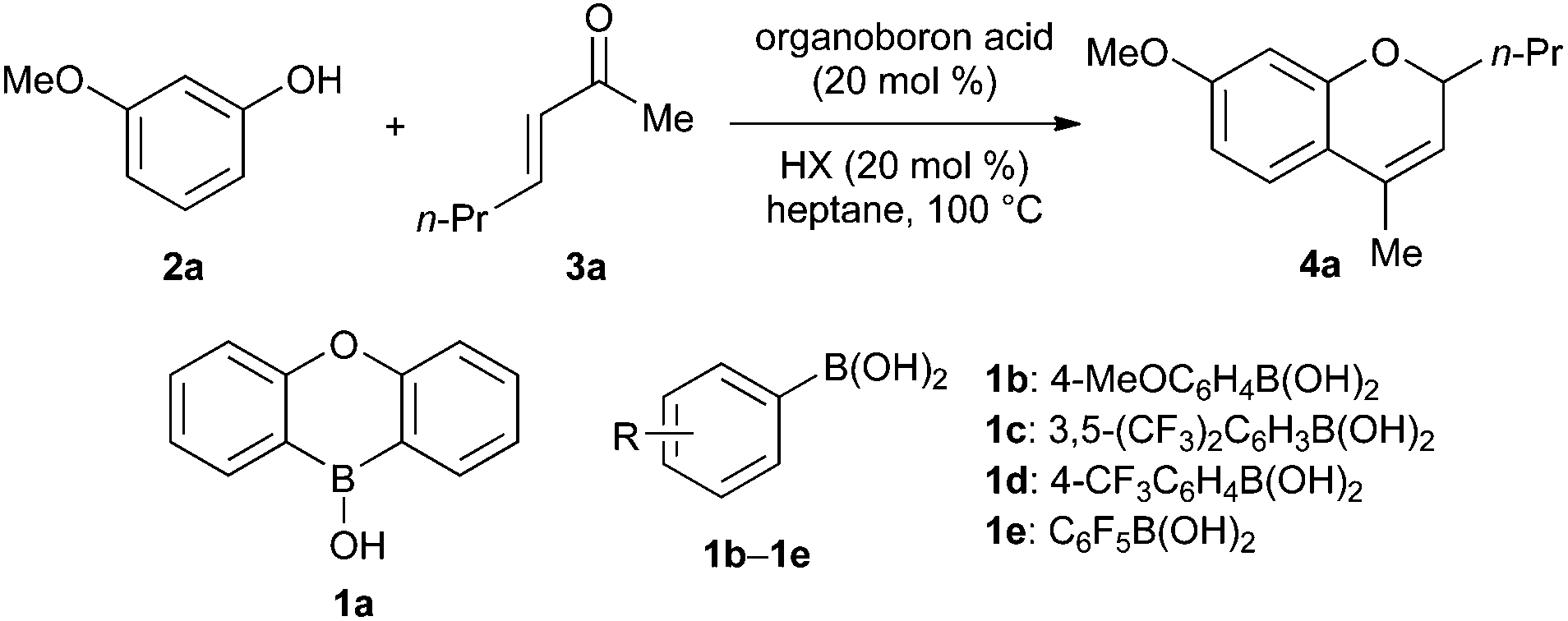
We began by testing diarylborinic acids R2BOH: these are more Lewis acidic than the corresponding boronic acids RB(OH)2, and are useful catalysts for several types of transformations, including carbon–carbon21 and carbon–heteroatom bond-forming reactions,22 dehydrations23 and oxidations.24 However, both Ph2BOH and oxaboraanthracene-derived borinic acid 1a showed only modest activity: yields of 4a were low (≤5%) in the absence of a Brønsted acid (entries 1 and 2), and were not significantly different from that obtained with PhB(OH)2 when benzoic acid was employed as a co-catalyst (entries 3–5). On the other hand, an improvement in yield was achieved using more Lewis acidic arylboronic acids 1c–1e (entries 7–9). Several Brønsted acids, including carboxylic, phosphinic and sulfonic acids, were then surveyed (entries 10–16). Diphenylphosphinic acid provided superior results to propionic and benzoic acid, whereas the use of the stronger acid camphorsulfonic acid (CSA) resulted in decomposition. Although further mechanistic study is needed to make a definitive conclusion on this point, we speculate that increasing the acidity of both the boronic acid and Brønsted acid components (relative to the original PhB(OH)2/carboxylic acid system) accelerates the addition of the phenol to the rather poorly electrophilic enone. It should be noted that the yields reported in Table 1 are for reactions carried out under an inert argon atmosphere. Inferior results were obtained when air was not excluded. However, azeotropic removal of water using a Dean-Stark apparatus was not required under this protocol.
Having identified the combination of pentafluorophenylboronic acid and diphenylphosphinic acid (20 mol% each) as being optimal, we investigated the scope of this process (Scheme 2). Enones 3a–3e were successfully coupled with 3-methoxyphenol and 1-naphthol, yielding 2,4-disubstituted and 2,2,4-trisubstituted 2H-chromene products. These were obtained in high levels of purity after column chromatography on silica gel, with the exception of products 4f and 4g, which were isolated along with small amounts (<10%) of the corresponding 4-methylenechromane regioisomers.
