
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of influenza A viral neuraminidase candidate inhibitors based on a bicyclo[3.1.0]hexane scaffold†
Cinzia
Colombo
ab,
B. Mario
Pinto
a,
Anna
Bernardi
b and
Andrew J.
Bennet
*a
aDepartment of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, British Columbia, Canada V5A 1S6. E-mail: bennet@sfu.ca; Fax: +1-778-782-3765; Tel: +1-778-782-8814
bUniversità degli Studi di Milano, Dipartimento di Chimica, Via Golgi 19, I-20133 Milano, Italy
First published on 7th June 2016
Abstract
This manuscript describes a novel class of derivatives based on a bicyclo[3.1.0]hexane scaffold, proposed as mimics of sialic acid in a distorted boat conformation that is on the catalytic pathway of neuraminidases (sialidases). A general synthetic route for these constrained-ring molecules was developed using a photochemical reaction followed by a Johnson–Corey–Chaykovsky cyclopropanation. Functionalization with the goal of occupying the 150-cavity was also exploited. Inhibition assays demonstrated low micromolar inhibition against both group-1 (H5N1) and group-2 (H9N2) influenza neuraminidase subtypes, indicating good affinity for the alpha and beta sialic acid mimics and 150-cavity-targeted derivatives. These results provide a validation of a bicyclo[3.1.0]hexane scaffold as a mimic of a distorted sialic acid bound in the neuraminidase active site during catalysis.
Introduction
Influenza neuraminidase (NA) is the enzyme expressed on the surface of the influenza virus that catalyzes the release of progeny virions from infected cells by cleaving the sialic acid receptor on host cells. Thus, it plays a fundamental role in the influenza infection process.1 To date, the most successful anti-influenza drugs target NA activity. Fig. 1 shows oseltamivir 1,2 zanamivir 2,3 and peramivir 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4,5 that were developed based on the structure of sialic acid bound in the active site of NA. The emergence of resistant influenza NA strains, especially in the case of oseltamivir, underscores the growing demand for the development of new antiviral drugs with novel structural motifs and/or substitution patterns.6
4,5 that were developed based on the structure of sialic acid bound in the active site of NA. The emergence of resistant influenza NA strains, especially in the case of oseltamivir, underscores the growing demand for the development of new antiviral drugs with novel structural motifs and/or substitution patterns.6
 | ||
| Fig. 1 Structure of oseltamivir 1, zanamivir 2 and peramivir 3 and related compounds designed to target the NA 150-cavity. | ||
Phylogenetically, the nine NA subtypes can be divided into two groups: group 1 (N1, 4, 5 and 8), and group 2 (N2, 3, 6, 7 and 9).7 Structural characterization of various NAs led to the discovery of a potential binding pocket close to the active site, which was named the ‘150-cavity’ because it is capped by a loop that contains residues 147–152 (150-loop).7 That is, within structures of apo-enzymes the 150-loop was reported to adopt an open conformation in group 1 enzymes and a closed conformation for group 2 neuraminidases.7
Some recent inhibitors have tried to exploit contacts in this region to increase affinity.8–10 The 150-cavity opens through the dynamics of residues 147–152 (the 150-loop). Movement of the 150-loop was initially thought to be restricted to group-1 NAs;7 however, MD simulations11–13 and crystallographic evidence of a partially open 150-loop in a group-2 NA14 show the flexibility of this loop, which implies that all NAs may retain the propensity for opening the 150-loop (Fig. 2).
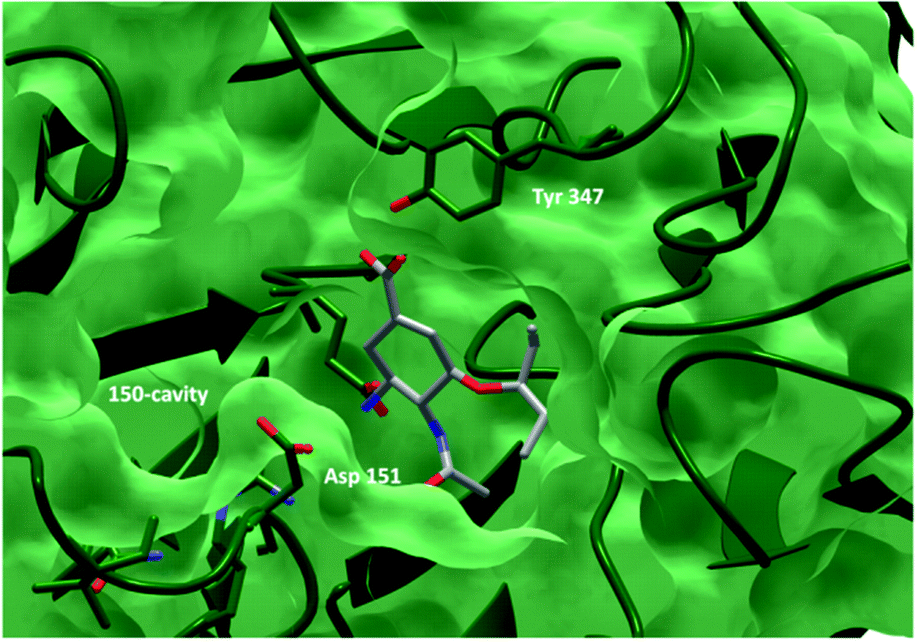 | ||
| Fig. 2 Molecular surface of N1 neuraminidase with the free acid of oseltamivir 1 bound (source: pdb 2HU0). The 150-cavity is accessible due to an open 150-loop conformation (residues 147–152). | ||
Recently, one of us reported a carbocyclic analogue of zanamivir in which the hydrophilic glycerol side chain is replaced with the 3-pentyloxy group of oseltamivir (4, Fig. 1) and a series of triazole-containing carbocycles to target both the catalytic site and the 150-cavity (5–7, Fig. 1).10,15,16 Compounds 4 and 5 displayed a much stronger affinity for an N1 NA than an N2 enzyme.15 Of note, second-generation oseltamivir-like compounds (6 and 7, Fig. 1), which reinstated the basic functionality on C-5, showed generally improved inhibition of virus replication in a cell-based assay, highlighting the importance of a basic group at C-5.17 These molecules showed strong inhibition of the HK1 (H3N2) strain but a slightly lower activity towards the PR8 (H1N1) strain, supporting the notion that the 150-cavity could be opened in both NA groups. Several compounds were then designed to exploit contacts in this region by appending substituents at various positions. The 3-(4-toluoyl)allyl derivative (8, Fig. 1), for instance, inhibited several N1 strains more strongly than a N2 strain (A/Paris/908/97; H3N2).8
Oseltamivir analogues bearing N-substituted guanidines (compounds of general formula 9–10, Fig. 1)9,18 and amines (compounds of general formula 11, Fig. 1)9 have also been made and tested. Only relatively small substituents appended to a terminal guanidino nitrogen atom (9, N-methyl and N-hydroxyl) had a beneficial effect on the activity while bulkier substituents (9, R > CH3) had a dramatically reduced activity.18 Of note, several alkyl derivatives of compounds with the general structures 10 and 11 had improved activities.9 While, p-phenylbenzyl amine 12 (Fig. 1) showed better IC50 values than oseltamivir carboxylate 1 against NAs from three H5N1 virus strains.9 All of these inhibitors were designed based on an understanding of the mechanism of neuraminidases, which are retaining glycosidases. The accepted mechanism for neuraminidases (sialidases) involves formation of a glycosylated enzyme intermediate19,20 in which both glycosylation and deglycosylation occur via transition states (TS) that have substantial oxacarbenium ion character and a distorted six-membered ring (Fig. 3).21–23
 | ||
| Fig. 3 Sialic acid ring distortion during catalysis and target compound described herein. | ||
The introduction of a double bond into the carbohydrate six-membered ring changes the ground state conformation and has been used as a general strategy to try to mimic the geometry of the enzymatic TS.2,10 Molecular modeling studies suggest that the Michaelis complex between N1 and substrate forces the pyran ring into a 4S2 or a B2,5 conformation,24 whereas that in a bacterial sialidase complex has been proposed to be a 6S2 skew-boat.23 If the substrate is bound in a skew-boat conformation and the TS is located between the Michaelis complex and the enzyme-bound intermediate,25 then the bicyclo[3.1.0] analogue 13 likely mimics the TS structure (Fig. 3).
