
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biosynthesis of isoxazolin-5-one and 3-nitropropanoic acid containing glucosides in juvenile Chrysomelina†
Tobias
Becker
,
Kerstin
Ploss
and
Wilhelm
Boland
*
Max Planck Institute for Chemical Ecology, Bioorganic Chemistry, Hans-Knoell-Straße 8, D-07745, Jena, Germany. E-mail: boland@ice.mpg.de
First published on 27th May 2016
Abstract
Stable-isotope-labeled precursors were used to establish the biosynthetic pathway leading from β-alanine towards isoxazolin-5-one glucoside 1 and its 3-nitropropanoate (3-NPA) ester 2 in Chrysomelina larvae. Both structural elements originate from sequestered plant-derived β-alanine or from propanoyl-CoA that is derived from the degradation of some essential amino acids, e.g. valine. β-Alanine is converted into 3-NPA and isoxazolinone 5 by consecutive oxidations of the amino group of β-Ala. Substituting the diphospho group of α-UDP-glucose with 5 generates the isoxazolin-5-one glucoside 1, which serves in the circulating hemolymph of the larva as a platform for esterification with 3-nitropropanoyl-CoA. The pathway was validated with larvae of Phaedon cochleariae, Chrysomela populi as well as Gastrophysa viridula.
Introduction
Leaf beetles of the subtribe Chrysomelina1–8 and a number of legume plants9–12 produce the glucosides 1 and 2 (Fig. 1). The latter serves as a pre-toxic storage compound for the actual poison 3-nitropropanoic acid (3-NPA). 3-NPA derived compounds provide a second defensive line in Chrysomelina, parallel with and independent of larval defensive secretions released from nine paired dorsal glands.13,14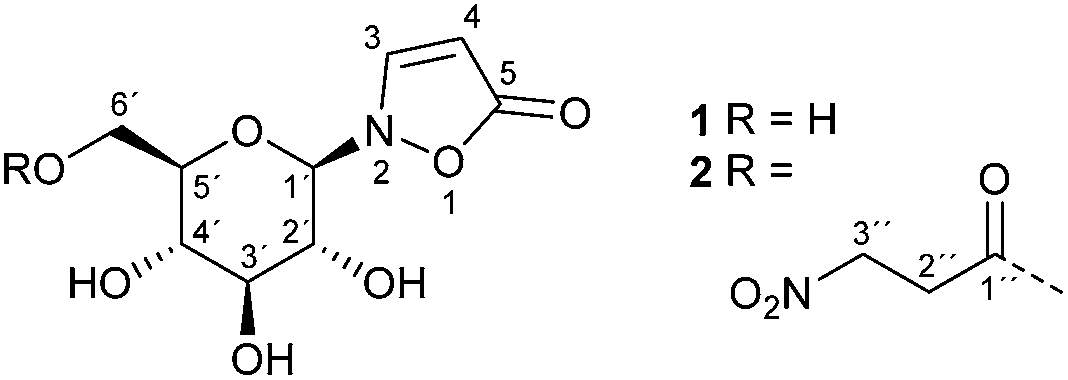 | ||
| Fig. 1 Isoxazolin-5-one glucosides in Chrysomelina larvae. | ||
Free 3-NPA was found in low concentration in the hemolymph of Chrysomelina, but was observed in significant amounts in adult secretions of these beetles as well as in some plants, e.g. Corynocarpus laevigatus, and fungi, for example in Penicillium atrovenetum.15–19 The toxic effect of this compound is due to its isoelectronic character to succinic acid, leading to a covalent addition product with mammalian succinate dehydrogenase.20,21 Thus, mitochondrial respiration is inhibited in these animals. As this effect is most relevant to nerve cells22 significant economic damage is caused to cattle feeding on 3-NPA containing food plants.23
Aspects of the biosynthesis of 3-NPA have been characterized in Penicillium atrovenetum using stable-isotope-labelled precursors, e.g. [2-13C,15N]-asp and 18O2.17,18,24 In plants, the biosyntheses of isoxazolin-5-one derivatives (glucosides and non-glucosides)9,25,26 as well as of the 3-NPA moiety24,27 have been examined using 14C-labeled compounds or with in vitro assays. In Indigofera spicata 3-NPA derives from malonate and malonyl monohydroxamate.27 The biosynthesis might involve β-alanine as an intermediate.28
Adult leaf beetles of Chrysomela tremulae feeding on host plant leaves impregnated with [14C4]-aspartate demonstrated incorporation of radioactivity into compounds 1 and 2.8 This experiment indicates the ability for de novo production of compounds 1 and 2 in Chrysomelina. Since no evidence was provided for any suggested intermediate, a contiguous metabolic pathway for the biosynthesis of compounds 1 and 2 in Chrysomelina leaf beetles has not yet been elucidated.6,8