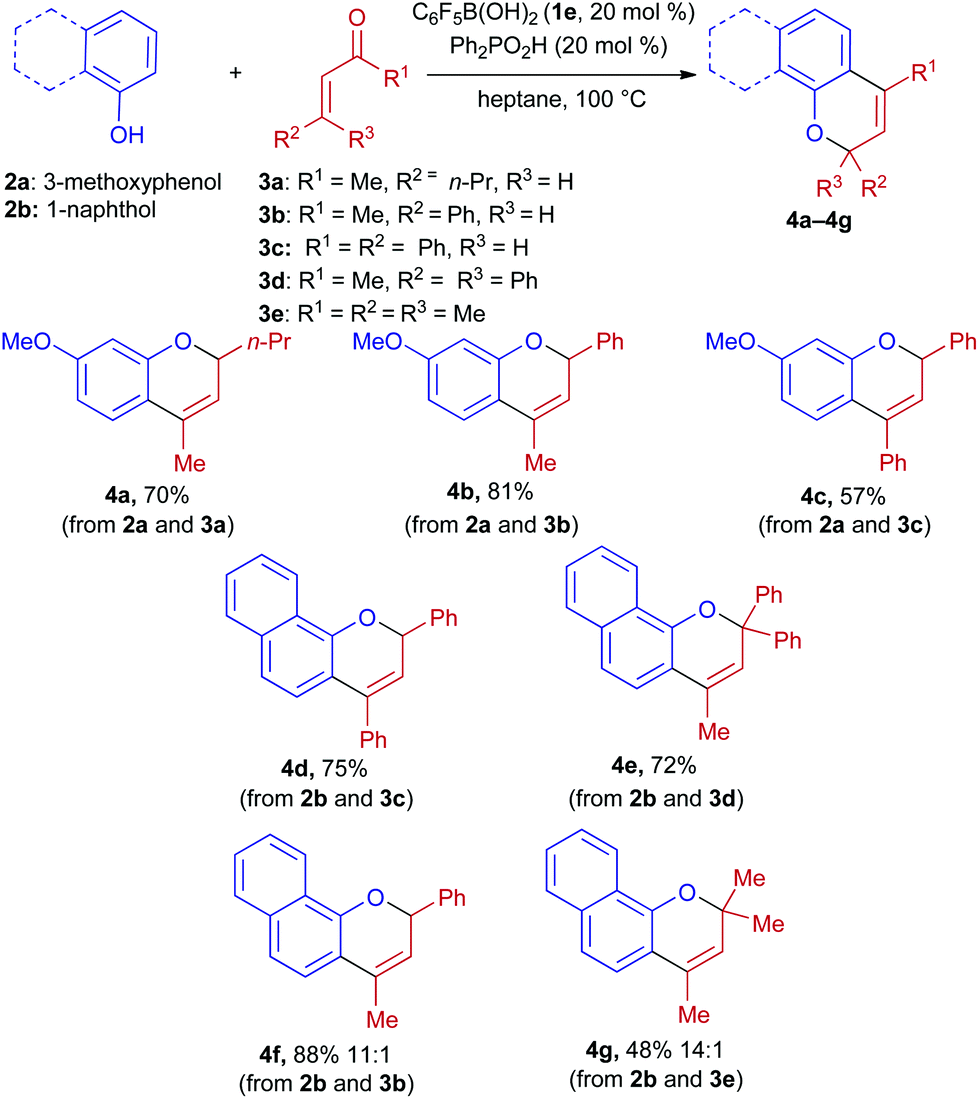 | ||
| Scheme 2 Condensations of α,β-unsaturated ketones with phenol derivatives. Reaction conditions: phenol (0.2 mmol), enone (0.4 mmol), 1e (20 mol%), Ph2PO2H (20 mol%) in heptane (1 mL) at 100 °C for 3 h under argon. Isolated yields after purification by chromatography on silica gel are listed. Products 4f and 4g were obtained as mixtures of endocyclic and exocyclic olefin isomers. | ||
Condensations of α,β-unsaturated aldehydes with phenols were also investigated under boronic acid/Brønsted acid co-catalysis (Scheme 3). Unlike the aforementioned reactions with enones, syntheses of 2H-chromenes from α,β-unsaturated aldehydes did not require an argon atmosphere, and benzoic acid (rather than Ph2PO2H) was found to be the most suitable Brønsted acid catalyst. Variation in the identity of the optimal boronic acid was also observed. The parent PhB(OH)2 was able to promote condensations of activated phenols, and provided superior results in cases where electron-deficient 1c promoted decomposition of the phenol (2a) or aldehyde (5b) component. On the other hand, 1c displayed higher activity than Ph2BOH for condensations of less nucleophilic phenols (2b and 2g–2i). When unsubstituted phenol was subjected to these conditions, little to no product was observed.
