
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA regioselective double Stille coupling reaction of bicyclic stannolanes†
Akio
Kamimura
*,
Toshiyuki
Tanaka
,
Masahiro
So
,
Tomoyuki
Itaya
,
Kantaro
Matsuda
and
Takuji
Kawamoto
Department of Applied Molecular Bioscience, Graduate School of Medicine, Yamaguchi University, Ube 755-8611, Japan. E-mail: ak10@yamaguchi-u.ac.jp
First published on 1st August 2016
Abstract
A regioselective double Stille coupling reaction was explored using bicyclic stannolanes that were easily prepared from the radical cascade reaction of β-amino-α-methylene esters. Various 1-bromo-2-iodoarenes underwent the double coupling reaction to afford benzoisoindole derivatives in a regioselective manner, where the carbon attached to the iodine selectively coupled with the vinylic carbon, and then the carbon attached to bromine coupled with the alkyl carbon. The combination of intra- and intermolecular coupling reactions provided hexahydroindeno[1,2-b]pyrrole derivatives in good yields. The yields were further improved in the presence of excess amounts of CsF. An attempt to identify the reaction intermediate was made wherein the decomposition of the stannolanes with aqueous HCl and HBr afforded trigonal bipyramidal (TBP) pentacoordinated tin complexes, as confirmed by microanalyses and 119Sn NMR. Using DCl for the decomposition selectively introduced a deuterium to the E-position of the exomethylene unit. The complexes smoothly underwent the intramolecular Stille coupling reaction in the presence of both a palladium catalyst and DABCO, affording hexahydroindeno[1,2-b]pyrroles in good yields. These results suggest that the double coupling reaction progresses through a TBP tin complex, promoting the second intramolecular coupling reaction between the aryl halide and Csp3–tin bond.
Introduction
Palladium-catalysed coupling is a key reaction in organic synthesis.1 Organotin compounds are frequently used as a coupling partner in the Migita–Kosugi–Stille coupling reaction.2,3 Vinylic and aromatic groups on tin compounds are frequently employed as a coupling partner, whereas alkyl groups on tin compounds are usually inert and rarely used in the reaction unless an activating unit is present on the tin compound.4 When two or more carbon–tin bonds exist in one organotin compound, these compounds may be used in a one-pot multicoupling reaction, which is recognized as a useful domino method in organic synthesis.5 For example, this is a useful strategy for preparing polycyclic aromatic hydrocarbons (PAHs), which are the compounds of interest in the development of organoelectronic devices.6 However, this double coupling strategy is limited to the coupling between Csp2–Csp2 type species, and there are only rare reports on the double coupling between Csp2–Csp3 species.7 Thus, the quantitative difference between the reactivities of the sp2 carbon–tin bond and the ordinary sp3 carbon–tin bond in this double coupling reaction remains unclear.Recently, we developed a one-step synthesis of a bicyclic stannolane through a highly cumulated cascade radical reaction wherein a radical addition–cyclization–substitution occurred in one pot.8 This process prompted us to sequentially develop a new type of double coupling reaction.9 In this study, we report the details of a regioselective double coupling reaction of stannolanes. We also explored the substrate scope of aryl halides and pseudohalides. With our methodology, multicyclic benzoisoindoles10 and hexahydroindeno[1,2-b]pyrroles11 are readily prepared in a few steps from simple compounds. These structures are known to feature in biologically active compounds.12,13 The identity of the intermediate in the double coupling reaction was explored through acidic hydrolysis of the stannolanes to afford pentacoordinated trigonal bipyramidal (TBP) tin complexes.
Results and discussion
Double Stille coupling reaction with various 1,2-substituted benzenes
We first examined various 1,2-disubstituted benzenes for the double coupling reaction with stannolane 1a. The results are summarized in Table 1.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Y | Additivesa | Time (h) | Temp. (°C) | 2a; yieldb (%) |
| a Equivalents in parentheses. b Isolated yield. | ||||||
| 1 | Cl | Cl | DABCO (3) | 20 | 100 | 0 |
| 2 | OTf | OTf | DABCO (3), LiCl (2.3) | 24 | 100 | 18 |
| 3 | OTf | OTf | DABCO (3), CuI (2) | 24 | 80 | 21 |
| 4 | Br | Br | DABCO (3) | 20 | 100 | 76 |
| 5 | Br | I | DABCO (3), CsF (5) | 24 | 100 | 44 |
| 6 | I | I | DABCO (0.2), CsF (3) | 24 | 100 | 79 |
The use of dichlorobenzene did not give any double coupling product 2a (entry 1).14 Ditriflate underwent a slow and sluggish reaction to give 2a in only 18% yield. The yield of 2a was improved to 21% when CuI was used as an additive (entry 3).15 Dibromobenzene gave the coupling product 2a in 76% yield (entry 4) and iodobromobenzene also afforded 2a in 44% yield (entry 5). Diiodobenzene underwent a smooth double coupling reaction to give 2a in 79% yield (entry 6). The addition of CsF was necessary to obtain 2a in good yield. Thus, iodoarenes and bromoarenes are the best candidates for the double coupling reaction.
To investigate the reactivity of the sp2 and sp3 carbon–tin bonds, we attempted to introduce two different aromatic groups to 1a using two equivalents of iodobenzene (Scheme 1). However, we could not obtain 3 under these conditions. This is in contrast to the result that the double coupling product 4a was obtained in 32% yield when 1b was used as the coupling partner. These results clearly indicate that the sp3 carbon–tin bond is less reactive and produces no coupling product with any intermolecular coupling partner. The yield of 4a was improved to 80% when three equivalents of CsF were used in the reaction. With this improvement, the amount of DABCO might be reduced to 20 mol%. Thus, the presence of the fluoride anion is effective in promoting the intramolecular coupling reaction with the sp2 carbon–tin bond as well as the sp3 carbon–tin bond.16
 | ||
| Scheme 1 Inter- and intramolecular double coupling reactions. | ||
Intermolecular and inter- and intramolecular regioselective double coupling reactions
We next examined various iodobromobenzenes for the coupling reaction with stannolane 1. The results are summarized in Table 2.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | Ar | R1 | R2 | 2; yielda (%) | Regioisomeric ratiob |
| a Isolated yield. b Determined by integration of 1H NMR spectra. | ||||||
| 1 | 1a | Ph | Cl | H | 2b; 68 | 94/6 |
| 2 | 1a | Ph | H | Cl | 2c; 38 | 95/5 |
| 3 | 1a | Ph | Me | H | 2d; 53 | 95/5 |
| 4 | 1a | Ph | MeO | H | 2e; 44 | >99/1 |
| 5 | 1a | Ph | H | OMe | 2f; 51 | >99/1 |
| 6 | 1a | Ph | NO2 | H | 2g; 63 | 94/6 |
| 7 | 1a | Ph | H | NO2 | 2h; 38 | 83/17 |
| 8 | 1a | Ph | CF3 | H | 2i; 58 | 96/4 |
| 9 | 1a | Ph | H | CF3 | 2j; 54 | 90/10 |
| 10 | 1c | 2-MeC6H4 | Me | H | 2k; 50 | 95/5 |
| 11 | 1d | 3-MeOC6H4 | H | NO2 | 2l; 55 | 84/16 |
| 12 | 1c | 2-MeC6H4 | H | CF3 | 2m; 19 | 98/2 |
| 13 | 1c | 2-MeC6H4 | Cl | H | 2n; 28 | 87/13 |
| 14 | 1c | 2-MeC6H4 | H | F | 2o; 35 | 94/6 |
| 15 | 1d | 3-MeOC6H4 | H | Cl | 2p; 26 | 80/20 |

The double coupling reaction progressed smoothly and products 2 were isolated in moderate to good yields. For example, 1-bromo-4-chloro-2-iodobenzene underwent the double coupling reaction to give 2b in 68% yield (entry 1), whereas the double coupling of 1a with 2-bromo-4-chloro-1-iodobenzene afforded its regioisomer 2c in 38% yield (entry 2). The regioselectivity was estimated by 1H NMR integration to be 94/6 in the formation of 2b and 95/5 in the formation of 2c. Other double couplings with various substituted iodobromobenzenes provided the regioselective formation of benzoisoindole 2. The selectivity ranged from 83/17 to 99/1 and nearly complete regioselective double coupling was achieved. The major and minor isomers formed in the reaction shown in entry 7, 2h and 2g, were inseparable by usual chromatographic methods. The regioselectivity was determined by the first coupling between haloarenes and the sp2-carbon–tin bond site. We expected that the aryl carbon-iodine bond would react faster than the aryl carbon-bromine bond. When the substituent of the dihaloarene is located at the m-position to the aryl–iodine bond site, high regioselectivity (>95/5) was achieved (entries 1, 3, 4, 6, 8, and 10). Conversely, a substituent at the p-position to the aryl–iodine bond affected the selectivity very much. For example, the presence of an electron-withdrawing group such as a nitro or a trifluoromethyl group at the para position to the carbon–iodine bond caused the regioselectivity to slightly decrease to 90/10 or 83/17 (entries 7, 9 and 11), whereas an electron-donating substituent at this position maintained high regioselectivity (entry 5). Thus, the regioselectivity is partially affected by the electronic effect of the substituents on the dihaloarenes.
The number of synthetic methods to prepare the multi-substituted benzoisoindole 2 was limited because it is usually difficult to control the position of substituents during the synthesis of such heterocyclic compounds. However, in the present synthesis, the starting materials 1 were prepared in an optically pure form from simple Michael/Mannich reactions followed by a radical cascade reaction. Thus, this methodology provides a new convenient preparation of benzoisoindoles in a few steps in a highly regioselective manner.
The inter- and intramolecular double coupling reactions of 1b and 1e were examined using various iodobenzenes. The results are summarized in Table 3.