Other conformationally restricted bicyclic compounds have been shown to be good glycosidase inhibitors.26,27 Herein, we describe the synthesis and evaluation of compounds that incorporate a bicyclo[3.1.0]hexane, to provide the ring distortion required to mimic the TS (compounds 13–17, Fig. 4). These possible transition state analogues, which contain a new structural motif, may display a reduced propensity for eliciting resistant strains. Inhibition studies of influenza A sialidases by 13–17 revealed interesting similarities with the more conformationally flexible phosphonate mimics of sialic acid 18 and 19 (Fig. 4), previously reported in the literature.28
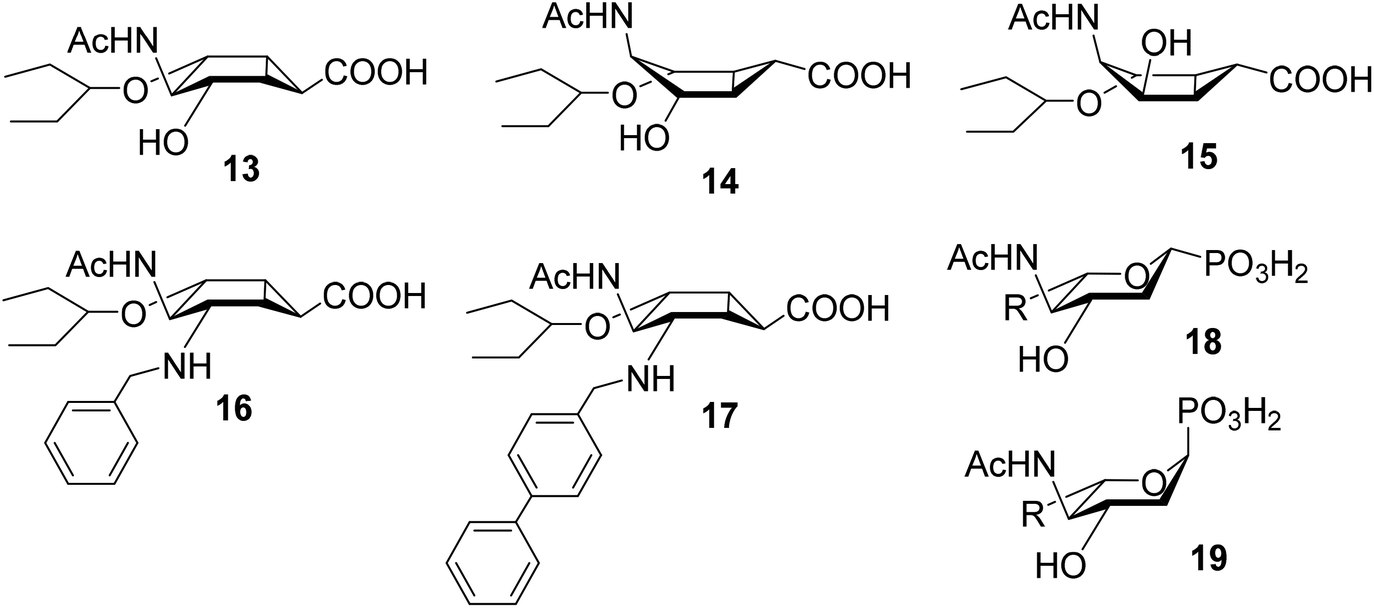 | ||
| Fig. 4 Constrained sialic acid analogues (13–17) described in this manuscript and α- and β-phosphonate analogues 18 and 19.39 Bicyclo[3.1.0]hexanes are drawn in their intrinsically preferred boat-like conformation. | ||
Results and discussion
Synthesis
Retrosynthetically, compound 13 can be made conveniently from pyridine, as shown in Scheme 1, through a photoinduced cyclization of a pyridinium cation with concomitant stereocontrolled nucleophile addition to produce aziridine 22. Enone 21 could then be obtained by regioselective ring opening of aziridine 22 followed by protecting group manipulation. Finally, cyclopropanation of cyclopentenone 21 should generate the bicyclo[3.1.0] framework of 20, which, after functional group interconversion and ester hydrolysis, should yield the bicyclo[3.1.0]hexane scaffold 13.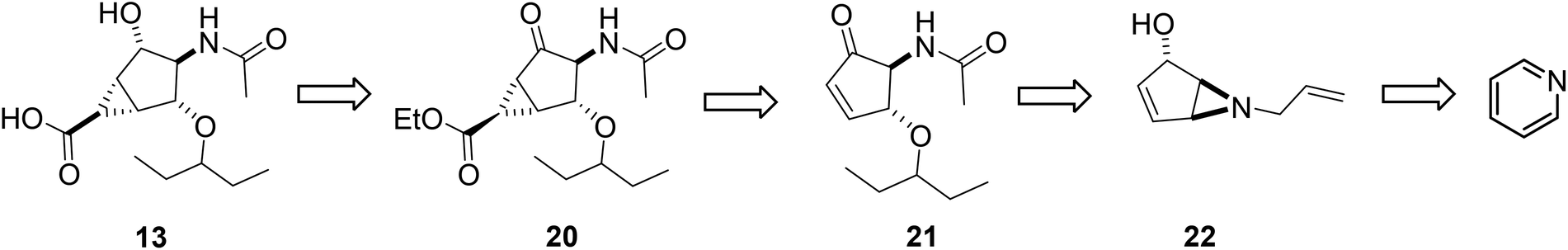 | ||
| Scheme 1 Retrosynthetic analysis for compound 13. | ||
The starting five-membered ring derivative 22 was indeed obtained through a photochemical reaction of the pyridinium cation 23 (Scheme 2).
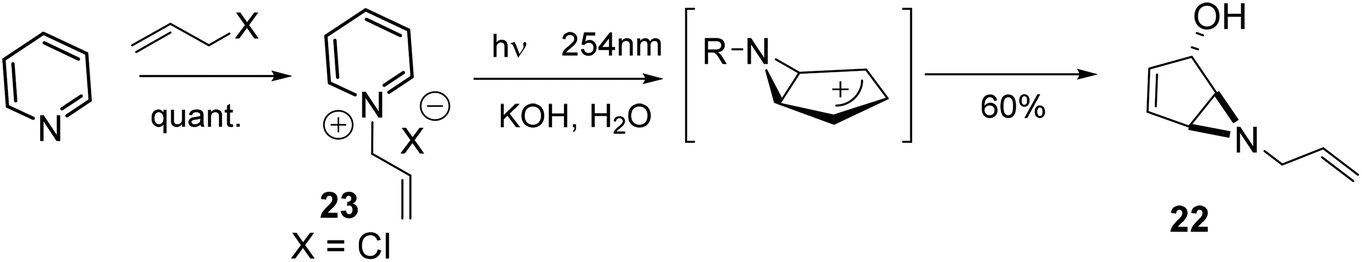 | ||
| Scheme 2 N-Allylpyridinium salt photoirradiation. | ||
Initially reported in 1972 by Wilzbach and co-workers29 for the conversion of N-methylpyridinium chloride to a bicyclic aziridine, photoirradiation of a pyridinium ion has been widely used with both functionalized pyridines and those containing a variety of N-alkyl substituents30 for the synthesis of regiochemically and stereochemically substituted cyclopentanes.31–33 Irradiation of N-allyl-pyridinium perchlorate salts without isolation of the aziridine intermediate prior to methanolysis has been reported.34 Using a similar strategy, we prepared aziridine 22 through the N-allylpyridinium chloride 23. The intermediate allylic cation, generated in the photochemical reaction is trapped by H2O, reacting trans to the aziridine ring to yield 22 as a racemate in 60% yield (Scheme 2). Optimization of reaction conditions on a small scale (1 mmol) gave isolated yields ranging from 64 to 58%, depending on the counter-ion (X− = Br−, 64%; X− = Cl−, 60%; X− = BF4−, 58%). However, in general, scale-up to a 7–10 mmol scale produced lower yields (30–55%), due to the lability of the aziridine, although the reaction with N-allylpyridinium chloride 23 produced noticeably fewer impurities (55% isolated yield). Following benzoylation of 22 (95% yield), several conditions were examined for the opening of aziridine 24 by 3-pentanol (Scheme 3).35–37
 | ||
| Scheme 3 Synthetic route to the intermediates 20 and 30. | ||
The conditions reported by Trost and Zhang,38 designed to generate strictly anhydrous conditions, with 3-pentanol as solvent and boron trifluoride etherate as the Lewis acid, however, only yielded compound 25 in 35% yield. Azeotropically drying the starting material 24 with toluene and using molecular sieves did not improve the conversion significantly, nor did the use of weaker Lewis acids such as copper triflate and trimethylsilyl triflate. We then observed that 25 formed more slowly but more efficiently at lower temperatures. That is, addition of substrate 24 (0.15 M) to boron trifluoride etherate in 3-pentanol at 0 °C, followed by overnight reaction at room temperature, resulted in the formation of 25 in 63% yield (Scheme 3). Removal of the N-allyl group from amine 25 was unsuccessful under a variety of standard conditions, including the use of Wilkinson's catalyst,39 Pd/C in ethanolamine,40 or Pd(dba)2/DPPB (1:1) 5 mol% with 1.1 equivalent of 2-thiobenzoic acid.41 We speculate that metal-catalyzed removal of the N-allyl group42 most likely competes with the cross-reactivity of the cyclopentene double bond and this leads to decomposition of the substrate. Next, we tried to remove the N-allyl group using potassium tert-butoxide in DMSO.43 However, in addition to removing the benzoate group, the N-allyl group remained intact even upon treatment with t-BuOK in DMSO at room temperature for 48 h or at 100 °C for 6 h. To circumvent this problem the acetamide 26 was prepared by acylation of 25 (Scheme 3). Again, neither Pd/C nor Pd(OCOCF3)2:DPPP 1:1 (10 mol%)44 gave more than a 10% conversion to the desired enamine isomer 31 (Scheme 4).
 | ||
| Scheme 4 N-Deallylation reaction: synthesis of 27. | ||
Unexpectedly, cleavage of enamine 31 to give 32 occurred spontaneously in CDCl3, as observed during 1H-NMR analysis (Fig. S1†). Finally, treatment of 26 with potassium tert-butoxide in DMSO afforded the desired allylic isomerization along with removal of both the amide and the benzoate groups (Scheme 4) to give enamine 33, which also cleaved in CDCl3. Compound 27 was therefore obtained by the following procedure: 33 was converted to 34 by dissolving it in CDCl3 and storing it at room temperature for 15 days, while monitoring the reaction by NMR spectroscopy (Fig. S2†). The reaction was ascribed to solvent residual acidity and did not occur in CHCl3. Extensive efforts were made to optimize the conditions using H2SO4 in either CHCl3 or acetone, and HCl in MeOH. However, these reactions resulted in partial decomposition as well as rearrangement of the endocyclic double bond. Observations reveal the lability of the free amino hydroxyl derivative 34 under acidic conditions. Although not ideal, cleavage in deuterated chloroform represents a mild solution, avoiding product decomposition and endocyclic double bond isomerization.
As the amino alcohol 34 could not be isolated without extensive decomposition, it was directly acetylated (acetic anhydride, pyridine) to give 27 (31% yield from 26). Direct cyclopropanation of 27 was attempted using ethyl diazoacetate with various rhodium and copper catalysts,45 but without success. Thus, ketone 21 was prepared by O-deacetylation (MeONa, MeOH) and Dess–Martin oxidation (Scheme 3), and it was subjected to a Johnson–Corey–Chaykovsky cyclopropanation by addition of a nucleophilic sulfur ylide.46 In particular, reaction of the unsaturated ketone 21 with the sulfonium ylide, formed in situ by treatment of the sulfonium salt 29 with DBU (1,8-diazobicyclo[5.4.0]undec-7-ene), gave the diastereomers 20 and 30 (1:1 ratio, Scheme 3), which were separated by flash chromatography. Stereochemical assignment for 20 and 30 was based on 1H-NMR coupling constants and chemical shifts, values which matched those reported for substituted bicyclo[3.1.0]hexane derivatives (for a full discussion of assignments see the ESI and Fig. S3†).47,48 Ketones 20 and 30 were then subjected to reduction with polymer-supported borohydride on Amberlite® to produce the corresponding alcohols (Scheme 5).
 | ||
| Scheme 5 Reduction of ketones 20 and 30 and ester hydrolysis. Reaction conditions: (a) polymer-supported borohydride, MeOH, r.t., 5 h. (b) NaOH, MeOH, water, 4 °C, overnight. | ||
Reduction of 20 afforded 35 in 90% yield by preferential hydride attack from the top, less hindered face. In contrast, hydride addition to substrate 30 led to two diastereoisomers 36 and 37 in ∼2:1 ratio (1H NMR), in 34% and 21% isolated yields, respectively.