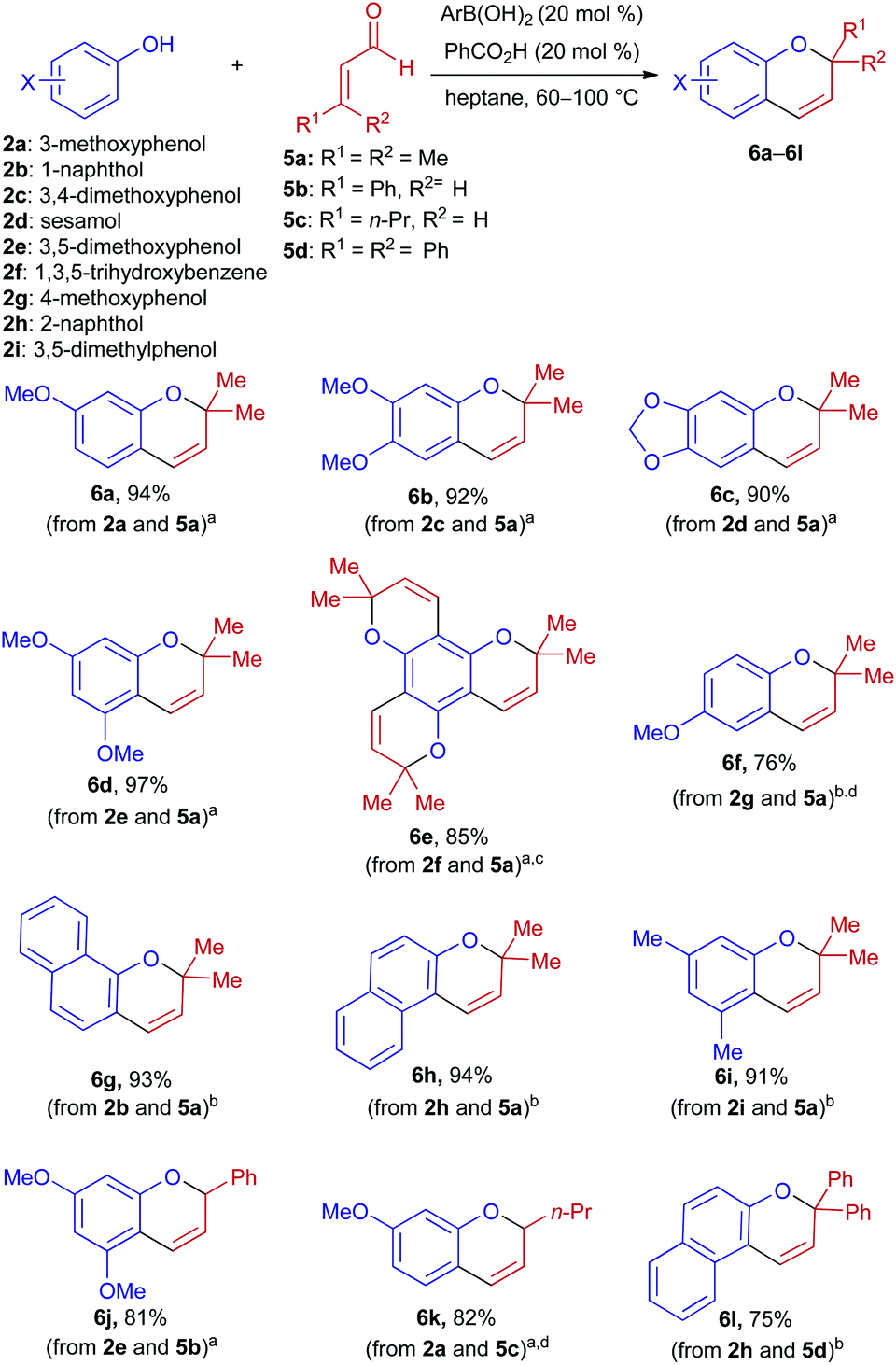 | ||
Scheme 3 Condensations of α,β-unsaturated aldehydes with phenol derivatives. Reaction conditions: phenol (0.5 mmol), aldehyde (1.0 mmol), ArB(OH)2 (5–20 mol%), PhCO2H (20 mol%), in heptane (2.5 mL) at 60–100 °C for 17 h. For details, see the Experimental section. Isolated yields after purification by chromatography on silica gel are listed. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PhB(OH)2 was used as the catalyst. b1c was used as the catalyst. cThe reaction was carried out using 6.0 equiv. of 5a. dPh2PO2H (10 mol%) was used as the Brønsted acid co-catalyst. PhB(OH)2 was used as the catalyst. b1c was used as the catalyst. cThe reaction was carried out using 6.0 equiv. of 5a. dPh2PO2H (10 mol%) was used as the Brønsted acid co-catalyst. | ||
To establish the preparative utility of this method, we undertook the synthesis of naphthopyran 6g on gram scale (5 mmol). For this larger-scale experiment, we took advantage of the ‘phase-switching’ protocol developed by Hall and co-workers.25 This technique uses simple liquid–liquid extractions to separate boronic acids from other organic compounds, based on the pH-switchable solubility of boronic acids in aqueous sorbitol solution (increased solubility at high pH due to formation of a tetracoordinate boronate ester). In the present case, working up the reaction mixture by extraction with basic aqueous sorbitol solution resulted in a straightforward separation of the product (organic phase) from the arylboronic acid and carboxylic acid components (aqueous phase). The product was purified by elution through a short plug of silica gel, while the catalyst mixture was recovered after acidification and extraction of the aqueous sorbitol solution (Scheme 4). The recovered catalyst mixture was re-used without further purification in a second gram scale synthesis, affording 6g in 70% yield.