Results and discussion
Along with previous identifications of 3-NPA in the defensive secretions of adult leaf beetles6,8 the first biosynthetic experiments with [14C4]-aspartate were published claiming aspartate as the ultimate precursor.8 As shown in Fig. 2, feeding of [13C415N]-Asp, surface-impregnated on leaves of B. rapa pekinensis and fed to larvae of Phaedon cochleariae (see Experimental) results in a cluster of ions in the area of the quasimolecular ion of 2 (formate adduct; m/z = 393 to 401) which is composed of labeled and randomly re-assembled fragments of [13C415N]-Asp. In contrast, administration of [13C315N]-β-Ala displays a distinct pattern of isotopomers consistent with the incorporation of an intact C3-segment and the 15N of the fed [13C315N]-β-Ala (m/z = 397) into 1. The 3-NPA ester 2 comprises two contiguous units of [13C315N]-β-Ala as is obvious from the fragment at m/z = 401. Peaks arising at m/z = 396 or 400 most likely result from loss of the nitrogen atom during transamination or from incomplete labelling of the commercial [13C315N]-β-Ala. Since β-alanine can be sequestered or originate from essential amino acids such as Val, Thr, Met, or Ile,29–31via propanoate as an intermediate, its origin was further addressed by feeding [13C3]-propanoate and [13C515N]-valine (Table S1†). In both cases the distinct fragment pattern from the incorporation of an intact carbon skeleton of the administered precursors was maintained. In the case of [13C3]-propanoate two fragments at m/z = 396 and 399 support incorporation of one intact propanoate moiety into 1 and up to two units into 2. The same pattern is observed after feeding of [13C515N]-valine, while the 15N of the precursor amino acid is lost. This loss of the nitrogen atom together with consecutive incorporation by later transamination can lead to either [M + 4]- and [M + 5]-peaks ([M + 7]- and [M + 8]-peaks respectively), when the nitrogen is incorporated into 13C-labelled precursors, or [M + 1]- and [M + 2]-peaks, if the labelled nitrogen is incorporated into natural unlabeled precursors.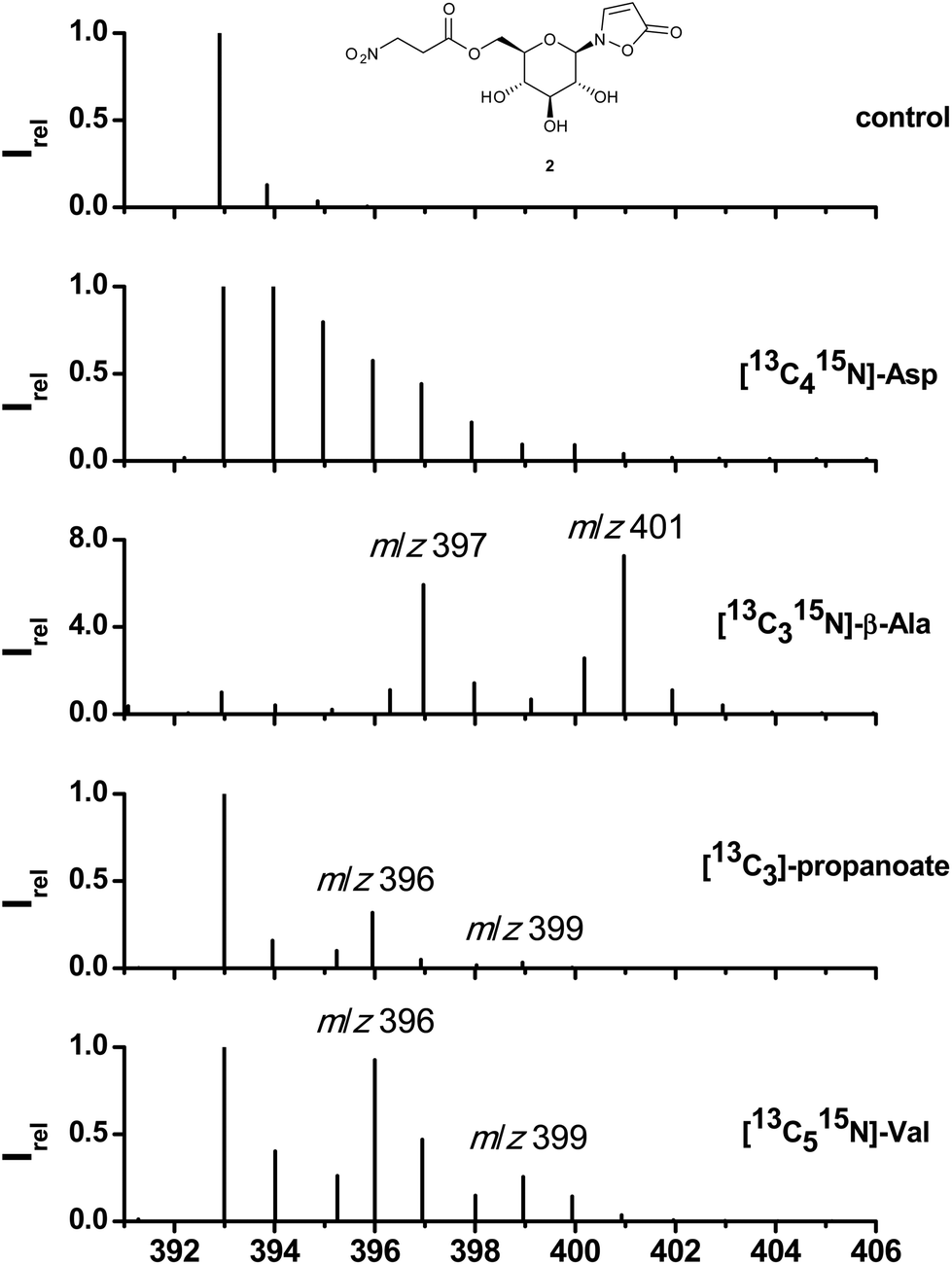 | ||
Fig. 2 Representative mass spectra of compound 2 after LC separation of larval extracts (MeCN/H2O, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) from P. cochleariae after feeding on different diets for 10 d; diets consisted of B. rapa pekinensis leaves, impregnated with KH2PO4/K2HPO4 buffered solutions of the compounds given above; as a control only blank buffer was used. :1) from P. cochleariae after feeding on different diets for 10 d; diets consisted of B. rapa pekinensis leaves, impregnated with KH2PO4/K2HPO4 buffered solutions of the compounds given above; as a control only blank buffer was used. | ||
The HR-MS analysis of hydrolytically cleaved free 3-NPA from 2 confirms these findings. Feeding of [13C315N]-β-Ala generates a molecular ion at m/z = 122.021471 (13C3H415NO4, calc. for 122.021681, [M − H]−; Fig. S4†). The carbon skeletons of [13C415N]-Thr, [13C315N]-Ala, and [13C2]-malonate were not incorporated at all into either 1 or 2 (Table S1†).