Stereochemical assignments were confirmed by 1H-NMR spectroscopy (Fig. 5) using typical coupling-constant data for bicyclo[3.1.0]hexane derivatives systems.47,48 Compound 35 (Fig. 5, top panel) showed coupling constants consistent with two cis relationships (J1–2 = 6.6 Hz, J4–5 = 4.2 Hz). The coupling constant values for compound 36 (Fig. 5, middle panel) are in agreement with both trans relationships (J1–2 = J4–5 = ∼0 Hz). Compound 37 (Fig. 5, bottom panel) showed a cis H1/H2 relationship (J1–2 = 4.6 Hz) and no coupling constant for trans H4/H5. In addition, significant chemical shift differences were noted for protons shielded or deshielded by the cyclopropyl ring, especially H2 and H3 (Fig. 5).
 | ||
| Fig. 5 1H-NMR spectra of compound 35 (top panel), 36 (middle panel), 37 (bottom panel). The proton spectral assignments are also shown for all compounds. | ||
Stereochemical assignments were verified through observation of NOE contacts between non-vicinal protons. A striking example is provided by the contact between H6 on the cyclopropyl ring and H2 and/or H4 on the cyclopentyl ring, which showed strong contacts for a cis orientation (e.g.36) but only weak contacts for a trans orientation (e.g.35, 37) (Fig. 6), consistent with literature data.49
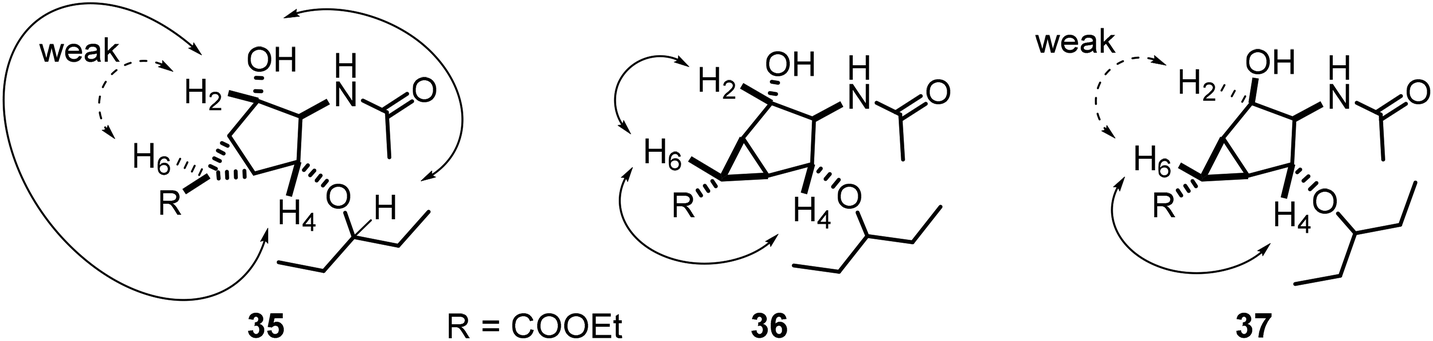 | ||
| Fig. 6 NOEs of compounds 35, 36, 37. | ||
Hydrolysis of the ethyl esters in 35–37 was accomplished with sodium hydroxide, in MeOH:H2O 5:1 at 4 °C, to give compounds 13–15 (Scheme 5).
To synthesize amines 16 and 17 (Scheme 6), reductive amination of 20 with sodium triacetoxyborohydride and glacial AcOH in THF was used, following the procedure reported by Abdel-Magid et al.50 A single isomer was generated, resulting from addition of hydride from the less hindered face, which led to the formation of the benzylamine derivative 16 and the 4-phenylbenzylamine derivative 17 in 59% and 60% yields, respectively, after ethyl ester removal under basic conditions (Scheme 6). Attempts to perform reductive amination with derivative 30 gave a complex mixture of products.
 | ||
| Scheme 6 Reductive amination of 20 and ester hydrolysis of 38 and 39. Reaction conditions: (a) Na(CH3COO)3BH, AcOH, dry THF, amine, 8 h (b) NaOH, MeOH, water, 4 °C, overnight. | ||
Enzyme inhibition
We note that all compounds made by us are racemates, as a result we expect that the IC50 values for the enantiomer that resembles D-sialic acid will be one half of that measured. Nevertheless, we report the measured values for the racemic mixtures. Of note, compounds 13, 16, and 17 share the stereochemical features of α-sialic acid, while compounds 14 and 15 (epi-C4) possess the stereochemistry associated with β-sialic acid.Compounds 13–17 were each evaluated using an enzyme inhibitory assay that involves measuring the hydrolysis of the fluorogenic substrate 2-(4-methylumbelliferyl) α-D-acetylneuraminide sodium salt hydrate (MUNANA) over a period of time to check for “slow-binding” inhibition. As a result, such inhibition assays often involve a preincubation step of the inhibitor with the enzyme prior to substrate addition, which initiates the enzymatic reaction that is monitored by measuring the fluorescence of the cleaved substrate. The use of different protocols with different preincubation times (as well as buffer, pH and temperatures) can affect the IC50 values and has resulted in a plethora of literature values, which makes valid comparisons difficult to achieve. Indeed, it is now well known that influenza neuraminidase inhibitors zanamivir, oseltamivir and peramivir are “slow-binding” inhibitors that show a slow equilibrium formation of a tight inhibitor–enzyme complex.51 Specifically, if the enzymatic reaction is initiated before equilibrium has been established, the resulting activity data will not yield accurate Ki values. A real-time kinetic assay recently reported by Barrett et al.52 has been shown to be suitable for the evaluation of time-dependent changes in IC50 values, and thus we used this method for the measurement of compound activity as well as to determine if our compounds show “slow-binding” kinetics. All compounds were tested as inhibitors against two neuraminidases from influenza type A, one representing group-1 (H5N1) and one group-2 (H9N2). Compound 4 (Fig. 1), a known nanomolar inhibitor of influenza A neuraminidase10 (Ki = 0.46 nM; H5N1), was used as a positive control in this real-time assay. IC50 values were calculated for consecutive 10 min intervals, using a range of concentrations, for each inhibitor over a period of 60 minutes.
First, the data show that the enzymatic activity changes significantly during the experimental time course for all of our compounds, including 4 (Table 1, initial slope and final slope columns) with both enzymes. An example of this behaviour is shown in Fig. 7 (left panel) for the inhibition of the H9N2 neuraminidase by compound 16, which clearly shows non-linear enzyme activity versus time.
 | ||
| Fig. 7 Comparison of preincubation or no preincubation curves with inhibitor 16 on the enzyme activity of A/Chicken/HongKong/G9/1997 H9N2 (left panel: no preincubation, 0–60 min acquisition. Right panel: 45 min preincubation, 0–10 min acquisition). | ||
b
| Compound | N2 | N1 | ||||
|---|---|---|---|---|---|---|
| IC50 (μM) initial slopec | IC50 (μM) final slopec | IC50 (μM) incubationd | IC50 (μM) initial slopec | IC50 (μM) final slopec | IC50 (μM) incubationd | |
| a Initial slope refers to the first time interval of constant slope (0–10 min or 0–5 min generally, depending on the inhibitor curves). The interval is fixed for each series of concentrations for that specific inhibitor. Final slope refers to the last time interval of constant slope (50–60 min generally). Values represent the average of duplicate experiments. The standard deviations were all <15%. b Rates in FU min−1 were calculated for each concentration and for each time interval. The IC50 value is the inhibitor concentration that inhibits the rate of the uninhibited control by 50%. This approach separates each reaction time so it is independent of the rates of preceding time intervals. c No preincubation. d Preincubation with inhibitor for 45 min. e Values reported to underline a significant variation of slope during the first minutes of analysis, affecting the relative IC50 values. f Compound 4 (Fig. 1) was used as positive control. | ||||||
| 13 | 717 | 105 | 40 | 452 | 65 | 64 |
| 14 | 453 | 35 | 23 | 813 | 188 | 48 |
| 15 | 1080 | n.d. | 329 | 840 | 452 | n.d. |
| 16 | 591 (0–5 min)e 208 (5–10 min)e | 33 | 23 | 2895 | 151 | 49 |
| 17 | 388 (0–3 min)e 215 (3–10 min)e | 22 | 11 | 661 | 53 | 10 |
|
4f |
0.05 | 5.7 × 10−4 | 4.5 × 10−4 | 0.108 | 1.65 × 10−3 | 7.8 × 10−4 |
In contrast, when the same experiment is performed after a 45 min preincubation of inhibitor 16 and protein, a linear response is obtained (Fig. 7, right panel; see ESI† for kinetics observed with the other compounds). Therefore, IC50 values were calculated from the measurements made after the neuraminidase and each inhibitor had been pre-incubated together for 45 minutes before addition of substrate. These values are listed in Table 1 (incubation column).6,51–53 Of note, the IC50 values obtained after preincubation of 13–17 (Table 1) are uniformly lower than those measured for reactions without preincubation. Our measured value of 0.78 nM (IC50) for 4 with the H5N1 enzyme is consistent with the literature Ki value,10 which justifies the use of the current kinetic protocols.