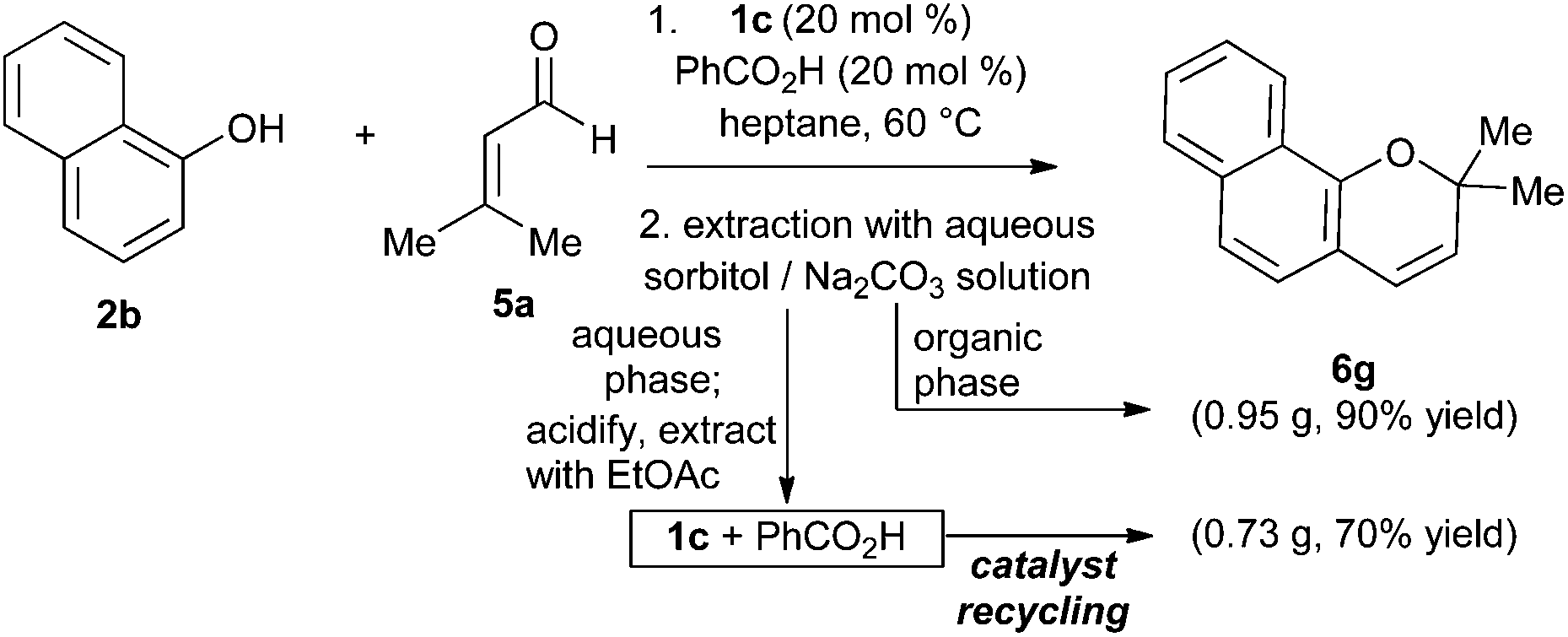 | ||
| Scheme 4 Separation of co-catalysts from naphthopyran product by phase-switching. | ||
Hall and co-workers have shown that electron-deficient boronic acids catalyze the Meyer–Schuster rearrangement of propargylic alcohols to α,β-unsaturated compounds.26 We envisioned that an in situ rearrangement of this type could enable the synthesis of 2H-chromene derivatives from propargylic alcohols under our optimized reaction conditions. To test this idea, we investigated condensations of 2-naphthol with readily available 1,1-diarylpropyn-1-ol derivatives as a way to access gem-diaryl-substituted naphthopyran products (Scheme 5). The latter have been studied extensively for applications in ophthalmic lenses due to their useful photochromic properties.2 Using 20 mol% each of 1c and benzoic acid, naphthopyran 6l was generated from 2-naphthol and propargylic alcohol 7a. The yield of this process was comparable to that obtained from enal 5d (Scheme 3), and the isomerization of 7a to 5d was evident upon monitoring the condensation reaction by thin layer chromatography. In a similar way, the photochromic and solvatochromic naphthopyran 6m was synthesized from 7b.27 As mentioned in the introduction, McCubbin and co-workers have also developed condensations of 2-naphthol with propargylic alcohols, using pentafluorophenylboronic acid and 4 Å molecular sieves in dichloromethane at room temperature. Propargylic alcohols having a (trimethylsilyl)alkynyl or phenylalkynyl group, rather than a terminal alkynyl group, were employed. The proposed mechanism involved Friedel–Crafts allenylation followed by cyclization through addition of the phenol group across the terminal carbon–carbon double bond. Subjecting compound 7a and 2-naphthol to these conditions resulted in appreciable consumption of 7a (approximately 55% conversion), but only a 10% yield of naphthopyran 6l (conversion and yield were determined by 1H NMR with an internal standard). Thus, these superficially similar protocols appear to differ in scope, and may proceed by different mechanisms.
