
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heat-enhanced peptide synthesis on Teflon-patterned paper†
Frédérique
Deiss‡§
,
Yang
Yang§
,
Wadim L.
Matochko¶
and
Ratmir
Derda
*
Department of Chemistry and Alberta Glycomics Centre, University of Alberta, 11227 Saskatchewan Drive, Edmonton, AB T6G 2G2, Canada. E-mail: ratmir@ualberta.ca
First published on 3rd May 2016
Abstract
In this report, we describe the methodology for 96 parallel organic syntheses of peptides on Teflon-patterned paper assisted by heating with an infra-red lamp. SPOT synthesis is an important technology for production of peptide arrays on a paper-based support for rapid identification of peptide ligands, epitope mapping, and identification of bio-conjugation reactions. The major drawback of the SPOT synthesis methodology published to-date is suboptimal reaction conversion due to mass transport limitations in the unmixed reaction spot. The technology developed in this report overcomes these problems by changing the environment of the reaction from static to dynamic (flow-through), and further accelerating the reaction by selective heating of the reaction support in contact with activated amino acids. Patterning paper with Teflon allows for droplets of organic solvents to be confined in a zone on the paper array and flow through the paper at a well-defined rate and provide a convenient, power-free setup for flow-through solid-phase synthesis and efficient assembly of peptide arrays. We employed an infra-red (IR) lamp to locally heat the cellulosic support during the flow-through delivery of the reagents to each zone of the paper-based array. We demonstrate that IR-heating in solid phase peptide synthesis shortened the reaction time necessary for amide bond formation down to 3 minutes; in some couplings of alpha amino acids, conversion rates increased up to fifteen folds. The IR-heating improved the assembly of difficult sequences, such as homo-oligomers of all 20 natural amino acids.
Introduction
Arrays of molecules synthesized in parallel on a planar support are a very attractive technology for testing molecular interactions in a rapid and parallel fashion. One of the most notable examples is parallel synthesis of DNA on a planar support developed by Fodor, Brown and others.1–4 DNA microarrays were the main tool for gene expression profiling for two decades.5 Arrays of peptides synthesized on a planar cellulose support have been used extensively for mapping protein–peptide interactions,6 identification of peptide-based epitopes for antibodies,7 identification of cell-binding peptides,8–12 small molecules that modulate quorum sensing,13 discovery of anti-microbial peptides,14–17 discovery of substrates for kinases,18,19 and other enzymes,20–22 design of synthetic proteins,23–26 discovery of small-molecule fluorescent dyes,27 and de novo discovery of bio-orthogonal reactions for modification of proteins.28 A more comprehensive overview of the applications of SPOT synthesis can be found in recent reviews.29–32 Many other classes of molecular arrays can also be generated via step-wise synthesis of small molecules33 or via site specific conjugation of pre-synthesized molecules.34–37 In every array, chemical reactions on the solid support have to be optimized to maximize the yield and purity of the molecules displayed on these arrays. In this report, we focus on peptide arrays on a planar porous support (paper) and we develop heat-acceleration of chemical reactions on these arrays.Heat-acceleration is popular in solid phase peptide synthesis (SPPS) because it shortens the time of the reaction and enables the synthesis of “difficult” sequences that are not accessible by traditional reactions at room temperature.38–40 It is now routinely employed in many commercial peptide synthesizers. SPOT synthesis is a powerful variant of the SPPS, which permits the parallel synthesis of arrays of peptides on a cellulose support.41,42 Although SPOT-synthesis has been developed over 25 years ago43 and used in multiple commercial technologies, there are no reports of heat acceleration for SPOT peptide synthesis technology to date. Blackwell and co-workers reported a microwave-assisted synthesis of small molecules on cellulose generating libraries of chalcone-derived molecules;44 using a microwave reactor, they reduced the time of most of the synthesis steps from 5–48 hours to 10–30 minutes. There are still fundamental limitations for heat-assisted peptide synthesis: SPOT-array is an open system operating with a limited amount of solvent; application of controlled heat to this system leads to fast evaporation of the solvents from the reagent-laden spots thus hampering the reactions. Recently, we have developed a technology for patterning paper with Teflon to create solvophobic barriers resistant to a broad range of solvent.11 These barriers confine organic solvents during SPOT-synthesis and permit exposing the planar solid support corralled by Teflon-barriers to 10–20-fold excess of solvents per unit area. We hypothesized that the excess of solvent can overcome the deleterious effect resulting from the evaporation of the solvent and permit applying heat to accelerate the coupling in the synthesis of peptide arrays on paper.
Although examples of heat accelerated peptide bond formation are limited in SPOT synthesis, we note that heat-accelerated peptide synthesis on polystyrene beads is well-established: it was first reported by the groups of Tam45 and Tsegenidis46 in the late 1980s. Twenty years later, microwave irradiation has been demonstrated to also be beneficial to peptide coupling.47,48 Raising the temperature of the support decreased the time required for the reaction to reach completion and improves the purity of peptides by limiting the contribution of kinetically slower side reactions;49 moderate heating in such reactions is known to produce no unwanted epimerization of the amino acids during the coupling. Side reactions can be further suppressed by repetitive coupling with freshly prepared reagents or by applying a continuous flow of reagents through a heated solid support.50 Integration of Teflon patterns into paper allows adapting both the concepts of localized heating of the support and gravity-driven flow-through delivery of the reagents to this support. In short, convex droplets of the solutions of reagents deposited atop patterned paper are confined by solvophobic barriers and cannot spread laterally; instead they flow through the paper at a well-defined rate (Fig. 1a).11 Heating the support from the bottom using an IR lamp allows increasing the temperature of the porous support, while maintaining the bulk solution of the reagents atop this support at room temperature. Such heating would not be possible in conventional SPOT synthesis and it would be hampered by rapid evaporation of solvents. While the technology described in this report focuses on acceleration of acylation chemistry, we anticipate that combination of IR heating and flow-through mixing will enable acceleration of numerous organic reactions that are compatible with cellulose supports, including aldol condensations,44 heterocycle-forming reactions,13,27,51 Ugi four-component reactions,52,53 and metal-catalysed organic transformations, such as Suzuki–Miyaura and copper-catalyzed azide–alkyne cycloaddition54 (for in depth reviews see ref. 33, 55). The key to adaptation and automation is simplicity; and a simple solution like an IR lamp can be envisaged as a trivial technical upgrade to the current state of the art robotic synthesis of arrays to enhance the quality of the arrays synthesized.