Second, alcohols 13 and 14 inhibit both enzymes with similar IC50 values that are around 20–60 μM, whereas the C4-epimer (15) is totally inactive. A comparison between 13 and 14 shows that the β-sialic acid mimic 14 binds tighter to the enzyme active sites. A similar observation was reported for the phosphonate inhibitor ePANA 18, which binds more tightly relative to aPANA 19 (Fig. 3).28 This observation is consistent with that made by Newstead et al. for nucleophilic mutant neuraminidases where the chair form of β-sialic acid binds preferentially,54 a result of lower steric strain. Whereas, in native neuraminidases only the α-sialic acid can bind, albeit in a boat conformation.
Third, replacing the hydroxyl group of 13 with p-phenylbenzyl amine in 17 led to tighter binding to the N2 (IC50 = 11 μM) and N1 (IC50 = 10 μM) enzymes. Unfortunately, with the synthetic approaches used so far we could not access the amines corresponding to framework 14. The slight improvement of 17 may conceivably arise from replacement of the hydroxyl group of 13 with a basic nitrogen and/or additional interactions of the lipophilic group within the 150-cavity. It is well known that replacement of the natural C-4 OH group on the sialic acid glycal framework by either an amino or guanidino (zanamivir, 2) group enhances binding by factors of 100 and 500, respectively.25 The minor change of activity on going from 13 to 17 clearly indicates that interaction of the current inhibitors with the neuraminidase active site still needs to be optimized. Also, there is no reason to suppose that the optimal ether side chain for the bicyclo[3.1.0]hexyl system will be the 3-pentyl group, which was originally optimized on the cyclohexene framework of oseltamivir (1).2 Indeed, it is known that 4 is less active than zanamivir 2,10 most likely because the cyclohexene framework of 4 does not optimally place the 3-pentyl side chain and the guanidino group within the neuraminidase active site. Clearly, given that all of our bicyclo[3.1.0]hexane inhibitors were at least four orders of magnitude less active than the control, this rigid carbon skeleton does not produce the same set of interactions as the cyclohexene or glycal frameworks used so far. However, it is noticeable that the locked boat-like conformation of the [3.1.0] fused system is showing a viable novel framework for neuraminidase inhibition. Further studies are currently underway to understand this aspect and improve interactions with future inhibitors.
Last, it is interesting that all the bicyclic inhibitors displayed the hallmark signs of “slow binding” inhibition. In general, key factors related to time-dependent binding kinetics have been proposed to be due to ligand exchange, hydrogen-bond rearrangement and the local-conformation change of the receptor to accommodate the ligand toward formation of the Michaelis complex, which would take the so-called “target-resident time” to reach the optimal binding mode.6,52 For instance, the slow binding mode of zanamivir 2 has been rationalized by the observation that a water molecule must be displaced before the guanidinium group can bind tightly in the active site.51 That can also explain the slow binding behavior of compound 4 (Table 1). In contrast, slow-binding of oseltamivir has been ascribed to the mandatory side chain rotation of E276 (N2 numbering) in the neuraminidase active site to accommodate the 3-pentyl ether side chain.53 We note that observation of time-dependent changes or so-called ‘slow binding’ competitive inhibition generally arises from one of two main sources, (i) a low concentration of inhibitor used to determine its IC50; or (ii) a slow conformational change in the enzyme from a weak binding to a tight binding complex.55 In the current case, we attribute the changes in IC50 values to a slow conformational change, because the inhibitor IC50 values of compounds 13–17 are in the low micromolar range.
Conclusion
In conclusion, we have successfully synthesized constrained oseltamivir analogues, based on a bicyclo[3.1.0]hexane scaffold using a photochemical reaction and a Johnson–Corey–Chaykovsky cyclopropanation. These new candidate inhibitors were designed to target the catalytic site and possibly to mimic a distorted boat conformation that occurs during transformation of the Michaelis complex to the glycosyl-enzyme intermediate with the goal of mimicking the glycosylation transition state structure. Two of the compounds, analogues of α- and β-sialic acid, showed promising inhibitory activity against N1 and N2 viral neuraminidases and the kinetic analyses showed ‘slow-binding’, time-dependent inhibition. Two amino derivatives were also synthesized with the aim of occupying the adjacent 150-cavity. In particular, the 4-phenylbenzyl derivative displayed an increased binding affinity. These results provide insight into the requirements for the configuration of constrained cyclopropyl sialic analogues and lay the groundwork for further development of novel constrained inhibitors that are effective against influenza A neuraminidases through additional cavity occupation.Experimental
General information
All chemicals were of analytical grade or better and were purchased from Sigma-Aldrich unless noted otherwise. 1H- and 13C-NMR spectra were acquired on a Bruker instrument and recorded at 600 or 400 MHz and 150 or 100 MHz, respectively. Spectra are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, bs = broad singlet), coupling constants (Hz). All assignments were confirmed with the aid of two-dimensional 1H–1H (COSY), 1H–13C (HSQC) and/or 1H–13C (HMBC) experiments using standard pulse programs. Processing of the spectra was performed using MestReNova software. Product numbering for spectral assignment is reported in the ESI† and is shown in Fig. 5. Analytical thin-layer chromatography (TLC) was performed on aluminum plates pre-coated with silica gel 60F-254. The developed plates were air dried, exposed to UV light, and/or sprayed with a solution containing molybdic reagents or permanganate reagents, and heated. Column chromatography was performed with an automated flash chromatography system. High resolution mass spectra were obtained by the electrospray ionization method, using a TOF LC/MS high-resolution magnetic sector mass spectrometer. Preparative photochemical reactions were conducted using a Rayonet photochemical chamber reactor (RPR-100) and a bank of 254 nm lamps. The photolysis solutions were purged with N2 both before and during irradiation. The progress of preparative photochemical reactions was monitored by 1H NMR spectroscopy.NA enzyme inhibitory assay
A/Chicken/HongKong/G9/1997 H9N2 and A/Anhui/1/2005 H5N1 were purchased from SinoBiochemical. The NA inhibition assay was performed according to a standard method,56 with minor modification as reported by Barrett et al.52 Accordingly, the substrate 2-(4-methylumbelliferyl) α-D-acetylneuraminic acid sodium salt hydrate (MUNANA; Sigma M8639) is cleaved by the enzyme to yield a quantifiable fluorescent product, measured during the assay. The fluorescence was assessed using an excitation wavelength of 365 nm and an emission wavelength of 450 nm and substrate blanks were subtracted from the sample readings. Stock solutions of influenza viruses (H5N1 and H9N2) were freshly prepared by dissolution in MES buffer (32.5 mM 2-(N-morpholino)ethanesulfonic acid, 4 mM CaCl2, pH 6.5) at a concentration of 10 U mL−1. Enzyme activity was titrated for both viruses to ensure linearity of the enzymatic reaction over time. Inhibitors were dissolved in DMSO at 20 mM initial concentration. Dilutions of inhibitors were performed in MES buffer, ranging from 1 mM to 1 μM and 5–7 serial dilutions were used; the concentrations depended on the appropriate range for IC50 determination, identified for each inhibitor.No preincubation assay
86 μL of inhibitor solution, 2 μL of 0.2 M of CaCl2, and 2 μL of 10 mM MUNANA were incubated for 5 min at 37 °C. Then 10 μL of stock solution was added to start the reaction. Fluorescence was measured for 60 min after the addition of enzyme, and linear slopes were determined for each consecutive 10 minute time interval. Graphs of log10 concentration of inhibitor versus percent enzyme inhibition compared to the control were plotted after 10, 20, 30, 40, 50 and 60 min, to calculate IC50 for each interval. The IC50 is the ligand concentration leading to 50% inhibition of the rate of the reaction, compared to the control rate with no inhibitor, calculated using a nonlinear regression function in GraphPad Prism.Preincubation assay
86 μL of inhibitor solution and 10 μL of neuraminidase stock solution were preincubated for 45 min at 37 °C, in the presence of CaCl2. The reaction was initiated by the addition of the substrate (MUNANA) and fluorescence was measured for 10 min.:acetone 1:1 and filtered. The organic phase was evaporated and the yellow oil obtained was then purified over basic alumina (Rf = 0.37; 6:4 acetone:CHCl3, KMnO4 stain) to afford compound 22 as an oil (630 mg, 60% yield). 1H NMR (400 MHz, CDCl3) δ 6.26 (dd, J1–5 = 5.6 Hz, J1–3 = 0.8 Hz, 1H, H1), 5.95–5.80 (m, 2H, H5, H7), 5.18 (dd, J8a–7trans = 17.2, J6–8a = 1.6 Hz, 1H, H8a), 5.11 (dd, J8b–7cis = 10.4, J6–8b = 1.6 Hz, 1H, H8b), 4.47 (bs, 1H, H2), 2.96 (dd, J6a–6b = 14.1, J6a–7 = 5.7 Hz, 1H, H6a), 2.88 (dd, J = 14.1, J6b–7 = 5.7 Hz, 1H, H6b), 2.54 (m, 2H, H4, OH), 2.48 (dd, J3–4 = 4.3 Hz, J3–5 = 1.7 Hz, 1H, H3). 13C NMR (100 MHz, CDCl3) δ 137.56 (C1), 135.52 (C5), 134.64 (C7), 116.78 (C8), 74.95 (C2), 60.22 (C6), 50.50 (C3), 46.78 (C4). HRMS: (ESI) m/z calculated for [C8H12NO]+ 138.09123; found 138.09134.
:4 acetone:CHCl3, Rf = 0.92, KMnO4 stain). The reaction mixture was diluted with CH2Cl2 and washed with 0.1 M NaHSO4 and saturated NaHCO3, (three times each alternating acid and basic wash), then with saturated NaCl. The organic phase was then dried with sodium sulfate. The solvent was evaporated and the crude material was filtered on basic alumina, eluting with CHCl3 to afford 24 (3.13 g; 95% yield). 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 7.9 Hz, 2H, Ar-H), 7.53 (t, J = 7.4 Hz, 1H, Ar-H), 7.41 (t, J = 7.7 Hz, 2H, Ar-H), 6.42 (d, J1–5 = 5.6 Hz, 1H, H1), 5.91 (m, 2H, H5, H7), 5.69 (s, 1H, H2), 5.23 (dd, J8–7trans = 17.2 Hz, J6–8a = 1.4 Hz, 1H, H8a), 5.14 (d, J8–7cis = 10.4 Hz, 1H, H8b), 2.98 (d, J6–7 = 5.5 Hz, 1H, H6), 2.70–2.67 (m, 1H, H4), 2.64 (m, 1H, H3). 13C NMR (125 MHz, CDCl3) δ 165.81 (CO), 138.02 (C1), 134.49 (C7), 133.86 (C5), 133.13 (CAr), 129.72 (CAr), 128.40 (CAr), 116.80 (C8), 77.24 (C2), 60.20 (C6), 47.79 (C3), 46.84 (C4). HRMS: (ESI) m/z calculated for [C15H16NO2]+ 242.11789; found 242.11755.