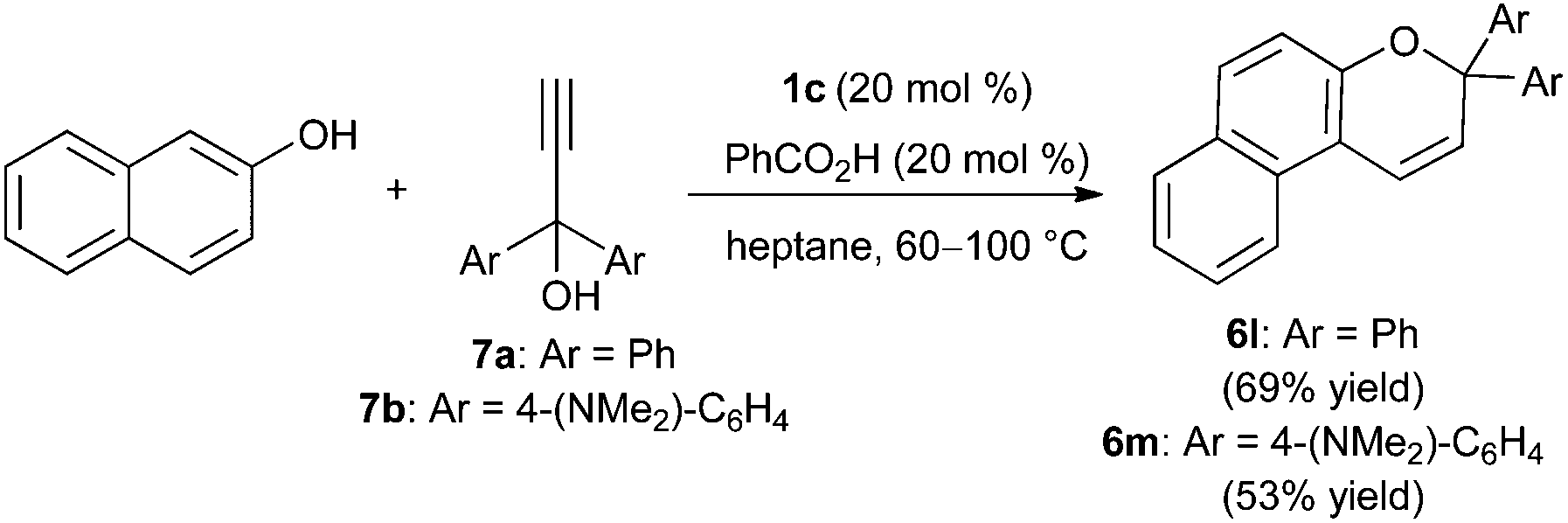 | ||
| Scheme 5 Synthesis of naphthopyrans from propargylic alcohols. | ||
Conclusions
Through variation of the arylboronic acid and Brønsted acid components, we have identified combinations that show optimal catalytic activity for 2H-chromene synthesis via condensations of phenols and α,β-unsaturated carbonyls. The use of an electron-deficient boronic acid (C6F5B(OH)2 or 3,5-(CF3)2-C6H3B(OH)2), in concert with diphenylphosphinic acid, is particularly effective for condensations of less reactive substrates such as α,β-unsaturated ketones or non-activated phenols. Hall's ‘phase-switching’ protocol has been used to achieve a convenient separation of both catalyst components from unpurified reaction mixtures by a simple liquid–liquid extraction.Experimental
General methods
Reactions were carried out without effort to exclude air or moisture, unless otherwise indicated. Stainless steel needles and syringes were used to transfer air- and moisture-sensitive liquids. Flash chromatography was carried out using neutral silica gel (60 Å, 230–400 mesh, Silicycle). Analytical thin layer chromatography was carried out using aluminum-backed silica gel 60 F254 plates (EMD), and compounds were visualized through the use of UV light or basic KMnO4 stain. HPLC grade THF was dried and purified using a solvent purification system equipped with columns of activated alumina, under nitrogen (Innovative Technology, Inc.). Anhydrous heptane was purchased from Sigma Aldrich. Distilled water was obtained from an in-house supply. All other solvents and reagents were purchased from Sigma Aldrich or Alfa Aesar and used without further purification. Screw cap test tubes were purchased from Pyrex® (13 mm × 100 mm, mfr. no. = Corning, 9825-13). Nuclear magnetic resonance (NMR) solvents were purchased from Cambridge Isotope Laboratories or from Sigma Aldrich. 1H and 13C NMR spectra were recorded in CDCl3 or C6D6 using either a Bruker Avance III 400 MHz, Varian Mercury 400 MHz or Agilent 500 MHz spectrometer. 1H NMR are reported in parts per million (ppm) relative to tetramethylsilane and referenced to residual protium in the solvent. Spectral features are tabulated in the following order: chemical shift (δ, ppm); multiplicity (s-singlet, d-doublet, t-triplet, q-quartet, m-complex multiplet); number of protons; coupling constant(s) (J, Hz). High-resolution mass spectra (HRMS) were obtained on a JEOL AccuTOF JMS-T1000LC mass spectrometer equipped with a DART (direct analysis in real time) ion source, and are not corrected for the mass of the electron. Infrared (IR) spectra were obtained on a Perkin-Elmer Spectrum 100 instrument equipped with a single-bounce diamond/ZnSe ATR accessory as solids or thin films, as indicated. Spectral features are tabulated as follows: wavenumber (cm−1); intensity (s-strong, m-medium, w-weak).:1, 1H NMR) (47.8 mg, 88%). 