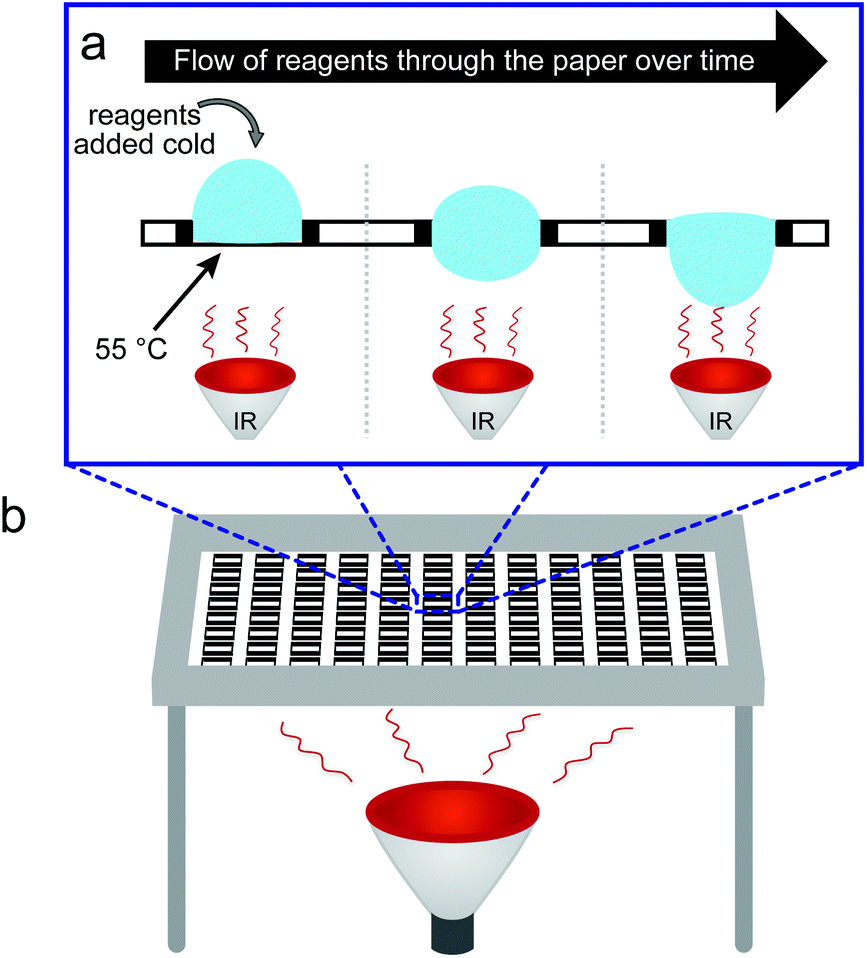 | ||
| Fig. 1 Set-up of synthesis on Teflon-patterned paper heated by using an infra-red lamp: the cold activated solution flows through the paper over time (a). The 96-zone array is clamped in a frame and maintained at 20 cm over the infra-red heating lamp (b). | ||
Results and discussion
The experimental setup for heat-accelerated SPOT synthesis is described in Fig. 1. Teflon-patterned paper arrays were prepared as described previously11 and clamped with a custom holder positioned on top of the IR lamp (Fig. 1 and S1a†). We found that it is necessary to use aluminum foil (or other reflective material) to confine the light and prevent excessive dissipation of heat. In this setup, we tested the uniformity of the heating of the support by using an infra-red thermometer: the temperature of the support was 55 ± 5 °C (Fig. S1b–e†). We have found that the small variations in temperature (±5 °C) observed for replicates at different positions of the array have negligible effect on the amount of Fmoc measured at each coupling step. The data does not exhibit any systematic drift in the measured Fmoc values (Fig. S7†) that would be expected otherwise for non-uniform irradiation. Unless noted otherwise, we used this temperature in all our experiments. The functionalization of the paper with a standard two-beta-alanine linker42 (“βAla–βAla”, Fig. 2a) consists of 3 steps: (i) esterification of the cellulose with the carboxyl group of the Fmoc-protected β-alanine, (ii) Fmoc-deprotection and (iii) coupling of the second Fmoc-protected β-alanine. We first tested whether heating can accelerate these steps. All reagents for these steps were adapted from previous reports;42 capping by acetic anhydride was used after all coupling reactions described in this report. The yields of all reactions were estimated by spectrophotometric measurements of the amount of the adduct dibenzofulvene–piperidine produced during the Fmoc-deprotection by piperidine (Fig. S2†). It is important to note that quantification of Fmoc is a destructive method and therefore we are unable to follow the same spot for each coupling step. Each step was analysed at another spot on the paper. As most of the characterization in this manuscript is based on the Fmoc quantification, we characterized the variability of the Fmoc-quantification assay itself. Fig. S8† describes the measurement of Fmoc loading in 96 spots across two arrays. We observed no systematic drift and only a 5–6% variability for the Fmoc measuring process under conditions that yield uniform loading (here: mild shaking of array submerged in its activating solution). In contrast, if the experiment had an intentional systematic difference under chemical conditions across the array, such as loading under non-uniform submersion (non-shaking) of the array, such an experiment would exhibit an increase in the standard deviation and a systematic drift across the array.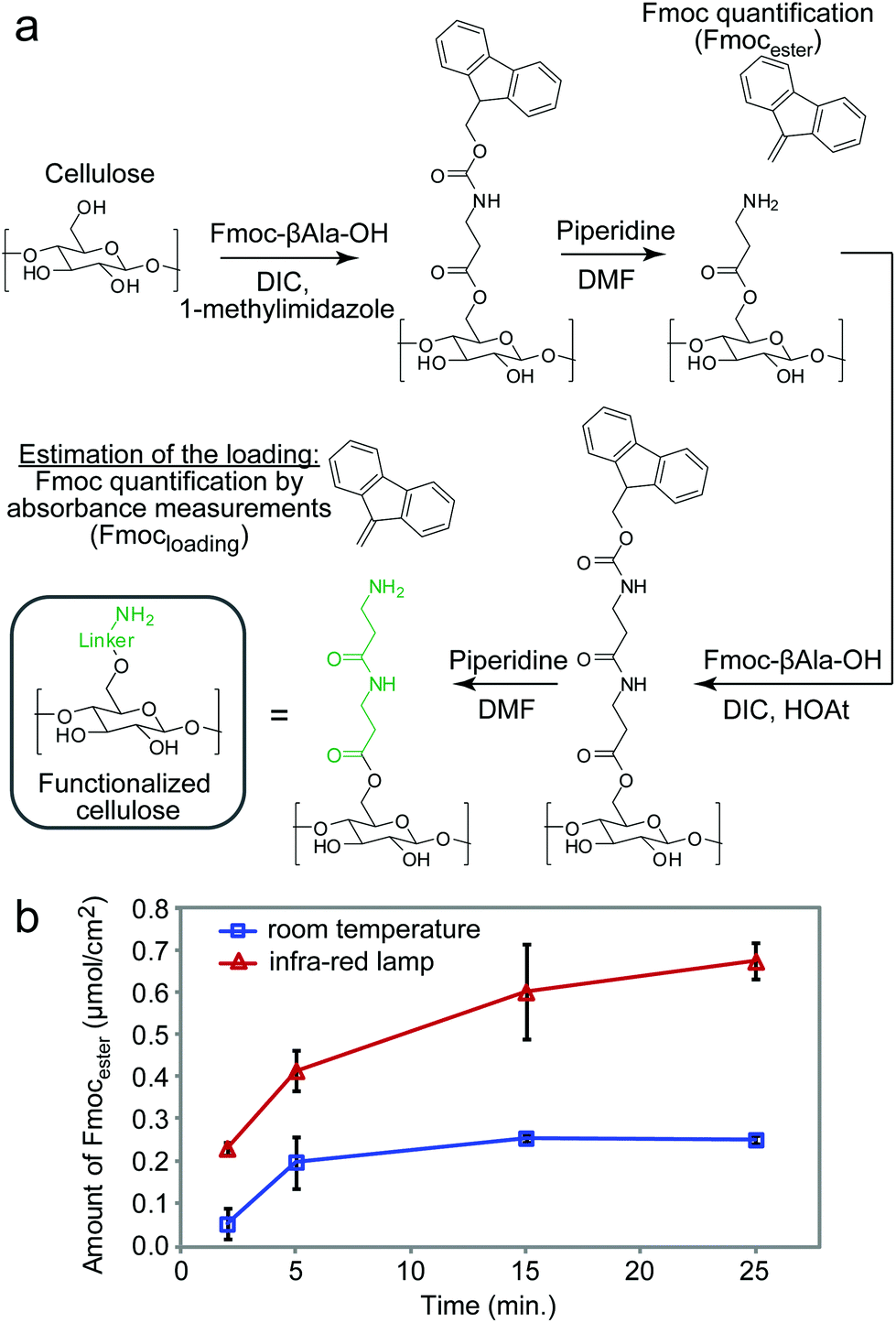 | ||