:1 CHCl3:acetone to obtain compound 25 (1.24 g; 63% yield). 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 7.3 Hz, 2H, Ar-H), 7.56 (t, J = 7.4 Hz, 1H, Ar-H), 7.44 (t, J = 7.7 Hz, 2H, Ar-H), 6.07 (d, J1–5 = 5.9 Hz, 1H, H1), 5.98–5.87 (m, 2H, H5, H7), 5.55 (bs, 1H, H2), 5.20 (d, J8–7trans = 17.2 Hz, 1H, H8a), 5.09 (d, J8–7cis = 10.4 Hz, 1H, H8b), 4.26 (m, 1H, H4), 3.44 (t, J2–3 = J3–4 = 3.9 Hz, 1H, H3), 3.41–3.31 (m, 2H, H9, H6), 1.55 (quint, J10–11 = 7.3 Hz, 4H, H10), 0.93 (td, J10–11 = 7.3, 4.4 Hz, 6H, H11). 13C NMR (125 MHz, CDCl3) δ 166.68 (CO), 136.56 (C7), 135.98 (C1), 133.21 (CAr), 130.88 (C5), 130.19 (CAr), 129.86 (CAr), 128.50 (CAr), 116.24 (C8), 86.47 (C4), 83.33 (C2), 81.74 (C9), 71.62 (C3), 50.96 (C6), 26.62 (C10), 26.47 (C10), 10.00 (C11), 9.62 (C11). HRMS: (ESI) m/z calculated for [C20H28NO3]+ 330.20636; found 330.20637. m/z calculated for [C20H27NNaO3]+ 352.18802; found 352.18831.
:2 Hex:EtOAc Rf = 0.36, UV and KMnO4 stain). The reaction mixture was diluted with CH2Cl2 and washed with 0.1 M NaHSO4, saturated NaHCO3 and with saturated NaCl. The organic phase was dried with sodium sulfate. The solvent was evaporated and the crude material was purified by flash chromatography (from 95:5 Hex:EtOAc to 8:2 Hex:EtOAc) to afford 26 (769 mg; 90% yield) (1:0.45 mixture of isomers around the amide bond A and B). 1H NMR (600 MHz, CDCl3) δ 8.06–7.94 (m, 4H, Ar-H), 7.61–7.51 (m, 2H, Ar-H), 7.48–7.39 (m, 4H, Ar-H), 6.23 (d, J2A–3A = 5.1 Hz, 1H, H2A), 6.11–6.06 (m, 2H, H1A, H1B), 6.02 (d, J1B–5B = 6.2 Hz, 1H, H5B), 5.98 (d, J1A–5A = 6.1 Hz, 1H, H5A), 5.96–5.89 (m, 1H, H7B), 5.85–5.73 (m, 1H, H7A), 5.19 (d, J8–7trans = 17.3 Hz, 1H, H8A), 5.18–5.09 (m, 2H, H8B), 5.07 (d, J8–7cis = 10.3 Hz, 1H, H8A), 4.90 (d, J3A–4A = 4.9 Hz, 1H, H4A), 4.55 (t, J3B–4B = 5.9 Hz, 1H, H3B), 4.48 (d, J = 5.9 Hz, 1H, H4B), 4.09–3.98 (m, 2H, H6A, H6B), 3.93 (dd, J6–6 = 17.0, J6–7 = 6.2 Hz, 2H, H6A, H6B), 3.78 (t, J = 5.1 Hz, 1H, H3A), 3.29 (dt, J = 11.8, 5.8 Hz, 2H, H9A, H9B), 2.28 (s, 3H, HAcB), 2.13 (s, 3H, HAcA), 1.58–1.45 (m, 4H, H10A, H10B), 0.90 (dt, J = 13.7 Hz, J = 7.5 Hz, 12H, H11A, H11B). 13C NMR (150 MHz, CDCl3) δ 171.48 (COAcA), 171.25 (COAcB), 166.32 (COBzA), 166.19 (COBzB), 135.85 (C1A), 134.17 (C7B), 133.57 (C7A), 133.52 (CArB), 133.08 (CArA), 131.09 (C1B), 130.95 (C5A, C5B), 130.34 (CArA), 129.81 (CArB), 129.77 (CAr), 129.74 (CAr), 129.65 (CArA), 128.67 (CArB), 128.55 (CArB), 128.45 (CArA), 117.64 (C8A), 116.34 (C8B), 82.78 (C9B), 82.51 (C4A), 82.23 (C9A), 81.17 (C4B), 79.03 (C2A), 77.77 (C2B), 73.92 (C3A), 71.98 (C3B), 54.69 (C6A), 46.14 (C6B), 26.57 (C10A), 26.49 (C10A), 26.44 (C10B), 26.37 (C10B), 22.75 (CAcA), 22.18 (CAcB), 9.86 (C11A), 9.74 (C11B), 9.64 (C11A), 9.50 (C11B). HRMS: (ESI) m/z calculated for [C22H30NO4]+ 372.21630; found 372.21629. m/z calculated for [C22H29NNaO3]+ 394.19852; found 394.19887.
:6 Hex:EtOAc compound 34Rf = 0.12, KMnO4 stain). Compound 331H NMR (400 MHz, CDCl3) δ 6.12 (dq, J6–7 = 7.7 Hz, J6–8 = 1.7 Hz, 1H, H6), 5.90 (s, 2H, H1, H5), 5.76–5.65 (m, 1H, H7), 4.66–4.55 (m, 2H, H4, H2), 4.25 (t, J1–5 = 5.8 Hz, 1H, H5), 3.32–3.16 (m, 1H, H9), 1.69 (dd, J8–8 = 7.0 Hz, 1.7 Hz, 3H, H8), 1.58–1.39 (m, 4H, H10), 0.88 (t, J10–11 = 7.4 Hz, 6H, H11). Compound 341H NMR (400 MHz, CDCl3) δ 5.98–5.90 (m, 2H, H1, H5), 4.57 (bs, 2H, NH2), 4.54 (d, J3–4 = 5.1 Hz, 1H, H4), 4.37 (d, J2–3 = 6.4 Hz, 1H, H2), 3.88–3.73 (m, 1H, H3), 3.33 (quint, J9–10 = 6.0 Hz, 1H, H9), 1.68–1.43 (m, 4H, H10), 1.00–0.86 (m, 6H, H11). 13C NMR (150 MHz, CDCl3) δ 172.63 (COAc), 134.37 (C5), 131.85 (C1), 84.43 (C4), 81.29 (C9), 79.62 (C2), 68.92 (C3), 26.83 (C10), 26.69 (C10), 9.84 (C11), 9.80 (C11). Compound 34 was not isolated due to its instability, the reaction mixture was concentrated to 10 mL, then 6 mL of CHCl3 was added to a final concentration of 0.15 M. Pyridine (0.65 mL, 8 mmol, 3.2 equiv.) and acetic anhydride (0.71 mL, 7.5 mmol, 3 equiv.) were added under nitrogen atmosphere. 4-Dimethylaminopyridine (DMAP) (61 mg, 0.44 mmol, 0.2 equiv.) was added and the resulting mixture was stirred for 8 h at room temperature (TLC 4:6 Hex:EtOAc Rf = 0.28, KMnO4 stain). The solvent was evaporated and the crude material was purified by flash chromatography (from 7:3 Hex:EtOAc to 0:100 Hex:EtOAc in 15 CV (column volume)) to afford 27 (466 mg; 80% yield) (based on compound 26). Compound 271H NMR (500 MHz, CDCl3) δ 6.12 (d, J3-NH = 7.8 Hz, 1H, NH), 6.01 (d, J1–5 = 6.0 Hz, 1H, H5), 5.85 (d, J = 6.0 Hz, 1H, H1), 5.55 (d, J2–3 = 4.3 Hz, 1H, H2), 4.48 (d, J3–4 = 4.2 Hz, 1H, H4), 3.98–3.93 (m, 1H, H3), 3.34 (quint, J9–10 = 5.8 Hz, 1H, H9), 2.03 (s, 3H, CH3NHAc), 1.97 (s, 3H, CH3OAc), 1.52–1.42 (m, 4H, H10), 0.88 (dt, J = 12.1 Hz, J10–11 = 7.4 Hz, 6H, H11). 13C NMR (125 MHz, CDCl3) δ 171.00 (CONHAc), 170.39 (COAc), 136.09 (C5), 130.89 (C1), 83.98 (C4), 81.93 (C9), 80.43 (C2), 63.89 (C3), 26.67 (C10), 26.37 (C10), 23.46 (CNHAc), 21.17 (CAc), 9.85 (C11), 9.54 (C11). HRMS: (ESI) m/z calculated for [C14H24NO4]+ 270.17012; found 270.16998. m/z calculated for [C14H23NNaO4]+ 292.15202; found 292.15193.
:8 Hex:EtOAc Rf (27) = 0.5, Rf (28) = 0.3 KMnO4 stain) showed total consumption of the starting material. A 2 M solution of NaHSO4 in water (204 μL, 0.408 mmol, 0.4 equiv.) was added. The white precipitate was filtered and washed with MeOH; the organic phase was evaporated to obtain compound 28 (220 mg, quant.), which was used without further purification in the next reaction. 1H NMR (600 MHz, CDCl3) δ 5.97–5.84 (m, 2H, H1, H5), 4.54 (s, 1H, NH), 4.51 (d, J3–4 = 4.5 Hz, 1H, H4), 4.38–4.30 (m, 1H, H2), 3.78 (m, 1H, H3), 3.30 (quint, J9–10 = 5.8 Hz, 1H, H9), 2.07 (s, 3H, CH3NHAc), 1.56–1.43 (m, 4H, H10), 0.97–0.86 (m, 6H, H11). 13C NMR (150 MHz, CDCl3) δ 172.64 (COAc), 134.36 (C5), 131.87 (C1), 84.42 (C4), 81.29 (C9), 76.95 (C2), 68.91 (C3), 26.83 (C10), 26.69 (C10), 23.23 (CNHAc), 9.84 (C11), 9.80 (C11). HRMS: (ESI) m/z calculated for [C12H22NO3]+ 228.15922; found 228.15942. m/z calculated for [C12H21NNaO3]+ 250.14118; found 250.14136. m/z calculated for [C12H21NKO3]+ 266.11486; found 266.11530.