To explore the probability of the incorporation (%, see Experimental) of plant-derived β-alanine versus the de novo synthesis of β-Ala from essential precursor amino acids,29–31 defined amounts of [13C415N]-Asp, [13C3]-propanoate, and [13C315N]-β-Ala were injected into the hemolymph of P. cochleariae larvae (see Experimental; Table S1†) and the products 1 and 2 were analyzed by mass spectrometry. In the case of Asp no significant incorporation into 1 or 2 was observed (0.019 ± 0.241%), while average values of 2.3 ± 1.5 and 17.4 ± 11.2% were determined for labelled propanoate and β-Ala, respectively. Altogether, these results indicate that aspartate catabolism plays no significant role in the biosynthesis of compounds 1 and 2 although Asp is abundantly present in food plants (Fig. S13†). Altogether, we conclude that 1 and 2 are produced from both, sequestered and de novo produced β-Ala from degradation of the appropriate essential amino acids present in the food plant.
To investigate the later steps of the metabolic route from β-alanine to the isoxazolinone glucoside 1 and to its 3-NPA ester 2, potential intermediates such as [1-13C15N]-3 and [1-13C15N]-4 were synthesized and injected into the larvae (Table S1†). Accordingly, a stepwise oxygenation at the nitrogen atom of β-alanine first produces (N-hydroxyamino)propanoic acid 3 and (N-hydroxyimino)propanoic acid 4 (Fig. 3) after elimination of water from the postulated and unstable N-dihydroxy precursor. Cyclization of 4 generates the isoxazolinone 5 that is condensed with activated α-UDP-glucose to the isoxazolinone glucoside 1. Evidence for this reaction was provided by 1H NMR measurements after incubation of fat body samples of all three investigated species together with the substrates (Fig. S2†). The isoxazolinone glucoside 1 circulates in the hemolymph and serves as a platform for acylation with activated 3-NPA.
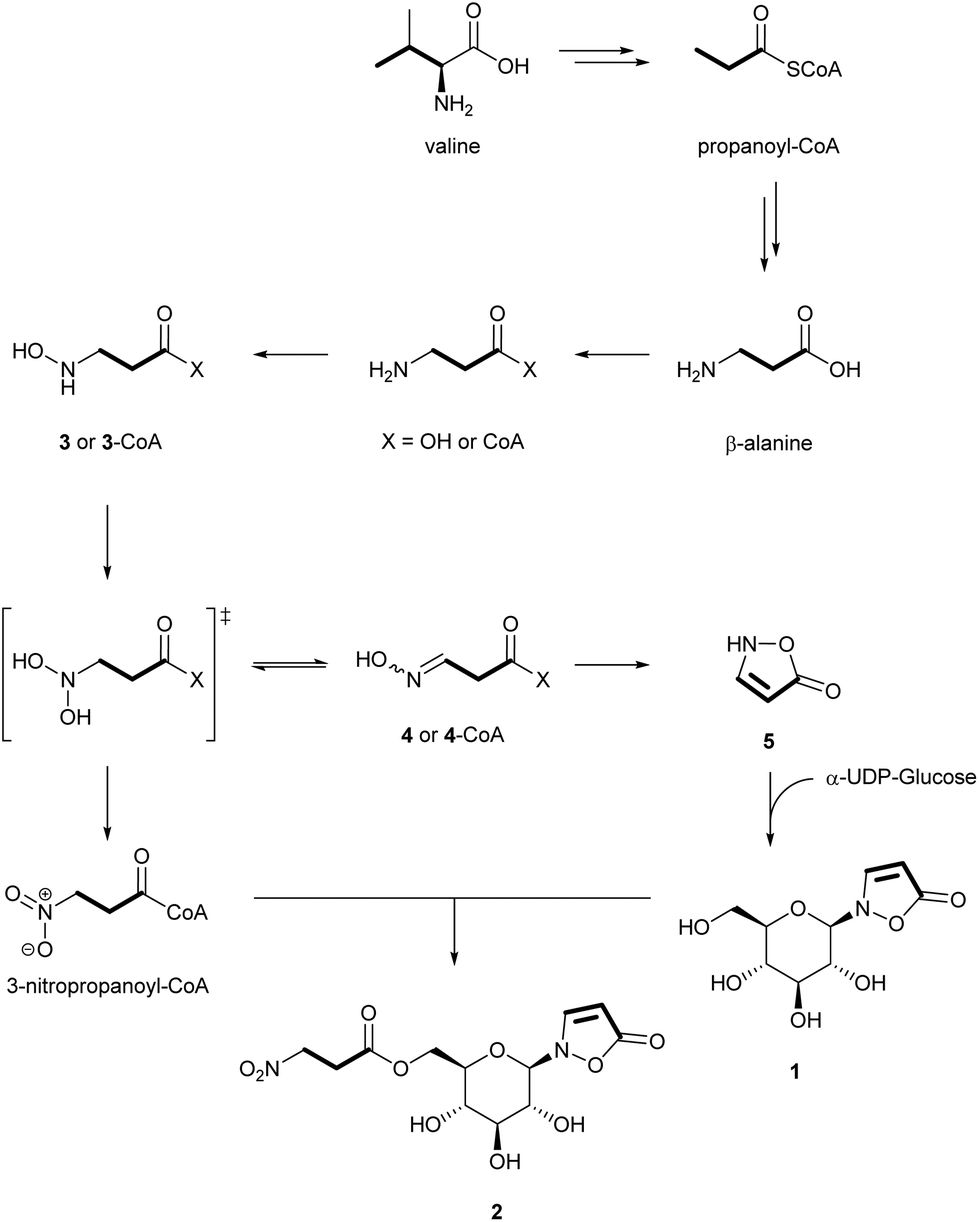 | ||
| Fig. 3 Proposed biosynthesis of compounds 1 and 2 in Chrysomelina larvae. | ||
Further oxidation of the geminal dihydroxy intermediate of β-Ala generates 3-NPA that is subsequently bound to the C(6′)-hydroxy group of the isoxazolinone glucoside 1. Activation of 3-NPA is achieved as a CoA-ester and requires ATP (Fig. S3†). The increase of the glucoside ester 2 in later stages of larval development is in line with a decrease of 1 suggesting a tight control of the individual steps of the biosynthesis of 1 (Fig. S6–S11†). A similar trend was observed for 1 and 2 in the hemolymph.1 Only very small amounts of free 3-NPA were detected in fresh hemolymph samples.1 Furthermore, neither free β-Ala nor other intermediates, such as 3 to 5 could be detected after silylation and GC-MS analysis of hemolymph samples or whole larval extracts.
Conclusion
Beetles produce 3-NPA- and the isoxazolinone-moiety from plant-derived β-alanine or by degradation of the appropriate amino acids such as L-Val via propanoate to β-Ala. The extent of these alternative routes most likely depends on the contents of the required amino acids in the food plants (Fig. S12 and S13†). Since aspartate is not a precursor of 1 or 2, this excludes a hidden contribution from the gut microbiome since the decarboxylation of aspartate to β-alanine is known from microbial metabolism.32 It is important to note that the oxidation products of β-alanine serve as