1H NMR (500 MHz, C6D6): δ 8.45–8.41 (m, 1H), 7.63–7.58 (m, 1H), 7.43–7.39 (m, 2H), 7.31 (d, J = 8.5 Hz, 1H), 7.26–7.17 (m, 4H), 7.15–7.07 (m, 3H), 7.08–7.03 (m, 1H), 5.86 (dq, J = 3.6, 1.8 Hz, 1H), 5.32 (dq, J = 3.2, 1.5 Hz, 1H), 1.83 (dd, J = 1.7, 1.7 Hz, 3H). 13C NMR (126 MHz, C6D6): δ 149.4, 142.2, 135.1, 130.3, 128.8, 128.2, 127.9, 127.0, 126.7, 125.9, 125.2, 122.9, 121.8, 120.6, 120.6, 117.9, 77.8, 18.3. IR (neat, cm−1): 3059 (w), 3030 (w), 2970 (w), 2915 (w), 2854 (w), 1653 (w), 1620 (w), 1564 (m), 1465 (w), 1453 (w), 1389 (m), 1377 (m), 1259 (m), 1205 (m), 1092 (m), 1074 (m), 947 (m), 811 (s), 739 (s), 695 (s), 678 (m). HRMS (DART-TOF+, m/z): Calculated for C20H17O1 [(M + H)+]: 273.1279. Found: 273.1276. Selected NMR data for the minor regioisomer: 1H NMR (500 MHz, C6D6): δ 5.51 (d, J = 2.1 Hz, 1H), 4.96 (dd, J = 11.5, 2.7 Hz, 1H), 4.76 (ddd, J = 2.1, 0.8, 0.7 Hz, 1H), 2.71 (dddd, J = 14.8, 11.5, 2.1, 2.1 Hz, 1H), 2.52 (ddd, J = 14.8, 2.7, 1.0 Hz, 1H).
:1, 1H NMR) (21.4 mg, 48%). 1H NMR (500 MHz, C6D6): δ 8.51–8.49 (m, 1H), 7.66–7.63 (m, 1H), 7.34–7.29 (m, 2H), 7.26 (ddd, J = 8.1, 6.8, 1.4 Hz, 1H), 7.23 (d, J = 8.5 Hz, 1H), 5.12 (q, J = 1.5 Hz, 1H), 1.84 (d, J = 1.5 Hz, 3H), 1.35 (s, 6H). 13C NMR (126 MHz, C6D6): δ 148.9, 135.0, 128.6, 127.9, 126.5, 126.0, 125.8, 125.6, 122.9, 121.8, 120.0, 117.4, 76.6, 28.0, 18.3. IR (neat, cm−1): 3056 (w), 2973 (w), 2923 (w), 2855 (w), 1657 (w), 1619 (w), 1565 (w), 1508 (w), 1459 (w), 1377 (s), 1358 (m), 1279 (m), 1208 (m), 1158 (m), 1146 (m), 1077 (s), 951 (m), 924 (m), 815 (s), 799 (s), 744 (s), 685 (m). HRMS (DART-TOF+, m/z): Calculated for C16H17O1 [(M + H)+]: 225.1279. Found: 225.1279. Selected NMR data for the minor regioisomer: 1H NMR (500 MHz, C6D6): δ 5.53 (dt, J = 2.3, 1.3, 1.3 Hz, 1H), 4.79 (dt, J = 2.5, 1.5, 1.5 Hz, 1H), 2.24 (dd, J = 1.4, 1.4 Hz, 2H), 1.20 (s, 6H).
:Na2CO3 (1 M:1 M) solution. The two layers were hand shaken for 10 minutes, separated and the organic layer was washed with H2O, and then saturated aq. NaCl. The organic phase was then dried with MgSO4, filtered and concentrated in vacuo to afford a brown oil. This crude oil was then loaded onto a 1.5 × 1.5′′ silica gel column, eluted with 2.5% EtOAc/hexanes (300 mL) and concentrated in vacuo to give the title compound as a yellow-orange oil (947.5 mg, 90%). Catalyst Recovery: The sorbitol layer was acidified with 4 M HCl (pH 2) and the catalyst mixture extracted three times with EtOAc, washed with H2O and then dried over MgSO4. The organic solution was then filtered and concentrated to afford a ∼1:1 mixture of boronic: benzoic acid as a light brown solid (356.5 mg, 94%). When this recovered mixture was used in the same reaction described above, the titled compound was isolated in 70% yield (728.2 mg). Workup procedure adapted from ref. 25.