The inter- and intramolecular double coupling reactions progressed smoothly in the presence of iodobenzene and 10 mol% of Pd(PtBu3)2, affording hexahydroindeno[1,2-b]pyrroles 4 in good yields. For example, 4-methoxyiodobenzene underwent the coupling reaction at the vinylic carbon–tin bond site, giving a double coupling product 4b in 68% yield (entry 1). Various substituted iodobenzenes smoothly reacted to give compound 4, except for 4-nitroiodobenzene, which gave a complex mixture (entry 4). Note that all aryl groups derived from iodobenzene were selectively introduced to the E-position of the alkylidene group. Thus, the intermolecular coupling progressed selectively with the retention of the vinylic tin unit configuration. Dimethoxy-substituted stannolane 1e also acted as a good double coupling donor to afford compound 4 in good yield (entries 5–9).
We explored this strategy for the synthesis of tetrasubstituted alkenes from 1f; however, we only obtained compound 4k in 59% yield, and not the desired compound 4l that was expected. This is probably because of the steric hindrance caused by the methyl group at the vinylic position, which prevents the Stille coupling reaction with iodotoluene (Scheme 2).
 | ||
| Scheme 2 Double coupling reaction of 1f. | ||
Attempt to identify the reaction intermediate
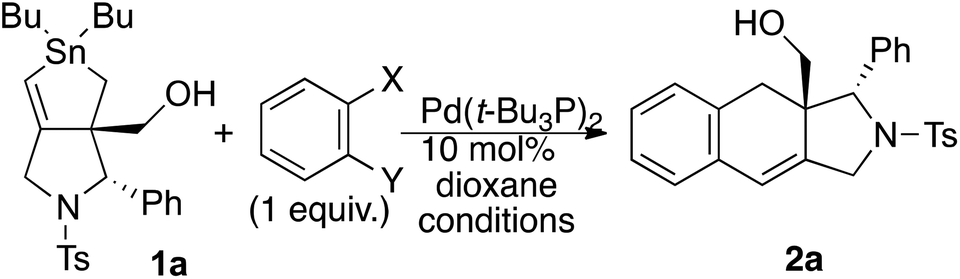
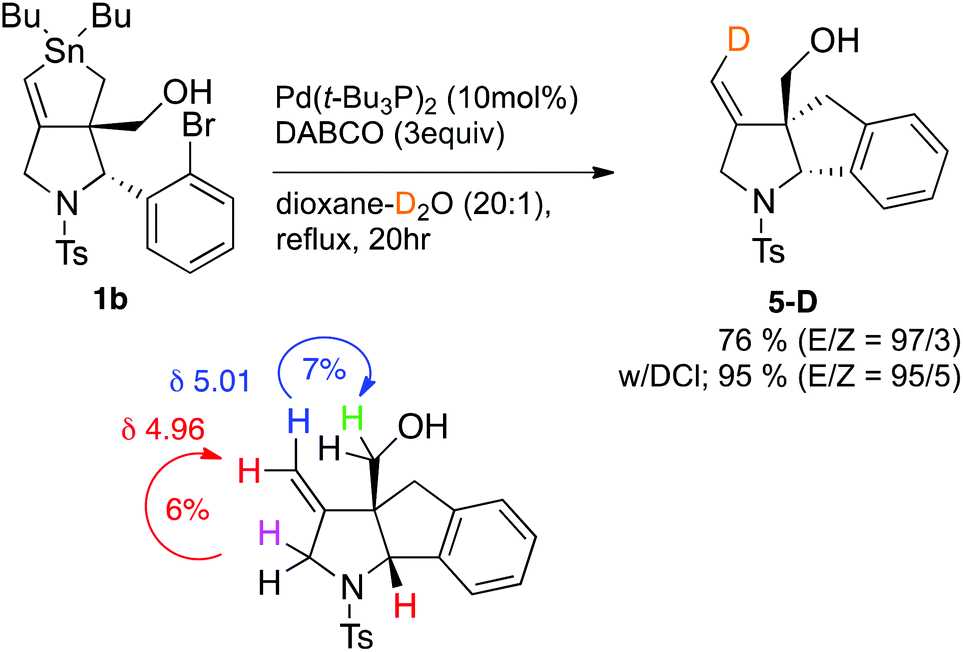
We performed several experiments to identify the reaction intermediate. As we previously reported,9 compound 1b gave exomethylene hexahydroindeno[1,2-b]pyrrole 5 when treated in the absence of aryl halide (Scheme 3). Thus, the vinylic tin unit was replaced by hydrogen during the reaction. To identify the origin of the hydrogen, we performed the reaction with dry dioxane containing small amounts of D2O (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). As expected, compound 5-D instead of 5 was isolated in 76% yield. Note that the deuterium atom was selectively installed at the E-position of the exomethylene unit with 93/7 selectivity. The D position in the alkene unit of 5D was determined by NOE experiments wherein 6% and 7% NOE enhancements were observed when the TsN-CH2 protons at δ = 5.01 and CH2OH protons at δ = 5.96 were irradiated, respectively. We concluded that the water in dioxane is the origin of the hydrogen atom introduced to the vinyl tin unit in the substitution reaction. Note that the yield of 5-D was greatly improved to 95% without any change in the E/Z selectivity or D content when the reaction was performed in the presence of DCl.
:1). As expected, compound 5-D instead of 5 was isolated in 76% yield. Note that the deuterium atom was selectively installed at the E-position of the exomethylene unit with 93/7 selectivity. The D position in the alkene unit of 5D was determined by NOE experiments wherein 6% and 7% NOE enhancements were observed when the TsN-CH2 protons at δ = 5.01 and CH2OH protons at δ = 5.96 were irradiated, respectively. We concluded that the water in dioxane is the origin of the hydrogen atom introduced to the vinyl tin unit in the substitution reaction. Note that the yield of 5-D was greatly improved to 95% without any change in the E/Z selectivity or D content when the reaction was performed in the presence of DCl.
 | ||
| Scheme 3 Introduction of deuterium and elucidation of stereochemistry of 5. | ||
We then attempted to identify the reaction intermediate of the double coupling reaction. The exposure of stannolane 1g to concentrated HCl resulted in the formation of a pentacoordinated trigonal bipyramidal (TBP) tin complex 6a in 72% yield (Scheme 4).8a,17 We exposed the TBP complex 6a to the standard coupling reaction conditions in the presence of five equivalents of CsF, affording the intermolecular coupling product 7 in 87% yield. To confirm the structure of 7, reduction was performed in the presence of DIBAL-H to give compound 5 in almost quantitative yield. Note that the intramolecular coupling progressed only when CsF was employed in the reaction.
 | ||
| Scheme 4 Intramolecular coupling reaction through the TBP complex of 1g. | ||
The acidic treatment of 1b also yielded the corresponding TBP complex 6b in 91% yield (Scheme 5). This is supported by the fact that a signal for the OH proton in 6b was observed in the 1H NMR at δ 2.84 (dd, J = 7.9, 3.8 Hz). Microanalysis data indicated that a chlorine atom is contained in 6b. Compound 6b afforded the intramolecular coupling product 5 in 62% yield by treatment with the catalytic amounts of Pd(tBu3P)3 in the presence of DABCO. We believe that the coordination of an oxygen atom to the tin atom promoted the coupling reaction of the alkyl tin unit; however, some basic additive, such as CsF, is necessary to achieve an efficient reaction.9
 | ||
| Scheme 5 Intramolecular coupling reaction through the TBP complex of 1b. | ||
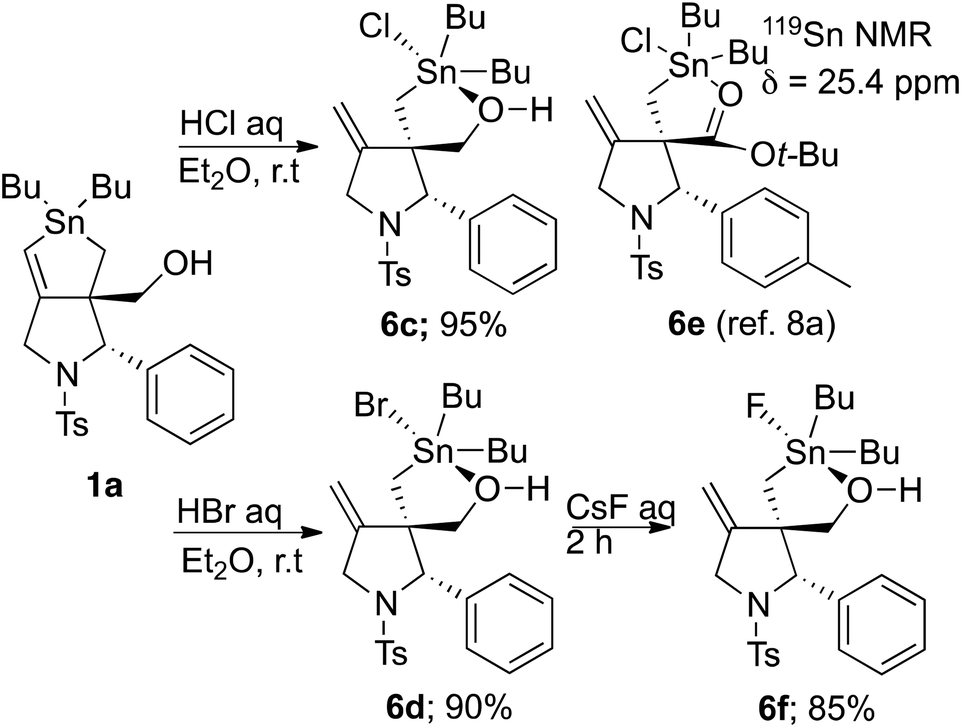
To obtain further information on the TBP complex, we performed simple acidic decomposition of 1a and analysed the products using NMR and microanalytical studies (Scheme 6). Treatment of 1a with HCl and HBr gave the corresponding products 6c and 6d, respectively, in good yields. There are several observations that should be noted for these compounds. Again, the OH proton signal was observed in the 1H NMR spectra for these compounds. Microanalyses of these compounds indicated that compounds 6c and 6d included a halogen atom. Furthermore, the 119Sn NMR signals of 6c and 6d in CDCl3 appeared at 33.3 and 28.8 ppm, respectively, which are highly up-field shifted compared with those of trialkylchlorotin compounds, such as Bu3SnCl (152.8 ppm in CDCl3),18 but close to the chemical shift of TBP tin complex 6e, which shows a 119Sn NMR peak at 25.4 ppm.8a These results clearly suggested that the hydroxyl groups in compounds 6c and 6d coordinate to the tin atom to form a pentacoordinated tin complex, such as 6e. Unfortunately we have not obtained any crystalline form of compounds 6b to 6d to date, and no further evidence for the complex is available. Note that the bromine atom in compound 6d was replaced by a chlorine or a fluorine atom by treatment with aqueous NaCl or CsF solution. For example, compound 6f was obtained in 85% yield by the simple stirring of a biphasic mixture of 6d in ether and aqueous CsF. We examined the intermolecular Stille coupling reaction of compound 6f with an excess of bromobenzene (10 equiv.). However, a complex mixture was obtained, and no expected products were observed.