precursors for both 3-NPA and the isoxazolinone 1. As the production of the isoxazolinone glucoside 1 during the very early larval stages precedes the formation of 3-NPA, most likely different oxidases are involved that are tightly regulated. The larval enzymes are still unknown, but show mechanistic similarities to recently described microbial enzymes.28 The isoxazolinone glucoside 1 circulates in the hemolymph of the insect and serves as a platform for acylation with activated 3-NPA (as CoA ester) as was demonstrated by injection of labelled free 3-NPA into the hemolymph of the larvae, which was then rapidly bound1 to the glucoside carrier 1. The steric and electronic features of N-glucoside prevent an export of this compound from the hemolymph into neighbouring tissue or the defensive secretion of the insects, although S- and O-glucosides, for example plant-derived salicin, are sequestered by the larvae and channeled via the hemolymph through the whole body into the glandular system for defense production. Both, the O- and the N-glucosides share the hemolymph as a common carrier medium, but only the natural O-glucosides and their synthetic S-analogs are allowed to pass the separating membranes.33 Therefore, the isoxazolinone glucoside 1 has never been found outside the hemolymph of larval systems. This is different in adults, which secrete several 3-NPA esters of 1 on their elytra15,34 when endangered by a predator. We assume that after ingestion of a larva, the 3-NPA esters of 1 are rapidly hydrolyzed in the gut of the predator leading to intoxication. In a living larva, release of free 3-NPA is cured by activation as a CoA-ester and re-esterification to the C(6′) of isoxazolinone glucoside.1 The identification and cloning of the enzymes catalyzing the transformation of the amino group of β-alanine to the functionally relevant nitro group are the next and urgent steps to understand the underlying mechanisms and the regulation of defense production in leaf beetle larvae.Experimental
Insect collection and rearing
The procedures were adapted from already described methods.1Chrysomela populi was collected near Dornburg, Germany (latitude 51.015, longitude 11.64), on Populus canadensis. The beetles were propagated using a light/dark cycle of 16 h light and 8 h darkness (LD 16/8), at 18 ± 2 °C in light and 13 ± 2 °C in darkness. Gastrophysa viridula was collected in Jena (latitude 50.929, longitude 11.597). Phaedon cochleariae (F.) was reared on Brassica rapa subsp. pekinensis (B. rapa pekinensis) “Cantonner Witkrop” (Quedlinburger Saatgut, Quedlinburg, Germany) and Gastrophysa viridula was reared on Rumex obtusifolius in a Snijder chamber (Snijders Scientific, Tilburg, Netherlands) in a light/dark cycle of 16 h light and 8 h darkness (LD 16/8) and 13 °C/11 °C ± 1 °C.Insect sample preparation
150 μl of MeCN/H2O 1:1 were added to individual larvae in grinding tubes equipped with 2 steel beads (diameter/bead = 4.5 mm). The samples were ground with a geno grinder (1210 rpm, 1 min) at rt for each larva. Then the mixtures were centrifuged at 40 °C and 13000 rcf for 30 min. 110 μl of the supernatant was transferred to a second tube. The samples were stored at −25 °C and vortexed for 15 s directly before analysis by HPLC/MS. All in vivo measurements are single-larva analyses. All replicates are biological replicates of individual larvae.