| Fig. 2 Functionalization of the cellulose with the canonical β-Ala–β-Ala linker by esterification of the cellulose with the carboxyl group of the first Fmoc-protected β-alanine, followed by Fmoc-deprotection and coupling of the second Fmoc-protected β-Ala (a). Comparison of the amount of Fmoc ester of dibenzofulvene scavenged by piperidine after esterification of paper over the infra-red lamp (∼55 °C) and at room temperature as a control (b). | ||
IR-heating accelerated the esterification of the cellulose by DIC-activated Fmoc-βAla-OH (Fig. 2a). The amount of Fmoc removed after the coupling of the 1st βAla to the paper (Fmocester in Fig. 2b), reached 0.2 μmol cm−2 on the IR-heated support in two minutes. In the control reaction at room temperature, a similar loading was achieved in over five minutes. Continuous heating of the reaction resulted in a loading of 0.7 μmol cm−2 after twelve minutes. In contrast, even after prolonged esterification at room temperature for 30 minutes, the loading of the 1st βAla plateaued at 0.2 μmol cm−2 (Fig. 2b). Due to evaporation of the solutions, it was not possible to extend the IR-heated coupling beyond 10–12 minutes: 70% of the zones of the arrays were dry after 15 minutes and 100% of the zones were dry after 25 minutes. The above results were obtained by spotting 15 μL per zone of the activated solution (96 × 15 = 1440 μL total), whereas a conventional method for esterification uses submersion of paper into a large excess (15 mL) of the DIC/HOAt-activated βAla solution for three hours.42 Under these conditions, the final loading of “βAla–βAla” can reach up to 1.5 μmol cm−2. IR-promoted esterification with droplets of reagents, thus, can be used as a rapid, reagent-economical method, while prolonged submersion-based modification can be used to maximize the loading.
We previously observed the increase in the rate of amide bond coupling on Teflon-patterned paper when compared to the canonical SPOT peptide synthesis on unpatterned paper.11 This improvement was the result of the gravity-driven flow of the activated solutions through the paper. To test whether heating can further accelerate the coupling of Fmoc-protected alanine (Fmoc-Ala) to the βAla–βAla linker (Fig. 3a) on paper in the flow-through setting, we interrupted the reaction at 30–120 s intervals for up to 24 min and measured the amount of Fmoc-Ala loaded on the paper (Fig. 3b–d). To relate the increase in the rate of coupling to the rate of flow of the reagents through paper, we used two types of paper with different porosities: Whatman filter paper grade 50 (pore size of 2.7 μm, thickness of 115 μm, low flow rate) and grade 1 (pore size of 11 μm, thickness of 180 μm, high flow rate). On ‘Whatman 50’ paper, IR heating accelerated the rate of the coupling of Fmoc-Ala by a factor of five when compared to the analogous reaction at room temperature (Fig. 3c and d) IR heating also accelerated the coupling of Fmoc-Ala to modified Whatman 1 paper, but the improvement was not as significant as that observed on paper of lower porosity (Fig. S3a and b†). On the other hand, the effect of heating in Whatman 1 paper was more pronounced in the coupling of bulkier amino acids such as tryptophan (Fig. S3e and f†).