Acknowledgements
This work was supported by NSERC (Discovery Grants and Canada Research Chairs Programs, Graduate Fellowship to T. S.), the Canada Foundation for Innovation (projects #17545 and #19119), and the Ontario Research Fund.Notes and references
- K. C. Nicolaou, J. A. Pfefferkorn, A. J. Roecker, G.-Q. Cao, S. Barluenga and H. J. Mitchell, J. Am. Chem. Soc., 2000, 122, 9939–9953 CrossRef CAS.
- B. van Gemert, Organic Photochromic and Thermochromic Compounds, ed. J. C. Crano and R. Guglielmetti, Plenum Press, New York, 1999, vol. 1, pp. 111–140 Search PubMed.
- J. D. Hepworth, Comprehensive Heterocyclic Chemistry, ed. A. R. Katrizky and C. W. Rees, Pergamon Press, Oxford, 1984, vol. 3, pp. 737–882 CrossRef CAS; A. Levai, T. Timar, P. Sebok and T. Eszenyi, Heterocycles, 2000, 53, 1193–1203 CrossRef CAS; N. Majumdar, N. D. Paul, S. Mandal, B. de Bruin and W. D. Wulff, ACS Catal., 2015, 5, 2329–2366 CrossRef.
- S. J. Pastine, S. W. Youn and D. Sames, Org. Lett., 2003, 5, 1055–1058 CrossRef CAS PubMed; R. S. Menon, A. D. Findlay, A. C. Bissember and M. G. Banwell, J. Org. Chem., 2009, 74, 8901–8903 CrossRef PubMed; C. Efe, I. N. Lykakis and M. Stratakis, Chem. Commun., 2011, 47, 803–805 RSC; I. N. Lykakis, C. Efe, C. Gryparis and M. Stratakis, Eur. J. Org. Chem., 2011, 2334–2338 CrossRef; A. Arcadi, F. Blesi, S. Cacchi, G. Fabrizi, A. Goggiamani and F. Marinelli, Org. Biomol. Chem., 2012, 10, 9700–9708 Search PubMed; H. Murase, K. Senda, M. Senoo, T. Hata and H. Urabe, Chem. – Eur. J., 2014, 20, 317–322 CrossRef PubMed.
- A. Aponick, B. Biannic and M. R. Jong, Chem. Commun., 2010, 46, 6849–6851 RSC; B.-S. Zeng, X. Yu, P. W. Siu and K. A. Scheidt, Chem. Sci., 2014, 5, 2277–2281 RSC; L. Calmus, A. Corbu and J. Cossy, Adv. Synth. Catal., 2015, 357, 1381–1386 CrossRef CAS.
- J. P. A. Harrity, D. S. La, D. R. Cefalo, M. S. Visser and A. H. Hoveyda, J. Am. Chem. Soc., 1998, 120, 2343–2351 CrossRef CAS; S. Chang and R. H. Grubbs, J. Org. Chem., 1998, 63, 864–866 CrossRef PubMed.
- J. J. Talley, Synthesis, 1983, 845–847 CrossRef CAS; M. Rueping, U. Uria, M.-Y. Lin and I. Atodiresei, J. Am. Chem. Soc., 2011, 133, 3732–3735 CrossRef PubMed.
- F. Bigi, S. Carloni, R. Maggi, C. Muchetti and G. Sartori, J. Org. Chem., 1997, 62, 7024–7027 CrossRef CAS; W. Zhao and E. M. Carreira, Org. Lett., 2003, 5, 4153–4154 CrossRef PubMed.
- H. Zeng, J. Ju and R. Hua, Tetrahedron Lett., 2011, 52, 3926–3928 CrossRef CAS; S. Madabhushi, R. Jillella, K. R. Godala, K. K. R. Mallu, C. R. Beeram and N. Chinthala, Tetrahedron Lett., 2012, 53, 5275–5279 CrossRef.
- W.-W. Qiu, K. Surendra, L. Yin and E. J. Corey, Org. Lett., 2011, 13, 5893–5895 CrossRef CAS PubMed; K. Surendra and E. J. Corey, J. Am. Chem. Soc., 2014, 136, 10918–10920 CrossRef PubMed; L. Alonso-Marañon, M. M. Martínez, L. A. Sarandeses and J. P. Sestelo, Org. Biomol. Chem., 2015, 13, 379–387 Search PubMed.
- J. D. Chambers, J. Crawford, H. W. R. Williams, C. Dufresne, J. Schegetz, M. A. Bernstein and C. K. Lau, Can. J. Chem., 1992, 70, 1717–1732 CrossRef CAS; S. Bissada, C. K. Lau, M. A. Bernstein and C. Dufresne, Can. J. Chem., 1994, 72, 1866–1869 CrossRef.
- B. A. Chandler, C. C. Lopes, R. S. C. Lopes, A. J. M. da Silva and V. Snieckus, Synthesis, 1998, 279–282 Search PubMed.
- H. G. Peer, Recl. Trav. Chim. Pays-Bas, 1960, 79, 825–835 CrossRef CAS; W. Nagata, K. Okada and T. Aoki, Synthesis, 1979, 365–368 CrossRef; H. Zheng and D. G. Hall, Tetrahedron Lett., 2010, 51, 4256–4259 CrossRef; K. S. Barbato, Y. Luan, D. Ramella, J. S. Panek and S. E. Schaus, Org. Lett., 2015, 17, 5812–5815 CrossRef PubMed.