Insect dissection
The larvae were immersed in liquid nitrogen for 30 s. Then the fat body was isolated after cutting the cuticles of the larvae with micro scissors. The tissue was stored at −80 °C prior to use.Statistical evaluation
The results of the treated larvae were compared to the results of larvae of the control groups, in order to determine the statistical significance of the difference of the mean values. First, the data were analyzed by the Shapiro–Wilk normality test. For normally distributed datasets, the t-test was applied. For differently distributed datasets, the Mann–Whitney rank sum test was applied. Statistical difference is defined by a confidence level of at least 95% (* ≙95% ≙0.05, ** ≙0.01, *** ≙0.001).HPLC/MS analysis
The analysis of compounds 1 and 2 was done by modifying the procedure that appears in the literature.1 Measurements were carried out on an Agilent HP1100 HPLC system equipped with an OH-endcapped RP-C18 column (RP-C18e), LiChroCART (250 × 4 mm, 5 μm; Merck KGaA, 64271, Darmstadt, Germany) connected to a Finnigan LTQ (Thermo Electron Corp., Dreieich, Germany) ion trap mass spectrometer operating in the APCI mode (vaporizer temperature: 500 °C, capillary temperature 300 °C). Standard compounds for identification were either purchased (Sigma-Aldrich (St Louis, MO, USA)) or synthesized. 2 to 5 μl of the sample volume was injected, depending on the larval size (up to 20 mg larval fresh weight: 5 μl; m > 20 mg: 2 μl). The following parameters were used: flow rate = 0.5 ml min−1 at rt: 90% solvent A (H2O + 0.1% v/v HCO2H) and 10% solvent B (MeCN + 0.1% v/v HCO2H) for 5 minutes, linear gradient to 100% solvent B in 5 min, then 100% B for 2 min, linear gradient to 10% B in 5 min and further elution with 10% B for 5 min. For identification and quantification, the signals of the formic acid adducts [M + HCO2H − H]− were analyzed (m/z 292 for 2-(β-D-glucopyranosyl)-3-isoxazolin-5-one (1) and m/z 393 for 2-[6′-(3′′-nitropropanoyl)-β-D-glucopyranosyl]-3-isoxazolin-5-one (2)). The column was washed for 20 h at 40 °C with 2-propanol/MeCN 1:1 and then equilibrated to H2O/MeCN 9:1 prior to use.
GC/MS analysis
The measurement parameters were similar to the literature protocols.35 1 μl of each sample was injected at a split ratio of 1:25 into a GC/MS system equipped with an A 200S autosampler, a GC 2000 gas chromatograph, and a Voyager quadrupole mass spectrometer including a dynode/phosphor/photomultiplier detector (all ThermoQuest, Manchester, UK). Tris(perfluorobutyl)amine (CF43) was used as a reference gas for tuning. Mass spectra were recorded from m/z 50 to 622 at 0.53 s scan−1 for trimethylsilylated samples (TMS). For quantifications, the parameters were as follows: an injection temperature of 230 °C was chosen, the interface temperature was adjusted to 250 °C, and the ion source temperature was 200 °C. Helium flow was 1.5 mL min−1. After a 5 min solvent delay at 70 °C, the oven temperature was increased by 5 °C min−1 to 140 °C, then by 40 °C min−1 to 310 °C; the temperature was constant for 1 min, then cooled to 70 °C and equilibrated for 5 min. Ion trace integration was performed manually over the complete intensities of the signals.
NMR and HRMS analysis
NMR spectra were recorded using a Bruker-Spektrospin AVANCE 400 UltraShield spectrometer operating at 400 MHz (1H) and 100 MHz (13C). Chemical shifts (δ) are quoted in parts per million (ppm) and are referenced to the signals of residual protonated solvents in 1H spectra (CHCl3 at δ 7.26 ppm; HDO at δ 4.79 ppm) and deuterated chloroform in 13C spectra (CDCl3 at δ 77.16 ppm). Acetonitrile was added as a reference for 13C NMR spectra in D2O (H3CCN at δ 1.47 ppm).36 The multiplicities are given as follows: s, singlet; d, doublet; t, triplet, dd, doublet of doublets; dt, doublet of triplets; q, quartet; dq, doublet of quartets; m, multiplet. High-resolution mass spectra were recorded on a Bruker Maxis UHR-qTOF mass spectrometer.Syntheses
Modified literature protocols were used to synthesize [1′,2′,3′,4′,5′,6′-13C6]-2-(β-D-glucopyranosyl)-3-isoxazolin-5-one [1′,2′,3′,4′,5′,6′-13C6]-1,37 [1′,2′,3′,4′,5′,6′-13C6]-2-[6′-(3′′-nitropropanoyl)-β-D-glucopyranosyl]-3-isoxazolin-5-one [1′,2′,3′,4′,5′,6′-13C6]-2,38 2,2,2-trichloroethyl-3-nitropropanoate (6),38 isoxazolin-5(2H)-one (5),39 [1-13C15N]-3-(hydroxyamino)propanoate (3)18,40 and [1-13C15N]-3-(hydroxyimino)propanoate (4).39 The details of the synthetic protocols as well as the analytical data are presented in the ESI (S5–S16).†In vitro assays
To show the incorporation of isoxazolin-5-one 5 into compound 1, 10 mg of the isolated fat body tissue was suspended in 400 μl H2O. In addition, one solution containing 2.6 mg of isoxazolin-5-one 5 and one solution containing 18.7 mg of commercial α-UDP-glucose each in 200 μl were prepared. Solutions of compound 5 and α-UDP-Glc were mixed and split again into two solutions of 200 μl. To one solution, 200 μl of the fat body suspension was added; to the residual solution, 200 μl of buffer (KH2PO4/K2HPO4, 100 mM) was added. After 1 d of incubation at 30 to 40 °C, 400 μl of D2O was added to each solution, including the fat body suspension, after which the mixtures were centrifuged and analyzed by 1D 1H NMR experiments (512 scans each).The ATP/CoA-dependent incorporation of 3-NPA into compound 2 was shown according to procedures in the literature41 Solutions of ATP (c = 125 mM), CoA (c = 5 mM), 3-NPA (10.5 mM), compound 1 (10.5 mM) and the isolated fat body tissue (m = 30 mg) in 2 ml of buffer (Tris-base 50 mM, sucrose 250 mM, MgCl2 2 mM, 1 μl ml−1 dithiothreitol solution, 10 μl ml−1 protease inhibitor mix) were prepared. The fat body mixture was homogenized using a geno grinder (1200 rpm, 30 s). Then five mixtures with an individual volume of 1 ml were prepared from these solutions as follows: solution 1 containing ATP, CoA, 3-NPA, compound 1, fat body; solution 2 (CoA, 3-NPA, compound 1, fat body, buffer); solution 3 (ATP, CoA, 3-NPA, compound 1, buffer); solution 4 (ATP, 3-NPA, compound 1, fat body, buffer) and solution 5 (ATP, CoA, fat body, 2 × buffer). After 1 d of incubation at 30 °C, 200 μl was taken and centrifuged (13000 rcf, 15 min), and the supernatant was directly analyzed by HPLC/MS.