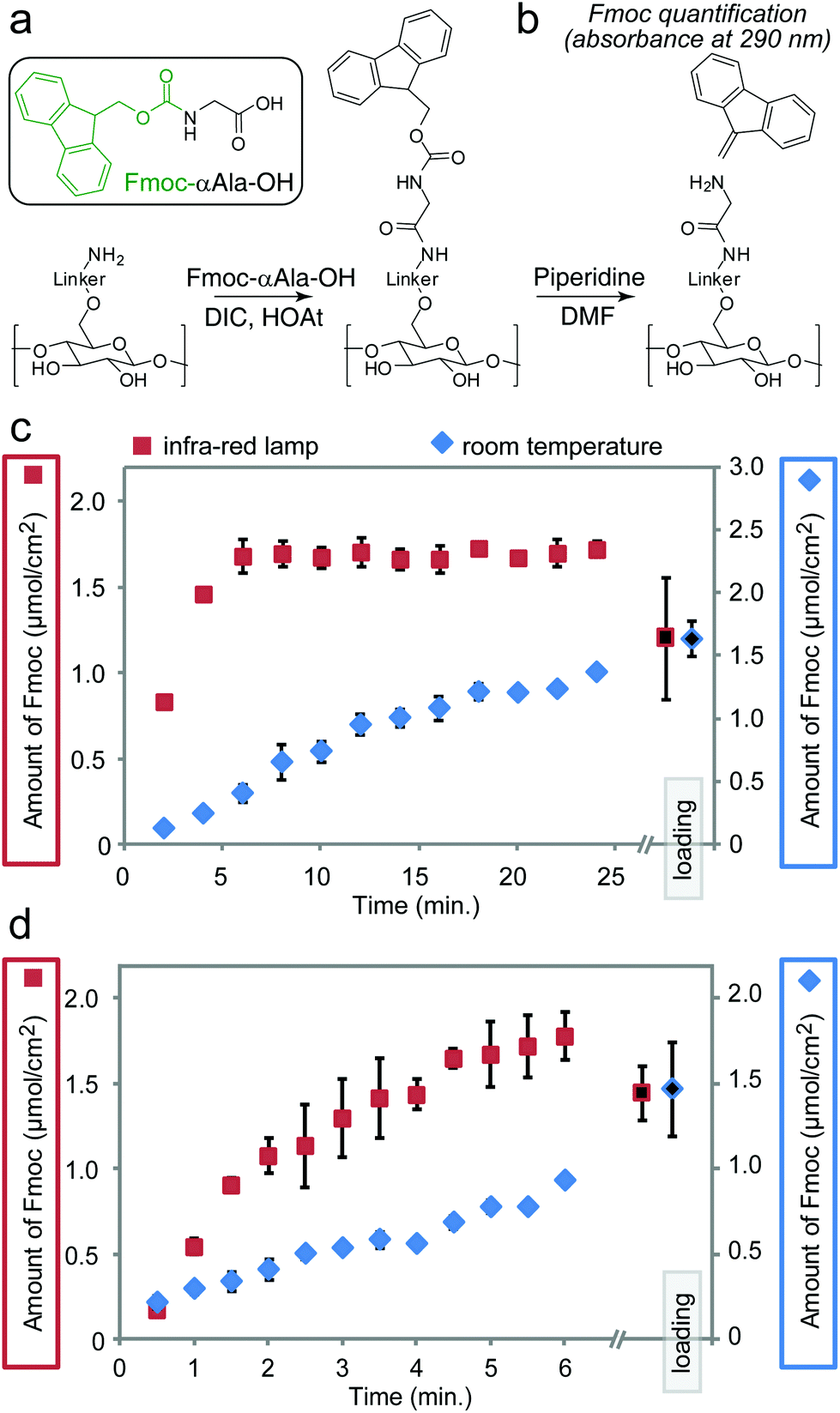 | ||
| Fig. 3 Coupling of α-alanine onto functionalized paper (a) followed by Fmoc-deprotection and quantification of dibenzofulvene–piperidine. (b) Fmoc quantification monitored the increase in the amount of alanine coupled onto the functionalized paper at room temperature and in the IR-accelerated reaction. We examined 12 reactions in which the coupling times varied between 2 and 24 minutes (c) and 12 reactions in which the coupling times varied between 0.5 and 6 minutes (d). The experiments in (c) and (d) were performed independently. All reactions were performed in triplicates; the error bars are 2 × (standard deviation) as calculated from replicates within the same array. | ||
To investigate the variability in coupling efficiency between different amino acids, we synthesized peptides with six identical residues (homo-hexapeptides) for all 20 amino acids (Table S1 and Fig. S4†). To detect both the increase and decrease in coupling efficiency, in all synthetic steps we used a 2-minute coupling, which was the time necessary to achieve ∼50% conversion in amide bond coupling (Fig. 2b). The first two reactions—coupling of the second βAla and the first α-amino acid—were the most critical in determining the amount of the final hexapeptide. A 2-minute coupling made it possible to make some hexapeptides (e.g., Asp6, Glu6, Met6) with high efficiency; the conversion rates were similar for all six amino acids. The peptides Cys6, Ile6, Asn6, Pro6 and Val6 exhibited decreasing conversion rates at every coupling, indicating that a 2 min coupling time is not sufficient for these amino acids. For Ala6, Ser6, and Thr6, we observed a decrease in the conversion in the 5th coupling and for Gly6 and Leu6 a decrease in the 4th coupling indicating the potential presence of “difficult” sequences at these steps. With the exception of Val6, all peptides produced in the IR-synthesis displayed a higher conversion rate than in the synthesis at room temperature (Table S1 and Fig. S5†). IR heating generally increases the purity of the final product, it is however not a universal answer to all difficulties associated with SPOT syntheses. In addition, while IR heating enables rapid coupling, it needs to be carefully controlled, as prolonged heating could potentially reach other reactive groups remaining on the peptides or the paper substrate.