:8 Hex:EtOAc Rf (28) = 0.3, Rf (21) = 0.4, KMnO4 stain) then quenched with sodium thiosulfate (saturated solution), extracted four times with EtOAc, washed with brine, dried with sodium sulfate, filtered and concentrated under vacuum. The product was purified by column chromatography (from 7:3 Hex:EtOAc to 0:100 Hex:EtOAc in 15 CV) to yield 21 (183 mg; 86% yield). 1H NMR (500 MHz, CDCl3) δ 7.59 (dd, J1–5 = 6.2 Hz, J3–5 = 1.9 Hz, 1H, H5), 6.57 (d, J3-NH = 6.8 Hz, 1H, NH), 6.44 (dd, J = 6.2 Hz, J1–3 = 1.2 Hz, 1H, H1), 5.00–4.94 (m, 1H, H4), 3.91 (m, 1H, H3), 3.56 (quint, J9–10 = 5.8 Hz, 1H, H9), 2.17 (s, 3H, CH3NHAc), 1.74–1.60 (m, 4H, H10), 1.05 (t, J10–11 = 7.4 Hz, 6H, H11). 13C NMR (125 MHz, CDCl3) δ 201.30 (C2), 170.75 (COAc), 159.39 (C1), 133.09 (C5), 82.44 (C4), 80.34 (C9), 63.37 (C3), 26.57 (C10), 26.17 (C10), 22.91 (CNHAc), 9.74 (C11), 9.60 (C11). HRMS: (ESI) m/z calculated for [C12H20NO3]+ 226.14379; found 226.14377. m/z calculated for [C12H19NNaO3]+ 248.12587; found 248.12571.
:8 Hex:EtOAc Rf (21) = 0.3, Rf (30) = 0.55, Rf (20) = 0.71, KMnO4 stain). The reaction mixture was diluted with CHCl3 (10 mL) and washed with 0.1 M NaHSO4, dried over sodium sulfate, filtered, and concentrated under vacuum. The residue was purified by column chromatography (from 70:30 Hex:EtOAc to 25:75 Hex:EtOAc in 20 CV) to yield 20 (87 mg; 32% isolated yield) and 90 mg of a fraction containing 30 and other impurities. This fraction was subjected to a second purification (from 100:0 CHCl3:MeOH to 92:8 CHCl3:MeOH in 13 CV) to afford pure 30 (77 mg; 30% isolated yield). Compound 201H NMR (600 MHz, acetone-d6) δ 7.44 (d, J3-NH = 7.7 Hz, 1H, NH), 4.51 (dd, J3–4 = 7.5 Hz, J4–5 = 5.1 Hz, 1H, H4), 4.19–4.10 (m, 2H, CH2Et), 3.86 (t, J = 7.6 Hz, 1H, H3), 3.42 (m, 1H, H9), 2.71 (ddd, J1–5 = 6.2 Hz, J4–5 = 5.1 Hz, J5–6 = 3.6 Hz, 1H, H5), 2.56 (t, J = 3.1 Hz, 1H, H6), 2.31 (ddd, J1–5 = 6.2 Hz, J1–6 = 2.7 Hz, 0.8 Hz, 1H, H1), 1.90 (s, 3H, CH3NHAc), 1.57–1.42 (m, 4H, H10), 1.24 (t, J = 7.1 Hz, 3H, CH3Et), 0.93 (t, J10–11 = 7.4 Hz, 3H, H11), 0.87 (t, J = 7.4 Hz, 3H, H11). 13C NMR (150 MHz, acetone-d6) δ 202.62 (C2), 170.69 (COEt), 169.98 (COAc), 81.22 (C9), 77.08 (C4), 61.75 (CH2Et), 58.65 (C3), 36.23 (C1), 31.45 (C5), 27.39 (C10), 27.23 (C10), 24.88 (C6), 22.56 (CAc), 14.46 (CH3Et), 10.19 (C11), 9.68 (C11). HRMS: (ESI) m/z calculated for [C16H26NO5]+ 312.18085; found 312.18054. m/z calculated for [C16H25NNaO5]+ 334.16318; found 334.16249. Compound 301H NMR (600 MHz, CDCl3) δ 5.75 (d, J3-NH = 8.0 Hz, 1H, NH), 4.43 (dd, J = 8.0 Hz, J3–4 = 5.1 Hz, 1H, H3), 4.25 (d, J = 5.1 Hz, 1H, H4), 4.17 (q, J = 7.1 Hz, 2H, CH2Et), 3.38–3.31 (m, 1H, H9), 2.64 (dd, J1–5 = 6.1 Hz, J5–6 = 3.8 Hz, 1H, H5), 2.38 (dd, J = 3.8 Hz, J1–6 = 2.5 Hz, 1H, H6), 2.34 (dd, J = 6.1, 2.5 Hz, 1H, H1), 2.04 (s, 3H, CH3NHAc), 1.59–1.41 (m, 4H, H10), 1.28 (t, J = 7.1 Hz, 3H, CH3Et), 0.90 (t, J10–11 = 7.4 Hz, 3H, H11), 0.85 (t, J = 7.4 Hz, 3H, H11). 13C NMR (150 MHz, CDCl3) δ 205.73 (C2), 170.27 (COAc), 169.32 (COEt), 81.61 (C9), 72.53 (C4), 61.73 (CH2Et), 53.62 (C3), 33.07 (C5), 31.50 (C1), 26.30 (C10), 26.12 (C6), 25.78 (C10), 22.99 (CAc), 14.13 (CH3Et), 9.76 (C11), 9.24 (C11). HRMS: (ESI) m/z calculated for [C16H26NO5]+ 312.18085; found 312.18055. m/z calculated for [C16H25NNaO5]+ 334.16318; found 334.16249.
:8 Hex:EtOAc Rf (20) = 0.71, Rf (35) = 0.12, KMnO4 stain), then filtered. The resin was washed with MeOH and the solution concentrated under vacuum. The product was purified by column chromatography (from 90:10 Hex:EtOAc to 0:100 Hex:EtOAc in 8 CV, then 0:100 Hex:EtOAc for 5 CV) to yield 35 (80 mg; 91% yield). 1H NMR (600 MHz, CDCl3) δ 5.63 (bs, 1H, NH), 5.11 (s, 1H, OH), 4.24 (dd, J1–2 = 6.6 Hz, J2–3 = 3.2 Hz, 1H, H2), 4.18–4.06 (m, 2H, CH2Et), 4.04 (dd, J3–4 = 7.6 Hz, J4–5 = 4.2 Hz, 1H, H4), 3.32–3.25 (m, 1H, H9), 3.25 (td, J = 7.6 Hz, 3.2 Hz, 1H, H3), 2.25–2.13 (m, 2H, H6, H1), 2.15 (dt, J1–5 = 6.7 Hz, 4.2 Hz, 1H, H5), 2.02 (s, 3H, CH3NHAc), 1.59–1.40 (m, 4H, H10), 1.24 (t, J = 7.1 Hz, 3H, CH3Et), 0.91 (dt, J = 16.7, J10–11 = 7.4 Hz, 6H, H11). 13C NMR (150 MHz, CDCl3) δ 173.13 (COAc), 172.73 (COEt), 80.77 (C9), 79.50 (C4), 74.71 (C2), 62.10 (C3), 60.89 (CH2Et), 30.14 (C1), 28.48 (C5), 26.98 (C10), 26.28 (C10), 23.20 (CAc), 18.85 (C6), 14.35 (CH3Et), 10.01 (C11), 9.69 (C11). HRMS: (ESI) m/z calculated for [C16H28NO5]+ 314.19495; found 314.19425. m/z calculated for [C16H27NNaO5]+ 336.17681; found 336.17833.
:8 Hex:EtOAc, Rf (30) = 0.55, Rf (36) = 0.33, Rf (37) = 0.42, KMnO4 stain), then filtered. The resin was washed with MeOH and the solution concentrated and dried under vacuum. The residue was purified by column chromatography (from 100:0 CHCl3:MeOH to 97:3 CHCl3:MeOH in 25 CV) to afford pure 36 (13 mg; 34% isolated yield) and 37 (8 mg; 21% isolated yield). Compound 361H NMR (600 MHz, CDCl3) δ 6.08 (d, J3-NH = 8.2 Hz, 1H, NH), 4.15–4.08 (m, 2H, CH2Et), 4.01 (dd, J2-OH = 9.6 Hz, J2–3 = 3.9 Hz, 1H, H2), 3.96 (d, J3–4 = 4.4 Hz, 1H, H4), 3.80–3.73 (m, 1H, H3), 3.30 (quimt, J9–10 = 5.6 Hz, 1H, H9), 2.18 (dd, J1–5 = 6.4 Hz, J5–6 = 2.9 Hz, 1H, H5), 2.15 (dd, J = 6.4 Hz, J1–6 = 3.1 Hz, 1H, H1), 2.10 (d, J = 9.6 Hz, 1H, OH), 2.00 (s, 3H, CH3NHAc), 1.59 (t, J = 3.0 Hz, 1H, H6), 1.57–1.44 (m, 4H, H10), 1.25 (t, J = 7.1 Hz, 3H, CH3Et), 0.92 (t, J10–11 = 7.4 Hz, 3H, H11), 0.86 (t, J = 7.5 Hz, 3H, H11). 13C NMR (150 MHz, CDCl3) δ 171.70 (COAc), 170.03 (COEt), 82.08 (C9), 76.83 (C4), 72.60 (C2), 61.12 (CH2Et), 52.50 (C3), 31.58 (C1), 30.24 (C5), 26.48 (C10), 25.82 (C10), 23.36 (CAc), 21.34 (C6), 14.30 (CH3Et), 10.01 (C11), 9.42 (C11). HRMS: (ESI) m/z calculated for [C16H28NO5]+ 314.19495; found 314.19620. Compound 371H NMR (600 MHz, CDCl3) δ 6.33 (d, J3-NH = 5.6 Hz, 1H, NH), 4.42 (dd, J2–3 = 7.1 Hz, J1–2 = 4.6 Hz, 1H, H2), 4.11 (qd, J = 7.1, 2.8 Hz, 2H, CH2Et), 4.01 (d, J3–4 = 5.9 Hz, 1H, H4), 3.60–3.66 (m, 1H, H3), 3.38–3.32 (m, 1H, H9), 2.25 (ddd, J1–5 = 6.7 Hz, J1–2 = 4.6 Hz, J1–6 = 3.1 Hz, 1H, H1), 2.07 (dd, J = 6.7 Hz, J5–6 = 3.1 Hz, 1H, H5), 2.01 (s, 3H, CH3NHAc), 1.91 (t, J = 3.1 Hz, 1H, H6), 1.61–1.46 (m, 4H, H10), 1.25 (t, J = 7.1 Hz, 3H, CH3Et), 0.94 (t, J10–11 = 7.4 Hz, 3H, H11), 0.91 (t, J = 7.5 Hz, 3H, H11). 13C NMR (150 MHz, CDCl3) δ 172.41 (COAc), 172.38 (COEt), 81.41 (C9), 76.17 (C4), 76.84 (C2), 61.00 (CH2Et), 58.60 (C3), 31.61 (C1), 29.97 (C5), 26.74 (C10), 25.84 (C10), 23.11 (CAc), 20.13 (C6), 14.34 (CH3Et), 10.10 (C11), 9.56 (C11). HRMS: (ESI) m/z calculated for [C16H28NO5]+ 314.19495; found 314.19620. m/z calculated for [C16H27NNaO5]+ 336.17681; found 336.17814.