In vivo injection experiments
The larvae were fixed with pincers manually upon ice under a light microscope. Then a dose of 20 to 40 nmol substance per mg larval fresh weight was injected with a thin glass capillary attached to a microinjector. Typically injection volumes of 200 nl containing solutions of compounds with a concentration of 0.5 mol l−1 dissolved in potassium phosphate (KH2PO4/K2HPO4) or potassium carbonate (K2CO3) buffers were applied. After the total volume was injected, the glass capillary was not removed from each larva for 1 min to prevent direct bleeding. Then the larvae were kept in plastic beakers covered with cardboard with a piece of their host plant leaf (100 to 400 mg) to enable air exchange at rt unless noted otherwise. The injected larvae were incubated for 24 h before being extracted with MeCN/H2O 1:1.
Feeding experiments
Whole leaves were impregnated with aqueous buffered (KH2PO4/K2HPO4, pH 7.4, 500 mmol l−1 + 300 μl of acetone, for reduction of surface tension) solutions of the isotopic-labeled compounds using a brush and given to P. cochleariae (L1 and L2) as food. The larvae were extracted after 7 to 10 d of feeding. Freshly impregnated leaves were added two or three times. The concentrations of the isotope-labeled compounds were as follows: [13C415N]-aspartate (c = 100 mmol l−1), [15N]-aspartate (c = 100 mmol l−1), [4-13C]-aspartate (c = 100 mmol l−1), [13C4]-aspartate (c = 100 mmol l−1), [1,3-13C2]-malonate, [13C315N]-α-L-alanine (c = 100 mmol l−1), [15N]-α-L-alanine (200 mmol l−1), [13C315N]-β-alanine (c = 100 mmol l−1), [13C3]-propanoic acid (c = 100 mmol l−1), [13C515N]-valine (200 mmol l−1) and [13C415N]-threonine (200 mmol l−1). An overview is presented in the ESI (Table S1†).Quantification and identification of amino acids in B. rapa pekinensis
The samples were analyzed according to procedures modified from the literature. 200–1000 mg FW of B. rapa subsp. pekinensis leaves were cut, transferred into a 2 mL tube, and mixed with 100 μl of [13C3, 15N]β-alanine (c = 0.6 mmol l−1). The samples were ground by using a geno grinder (1200 rpm, 3 × 5 min) and shaken at 40–60 °C for 3 h. 1 mL of methanol was added, and the samples were centrifuged for 15 min at 13000 rcf. The supernatant was pipetted into a screw-top glass tube, and 2.5 ml of MeCN was added. The tubes were equipped with a needle, and the solvents were removed in a desiccator under reduced pressure at rt. This procedure was repeated until the samples were completely dried. To each sample, N-methyl-N-trimethylsilyltrifluoroacetamide (MSTFA) was added, and the samples were shaken at 40 °C for 30 min. The clear solutions were directly analyzed by GC/MS. The peak areas of the heavy isotope signals corresponding to the standards and the areas of the normal isotopes were integrated. The ratio of the peak areas gave the molar content of the amino acid cm,AA,leaf in nmol (mg fresh weight)−1 according to eqn (1):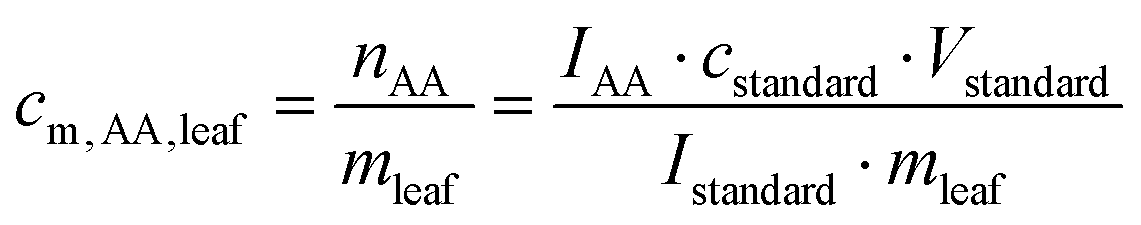 | (1) |
Quantification of compounds 1 and 2