Based on the results above, we selected three classes of sequences of varying degrees of synthetic difficulty—hexaalanine (Ala6, Fig. 4a and d), hexatryptophan (Trp6, Fig. 4b and e), and hexaarginine (Arg6, Fig. 4c and f)—and tested the effect of coupling time on the efficiency of synthesis. We have confirmed the projected purities predicted by Fmoc by characterizing all intermediates by LCMS (see Fig. 5 and 6). These syntheses were performed on the support with high loading of the “βAlaβAla”-linker (1.5–2 μmol cm−2). Small variations in the initial loading originate from batch-to-batch variation in loading between different arrays. Similarly to the observations above, we observed the most dramatic changes at the first coupling step. The conversion was high for Ala1 (60% vs. 100% with heat enhancement), medium for Trp1 (50% vs. 90% with heat-enhancement), and low for Arg1 (20% vs. 60% with heat enhancement) (see Fig. S6† for percent conversion at each step). Extending the reaction time from 3 to 10 minutes was beneficial at room temperature, but it had negligible effect at an elevated temperature. At room temperature and maximum coupling time (10 minutes), only 32% of the linker was converted to Trp6. In contrast, IR-enhanced synthesis and the coupling time of three minutes per step converted 75% of the linker to Trp6 (Fig. 4b and e). When compared to the control at room temperature the final amount of Ala6 obtained by heat-enhanced synthesis increased by a factor of four. Similarly Arg6, the most difficult of the three peptides to assemble, yielded a 30% conversion in heat-assisted synthesis with 3 or 10 minute coupling times, but significantly less (6% and 2%) at room temperature.
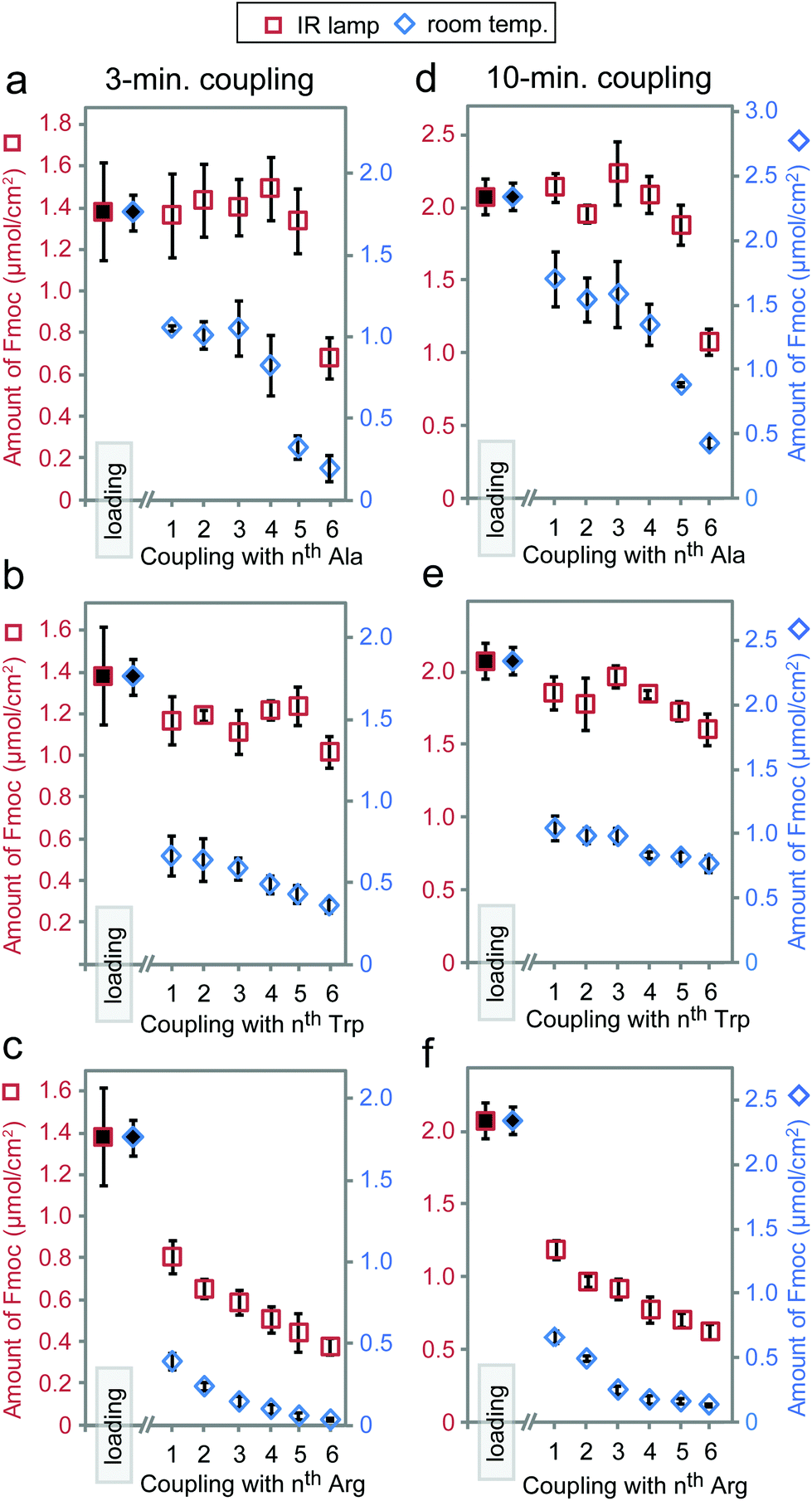 | ||
| Fig. 4 Amount of Fmoc loaded at each coupling step of the syntheses of Trp6 (a, d), Ala6 (b, e) and Arg6 (c, f) with coupling times of 3 minutes (a–c) and 10 minutes (d–f). Loading represents the averaged values over 12 replicates of the amount of Fmoc loaded after the functionalization of the paper with the βAla–βAla linker. The other data describe average values from four replicates; the error bars are 2 × (standard deviation). | ||
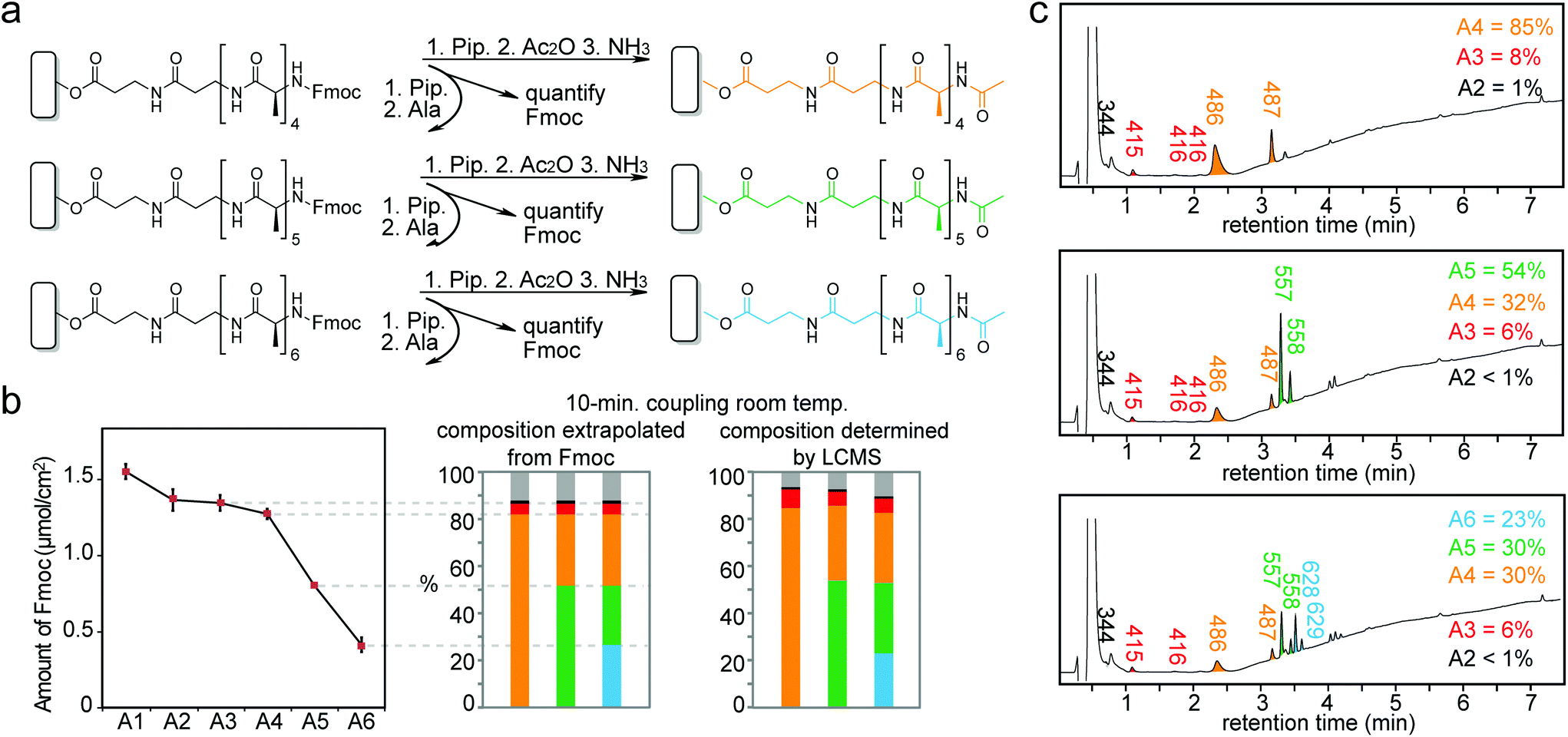 | ||
| Fig. 5 Fmoc and LCMS characterization of peptides formed in the last 3 coupling steps when synthesizing Ala6 are very similar. Syntheses from Ala4 to Ala6 (a). Composition of peptides calculated from Fmoc and LCMS data (b). Corresponding LCMS traces with labelled peaks and percent purity for the last 3 steps in the synthesis of Ala6 (c). | ||
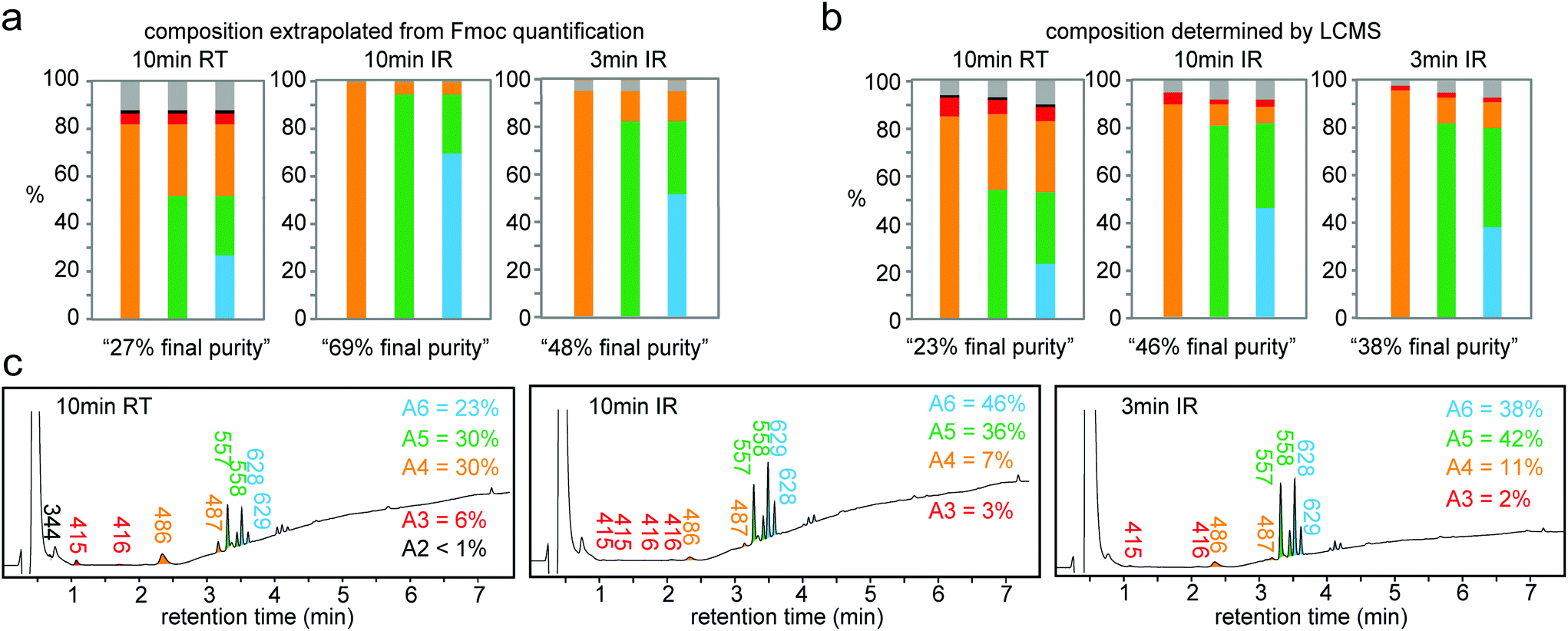 | ||
| Fig. 6 Both Fmoc and LCMS characterization confirm that IR-heating improved the quality of the synthesis. Peptide composition calculated from Fmoc (a) and LCMS (b) after coupling of 10 min at room temperature, 10 min and 3 min with IR heating. Corresponding LCMS traces and percent purity for the final step in the synthesis of Ala6 (c). | ||
Synthesis for the difficult sequence Arg6 can be further optimized by double coupling with freshly prepared reagents as well as reducing the βAla–βAla linker loading (see Fig. 7). The results demonstrated that double coupling and reducing initial loading can further boost the quality of synthesis giving an approximately 50% final purity, which represent nearly 10-fold improvement over traditional room temperature synthesis.