:2 Hex:EtOAc Rf = 0.11, KMnO4 stain). The reaction mixture was diluted with Et2O and washed twice with an aqueous saturated NaHCO3 solution. The Et2O extract was dried over Na2SO4 and the solution filtered, concentrated and dried under vacuum. The residue was purified by column chromatography (from 5:95 Hex:EtOAc to 100:0 Hex:EtOAc in 15 CV, then 5 CV 100:0 Hex:EtOAc) to afford pure 38 (50 mg; 60% isolated yield). 1H NMR (600 MHz, CDCl3) δ 7.34–7.27 (m, 5H, HBn, NHBn), 7.25–7.21 (m, 1H, HBn), 5.66 (bs, 1H, NHAc), 4.19–4.15 (m, 1H, H4), 4.15–4.03 (m, 2H, CH2Et), 3.92 (d, J = 13.5 Hz, 1H, CH2Bn), 3.82 (d, J = 13.6 Hz, 1H, CH2Bn), 3.45–3.35 (m, 2H, H2, H3), 3.20 (quint, J9–10 = 5.7 Hz, 1H, H9), 2.13–2.06 (m, 2H, H5, H6), 2.05–1.99 (m, 1H, H1), 1.93 (s, 3H, CH3NHAc), 1.49–1.39 (m, 4H, H10), 1.25 (t, J = 7.1 Hz, 3H, CH3Et), 0.88 (t, J10–11 = 7.4 Hz, 3H, H11), 0.84 (t, J = 7.4 Hz, 3H, H11). 13C NMR (150 MHz, CDCl3) δ 172.97 (COAc), 170.90 (COEt), 128.57 (CBn), 128.37 (CBn), 127.33 (CBn), 81.27 (C9), 79.72 (C4), 60.80 (CH2Et), 60.27 (C2), 58.50 (C3), 51.69 (CH2Bn), 28.97 (C1), 28.73 (C5), 26.99 (C10), 26.50 (C10), 23.65 (CAc), 18.66 (C6), 14.33 (CH3Et), 10.01 (C11), 9.54 (C11). HRMS: (ESI) m/z calculated for [C23H35N2O4]+ 403.25919; found 403.25899.
:2 CHCl3:MeOH to CHCl3:MeOH in 15 CV) to afford pure 16 (5 mg; 45% isolated yield). 1H NMR (400 MHz, CDCl3) δ 7.41–7.31 (m, 4H, HBn), 7.26 (m, 1H, HBn), 5.40 (bs, 1H, NHAc), 4.17 (dd, J3–4 = 7.9 Hz, J4–5 = 4.2 Hz, 1H, H4), 3.91 (d, J = 13.7 Hz, 1H, CH2Bn), 3.82 (d, J = 13.7 Hz, 1H, CH2Bn), 3.42–3.31 (m, 2H, H2, H3), 3.23 (dt, J = 11.5, 5.7 Hz, 1H, H9), 2.16–2.08 (m, 2H, H5, H6), 2.08–2.04 (m, 1H, H1), 1.98 (s, 3H, CH3NHAc), 1.43–1.38 (m, 4H, H10), 0.96–0.82 (m, 6H, H11). 13C NMR (125 MHz, CDCl3) δ 173.48 (COCOOH), 170.58 (COAc), 140.43 (CBn), 128.35 (CBn), 128.02 (CBn), 126.91 (CBn), 99.98 (CqBn), 81.06 (C9), 79.70 (C4), 60.31 (C2), 58.71 (C3), 51.77 (CH2Bn), 29.37 (C1), 28.83 (C5), 26.82 (C10), 26.25 (C10), 23.59 (CAc), 18.21 (C6), 9.85 (C11), 9.38 (C11). HRMS: (ESI) m/z calculated for [C21H31N2O4]+ 375.22828; found 375.22783. m/z calculated for [C21H30N2NaO4]+ 397.20975; found 397.20978.
:2 Hex:EtOAc Rf = 0.21, KMnO4 stain). The reaction mixture was diluted with Et2O and washed twice with an aqueous saturated NaHCO3 solution. The Et2O extract was dried over Na2SO4 and the solution filtered, concentrated and dried under vacuum. The residue was purified by column chromatography (from 100:0 CHCl3:MeOH to 70:30 CHCl3:MeOH in 18 CV) to afford pure 39 (20 mg; 59% isolated yield). 1H NMR (400 MHz, CDCl3) δ 7.61–7.50 (m, 4H, Ar-H), 7.48–7.37 (m, 4H, Ar-H), 7.33 (t, J = 7.3 Hz, 1H, Ar-H), 5.47 (bs, 1H, NHAc), 4.21–4.02 (m, 3H, H4, CH2Et), 3.95 (d, J = 13.7 Hz, 1H, CH2Bn), 3.84 (d, J = 13.7 Hz, 1H, CH2Bn), 3.47–3.32 (m, 2H, H2, H3), 3.27–3.17 (m, 1H, H9), 2.14–2.06 (m, 3H, H6, H1, H5), 1.96 (s, 3H, CH3NHAc), 1.55–1.40 (m, 4H, H10), 1.24 (t, J = 7.1 Hz, 3H, CH3Et), 0.89 (t, J10–11 = 7.4 Hz, 3H, H11), 0.85 (t, J = 7.4 Hz, 3H, H11). 13C NMR (150 MHz, CDCl3) δ 173.10 (COAc), 170.74 (COEt), 141.07 (CAr), 140.09 (CAr), 128.89 (CAr), 128.66 (CAr), 127.32 (CAr), 127.25 (CAr), 127.16 (CAr), 81.22 (C9), 79.94 (C4), 60.78 (CH2Et), 60.45 (C2), 58.86 (C3), 51.48 (CH2Bn), 28.97 (C1, C5), 27.02 (C10), 26.52 (C10), 23.76 (CAc), 18.66 (C6), 14.36 (CH3Et), 10.04 (C11), 9.58 (C11). HRMS: (ESI) m/z calculated for [C29H39N2O4]+ 479.29247; found 479.29043. m/z calculated for [C29H38N2NaO4]+ 501.27266; found 501.27238.
:2 CHCl3:MeOH to 70:30 CHCl3:MeOH in 12 CV) to afford pure 17 (4 mg; 70% isolated yield). 1H NMR (500 MHz, CD3OD) δ 7.60 (dd, J = 12.9, 7.8 Hz, 4H, Ar-H), 7.48–7.40 (m, 4H, Ar-H), 7.34 (t, J = 7.4 Hz, 1H, Ar-H), 4.10 (dd, J3–4 = 7.3 Hz, J4–5 = 4.5 Hz, 1H, H4), 3.87 (s, 2H, CH2Bn), 3.63 (t, J = 8.2 Hz, 1H, H3), 3.28–3.20 (m, 2H, H2, H9), 2.18 (t, J = 3.0 Hz, 1H, H6), 2.10 (dt, J1–5 = J5–4 = 7.3 Hz, J5–6 = 3.7 Hz, 1H, H5), 2.03 (dt, J = 7.3 Hz, J1–6 = 3.5 Hz, 1H, H1), 1.95 (s, 3H, CH3NHAc), 1.55–1.45 (m, 4H, H10), 0.92 (t, J10–11 = 7.4 Hz, 3H, H11), 0.88 (t, J = 7.4 Hz, 3H, H11). 13C NMR (150 MHz, CD3OD) δ 173.62 (COCOOH), 172.11 (COAc), 140.74 (CAr), 139.95 (CAr), 138.76, 128.48 (CAr), 128.43 (CAr), 126.87 (CAr), 126.61 (CAr), 126.45 (CAr), 81.35 (C9), 80.27 (C4), 60.56 (C2), 56.55 (C3), 50.62 (CH2Bn), 28.71 (C1), 28.58 (C5), 26.39 (C10), 25.98 (C10), 21.46 (CAc), 18.04 (C6), 8.72 (C11), 8.31 (C11). HRMS: (ESI) m/z calculated for [C27H35N2O4]+ 451.26074; found 451.25913. m/z calculated for [C27H34N2NaO4]+ 473.24340; found 473.24108.
Competing interest
The authors declare no competing financial interest.Acknowledgements
The research leading to these results received funding from the People Programme of the European Union's Seventh Framework Programme by a Marie Curie Outgoing Fellowship to C. C. under REA grant agreement no. PIOF-GA-2012-327579. AJB and BMP thank the Canadian Institutes of Health Research for an operating grant (MOP-259122).References
- J. Du, T. A. Cross and H. X. Zhou, Drug Discovery Today, 2012, 17, 1111–1120 CrossRef CAS PubMed.