Solutions of synthetic [1′,2′,3′,4′,5′,6′-13C6]-2-(β-D-glucopyranosyl)-3-isoxazolin-5-one [1′,2′,3′,4′,5′,6′-13C6]-1 (c = 17.33 mmol l−1, V = 10 μl) as well as [1′,2′,3′,4′,5′,6′-13C6]-2-[6′-(3′′-nitropropanoyl)-β-D-glucopyranosyl]-3-isoxazolin-5-one [1′,2′,3′,4′,5′,6′-13C6]-2 (c = 14.65 mmol l−1, V = 10 μl) in MeCN/H2O 1:1 were added to each larval extract (SIL-IS42,43). The peak areas of the heavy isotope signals in the chromatogram (m/z 298 for compound 1, m/z 399 for compound 2) were compared to the peak areas of the normal isotopes (m/z 292 for compound 1, m/z 393 for comp. 2). The molar contents cm,analyte,larva of compounds 1 and 2 in nmol (mg larval fresh weight)−1 were calculated using eqn (2): | (2) |
Incorporation of injected compounds
To demonstrate the incorporation of the injected compounds had taken place, the areas of the isotopic peaks of compounds 1 and 2 were divided by the areas of the non-isotopic peaks. These ratios were compared to the ratios determined in the control groups. In order to determine the percentile C of the incorporation of the injected compounds in % eqn (3) was used: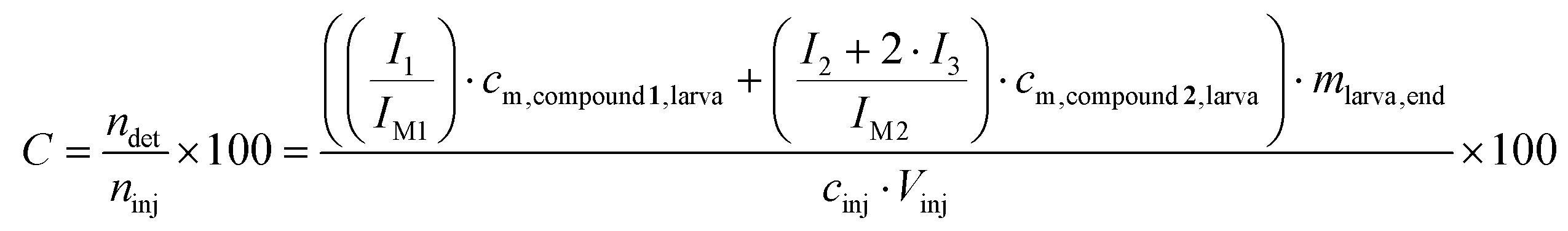 | (3) |
Author contributions
T. B. and W. B. planned and designed the project and experiments. T. B. synthesized the compounds. T. B. performed injection and feeding experiments. T. B. carried out the in vitro experiments. T. B. prepared the samples. T. B. measured and analyzed all NMR spectra. T. B. and K. P. performed HPLC/MS measurements. T. B. performed GC-MS measurements. T. B. analyzed GC- and LC-mass spectra. T. B. analyzed the statistical data. T. B. and W. B. wrote the manuscript.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We thank Emily Wheeler for her editorial assistance. We thank Dr Maritta Kunert and Dr Antje Burse for their help collecting samples. The help of Angelika Berg and Anja David for rearing the beetles is also acknowledged. We acknowledge René R. Gretscher for artwork of the graphical abstract. This work was supported by the Max Planck Society.Notes and references
- G. Pauls, T. Becker, P. Rahfeld, R. R. Gretscher, C. Paetz, J. M. Pasteels, S. H. von Reuss, A. Burse and W. Boland, J. Chem. Ecol., 2016, 42, 240 CrossRef CAS PubMed.
- J. M. Pasteels, D. Daloze and M. Rowell-Rahier, Physiol. Entomol., 1986, 11, 29 CrossRef CAS.
- J. M. Pasteels, J. C. Braekman, D. Daloze and R. Ottinger, Tetrahedron, 1982, 38, 1891 CrossRef CAS.
- J. M. Pasteels, D. Daloze and M. Rowellrahier, Physiol. Entomol., 1986, 11, 29 CrossRef CAS.
- M. Rowell-Rahier and J. M. Pasteels, J. Chem. Ecol., 1986, 12, 1189 CrossRef CAS PubMed.
- P. Laurent, J.-C. Braekman, D. Daloze and J. Pasteels, Eur. J. Org. Chem., 2003, 2733 CrossRef CAS.
- P. Laurent, J.-C. Braekman and D. Daloze, in The Chemistry of Pheromones and Other Semiochemicals II, ed. S. Schulz, Springer, Berlin, Heidelberg, 2005, vol. 240, p. 167 Search PubMed.
- T. Randoux, J. C. Braekman, D. Daloze and J. M. Pasteels, Naturwissenschaften, 1991, 78, 313 CrossRef CAS PubMed.
- I. Murakoshi, F. Ikegami, F. Kato, J. Haginiwa, F. Lambein, L. Van Rompuy and R. Van Parijs, Phytochemistry, 1975, 14, 1269 CrossRef CAS.
- L. van Rompuy, F. Lambein, R. De Gussem and R. van Parijs, Biochem. Biophys. Res. Commun., 1974, 56, 199 CrossRef CAS PubMed.
- F. Ikegami, F. Lambein, Y. H. Kuo and I. Murakoshi, Phytochemistry, 1984, 23, 1567 CrossRef CAS.