 | ||
| Scheme 6 Conversion to TBP complexes of 1a. | ||
Considering these results, we suggest that the reaction mechanism is as follows (Scheme 7): aryliodide reacts with the vinylic carbon–tin bond to afford arylalkene intermediate A, a TBP complex in which the hydroxy group coordinates to a tin atom.16 The aryl group is selectively introduced at the E-position of the exomethylene unit. This intermediate is sufficiently reactive toward the intramolecular reaction by activation in the presence of CsF or DABCO to give the double coupling adduct 2 from bromoiodoarenes or 4 from o-bromoaryl-substituted precursor 1. Conversely, in the intermolecular reaction, intermediate A is less reactive and does not give any intermolecular adduct.
 | ||
| Scheme 7 Plausible reaction mechanism. | ||
Conclusions
We have successfully developed a regioselective double Stille coupling reaction with stannolanes. 1,2-Bromoiodoarene served as a good partner for the double coupling reaction. When a bromoarene was installed in a suitable position on the stannolanes, an inter- and intramolecular double coupling reaction smoothly occurred. Since the stannolanes are readily obtained in good yields from simple aza-1,6-enynes, this methodology provides a facile preparation of heterocyclic compounds such as benzoisoindoles and hexahydroindeno[1,2-b]pyrroles.Experimental
General
All 1H and 13C NMR spectra were recorded on a JEOL lamda-500 or JNM-ECA 500 Delta2 (500 MHz for 1H and 126 MHz for 13C) spectrometer. Palladium complexes were purchased from Aldrich. Stannolanes 1 were prepared by using the previously reported method.8a All the reactions in this paper were performed under a nitrogen atmosphere unless otherwise mentioned. High resolution mass spectra (HRMS) were recorded on a JEOL JMS T-100LP LC-ESI mass spectrometer.:1 then 3:1) to give 2a in 76% yield as viscous oil (0.094 g, 0.22 mmol).
[α]D +33.6 (c 1.83, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.3 Hz, 2H), 7.18 (d, J = 4.2 Hz, 3H), 7.14 (d, J = 8.1 Hz, 2H), 7.07 (t, J = 7.4 Hz, 1H), 7.11–6.98 (m, 2H), 7.01 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 6.4 Hz, 1H), 6.94 (d, J = 6.1 Hz, 1H), 6.43 (s, 1H), 5.29 (s, 1H), 4.42 (dd, J = 15.1, 2.3 Hz, 1H), 4.33 (dd, J = 15.1, 1.3 Hz, 1H), 3.4–3.6 (br, 1H), 3.26 (d, J = 10.7 Hz, 1H), 3.10 (d, J = 10.7 Hz, 1H), 2.58 (d, J = 16.1 Hz, 1H), 2.35 (s, 3H), 2.08 (d, J = 16.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 143.4, 139.9, 139.3, 135.7, 133.2, 132.3, 129.5 (2C), 128.8, 128.5 (2C), 127.5, 127.4, 127.3 (2C), 126.8 (2C), 126.22, 126.21, 122.3, 69.1, 62.0, 53.0, 51.1, 31.8, 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C26H25NNaO3S 454.1453, found 454.1447.
:1 then 3:1) to give 2b in 68% yield as pale yellow viscous oil (0.0708 g, 0.152 mmol).
[α]D +47.2 (c 1.79, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 8.3 Hz, 2H), 7.22–7.17 (m, 3H), 7.15 (d, J = 7.9 Hz, 2H), 7.09–7.00 (m, 2H), 6.98 (dd, J = 8.0, 2.2 Hz, 1H), 6.94 (d, J = 2.1 Hz, 1H), 6.86 (d, J = 8.2 Hz, 1H), 6.37 (d, J = 2.0 Hz, 1H), 5.25 (s, 1H), 4.42 (dd, J = 15.3, 2.4 Hz, 1H), 4.33 (dd, J = 15.3, 1.7 Hz, 1H), 3.23 (dd, J = 10.6, 4.7 Hz, 1H), 3.08 (ddd, J = 10.6, 5.9, 1.5 Hz, 1H), 2.53 (d, J = 16.1 Hz, 1H), 2.36 (s, 3H), 1.99 (dd, J = 15.9, 1.5 Hz, 1H), 1.70–1.64 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 143.4, 141.1, 139.6, 135.6, 133.9, 132.2, 131.5, 129.9, 129.5 (2C), 128.5 (2C), 128.1–127.4 (br, 2C), 127.6, 127.3 (2C), 127.2, 126.0, 121.3, 69.0, 61.9, 53.1, 50.9, 31.2, 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C26H24ClNNaO3S 488.1063, found 488.1055.
:1 then 3:1) to give 2c in 38% yield as pale yellow viscous oil (0.0708 g, 0.085 mmol).
[α]D +6.6 (c 1.12, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 8.2 Hz, 2H), 7.24–7.18 (m, 3H), 7.16 (d, J = 7.7 Hz, 2H), 7.11–6.98 (m, 2H), 7.05 (dd, J = 8.1, 2.1 Hz, 2H), 6.94 (s, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.41 (s, 1H), 5.25 (s, 1H), 4.41 (dd, J = 15.3, 2.4 Hz, 1H), 4.34 (dd, J = 15.0, 1.2 Hz, 1H), 3.23 (dd, J = 10.6, 4.4 Hz, 1H), 3.07 (dd, J = 11.0, 5.3 Hz, 1H), 2.53 (d, J = 16.2 Hz, 1H), 2.37 (s, 3H), 2.04 (d, J = 16.9 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 143.4, 139.8, 139.6, 135.6, 135.1, 132.7, 130.8, 129.5 (2C), 128.9, 128.5 (2C), 128.0–127.4 (br, 2C), 127.6, 127.3 (2C), 127.2, 126.8, 121.4, 69.0, 61.9, 52.8, 50.9, 31.7, 21.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H25ClNO3S 466.1244, found 466.1253.
:1 then 3:1) to give 2d in 53% yield as viscous oil (0.0619 g, 0.140 mmol).
[α]D +12.0 (c 0.53, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.54–7.45 (m, 2H), 7.23–7.17 (m, 3H), 7.17–7.13 (m, 2H), 7.10–6.97 (m, 2H), 6.84–6.81 (m, 2H), 6.77 (d, J = 1.5 Hz, 1H), 6.39 (t, J = 2.0 Hz, 1H), 5.24 (s, 1H), 4.40 (dd, J = 15.1, 2.4 Hz, 1H), 4.32 (dd, J = 15.2, 1.6 Hz, 1H), 3.26 (dd, J = 10.7, 4.6 Hz, 1H), 3.07 (dd, J = 9.4, 5.2 Hz, 1H), 2.48 (d, J = 16.0 Hz, 1H), 2.36 (s, 3H), 2.23 (s, 3H), 2.09–1.95 (m, 1H), 1.54 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 143.3, 139.9, 139.2, 136.3, 135.7, 132.1, 130.0, 129.4 (2C), 128.6, 128.4 (2C), 128.1, 128.0–127.4 (br, 2C), 127.4, 127.3 (2C), 127.1, 122.4, 69.1, 62.1, 53.1, 50.9, 31.4, 21.6, 21.0; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C27H27NNaO3S 468.1609, found 468.1600.
:1 then 3:1) to give 2e in 44% yield as viscous oil (0.0638 g, 0.138 mmol).
[α]D −55.5 (c 1.62, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.2 Hz, 2H), 7.24–7.17 (m, 3H), 7.15 (d, J = 8.5 Hz, 2H), 7.12–7.01 (m, 2H), 6.85 (dd, J = 8.3, 1.0 Hz, 1H), 6.57 (dd, J = 8.2, 2.7 Hz, 1H), 6.53 (d, J = 2.7 Hz, 1H), 6.39 (t, J = 2.1 Hz, 1H), 5.24 (s, 1H), 4.40 (dd, J = 15.2, 2.4 Hz, 1H), 4.33 (dd, J = 15.1, 1.7 Hz, 1H), 3.72 (s, 3H), 3.26 (dd, J = 10.6, 4.4 Hz, 1H), 3.05 (dd, J = 11.0, 6.8 Hz, 1H), 2.46 (d, J = 15.9 Hz, 1H), 2.36 (s, 3H), 1.99 (dt, J = 15.7, 1.4 Hz, 1H), 1.54 (dd, J = 6.0, 4.7 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 158.5, 143.3, 140.1, 139.9, 135.7, 133.3, 129.5 (2C), 129.4, 128.4 (2C), 127.7, 127.5, 127.3 (2C), 125.0 (2C), 122.3, 112.3, 112.2, 69.1, 62.1, 55.4, 53.3, 50.9, 30.9, 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C27H27NNaO4S 484.1559, found 484.1562.
:1 then 3:1) to give 2f in 63% yield as viscous oil (0.059 g, 0.13 mmol).