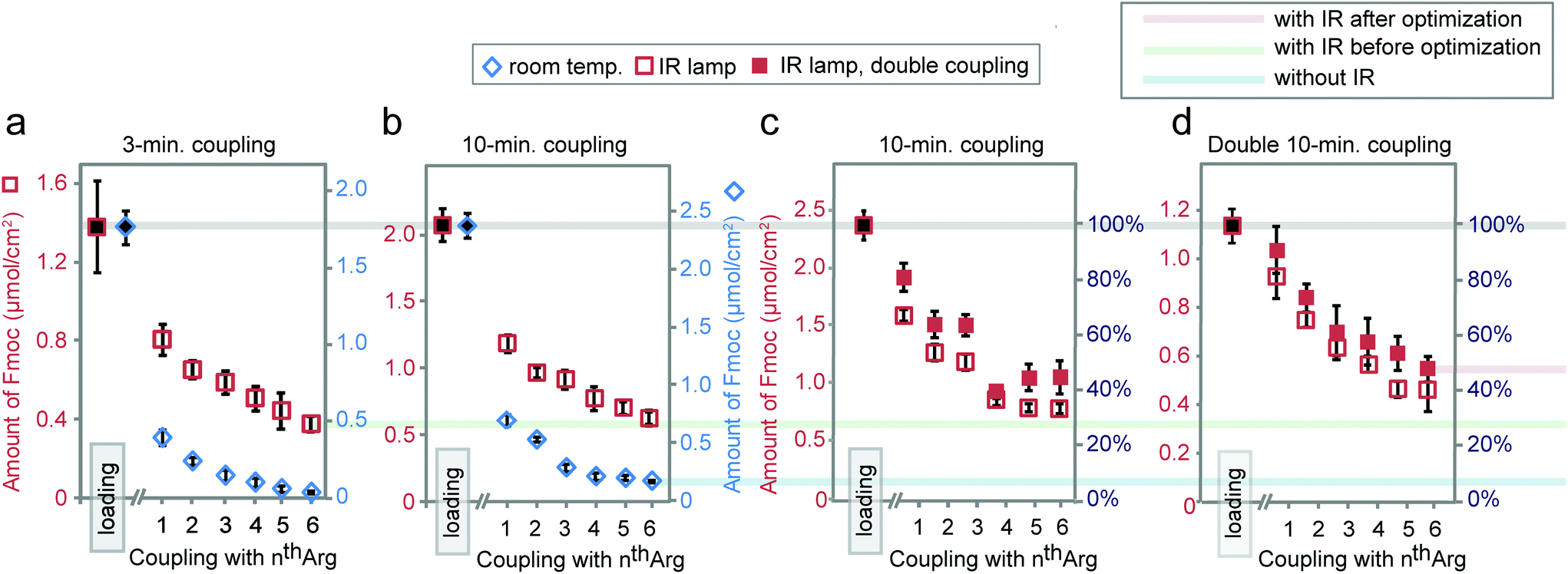 | ||
| Fig. 7 Synthesis of Arg6 with coupling times of 3 (a) or 10 minutes (b) at room temperature or under IR. Synthesis of Arg6via single or double 10-minute coupling under IR using arrays with normal (c) or reduced loading (d). Reduced loading was achieved by shortening the 1st βAla coupling time to 30 minutes. Data in (a) and Fig. 5b is exactly the same as in Fig. 4c and f. | ||
To date, no single strategy is capable of pushing the synthesis of all possible combinations of peptides of an array to a 100% purity. Nevertheless, even partial improvement of the synthesis (such as 5 to 50% increase in overall purity in Fig. 7) can improve the downstream assays. To illustrate, we focused on cell-binding assays, in which multivalent interactions between cellular receptors and multivalent display of ligands on the surface makes the assay sensitive to density of the ligands on the surface. We observed that replicates of arrays that were synthesized in one synthesis batch reproducibly support cell adhesion across the ten replicates of peptides (Fig. 8b). However, replicates of arrays that were synthesized on different days exhibit irreproducibility which originates from variability in the purity of the peptides on arrays. Fig. 9b demonstrates that variation in purity translates directly to variation in cell adhesion. Minor improvements in purity thus can have dramatic impact on the reproducibility of the downstream assays. IR-driven synthesis can therefore help minimize the variability by maximizing the purity of the peptides.