- C. U. Kim, W. Lew, M. A. Williams, H. T. Liu, L. J. Zhang, S. Swaminathan, N. Bischofberger, M. S. Chen, D. B. Mendel, C. Y. Tai, W. G. Laver and R. C. Stevens, J. Am. Chem. Soc., 1997, 119, 681–690 CrossRef CAS PubMed.
- M. von Itzstein, W. Y. Wu, G. B. Kok, M. S. Pegg, J. C. Dyason, B. Jin, T. V. Phan, M. L. Smythe, H. F. White, S. W. Oliver, P. M. Colman, J. N. Varghese, D. M. Ryan, J. M. Woods, R. C. Bethell, V. J. Hotham, J. M. Cameron and C. R. Penn, Nature, 1993, 363, 418–423 CrossRef CAS PubMed.
- Y. S. Babu, P. Chand, S. Bantia, P. Kotian, A. Dehghani, Y. El-Kattan, T. H. Lin, T. L. Hutchison, A. J. Elliott, C. D. Parker, S. L. Ananth, L. L. Horn, G. W. Laver and J. A. Montgomery, J. Med. Chem., 2000, 43, 3482–3486 CrossRef CAS PubMed.
- S. Jain and A. M. Fry, Clin. Infect. Dis., 2011, 52, 707–709 CrossRef PubMed.
- J. L. McKimm-Breschkin, Influenza Other Respir. Viruses, 2013, 7, 25–36 CrossRef CAS PubMed.
- R. J. Russell, L. F. Haire, D. J. Stevens, P. J. Collins, Y. P. Lin, G. M. Blackburn, A. J. Hay, S. J. Gamblin and J. J. Skehel, Nature, 2006, 443, 45–49 CrossRef CAS PubMed.
- S. Rudrawar, J. C. Dyason, M. A. Rameix-Welti, F. J. Rose, P. S. Kerry, R. J. M. Russell, S. van der Werf, R. J. Thomson, N. Naffakh and M. von Itzstein, Nat. Commun., 2010, 1, 1–7 CAS.
- Y. C. Xie, D. Q. Xu, B. Huang, X. L. Ma, W. B. Qi, F. Y. Shi, X. Y. Liu, Y. J. Zhang and W. F. Xu, J. Med. Chem., 2014, 57, 8445–8458 CrossRef CAS PubMed.
- S. Mohan, S. McAtamney, T. Haselhorst, M. von Itzstein and B. M. Pinto, J. Med. Chem., 2010, 53, 7377–7391 CrossRef CAS PubMed.
- R. E. Amaro, R. V. Swift, L. Votapka, W. W. Li, R. C. Walker and R. M. Bush, Nat. Commun., 2011, 2, 338 CrossRef PubMed.
- K. T. Greenway, E. B. LeGresley and B. M. Pinto, PLoS One, 2013, 8, e59873 CAS.
- R. E. Amaro, X. L. Cheng, I. Ivanov, D. Xu and J. A. McCammon, J. Am. Chem. Soc., 2009, 131, 4702–4709 CrossRef CAS PubMed.
- Y. Wu, G. R. Qin, F. Gao, Y. Liu, C. J. Vavricka, J. X. Qi, H. L. Jiang, K. Q. Yu and G. F. Gao, Sci. Rep., 2013, 3, 1551 Search PubMed.
- M. Niikura, N. Bance, S. Mohan and B. M. Pinto, Antiviral Res., 2011, 90, 160–163 CrossRef CAS PubMed.
- P. S. Kerry, S. Mohan, R. J. M. Russell, N. Bance, M. Niikura and B. M. Pinto, Sci. Rep., 2013, 3, 2871 Search PubMed.
- P. J. P. Adabala, E. B. LeGresley, N. Bance, M. Niikura and B. M. Pinto, J. Org. Chem., 2013, 78, 10867–10877 CrossRef CAS PubMed.
- C. A. Mooney, S. A. Johnson, P. T. Hart, L. Q. van Ufford, C. A. M. de Haan, E. E. Moret and N. I. Martin, J. Med. Chem., 2014, 57, 3154–3160 CrossRef CAS PubMed.
- J. N. Watson, V. Dookhun, T. J. Borgford and A. J. Bennet, Biochemistry, 2003, 42, 12682–12690 CrossRef CAS PubMed.
- A. G. Watts, I. Damager, M. L. Amaya, A. Buschiazzo, P. Alzari, A. C. Frasch and S. G. Withers, J. Am. Chem. Soc., 2003, 125, 7532–7533 CrossRef CAS PubMed.
- J. Chan, A. R. Lewis, M. Gilbert, M. F. Karwaski and A. J. Bennet, Nat. Chem. Biol., 2010, 6, 405–407 CrossRef CAS PubMed.
- J. Chan, A. R. Lewis, D. Indurugalla, M. Schur, W. Wakarchuk and A. J. Bennet, J. Am. Chem. Soc., 2012, 134, 3748–3757 CrossRef CAS PubMed.
- J. Chan, A. Lu and A. J. Bennet, J. Am. Chem. Soc., 2011, 133, 2989–2997 CrossRef CAS PubMed.
- M. Raab and I. Tvaroska, J. Mol. Model., 2011, 17, 1445–1456 CrossRef CAS PubMed.
- F. S. Shidmoossavee, J. N. Watson and A. J. Bennet, J. Am. Chem. Soc., 2013, 135, 13254–13257 CrossRef CAS PubMed.
- K. S. E. Tanaka, G. C. Winters, R. J. Batchelor, F. W. B. Einstein and A. J. Bennet, J. Am. Chem. Soc., 2001, 123, 998–999 CrossRef CAS PubMed.
- Y. Wang and A. J. Bennet, Org. Biomol. Chem., 2007, 5, 1731–1738 CAS.
- C. L. White, M. N. Janakiraman, W. G. Laver, C. Philippon, A. Vasella, G. M. Air and M. Luo, J. Mol. Biol., 1995, 245, 623–634 CrossRef CAS PubMed.
- L. Kaplan, J. W. Pavlik and K. E. Wilzbach, J. Am. Chem. Soc., 1972, 94, 3283–3284 CrossRef CAS.
- T. Damiano, D. Morton and A. Nelson, Org. Biomol. Chem., 2007, 5, 2735–2752 CAS.
- E. A. Acar, F. Glarner and U. Burger, Helv. Chim. Acta, 1998, 81, 1095–1104 CrossRef CAS.
- F. Glarner, B. Acar, I. Etter, T. Damiano, E. A. Acar, G. Bernardinelli and U. Burger, Tetrahedron, 2000, 56, 4311–4316 CrossRef CAS.
- R. Ling and P. S. Mariano, J. Org. Chem., 1998, 63, 6072–6076 CrossRef CAS PubMed.
- R. Ling, M. Yoshida and P. S. Mariano, J. Org. Chem., 1996, 61, 4439–4449 CrossRef CAS PubMed.
- J. F. Hayes, M. Shipman and H. Twin, J. Org. Chem., 2002, 67, 935–942 CrossRef CAS PubMed.
- M. J. Eis and B. Ganem, Tetrahedron Lett., 1985, 26, 1153–1156 CrossRef CAS.
- J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247–258 RSC.
- B. M. Trost and T. Zhang, Chem. – Eur. J., 2011, 17, 3630–3643 CrossRef CAS PubMed.
- H. Doi, T. Sakai, K. Yamada and K. Tomioka, Chem. Commun., 2004, 1850–1851 RSC.
- M. Karpf and R. Trussardi, J. Org. Chem., 2001, 66, 2044–2051 CrossRef CAS PubMed.
- S. Lemaireaudoire, M. Savignac, J. P. Genet and J. M. Bernard, Tetrahedron Lett., 1995, 36, 1267–1270 CrossRef CAS.
- F. Guibe, Tetrahedron, 1998, 54, 2967–3042 CrossRef CAS.
- R. Gigg and C. D. Warren, J. Chem. Soc., 1965, 2205–2210 RSC.
- N. Ohmura, A. Nakamura, A. Hamasaki and M. Tokunaga, Eur. J. Org. Chem., 2008, 5042–5045 CrossRef CAS.
- H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- J. L. G. Ruano, C. Fajardo, M. R. Martin, W. Midura and M. Mikolajczyk, Tetrahedron: Asymmetry, 2004, 15, 2475–2482 CrossRef.
- R. Zhang, A. Mamai and J. S. Madalengoitia, J. Org. Chem., 1999, 64, 547–555 CrossRef CAS.
- D. Romo and A. I. Meyers, J. Org. Chem., 1992, 57, 6265–6270 CrossRef CAS.
- A. Nakazato, T. Kumagai, K. Sakagami, R. Yoshikawa, Y. Suzuki, S. Chaki, H. Ito, T. Taguchi, S. Nakanishi and S. Okuyama, J. Med. Chem., 2000, 43, 4893–4909 CrossRef CAS PubMed.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- M. S. Pegg and M. Vonitzstein, Biochem. Mol. Biol. Int., 1994, 32, 851–858 CAS.
- S. Barrett, P. G. Mohr, P. M. Schmidt and J. L. McKimm-Breschkin, PLoS One, 2011, 6 Search PubMed.
- M. Z. Wang, C. Y. Tai and D. B. Mendel, Antimicrob. Agents Chemother., 2002, 46, 3809–3816 CrossRef CAS PubMed.
- S. Newstead, J. N. Watson, T. L. Knoll, A. J. Bennet and G. Taylor, Biochemistry, 2005, 44, 9117–9122 CrossRef CAS PubMed.
- A. Fersht, Structure and mechanism in protein science : A guide to enzyme catalysis and protein folding, W.H. Freeman, New York, 1999 Search PubMed.
- M. Potier, L. Mameli, M. Belisle, L. Dallaire and S. B. Melancon, Anal. Biochem., 1979, 94, 287–296 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures and tables containing 1H and 13C NMR spectra, kinetics data. See DOI: 10.1039/c6ob00999a |
| This journal is © The Royal Society of Chemistry 2016 |