[α]D −31.8 (c 0.02, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 5.5 Hz, 3H), 7.15 (d, J = 8.0 Hz, 2H), 7.11–6.95 (m, 2H), 6.88 (d, J = 8.3 Hz, 1H), 6.60 (dd, J = 8.3, 2.6 Hz, 1H), 6.53 (d, J = 2.5 Hz, 1H), 6.38 (t, J = 2.2 Hz, 1H), 5.26 (s, 1H), 4.39 (dd, J = 15.0, 2.4 Hz, 1H), 4.32 (dd, J = 15.0, 1.6 Hz, 1H), 3.69 (s, 3H), 3.26 (d, J = 10.6 Hz, 1H), 3.07 (d, J = 10.7 Hz, 1H), 2.53 (d, J = 16.1 Hz, 1H), 2.36 (s, 3H), 2.09–2.02 (m, 1H), 1.87 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 159.0, 143.3, 140.0, 136.3, 135.7, 135.0, 129.4 (2C), 128.5 (2C), 128.0–127.6 (br, 2C), 127.4, 127.3 (2C), 127.2, 125.4, 121.8, 115.0, 111.5, 69.1, 62.0, 55.3, 52.8, 50.9, 32.3, 21.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H28NO4S 462.1739, found 462.1728.
:1 then 1:1) to give 2g in 51% yield as viscous oil (0.0545 g, 0.114 mmol).
[α]D +56.4 (c 1.57, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.86 (dt, J = 8.4, 2.3 Hz, 1H), 7.80 (dd, J = 2.4, 1.3 Hz, 1H), 7.50 (d, J = 7.2 Hz, 2H), 7.25–7.18 (m, 3H), 7.15 (d, J = 8.5 Hz, 2H), 7.09 (d, J = 8.0 Hz, 1H), 7.07–6.97 (m, 2H), 6.51 (t, J = 1.9 Hz, 1H), 5.30 (s, 1H), 4.47 (ddd, J = 15.9, 2.5, 1.0 Hz, 1H), 4.43–4.30 (m, 1H), 3.21 (d, J = 10.6 Hz, 1H), 3.17 (d, J = 10.6 Hz, 1H), 2.75 (d, J = 16.6 Hz, 1H), 2.36 (s, 3H), 2.09 (d, J = 17.7 Hz, 1H), 1.90–1.76 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 147.1, 143.6, 142.7, 141.1, 139.2, 135.5, 133.6, 129.5 (2C), 129.4, 128.7 (2C), 127.8, 127.7–127.3 (br, 2C), 127.2 (2C), 122.3, 120.9, 120.6, 68.9, 62.1, 53.0, 50.8, 32.0, 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C26H24N2NaO5S 499.1304, found 499.1308.
:1 then 1:1) to give 2h in 38% yield as pale yellow viscous oil (0.0367 g, 0.077 mmol).
[α]D −16.6 (c 1.16, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.94 (dd, J = 8.4, 2.3 Hz, 1H), 7.83 (d, J = 2.2 Hz, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.24–7.19 (m, 3H), 7.16 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 8.5 Hz, 1H), 7.05–6.93 (m, 2H), 6.60–6.47 (m, 1H), 5.31 (s, 1H), 4.47 (dd, J = 15.9, 2.5 Hz, 1H), 4.39 (dd, J = 16.0, 1.6 Hz, 1H), 3.21 (d, J = 10.8 Hz, 1H), 3.13 (d, J = 10.8 Hz, 1H), 2.72 (d, J = 16.2 Hz, 1H), 2.36 (s, 3H), 2.09 (d, J = 16.3 Hz, 1H), 1.77–1.68 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 146.6, 145.2, 143.6, 139.2, 138.5, 135.5, 134.8, 129.5 (2C), 128.7 (2C), 127.8 (2C), 127.3 (2C), 127.4–127.1, 126.4, 123.8, 122.6, 121.1, 76.9, 68.9, 62.0, 52.9, 51.1, 31.7, 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C26H24N2NaO5S 499.1304, found 499.1294.
:1 then 1:1) to give 2i in 58% yield as pale yellow viscous oil (0.0622 g, 0.125 mmol).
[α]D +38.2 (c 1.45, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 7.6 Hz, 1H), 7.23–7.18 (br, 5H), 7.15 (d, J = 8.0 Hz, 2H), 7.05 (d, J = 7.8 Hz, 2H), 6.47 (s, 1H), 5.29 (s, 1H), 4.46 (d, J = 15.4 Hz, 1H), 4.35 (d, J = 15.3 Hz, 1H), 3.23 (d, J = 10.6 Hz, 1H), 3.15 (d, J = 10.7 Hz, 1H), 2.67 (d, J = 16.3 Hz, 1H), 2.36 (s, 3H), 2.18–2.14 (br, 1H), 2.08 (d, J = 16.3 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 143.5, 141.5, 139.5, 137.3, 135.6, 132.9, 129.5 (2C), 129.2 (d, J = 32.3 Hz), 129.0, 128.6 (2C), 127.7, 128.0–127.5 (br, 2C) 127.3 (2C), 124.1 (q, J = 272.0 Hz), 124.1 (q, J = 3.8 Hz), 122.7 (q, J = 3.8 Hz), 121.4, 68.9, 62.0, 52.9, 50.9, 31.7, 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C27H24F3NNaO3S 522.132, found 522.1339.
:1 then 1:1) to give 2j in 54% yield as pale yellow viscous oil (0.0596 g, 0.119 mmol).
[α]D +30.8 (c 1.93, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 8.3 Hz, 2H), 7.32 (ddd, J = 7.8, 1.9, 0.9 Hz, 1H), 7.24–7.17 (m, 5H), 7.16 (dd, J = 7.8, 0.9 Hz, 2H), 7.04 (d, J = 7.8 Hz, 2H), 6.48 (d, J = 1.9 Hz, 1H), 5.30 (s, 1H), 4.45 (d, J = 14.1 Hz, 1H), 4.37 (dd, J = 15.7, 1.9 Hz, 1H), 3.23 (dd, J = 10.7, 4.7 Hz, 1H), 3.11 (dd, J = 10.7, 5.5 Hz, 1H), 2.66 (d, J = 16.3 Hz, 1H), 2.36 (s, 3H), 2.26–2.11 (m, 1H), 2.07 (d, J = 16.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 143.5, 142.5, 139.5, 135.5 (2C), 133.9, 129.5 (2C), 129.2 (q, J = 32.2 Hz), 128.6 (2C), 127.8–128.5 (br, 2C), 127.7, 127.3 (2C), 126.2, 125.5 (q, J = 3.6 Hz), 124.1 (q, J = 272.1 Hz), 123.9 (q, J = 4.2 Hz), 121.4, 68.9, 61.9, 53.0, 51.0, 31.7, 21.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H25F3NO3S 500.1507, found 500.1513.
:1 then 3:1) to give 2k in 50% yield as viscous oil (0.0478 g, 0.104 mmol).
[α]D +63.2 (c 1.47, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.45 (d, J = 7.8 Hz, 2H), 7.08 (d, J = 8.0 Hz, 2H), 7.04 (d, J = 7.6 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 6.83 (t, J = 7.5 Hz, 1H), 6.75 (s, 2H), 6.70 (d, J = 6.1 Hz, 3H), 6.32 (s, 1H), 5.55 (s, 1H), 4.35 (d, J = 15.0 Hz, 1H), 4.26 (d, J = 15.0 Hz, 1H), 3.20 (d, J = 10.5 Hz, 1H), 3.02 (d, J = 10.5 Hz, 1H), 2.50 (d, J = 15.9 Hz, 1H), 2.39 (s, 3H), 2.35–2.30 (m, 1H), 2.29 (s, 3H), 2.15 (s, 3H), 2.01 (d, J = 15.9 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 143.3, 139.2, 138.4, 136.3, 135.9, 135.7, 132.1, 130.4, 130.0, 129.4 (2C), 128.6, 128.1, 127.7, 127.3 (2C), 127.1, 127.0, 126.2, 122.5, 64.8, 62.2, 53.6, 51.0, 30.2, 21.6, 21.0, 19.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C28H29NNaO3S 482.1766, found 482.1765.
:1 then 1:1) to give 2l in 55% yield as viscous oil (0.0568 g, 0.112 mmol).
[α]D −47.1 (c 0.28, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.93 (d, J = 8.2 Hz, 1H), 7.83 (s, 1H), 7.53 (d, J = 8.1 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 7.14–7.09 (m, 1H), 7.07 (d, J = 8.3 Hz, 1H), 6.95–6.40 (br, 1H), 6.73 (d, J = 8.4 Hz, 1H), 6.50 (s, 1H), 5.27 (s, 1H), 4.46 (d, J = 16.3 Hz, 1H), 4.36 (d, J = 15.8 Hz, 1H), 3.84–3.50 (m, 4H), 3.20 (d, J = 10.7 Hz, 1H), 3.13 (d, J = 10.7 Hz, 1H), 2.75 (d, J = 16.3 Hz, 1H), 2.36 (s, 3H), 2.15–2.06 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 159.7, 146.6, 145.2, 143.6, 140.7, 138.5, 135.4, 134.8, 133.2, 129.7, 129.5 (3C), 127.3 (2C), 126.4, 123.8, 122.5, 121.0, 112.8, 68.8, 61.9, 55.2, 53.0, 51.1, 31.5, 21.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H27N2O6S 507.1590, found 507.1596.
:1 then 3:1) to give 2m in 19% yield as viscous oil (0.0205 g, 0.04 mmol).
[α]D +60.7 (c 0.68, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 8.3 Hz, 2H), 7.26 (d, J = 8.8 Hz, 1H), 7.22–7.01 (m, 6H), 6.91 (t, J = 7.5 Hz, 1H), 6.74 (d, J = 7.8 Hz, 1H), 6.47 (s, 1H), 5.64 (s, 1H), 4.47 (dd, J = 15.4, 2.3 Hz, 1H), 4.36 (dd, J = 15.4, 1.3 Hz, 1H), 3.25 (d, J = 10.5 Hz, 1H), 3.16 (d, J = 10.4 Hz, 1H), 2.72 (d, J = 16.3 Hz, 1H), 2.46 (s, 3H), 2.36 (s, 3H), 2.13 (d, J = 16.2 Hz, 1H), 1.82 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 143.4, 141.5, 138.0, 137.2, 135.8, 132.8, 130.6, 129.4 (2C), 129.4, 129.1, 129.0, 127.5, 127.3, 127.2 (2C), 126.3, 124.6 (q, J = 273.3 Hz), 124.1, 122.7, 121.5, 64.7, 62.1, 53.3, 51.0, 30.6, 21.6, 19.7; HRMS (ESI-TOF) m/z [M + H]+ calcd for C28H27F3NO3S 514.1658, found 514.1663.