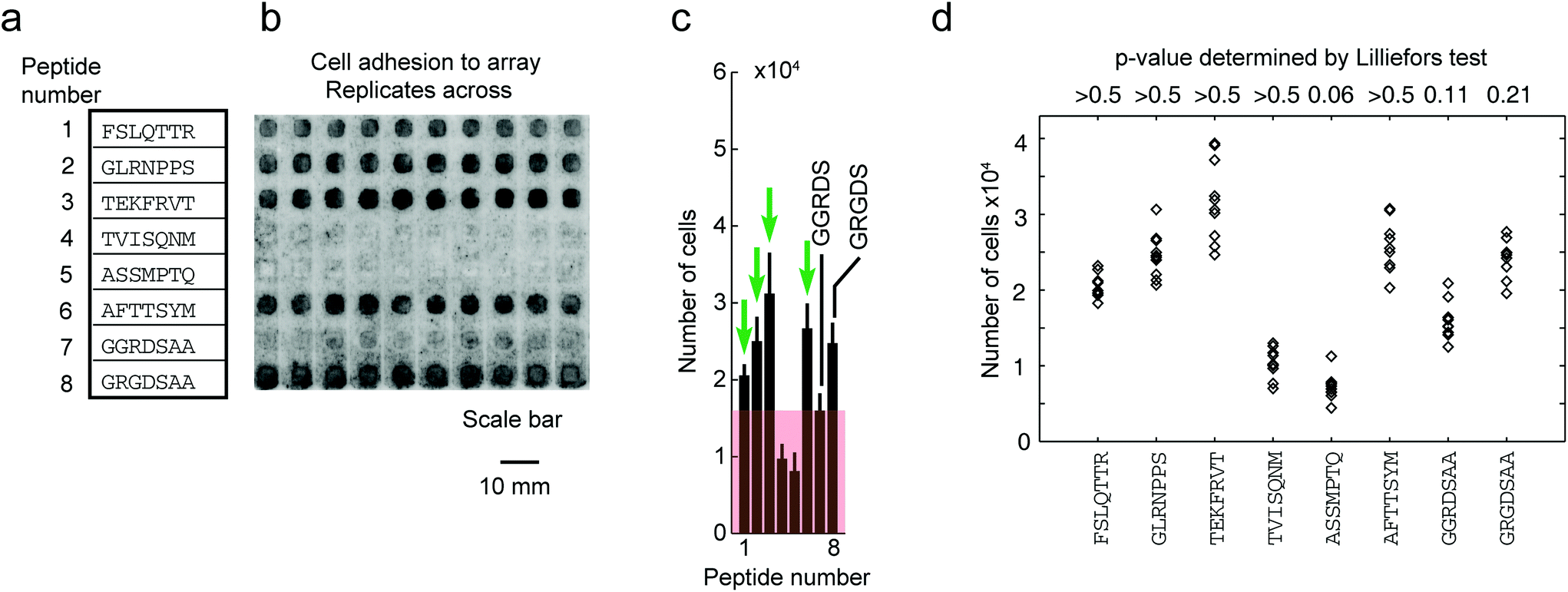 | ||
| Fig. 8 Adhesion of cells to peptide arrays synthesized in one synthesis is reproducible in 10 technical replicates. (a) Eight chosen peptides synthesized on the array. (b) Representative fluorescent gel scanner images of MDA-MB-231-GFP adhering to peptide arrays (after 3 hours). Grey-scale intensities were adjusted in this figure to simplify visualization (same level for all images). Original .gel files were used in processing without any adjustments to grey-scale intensities. (c) The average grey-scale intensity from each replicate was converted to the number of cells using a calibration curve (Fig. S9†). Data represent an average from 10 experiments; error bar is one standard deviation. Cell-binding hits were peptides that supported adhesion of significantly higher (p < 0.05) number of cells than the negative control (GGRDS, red box). Green arrows indicate four cell-binding hits. (d) Cell adhesion data for each peptide was tested for normality using the Lilliefors test. P-Values > 0.05 indicate data originating from a normally distributed population. | ||
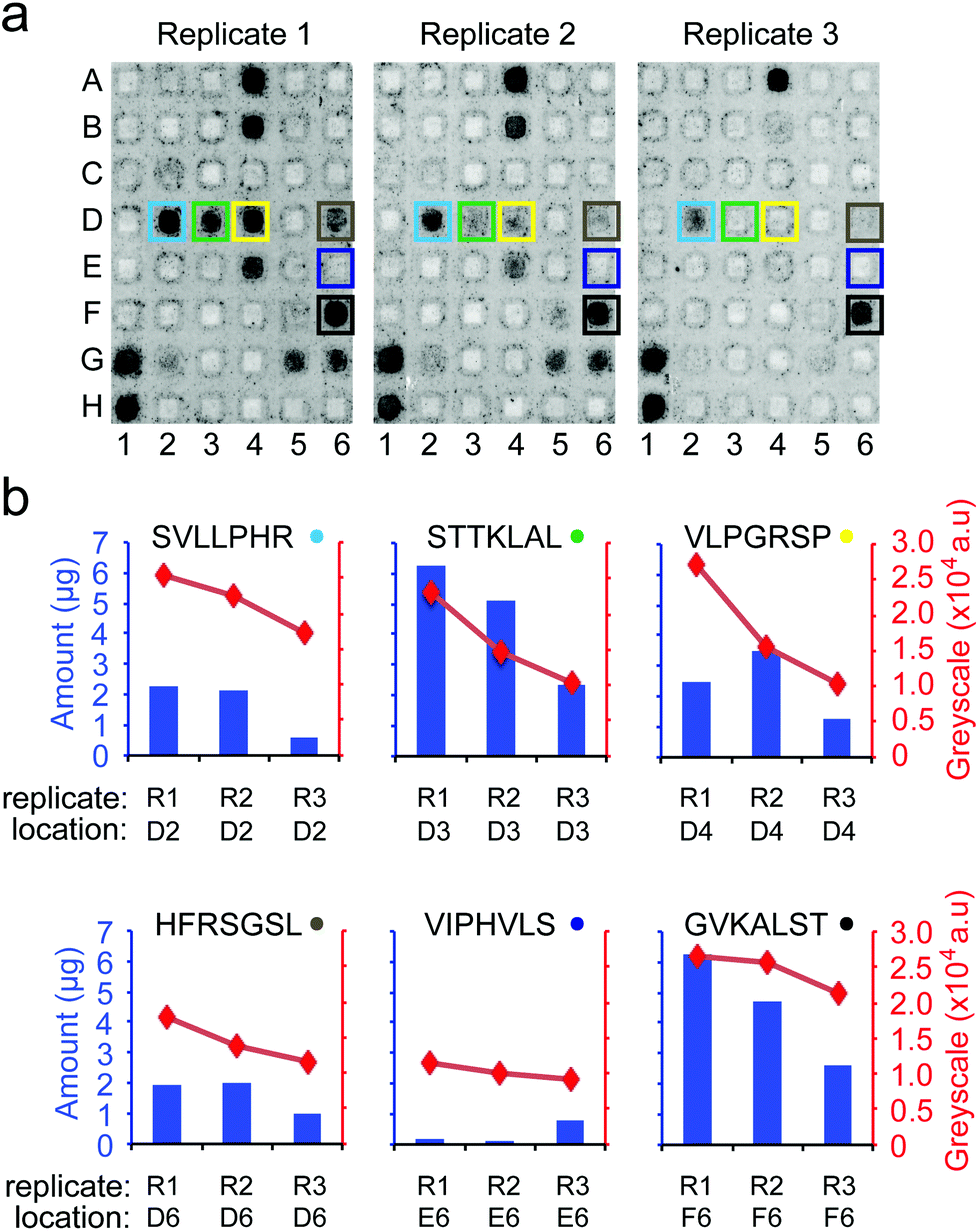 | ||
| Fig. 9 Variability in adhesion of cells to arrays can be related to the variability in peptide density on the surface. (a) Images of arrays from 3 independent synthetic replicates. Each array was tested for a short-term adhesion of MDA-MB-231 cells. The grey-scale intensities were adjusted to simplify visualization (same level for all images). (b) The grey-scale intensity of each peptide zone (red points, as determined using ImageJ) correlates with the amount of peptide displayed on this zone (blue bars). The amount of peptide per zone was determined after cell adhesion assay using LCMS as described in the Experimental section. | ||
Conclusions
Heating the paper with a simple IR-lamp accelerated the formation of the ester and amide bonds on the paper support. It reduced the reaction time and increased the quantity of peptides formed on the solid support even for problematic peptide sequences. We observed a 5-fold increase or more when converting the βAlaβAla linker on the paper to a peptide with six residues. Such an increase maximizes the loading of the desired final peptide and decreases the amount of truncated, acetyl-capped peptides. While IR-heating provided a significant advantage for all difficult coupling steps, it is not a universal remedy. Many IR-enhanced difficult coupling steps exhibited incomplete conversion. To maximize the conversion in difficult steps, one should employ other techniques for the optimization of coupling, such as different activator reagents or solvents.56 In this report, we only increased the temperature during the coupling steps. Heating, however, can accelerate other steps such as the Fmoc deprotection50 or the acetylation during the capping step and further reduce the cycle time and the time required for the overall synthesis. We also anticipate that combination of flow-through and IR-heating will be generally applicable to improve other classes of carbon–heteroatom and carbon–carbon bond forming reactions developed in cellulose-supported SPOT synthesis.13,27,44,51–55Experimental
Heating set-up
A custom-made aluminum platform to house four 96-zone paper arrays at a fixed distance of an infrared incandescent reflector lamp (Philips 175W PAR38) was manufactured at the Chemistry Machine Shop of the University of Alberta (see image in Fig. S1a†). With a distance between the lamp and the paper of 20 cm, the paper reached a temperature of 55 ± 5 °C (Fig. S1b†).Peptide synthesis on Teflon patterned paper
Teflon-patterned paper was prepared as described previously.11 The arrays were functionalized in batches performing the esterification of the cellulose by immersion of the array in 15 mL of the solution of Fmoc-βAla-OH (0.2 M), DIC (0.24 M), and 1-methylimidazole (0.38 M) in DMF for 3 hours. Alternatively, the same solution was spotted atop the Teflon patterned array (15 μL per zone) using a protocol previously reported.11Specific reagents and steps in the synthesis were adapted from a standard protocol with no modification to the solvents, reagents or their concentrations;15 adaptation of this protocol for the synthesis on a Teflon-patterned paper array was described previously.11 In this report, we only varied the time of the coupling reaction or the temperature of the reaction. As there were minor variations in the loading for each paper array, we excised 12 zones from each array and quantified the Fmoc loading as described below.
Assessment of coupling efficiency by quantification of Fmoc-deprotection