- F. Lambein, L. Van Rompuy, A. De Bruyn and R. Van Parijs, Arch. Int. Physiol. Biochim., 1974, 82, 187 CAS.
- A. Burse, S. Frick, S. Discher, K. Tolzin-Banasch, R. Kirsch, A. Strauss, M. Kunert and W. Boland, Phytochemistry, 2009, 70, 1899 CrossRef CAS PubMed.
- P. Rahfeld, W. Haeger, R. Kirsch, G. Pauls, T. Becker, E. Schulze, N. Wielsch, D. Wang, M. Groth, W. Brandt, W. Boland and A. Burse, Insect Biochem. Mol. Biol., 2015, 58, 28 CrossRef CAS PubMed.
- W. Sugeno and K. Matsuda, Appl. Entomol. Zool., 2002, 37, 191 CrossRef CAS.
- J. W. Hylin and H. Matsumoto, Arch. Biochem. Biophys., 1961, 93, 542 CrossRef CAS PubMed.
- R. L. Baxter, E. M. Abbot, S. L. Greenwood and I. J. Mcfarlane, J. Chem. Soc., Chem. Commun., 1985, 564 RSC.
- R. L. Baxter, A. B. Hanley, H. W. S. Chan, S. L. Greenwood, E. M. Abbot, I. J. Mcfarlane and K. Milne, J. Chem. Soc., Perkin Trans. 1, 1992, 2495 RSC.
- R. C. Anderson, W. Majak, M. A. Rassmussen, T. R. Callaway, R. C. Beier, D. J. Nisbet and M. J. Allison, J. Agric. Food Chem., 2005, 53, 2344 CrossRef CAS PubMed.
- L. S. Huang, G. Sun, D. Cobessi, A. C. Wang, J. T. Shen, E. Y. Tung, V. E. Anderson and E. A. Berry, J. Biol. Chem., 2006, 281, 5965 CrossRef CAS PubMed.
- T. A. Alston, L. Mela and H. J. Bright, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 3767 CrossRef CAS.
- C. Olsen, A. Rustad, F. Fonnum, R. E. Paulsen and B. Hassel, Brain Res., 1999, 850, 144 CrossRef CAS PubMed.
- M. C. Williams, Rev. Weed Sci., 1994, 6, 1 CAS.
- K. Francis, C. Smitherman, S. F. Nishino, J. C. Spain and G. Gadda, IUBMB Life, 2013, 65, 759 CrossRef CAS PubMed.
- Y.-H. Kuo, F. Ikegami and F. Lambein, Phytochemistry, 1998, 49, 43 CrossRef CAS.
- F. Lambein and Y.-H. Kuo, Int. J. Toxicol., Occup. Environ. Health, 1993, 2, 90 CAS.
- E. Candlish, L. J. La Croix and A. M. Unrau, Biochemistry, 1969, 8, 182 CrossRef CAS PubMed.
- R. Winkler and C. Hertweck, ChemBioChem, 2007, 8, 973 CrossRef CAS PubMed.
- D. Y. Boudko, J. Insect Physiol., 2012, 58, 433 CrossRef CAS PubMed.
- Y. Kaziro and S. Ochoa, in Adv. Enzymol. Relat. Areas Mol. Biol, John Wiley & Sons, Inc., 2006, p. 283 Search PubMed.
- P. A. Brindle, D. A. Schooley, L. W. Tsai and F. C. Baker, J. Biol. Chem., 1988, 263, 10653 CAS.
- P. Spiteller, in Amino Acids, Peptides and Proteins in Organic Chemistry, ed. A. B. Hughes, WILEY VCH, 2009, vol. 1, p. 119 Search PubMed.
- J. Kuhn, E. M. Pettersson, B. K. Feld, A. Burse, A. Termonia, J. M. Pasteels and W. Boland, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13808 CrossRef CAS PubMed.
- J. M. Pasteels, A. Termonia, D. Daloze and D. M. Windsor, in Proceedings of the Fifth International Symposium on the Chrysomelidae, Iguacu, 2000, ed. D. G. Furth, Pensoft Publishers, 2003, vol. 21, p. 261 Search PubMed.
- O. Fiehn, J. Kopka, R. N. Trethewey and L. Willmitzer, Anal. Chem., 2000, 72, 3573 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512 CrossRef CAS PubMed.
- T. Becker, P. Kartikeya, C. Paetz, S. H. von Reuss and W. Boland, Org. Biomol. Chem., 2015, 13, 4025 CAS.
- T. Becker, H. Görls, G. Pauls, R. Wedekind, M. Kai, S. H. von Reuss and W. Boland, J. Org. Chem., 2013, 78, 12779 CrossRef CAS PubMed.
- F. De Sarlo, G. Dini and P. Lacrimini, J. Chem. Soc. C, 1971, 86 RSC.
- C. Song, S. Tapaneeyakorn, A. C. Murphy, C. Butts, A. Watts and C. L. Willis, J. Org. Chem., 2009, 74, 8980 CrossRef CAS PubMed.
- A. Novoselov, T. Becker, G. Pauls, S. H. von Reuss and W. Boland, Insect Biochem. Mol. Biol., 2015, 63, 97 CrossRef CAS PubMed.
- S. Arrivault, M. Guenther, S. C. Fry, M. M. F. F. Fuenfgeld, D. Veyel, T. Mettler-Altmann, M. Stitt and J. E. Lunn, Anal. Chem., 2015, 87, 6896 CrossRef CAS PubMed.
- B. K. Matuszewski, J. Chromatogr., B, 2006, 830, 293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00899b |
| This journal is © The Royal Society of Chemistry 2016 |