:1 then 3:1) to give 2n in 28% yield as viscous oil (0.0205 g, 0.04 mmol).
[α]D +62.1 (c 0.89, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 7.6 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.98 (dd, J = 8.0, 2.0 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H), 6.91 (d, J = 7.6 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 6.74 (d, J = 7.7 Hz, 1H), 6.38 (s, 1H), 5.62 (s, 1H), 4.44 (dd, J = 15.4, 2.1 Hz, 1H), 4.34 (d, J = 15.5 Hz, 1H), 3.25 (d, J = 10.5 Hz, 1H), 3.13 (d, J = 10.3 Hz, 1H), 2.61 (d, J = 16.0 Hz, 1H), 2.46 (s, 3H), 2.36 (s, 3H), 2.04 (d, J = 16.0 Hz, 1H), 1.78 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 143.4, 141.2, 138.1, 135.8, 135.8, 133.8, 132.2, 131.4, 130.5, 129.9, 129.4 (2C), 127.5, 127.3, 127.2 (2C), 127.2, 126.3, 126.0, 121.5, 64.7, 62.0, 53.5, 51.0, 30.0, 21.6, 19.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C27H26ClNNaO3S 502.1220, found 502.1209.
:1 then 2:1) to give 2o in 35% yield as pale yellow oil (0.0323 g, 0.073 mmol).
[α]D +79.3 (c 1.13, CHCl3); 1H NMR (500 MHz, CDCl3) δ 77.51 (d, J = 8.2 Hz, 2H), 7.15 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 7.6 Hz, 2H), 7.06 (dd, J = 7.7, 6.9 Hz, 1H), 6.95–6.86 (m, 2H), 6.78–6.71 (m, 2H), 6.66 (d, J = 8.9 Hz, 1H), 6.41 (s, 1H), 5.61 (s, 1H), 4.42 (d, J = 15.0 Hz, 1H), 4.33 (d, J = 15.0 Hz, 1H), 3.26 (d, J = 10.5 Hz, 1H), 3.12 (d, J = 10.5 Hz, 1H), 2.61 (d, J = 16.1 Hz, 1H), 2.45 (s, 3H), 2.36 (s, 3H), 2.10 (d, J = 16.1 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 157.7, 143.3, 139.7, 138.6, 138.2, 135.8, 135.7, 130.5, 129.4 (2C), 127.6, 127.4, 127.3 (2C), 126.2, 124.2, 121.5, 116.1, 116.0, 113.3 (d, J = 21.7 Hz), 64.7, 62.0, 53.1, 50.9, 30.8, 21.6, 19.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C27H26FNNaO3S 486.1515, found 486.1506.
:1 then 1:1) to give 2p in 26% yield as pale yellow oil (0.0386 g, 0.078 mmol).
[α]D −5.81 (c 1.39, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.2 Hz, 2H), 7.34–7.24 (m, 1H), 7.21–7.11 (m, 1H), 7.16 (d, J = 8.0 Hz, 2H), 7.04 (d, J = 7.8 Hz, 1H), 6.94 (s, 1H), 6.87 (d, J = 8.0 Hz, 1H), 6.72 (d, J = 7.5 Hz, 1H), 6.67–6.57 (m, 1H), 6.39 (s, 1H), 5.22 (d, J = 12.1 Hz, 1H), 4.41 (d, J = 15.8 Hz, 1H), 4.31 (d, J = 15.0 Hz, 1H), 3.80–3.58 (br, 3H), 3.23 (d, J = 10.5 Hz, 1H), 3.10 (d, J = 10.9 Hz, 1H), 2.55 (d, J = 16.2 Hz, 1H), 2.37 (s, 3H), 2.07 (d, J = 16.2 Hz, 1H), 1.73–1.66 (br, 1H); 13C NMR (126 MHz, CDCl3) δ 143.4, 139.8, 135.7, 135.1, 132.8, 132.7, 130.8, 129.6, 129.4 (2C), 128.9, 127.3, 127.2 (2C), 126.8, 121.4, 120.4, 112.7, 101.5, 101.3, 68.9, 62.0, 55.1, 52.8, 51.0, 31.6, 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C27H26ClNNaO4S 518.1169, found 518.1181.
:1 then 3:1) to give 4a in 80% yield as viscous oil (0.0643 g, 0.14 mmol).
[α]D −19.6 (c 0.66, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 7.3 Hz, 1H), 7.36 (d, J = 8.1 Hz, 2H), 7.25 (dd, J = 18.0, 6.5 Hz, 1H), 7.21 (t, J = 6.6 Hz, 1H), 7.11 (d, J = 7.9 Hz, 2H), 7.02 (d, J = 7.9 Hz, 3H), 6.45 (s, 1H), 5.46 (s, 1H), 4.30 (dd, J = 15.1, 1.9 Hz, 1H), 4.09 (dd, J = 15.2, 2.2 Hz, 1H), 3.48 (d, J = 11.4 Hz, 1H), 3.18 (d, J = 16.5 Hz, 1H), 3.08 (d, J = 11.2 Hz, 1H), 2.87 (d, J = 16.4 Hz, 1H), 2.46 (s, 3H), 2.34 (s, 3H), 1.34–1.24 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 143.9, 141.7, 140.9, 140.6, 137.5, 135.9, 133.1, 129.9 (2C), 129.2 (2C), 128.7 (2C), 128.6, 128.0 (2C), 127.5, 126.6, 124.9, 124.5, 73.0, 64.4, 59.4, 56.0, 38.3, 21.7, 21.2. HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H28NO3S 446.1790, found 446.1798.
:1 then 2:1) to give 4b in 68% yield as viscous oil (0.0698 g, 0.15 mmol).
[α]D −64.1 (c 0.46, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.90–7.81 (m, 2H), 7.65 (d, J = 7.5 Hz, 1H), 7.35 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 7.5 Hz, 1H), 7.20 (t, J = 7.5 Hz, 1H), 7.06 (d, J = 9.3 Hz, 2H), 7.01 (d, J = 6.8 Hz, 1H), 6.83 (d, J = 8.6 Hz, 2H), 6.42 (s, 1H), 5.44 (s, 1H), 4.28 (dd, J = 15.0, 2.0 Hz, 1H), 4.08 (dd, J = 15.1, 2.0 Hz, 1H), 3.80 (s, 3H), 3.48 (d, J = 11.3 Hz, 1H), 3.18 (d, J = 16.4 Hz, 1H), 3.08 (d, J = 11.3 Hz, 1H), 2.87 (d, J = 16.4 Hz, 1H), 2.45 (s, 3H), 1.71–1.56 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 159.0, 143.9, 141.5, 140.8, 139.7, 135.7, 130.0 (2C), 129.8 (2C), 128.6, 128.2, 127.9 (2C), 127.4, 126.6, 124.6, 124.5, 113.8 (2C), 73.0, 64.4, 59.3, 56.1, 55.4, 38.1, 21.7; HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H28NO4S 462.1739, found 462.1736.
:1 then 2:1) to give 4c in 83% yield as a white solid (0.1053 g, 0.23 mmol).
Mp 138.5–139.5 °C; [α]D −37.7 (c 0.56, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 7.8 Hz, 2H), 7.65 (d, J = 7.4 Hz, 1H), 7.36 (d, J = 7.8 Hz, 2H), 7.28–7.19 (m, 3H), 7.01 (d, J = 7.2 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 6.71 (d, J = 7.6 Hz, 1H), 6.67 (s, 1H), 6.46 (s, 1H), 5.46 (s, 1H), 4.29 (d, J = 15.1 Hz, 1H), 4.08 (d, J = 15.2 Hz, 1H), 3.78 (s, 3H), 3.47 (d, J = 11.3 Hz, 1H), 3.15 (d, J = 16.3 Hz, 1H), 3.07 (d, J = 11.4 Hz, 1H), 2.87 (d, J = 16.3 Hz, 1H), 2.46 (s, 3H), 1.47–1.43 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 159.5, 143.9, 141.5, 141.3, 140.8, 137.4, 135.7, 129.8 (2C), 129.5, 128.6, 127.9 (2C), 127.4, 126.5, 124.7, 124.4, 121.1, 114.5, 112.8, 72.9, 64.5, 59.5, 55.9, 55.3, 38.3, 21.7; HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H28NO4S 462.1739, found 462.1729.
:1 then 1:1) to give 4d in 74% yield as viscous oil (0.0898 g, 0.18 mmol).
[α]D −13.8 (c 2.99, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 7.6 Hz, 2H), 7.65 (d, J = 6.3 Hz, 1H), 7.36 (d, J = 7.6 Hz, 2H), 7.25 (td, J = 7.2, 1.5 Hz, 1H), 7.21 (t, J = 7.3 Hz, 1H), 7.02 (dd, J = 7.5, 1.2 Hz, 1H), 6.65 (s, 1H), 6.57 (s, 1H), 6.38 (t, J = 1.7 Hz, 1H), 5.40 (s, 1H), 4.30 (dd, J = 15.0, 1.9 Hz, 1H), 4.11 (dd, J = 15.0, 2.0 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.33 (dd, J = 11.2, 4.3 Hz, 1H), 3.11 (d, J = 9.6 Hz, 1H), 3.03 (d, J = 16.4 Hz, 1H), 2.71 (d, J = 16.4 Hz, 1H), 2.45 (s, 3H), 1.92 (s, 3H), 1.61–1.55 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 148.4, 146.5, 143.8, 141.3, 141.0, 140.6, 135.9, 129.8 (2C), 128.7, 128.6, 128.0 (2C), 127.5, 127.2, 126.6, 124.3, 124.1, 113.0, 112.6, 72.7, 64.8, 59.4, 56.2, 55.9, 55.6, 38.6, 21.7, 19.5; HRMS (ESI-TOF) m/z [M + H]+ calcd for C29H32NO5S 506.2001, found 506.1997.