- G. B. C. Alves, R. S. C. Lopes, C. C. Lopes and V. Snieckus, Synthesis, 1999, 1875–1877 CrossRef CAS; J. Dai, D. Ma, C. Fu and S. Ma, Eur. J. Org. Chem., 2015, 5655–5662 CrossRef.
- K. K. Julich-Gruner, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Chem. – Eur. J., 2014, 20, 8536–8540 CrossRef CAS PubMed; C. Schuster, K. K. Julich-Gruner, H. Schnitzler, R. Hesse, A. Jäger, A. W. Schmidt and H.-J. Knölker, J. Org. Chem., 2015, 80, 5666–5673 CrossRef PubMed; C. Schuster, M. Rönnefahrt, K. K. Julich-Gruner, A. Jäger, A. W. Schmidt and H.-J. Knölker, Synthesis, 2016, 150–160 Search PubMed.
- J. H. Lee, H. B. Baeng, S. Y. Han and J.-G. Jun, Tetrahedron Lett., 2007, 48, 2889–2892 CrossRef CAS.
- J. D. Pettigrew, J. A. Cadieux, S. S. S. Ho and P. D. Wilson, Org. Lett., 2005, 7, 467–470 CrossRef CAS PubMed.
- J. A. McCubbin, C. Nassar and O. V. Krokhin, Synthesis, 2011, 3152–3160 CrossRef CAS.
- N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS PubMed; C. L. Ricardo, X. Mo, J. A. McCubbin and D. G. Hall, Chem. – Eur. J., 2015, 21, 4218–4223 CrossRef PubMed; X. Mo, J. Yakiwchuk, J. Dansereau, J. A. McCubbin and D. G. Hall, J. Am. Chem. Soc., 2015, 137, 9694–9703 CrossRef PubMed.
- H. Zheng and D. G. Hall, Aldrichimica Acta, 2014, 47, 41–51 Search PubMed; E. Dimitrijevic and M. S. Taylor, ACS Catal., 2013, 3, 945–962 CrossRef CAS.
- Y. Mori, K. Manaba and S. Kobayashi, Angew. Chem., Int. Ed., 2001, 40, 2816–2818 CrossRef CAS; D. Lee, S. G. Newman and M. S. Taylor, Org. Lett., 2009, 11, 5486–5489 CrossRef PubMed.
- Y. Tanaka, T. Hasui and M. Suginome, Synlett, 2008, 1239–1242 Search PubMed; D. Lee, C. L. Williamson, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2012, 134, 8260–8267 CrossRef CAS PubMed; E. Dimitrijević and M. S. Taylor, Chem. Sci., 2013, 4, 3298–3303 RSC; K. Tanveer, K. Jarrah and M. S. Taylor, Org. Lett., 2015, 17, 3482–3485 CrossRef PubMed; T. M. el Dine, D. Evans, J. Rouden and J. Blanchet, Chem. – Eur. J., 2016, 22, 5894–5898 CrossRef PubMed.
- K. Ishihara, H. Kurihara and H. Yamamoto, Synlett, 1997, 597–599 CrossRef CAS.
- K. Ishihara, H. Kurihara and H. Yamamoto, J. Org. Chem., 1997, 62, 5664–5665 CrossRef CAS.
- S. Mothana, J.-M. Grassot and D. G. Hall, Angew. Chem., Int. Ed., 2010, 49, 2883–2887 CrossRef CAS PubMed.
- H. Zheng, M. Lejkowski and D. G. Hall, Chem. Sci., 2011, 2, 1305–1310 RSC.
- G. Harié, A. Samat and R. Guglielmetti, Tetrahedron Lett., 1997, 38, 3075–3078 CrossRef.
- T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512–1513 CrossRef CAS PubMed.
- C. Pouget, C. Fagnere, J.-P. Basly, H. Leveque and A.-J. Chuila, Tetrahedron, 2000, 56, 6047–6052 CrossRef CAS.
- H. Lee and C. S. Yi, Eur. J. Org. Chem., 2015, 1899–1904 CrossRef CAS PubMed.
- L. E. Friedrich, N. de Vera and M. Hamilton, Synth. Commun., 1980, 10, 637–643 CrossRef CAS.
- P. Gandeepan, P. Rajamalli and C.-H. Cheng, ACS Catal., 2014, 4, 4485–4489 CrossRef CAS.
- P. Gawel, C. Dengiz, A. D. Finke, N. Trapp, C. Boudon, J.-P. Gisselbrecht and F. Diederich, Angew. Chem., Int. Ed., 2014, 53, 4341–4345 CrossRef CAS PubMed.
- X. Zhang, W. T. Teo and P. W. H. Chan, Org. Lett., 2009, 11, 4990–4993 CrossRef CAS PubMed.
- C. D. Gabbutt, B. M. Heron, A. C. Instone, D. A. Thomas, S. M. Partington, M. B. Hursthouse and T. Gelbrich, Eur. J. Org. Chem., 2003, 1220–1230 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c6ob01026a |
| This journal is © The Royal Society of Chemistry 2016 |