:1 then 2:1) to give 4f in 56% yield as viscous oil (0.0527 g, 0.10 mmol).
[α]D −71.4 (c 1.76, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 7.9 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 7.11 (s, 1H), 7.10 (d, J = 7.9 Hz, 2H), 7.02 (d, J = 7.9 Hz, 2H), 6.51 (s, 1H), 6.44 (s, 1H), 5.41 (s, 1H), 4.31 (d, J = 15.2 Hz, 1H), 4.18–4.07 (m, 1H), 3.87 (s, 3H), 3.80 (s, 3H), 3.47 (d, J = 11.2 Hz, 1H), 3.15 (d, J = 16.1 Hz, 1H), 3.04 (d, J = 11.2 Hz, 1H), 2.82 (d, J = 16.0 Hz, 1H), 2.45 (s, 3H), 2.33 (s, 3H), 1.74–1.61 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 150.0, 149.0, 143.8, 140.8, 137.4, 135.9, 133.2, 133.0, 132.2, 129.8 (2C), 129.1 (2C), 128.7 (2C), 127.9 (2C), 124.5, 108.4, 106.7, 73.6, 64.6, 60.5, 59.7, 56.1, 56.0, 38.6, 21.7, 21.2; HRMS (ESI-TOF) m/z [M + H]+ calcd for C29H32NO5S 506.2001, found 506.2002.
:1 then 1:1) to give 4g in 75% yield as pale yellow viscous oil (0.0748 g, 0.14 mmol).
[α]D −52.1 (c 2.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 7.9 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.11 (s, 1H), 7.07 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.2 Hz, 2H), 6.51 (s, 1H), 6.41 (s, 1H), 5.40 (s, 1H), 4.30 (d, J = 15.1 Hz, 1H), 4.11 (d, J = 14.9 Hz, 1H), 3.86 (s, 3H), 3.79 (s, 3H), 3.79 (s, 3H), 3.49 (d, J = 11.2 Hz, 1H), 3.16 (d, J = 16.0 Hz, 1H), 3.05 (d, J = 11.2 Hz, 1H), 2.83 (d, J = 16.0 Hz, 1H), 2.44 (s, 3H), 1.37–1.33 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 158.9, 149.9, 149.0, 143.8, 140.0, 135.9, 133.2, 132.3, 130.1 (2C), 129.8 (2C), 128.3, 127.9 (2C), 124.2, 113.8 (2C), 108.3, 106.7, 73.6, 68.2, 64.6, 59.6, 56.0, 56.0, 55.3, 38.3, 21.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C29H31NNaO6S 544.1770, found 544.1761.
:1 then 1:1) to give 4h in 74% yield as pale yellow viscous oil (0.0924 g, 0.18 mmol).
[α]D −51.4 (c 3.08, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.85 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 7.21 (t, J = 7.9 Hz, 1H), 7.09 (s, 1H), 6.79 (d, J = 9.2 Hz, 1H), 6.71 (d, J = 7.6 Hz, 1H), 6.67 (s, 1H), 6.51 (s, 1H), 6.44 (s, 1H), 5.41 (s, 1H), 4.30 (d, J = 15.3 Hz, 1H), 4.11 (d, J = 14.4 Hz, 1H), 3.85 (s, 3H), 3.79 (s, 3H), 3.77 (s, 3H), 3.46 (d, J = 11.2 Hz, 1H), 3.12 (d, J = 16.0 Hz, 1H), 3.04 (d, J = 11.1 Hz, 1H), 2.81 (d, J = 16.1 Hz, 1H), 2.44 (s, 3H), 1.51–1.43 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 159.4, 150.0, 149.0, 143.9, 141.7, 137.4, 135.9, 133.3, 132.3, 129.8 (2C), 129.4, 127.9 (2C), 124.4, 121.1, 114.5, 112.7, 108.4, 106.8, 73.5, 64.7, 60.5, 59.8, 56.0, 56.0, 55.3, 38.7, 21.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C29H31NNaO6S 544.1770, found 544.1774.
:1 then 1:1) to give 4i in 74% yield as pale yellow viscous oil (0.0958 g, 0.170 mmol).
[α]D −55.2 (c 3.19, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.86 (d, J = 7.7 Hz, 2H), 7.34 (d, J = 7.8 Hz, 2H), 7.10 (s, 1H), 6.64 (s, 1H), 6.57 (s, 1H), 6.51 (s, 1H), 6.34 (s, 1H), 5.35 (s, 1H), 4.29 (d, J = 15.1 Hz, 1H), 4.13 (d, J = 14.4 Hz, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.80 (s, 3H), 3.79 (s, 3H), 3.29 (d, J = 11.1 Hz, 1H), 3.06 (d, J = 11.8 Hz, 1H), 3.01 (d, J = 17.2 Hz, 1H), 2.68 (d, J = 16.0 Hz, 1H), 2.42 (s, 3H), 1.93 (s, 3H), 1.82–1.69 (m, 1H); 13C NMR (126 MHz, CHCl3) δ 150.0, 149.1, 148.4, 146.5, 143.7, 141.0, 136.0, 133.1, 132.5, 129.8 (2C), 128.7, 127.9 (2C), 127.3, 123.5, 113.1, 112.6, 108.5, 106.7, 73.3, 64.8, 60.5, 59.7, 56.2, 56.1, 56.0, 55.9, 55.6, 39.2, 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C31H35NNaO7S 588.2032, found 588.2022.
:1 then 1:1) to give 4j in 74% yield as pale yellow viscous oil (0.0846 g, 0.17 mmol).
[α]D −85.7 (c 2.82, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.86 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.29 (t, J = 7.4 Hz, 2H), 7.25 (d, J = 7.3 Hz, 1H), 7.12 (d, J = 7.6 Hz, 2H), 7.09 (s, 1H), 6.49 (s, 1H), 6.47 (s, 1H), 5.41 (s, 1H), 4.29 (d, J = 14.6 Hz, 3H), 4.12 (d, J = 15.2 Hz, 1H), 3.85 (s, 3H), 3.78 (s, 3H), 3.46 (d, J = 11.2 Hz, 1H), 3.11 (d, J = 16.1 Hz, 1H), 3.01 (d, J = 11.4 Hz, 1H), 2.80 (d, J = 16.1 Hz, 1H), 2.44 (s, 3H), 1.35 (s, 1H); 13C NMR (126 MHz, CHCl3) δ 150.0, 149.0, 143.9, 141.5, 136.0, 135.9, 133.2, 132.2, 129.8 (2C), 128.7 (2C), 128.4 (2C), 127.9 (2C), 127.5, 124.6, 108.3, 106.7, 77.4, 73.5, 64.7, 59.8, 56.1, 56.0, 38.6, 21.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C28H29NNaO5S 514.1664, found 514.1659.
:1 then 1:1) to give 4k in 59% yield as colourless viscous oil (0.0386 g, 0.104 mmol).
[α]D −23.2 (c 0.92, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 8.2 Hz, 2H), 7.67 (d, J = 6.5 Hz, 1H), 7.35 (d, J = 7.7 Hz, 2H), 7.27–7.20 (m, 2H), 7.10 (d, J = 6.8 Hz, 1H), 5.45 (qt, J = 6.8, 2.6 Hz, 1H), 5.24 (s, 1H), 4.07 (ddq, J = 15.1, 3.2, 1.7 Hz, 1H), 4.01 (ddd, J = 15.1, 2.5, 1.4 Hz, 1H), 3.07 (d, J = 16.0 Hz, 1H), 3.03 (d, J = 11.0 Hz, 1H), 2.96 (d, J = 16.0 Hz, 1H), 2.93 (d, J = 11.4 Hz, 1H), 2.45 (s, 3H), 1.54 (dt, J = 6.9, 1.6 Hz, 3H), 1.35–1.20 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 143.9, 141.4, 140.9, 140.5, 135.9, 129.9, 128.5, 127.5, 127.4, 126.4, 124.7, 118.8, 70.7, 65.7, 59.7, 50.4, 39.2, 21.7, 14.8; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C21H23NNaO3S 392.1296, found 392.1294.
:1 then 1:1) to give 5-D in 76% yield as colourless viscous oil (0.0984 g, 0.27 mmol).
[α]D +16.4 (c 3.28, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.81 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.3 Hz, 1H), 7.33 (d, J = 7.6 Hz, 2H), 7.29–7.14 (m, 2H), 7.10 (d, J = 8.8 Hz, 1H), 5.27 (s, 1H), 5.01 (t, J = 2.0 Hz, 1H), 4.13 (dd, J = 15.1, 2.3 Hz, 1H), 3.95 (dd, J = 15.1, 1.9 Hz, 1H), 3.11 (d, J = 15.7 Hz, 1H), 3.08 (d, J = 10.8 Hz, 1H), 2.99 (d, J = 16.5 Hz, 1H), 2.96 (d, J = 12.1 Hz, 1H), 2.43 (s, 3H), 1.75–1.68 (m, 1H); 13C NMR (126 MHz, CHCl3) δ 149.2, 144.0, 141.3, 140.8, 135.5, 129.9 (2C), 128.6, 127.6 (2C), 127.5, 126.4, 124.7, 108.3 (t, J = 23.7 Hz), 70.8, 65.2, 59.8, 52.9, 39.0, 21.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C20H20DNNaO3S 379.1203, found 379.1196.
:1 then 1:1) to give 5-D in 95% yield as pale colourless viscous oil (0.1104 g, 0.31 mmol).
:1 then 2:1) to give 6a in 72% yield (0.4903 g, 0.63 mmol) as colourless viscous oil.
[α]D +10.6 (c 3.13, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.47 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 8.2 Hz, 2H), 7.06–6.99 (m, 3H), 6.87 (t, J = 7.1 Hz, 1H), 6.56 (d, J = 6.8 Hz, 1H), 5.88 (s, 1H), 5.35 (s, 1H), 5.13 (s, 1H), 4.53 (d, J = 12.9 Hz, 1H), 4.11 (d, J = 12.9 Hz, 1H), 2.31 (s, 3H), 1.57 (s, 9H), 1.41–1.14 (m, 13H), 0.93–0.80 (m, 6H), 0.50 (d, J = 13.8 Hz, J119Sn–1H = 54.1 Hz, 1H); 13C NMR (126 MHz, CHCl3) δ 178.8, 147.1, 143.1, 137.7, 137.1, 132.7, 129.4, 129.1 (2C), 127.5, 126.9 (2C), 124.4, 112.6, 87.4, 69.2, 60.3, 52.9, 28.1, 28.0, 27.9 (3C), 27.8, 26.8, 26.7, 22.1, 21.7, 21.5, 19.3, 13.8, 13.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C32H45BrClNNaO4SSn 798.0840, found 798.0853.
:1 then 1:1) to give 6b in 91% yield (0.2108 g, 0.30 mmol) as colourless viscous oil.
[α]D +21.7 (c 0.16, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.53–7.45 (m, 3H), 7.15 (d, J = 7.9 Hz, 2H), 7.11–7.00 (m, 2H), 6.86–6.79 (m, 1H), 5.37 (s, 1H), 5.12 (s, 1H), 5.03 (s, 1H), 4.28 (dt, J = 13.6, 2.4 Hz, 1H), 4.16 (d, J = 13.7 Hz, 1H), 3.42 (dt, J = 10.5, 3.0 Hz, 1H), 3.31 (dd, J = 10.9, 7.6 Hz, 1H), 2.84 (dd, J = 7.9, 3.8 Hz, 1H), 2.37 (s, 3H), 1.72–1.46 (m, 4H), 1.46–1.03 (m, 9H), 0.91 (t, J = 7.3 Hz, 3H), 0.85 (t, J = 7.3 Hz, 3H), 0.54 (dd, J = 13.9, 2.5 Hz, J119Sn–1H = 56.7 Hz, 1H); 13C NMR (126 MHz, CHCl3) δ 147.2, 144.0, 138.8, 138.0, 132.9, 129.7 (2C), 129.4, 128.7, 127.8, 127.5 (2C), 124.3, 112.7, 77.2, 68.0, 67.2, 55.7, 51.7, 28.2, 28.2, 26.9, 26.8, 22.2, 21.6, 19.2, 19.1, 13.8, 13.6; HRMS (ESI-TOF) m/z [M − HCl + Na]+ calcd for C28H38BrNNaO3SSn 690.0675, found 690.0655.
:1 then 1:1) to give 6c in 95% yield (0.2471 g, 0.44 mmol) as colourless viscous oil.
[α]D −77.1 (c 3.18, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.16–7.09 (m, 4H), 6.99 (d, J = 8.1 Hz, 2H), 6.92 (d, J = 7.3 Hz, 2H), 5.26 (s, 1H), 5.15 (s, 1H), 4.80 (s, 1H), 4.55 (d, J = 3.3 Hz, 1H), 4.36 (d, J = 13.8 Hz, 1H), 3.97 (d, J = 13.8 Hz, 1H), 3.68 (dd, J = 9.9, 4.5 Hz, 1H), 3.37 (dd, J = 9.9, 2.8 Hz, 1H), 2.31 (s, 3H), 1.67–1.46 (m, 4H), 1.39–1.17 (m, 8H), 1.08 (d, J = 13.7 Hz, J119Sn–1H = 48.5 Hz, 1H), 0.92 (t, J = 8.0 Hz, 3H), 0.81 (t, J = 7.4 Hz, 3H), 0.73 (d, J = 13.6 Hz, J119Sn–1H = 70.2 Hz, 1H); 13C NMR (126 MHz, CHCl3) δ 148.0, 143.3, 137.3, 135.1, 129.2 (2C), 128.5 (2C), 128.2, 128.1 (2C), 126.9 (2C), 110.7, 69.6, 68.0, 55.4, 51.0, 28.1, 28.1, 26.9, 26.8, 21.5, 20.7, 20.4, 19.4, 13.7, 13.6; 119Sn NMR (186 MHz, CDCl3) δ 33.3; HRMS (ESI-TOF) m/z [M + H]+ calcd for C28H41ClNO3SSn 626.15176, found 626.15087; Anal. Calcd for C28H40ClNO3SSn: C, 53.82; H, 6.45; N, 2.24. Found: C, 53.76; H, 6.56; N, 2.22.
[α]D −83.6 (c 0.87, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.21 (t, J = 7.3 Hz, 1H), 7.15–7.11 (m, 4H), 6.99 (d, J = 7.9 Hz, 2H), 6.91 (d, J = 7.1 Hz, 2H), 5.27 (s, 1H), 5.16 (s, 1H), 4.81 (s, 1H), 4.68 (s, 1H), 4.37 (d, J = 13.8 Hz, 1H), 3.97 (d, J = 13.9 Hz, 1H), 3.73 (t, J = 8.6, 3.8 Hz, 1H), 3.37 (d, J = 9.8 Hz, 1H), 2.31 (s, 3H), 1.67–1.47 (m, 4H), 1.40–1.22 (m, 8H), 1.19 (d, J = 14.4 Hz, J119Sn–1H = 73.6 Hz, 1H), 0.92 (t, J = 7.3 Hz, 3H), 0.81 (t, J = 7.7 Hz, 3H), 0.78 (d, J = 14.4 Hz, J119Sn–1H = 50.2 Hz, 1H); 13C NMR (126 MHz, CHCl3) δ 147.9, 143.4, 137.3, 135.0, 129.2 (2C), 128.5 (2C), 128.2 (2C), 128.1, 126.9 (2C), 110.7, 69.5, 68.1, 55.8, 51.0, 28.5 (d, J13C −119Sn = 28.0 Hz), 28.4 (d, J13C–119Sn = 28.0 Hz), 26.8 (d, J13C–119Sn = 82.9 Hz, J13C–117Sn = 80.5 Hz), 26.7 (d, J13C–119Sn = 84.6 Hz, J13C–117Sn = 78.8 Hz), 21.5, 21.0 (d, J13C–119Sn = 435.6 Hz, J13C–117Sn = 416.4 Hz), 20.9 (d, J13C–119Sn = 428.2 Hz, J13C–117Sn = 410.8 Hz), 20.3 (d, J13C–119Sn = 400.0 Hz, J13C–117Sn = 381.4 Hz), 13.7, 13.6; 119Sn NMR (186 MHz, CDCl3) δ 28.8; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C28H40BrNNaO3SSn 692.08319, found 692.08200; Anal. Calcd for C28H40BrNO3SSn: C, 50.25; H, 6.02; N, 2.09. Found: C, 50.45; H, 6.13; N, 1.96.
[α]D −82.5 (c 0.08, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.19 (t, J = 7.4 Hz, 1H), 7.16–7.08 (m, 4H), 6.97 (d, J = 7.9 Hz, 2H), 6.91 (d, J = 7.7 Hz, 2H), 5.22 (s, 1H), 5.12 (s, 1H), 4.70 (s, 1H), 4.32 (d, J = 13.9 Hz, 1H), 3.95 (d, J = 14.0 Hz, 1H), 3.52 (d, J = 10.3 Hz, 1H), 3.23 (d, J = 10.4 Hz, 1H), 2.30 (s, 3H), 1.91 (s, 1H), 1.67–0.97 (m, 13H), 0.91 (t, J = 7.1 Hz, 3H), 0.85 (t, J = 7.7 Hz, 3H), 0.57 (d, J = 13.7 Hz, J119Sn–1H = 73.2 Hz, 1H); 13C NMR (126 MHz, CHCl3) δ 148.7, 143.1, 138.0, 135.8, 129.3 (2C), 128.5 (2C), 128.4 (2C), 128.2, 127.1 (2C), 110.4, 69.9, 67.9, 55.3, 51.3, 27.9 (2C), 27.1 (2C), 21.6, 19.0, 18.8, 16.6, 13.9 (2C); 119Sn NMR (186 MHz, CDCl3) δ 15.56 (d, J = 2081.5 Hz); 19F NMR (376 MHz, CDCl3) δ −183.8 (d, J = 2052.9 Hz); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C28H40FNNaO3SSn 632.1633, found 632.1626.
:1 then 8:1) to give 7 in 87% yield as pale yellow viscous oil (0.1172 g, 0.280 mmol).
[α]D +21.3 (c 0.45, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.83 (d, J = 7.5 Hz, 2H), 7.75–7.67 (m, 1H), 7.31 (d, J = 9.1 Hz, 2H), 7.28–7.23 (m, 2H), 7.12 (d, J = 6.1 Hz, 1H), 5.80 (s, 1H), 5.29 (s, 1H), 5.09 (s, 1H), 4.17 (dt, J = 14.2, 2.5 Hz, 1H), 3.90 (dt, J = 14.2, 1.8 Hz, 1H), 3.49 (d, J = 16.9 Hz, 1H), 3.22 (d, J = 16.4 Hz, 1H), 2.41 (s, 3H), 1.30 (s, 9H); 13C NMR (126 MHz, CHCl3) δ 160.8, 147.4, 143.5, 140.9, 139.7, 136.1, 129.7 (2C), 128.7, 127.9 (2C), 127.8, 126.5, 124.4, 109.8, 81.9, 72.7, 63.5, 53.0, 41.2, 27.7 (3C), 21.6; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C24H27NNaO4S 448.1559, found 448.1560.
Mp 165.0–166.0 °C; [α]D +15.8 (c 1.02, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.83 (d, J = 8.2 Hz, 2H), 7.68–7.62 (m, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.28–7.20 (m, 2H), 7.11 (d, J = 6.6 Hz, 1H), 5.27 (s, 1H), 5.08 (t, J = 2.2 Hz, 1H), 5.04 (s, 1H), 4.15 (ddd, J = 15.1, 3.2, 1.5 Hz, 1H), 4.00–3.93 (m, 1H), 3.14–3.06 (m, 2H), 3.00 (t, J = 10.4 Hz, 2H), 2.44 (s, 3H), 1.71–1.61 (m, 1H); 13C NMR (126 MHz, CHCl3) δ 149.4, 144.0, 141.3, 140.7, 135.6, 129.9 (2C), 128.6, 127.7 (2C), 127.5, 126.4, 124.7, 108.4, 70.8, 65.3, 59.9, 52.9, 39.1, 21.7; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C20H21NNaO3S 378.1140, found 378.1129.
:1 then 1:1) to give 5 in 62% yield as colourless viscous oil (0.0656 g, 0.185 mmol).
Acknowledgements
We are grateful to Dr Yousuke Oota and Dr Kyouhei Shingai, UBE Scientific Analysis Laboratory Inc., for the NMR analyses of several compounds.Notes and references
- Review: C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef PubMed.
- (a) M. Kosugi, K. Sasazawa, Y. Shimizu and T. Migita, Chem. Lett., 1977, 301 CrossRef CAS; (b) D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636 CrossRef CAS.
- (a) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef; (b) T. N. Mitchell, Synthesis, 1992, 803 CrossRef CAS; (c) V. Farina, V. Krishnamurthy and W. K. Scott, Org. React., 1997, 50, 1 CAS; (d) M. A. J. Duncton and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1999, 1235 RSC; (e) T. N. Mitchell, in Metal-Catalysed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH Verlag GmbH & Co., Weinheim, 2nd edn, 2004, ch. 3, vol. 1 Search PubMed.
- (a) D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1979, 101, 4992 CrossRef CAS; (b) J. W. Labadie and J. K. Stille, J. Am. Chem. Soc., 1983, 105, 6129 CrossRef CAS; (c) R. J. Linderman, D. M. Graves, W. R. Kwochka, A. F. Ghannam and T. V. Anklekar, J. Am. Chem. Soc., 1990, 112, 7438 CrossRef CAS; (d) R. K. Bhatt, D. S. Shin, J. R. Falck and C. Mioskowski, Tetrahedron Lett., 1992, 33, 4885 CrossRef CAS; (e) E. Vedejs, A. R. Haight and W. O. Moss, J. Am. Chem. Soc., 1992, 114, 6556 CrossRef CAS; (f) J. Ye, R. K. Bhatt and J. R. Falck, J. Am. Chem. Soc., 1994, 116, 1 CrossRef CAS; (g) Y. Y. Belosludtsev, R. K. Bhatt and J. R. Falck, Tetrahedron Lett., 1995, 36, 5881 CrossRef CAS; (h) S. Jarosz, Tetrahedron Lett., 1996, 37, 3063 CrossRef CAS; (i) J. R. Falck, R. K. Bhatt, K. M. Reddy and J. Ye, Synlett, 1997, 481 CrossRef CAS; (j) E. Fouquet, M. Pereyre and A. L. Rodriguez, J. Org. Chem., 1997, 62, 5242 CrossRef CAS; (k) M. S. Jensen, C. Yang, Y. Hsiao, N. Rivera, K. M. Wells, J. Y. L. Chung, N. Yasuda, D. L. Hughes and P. J. Reider, Org. Lett., 2000, 2, 1081 CrossRef CAS PubMed; (l) K. W. Kells and J. M. Chong, J. Am. Chem. Soc., 2004, 126, 15666 CrossRef CAS PubMed; (m) M. Goli, A. He and J. R. Falck, Org. Lett., 2011, 13, 344 CrossRef CAS PubMed; (n) L. Li, C. Y. Wang, R. Huang and M. R. Biscoe, Nat. Chem., 2013, 5, 607 CrossRef CAS PubMed.
- (a) N. B. Carter, R. Mabon, R. Walmsley, A. M. E. Richecœur and J. B. Sweeney, Synlett, 2006, 1747 CAS; (b) M. Shimizu and T. Hiyama, Eur. J. Org. Chem., 2013, 8069 CrossRef CAS; (c) M. Shimizu, I. Nagao, S.-i. Kiyomoto and T. Hiyama, Aust. J. Chem., 2012, 65, 1277 CrossRef CAS; (d) I. Nagao, M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2009, 48, 7573 CrossRef CAS PubMed.
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS PubMed; (b) A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066 CrossRef CAS PubMed.
- M. Shimizu, Y. Tomioka, I. Nagao and T. Hiyama, Synlett, 2009, 3147 CrossRef CAS.
- (a) A. Kamimura, S. Ishikawa, F. Noguchi, T. Moriyama, M. So, T. Murafuji and H. Uno, Chem. Commun., 2012, 42, 6592 RSC; (b) A. Kamimura, T. Yoshinaga, F. Noguchi, K. Miyazaki and H. Uno, Org. Chem. Front., 2015, 2, 713 RSC; (c) A. Kamimura, K. Miyazaki, T. Kawamoto and H. Uno, Tetrahedron, 2016, 72 DOI:10.1016/j.tet.2016.04.0782.
- A. Kamimura, M. So, S. Ishikawa and H. Uno, Org. Lett., 2013, 15, 1402 CrossRef CAS PubMed.
- R. Pedrosa, C. Andrés and J. Nieto, J. Org. Chem., 2002, 67, 782 CrossRef CAS PubMed.
- (a) A. Alizadeh, R. Ghanbaripour, M. Feizabadi, L.-G. Zhu and M. Dusek, RSC Adv., 2015, 5, 80518 RSC; (b) P. Zhou, W.-J. Hao, J.-P. Zhang, B. Jiang, G. Licd and S.-J. Tu, Chem. Commun., 2015, 51, 13012 RSC; (c) K. Pradhan, S. Paul and A. R. Das, RSC Adv., 2015, 5, 12062 RSC; (d) Y. Luo and J. Wu, Org. Lett., 2012, 14, 1592 CrossRef CAS PubMed; (e) F. Behler, F. Habecker, W. Saak, T. Klüner and J. Christoffers, Eur. J. Org. Chem., 2011, 4231 CrossRef CAS; (f) J. D. Harling and B. S. Orlek, Tetrahedron, 1998, 54, 14905 CrossRef CAS.
- For bioactive compounds of benzoisoindoles: (a) D. Middlemiss, Ger. Pat, 2259498 Search PubMed; Chem. Abstr. 1973 79 66171u Search PubMed; (b) R. Achini and W. Oppolzer, Ger. Pat, 2348593 Search PubMed; Chem. Abstr. 1974 81 13386c Search PubMed; (c) A. Achini, W. Oppolzer and E. Pfenninger, Swiss Pat, 611886, 1979 Search PubMed; Chem. Abstr. 1979 91 140720p Search PubMed; (d) A. Achini, W. Oppolzer and E. Pfenninger, Swiss Pat, 611884, 1979 Search PubMed; Chem. Abstr. 1980 92 41756u Search PubMed; (e) J. F. Debernardis, M. D. Meyer and K. B. Sippy, U.S. Pat, WO9006927, 1990 Search PubMed; Chem. Abstr. 1991 114 815704 Search PubMed; (f) R. M. Bowman and H. W. Gschwend, U.S. Pat, 4014899, 1977 Search PubMed; Chem. Abstr. 1977 87 53077h Search PubMed.
- For bioactive compounds of indenopyrroles: (a) B. R. Huck, L. Llamas, M. J. Robarge, T. C. Dent, J. Song, W. F. Hodnick, C. Crumrine, A. Stricker-Krongrad, J. Harrington, K. R. Brunden and Y. L. Bennani, Bioorg. Med. Chem. Lett., 2006, 16, 4130 CrossRef CAS PubMed; (b) L. Qiao, L.-Y. Zhao, S.-B. Rong, X.-W. Wu, S. Wang, T. Fujii, M. G. Kazanietz, L. Rauser, J. Savage, B. L. Roth, J. Flippen-Anderson and A. P. Kozikowski, Bioorg. Med. Chem. Lett., 2001, 11, 955 CrossRef CAS PubMed; (c) F. Behler, F. Habecker, W. Saak, T. Klüner and J. Christoffers, Eur. J. Org. Chem., 2011, 4231 CrossRef CAS; (d) W. Zhou, G. An, G. Zhang, J. Han and Y. Pan, Org. Biomol. Chem., 2011, 9, 583 Search PubMed.
- A. F. Littke, L. Schwarz and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 6343 CrossRef CAS PubMed.
- (a) V. Farina, S. B. Kapadia, C. Wang and I. Liebeskind, J. Org. Chem., 1994, 59, 5905 CrossRef CAS; (b) X. Han, B. M. Stoltz and E. J. Corey, J. Am. Chem. Soc., 1999, 121, 7600 CrossRef CAS; (c) S. P. H. Mee, V. Lee and J. E. Baldwin, Chem. – Eur. J., 2005, 11, 3294 CrossRef CAS PubMed.
- (a) A. García-Martínez, J. Osío Barcina, A. de Fresno Cerezo and L. R. Subramanian, Synlett, 1994, 1047 CrossRef; (b) E. Fouquet, M. Pereyre and A. L. Rodriguez, J. Org. Chem., 1997, 62, 5242 CrossRef CAS; (c) K. Fugami, S.-y. Ohnuma, M. Kameyama, T. Saotome and M. Kosugi, Synlett, 1999, 63 CrossRef CAS; (d) T. Okitsu, K. Iwastuka and A. Wada, Chem. Commun., 2008, 41, 6330 RSC.
- Crystallographic data (excluding structure factors) for the structure 6e have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 845991.
- M. Nádvorník, J. Holeček, K. Handlíř and A. Lyčka, J. Organomet. Chem., 1984, 275, 43 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR for compounds 2, 4, 5, 6, and 7, and an ORTEP chart for 6e. CCDC 845991. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01018k |
| This journal is © The Royal Society of Chemistry 2016 |