The standard Fmoc-deprotection step uses 4–6 minutes exposure of the paper support to 20% piperidine in DMF.50 We performed Fmoc-deprotection for 30 minutes instead as it was more convenient in our staggered kinetic assays. Specifically, we used a 3.1 mm diameter hole-puncher to excise each zone into a well of a polypropylene deep well plate. A volume of 200 μL of piperidine in DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) was added to each well, the reaction was allowed to proceed for 30 min, and 150 μL of the supernatant was the transferred to a custom-made 96 quartz-cup plate. The absorbance at 290 nm (A290) was measured by using a SpectraMax M2e plate reader (Molecular Devices). Since the sample was too concentrated for the linear range of the calibration curve (Fig. S2c†), 75 μL of the sample was transferred to the 96-quartz-cup plate along with 75 μL of piperidine in DMF (1:4). The calibration curve (Fig. S2d†) allowed converting the A290 to the quantity of peptide in μmol cm−2 of paper as represented throughout this report (Fig. 2b, 3c,d, 4a–f, S3, S5, and S6†).
:4) was added to each well, the reaction was allowed to proceed for 30 min, and 150 μL of the supernatant was the transferred to a custom-made 96 quartz-cup plate. The absorbance at 290 nm (A290) was measured by using a SpectraMax M2e plate reader (Molecular Devices). Since the sample was too concentrated for the linear range of the calibration curve (Fig. S2c†), 75 μL of the sample was transferred to the 96-quartz-cup plate along with 75 μL of piperidine in DMF (1:4). The calibration curve (Fig. S2d†) allowed converting the A290 to the quantity of peptide in μmol cm−2 of paper as represented throughout this report (Fig. 2b, 3c,d, 4a–f, S3, S5, and S6†).
Semi-automated synthesis of homo-hexapeptides
For the synthesis of the arrays described in Table S1† and Fig. 4, we used a fluid handling workstation (BioTek Precision XS). Briefly, we distributed solutions of amino acids (300 μL per well for four arrays of stock solution at 0.75 M in NMP) to the wells of a “source” deep-well plate. All wells were supplemented with solutions of HOAt (100 μL per well of stock solution at 2.25 M in NMP) and DIC (100 μL per well of stock solution at 1.68 M in NMP) and allowed to incubate for 10 minutes. A plate-to-plate transfer method was used to transfer 15 μL of the activated amino acid solutions from the “source” deep-well plate to the zones of the Teflon-patterned paper arrays. We examined up to six hexa-peptides simultaneously, for example, four replicate arrays with six different homo-hexapeptides per array (Fig. S5†). Two replicate arrays were allowed to react at room temperature and the other replicate arrays were placed over the IR lamp for the duration of the coupling reaction (2, 3 or 10 minutes).Quantification of peptides on modified cellulose support
Arrays were synthesized as described in our previous publication11 using no assistance with IR-heating. We used three separate instances of synthesis (new array, new reagents) on separate days to test the reproducibility of the synthesis and the influence of this non-reproducibility on downstream assays (cell adhesion, see below). Following cell adhesion assay, three replicates of a peptide array were treated with 50% TFA:DCM for 5 min to remove cells after the cell-adhesion assay. The arrays were washed with DCM (3 × 10 mL), methanol (3 × 10 mL) and air-dried. The middle of each peptide zone was punched out using a 2 mm hole puncher and the 2 mm paper zone was placed in a 2 mL vial and treated with NH3 gas overnight. The peptides were dissolved in 50 μL of H2O and analyzed by LC-MS (Agilent Technologies 6130 LCMS). The amount of each peptide was determined by comparing the peak areas of each peptide to the peak area of an internal standard peptide.
Adhesion of cells to arrays of peptides on paper
Arrays were tested in short-term cell adhesion assay as described in our previous publication11 with only minor modifications. Briefly, the peptide functionalized on paper was soaked in milliQ H2O for 30 min in a Nunc Omni-Tray. The paper was then washed twice with 13 mL of MEM media, followed by two washes with 13 mL of MEM media (2 × 5 min at 45 rpm). A custom made insert (for design of insert, see Fig. S10†) was added to hold the paper submerged and the paper and insert were washed twice with 13 mL of binding-media (0.5% BSA-MEM media). A suspension of live MDA-MB-231-GFP cells (0.3 × 105 cells per mL) in 25 mL of binding media was added to the array and incubated for 3 h at 37 °C in a CO2 incubator. The array was subsequently washed with MEM (3 × 13 mL) and imaged using a fluorescent gel scanner (GE Healthcare, Typhoon FLA9500). Images of cells were confirmed by using a confocal fluorescent microscope (Zeiss LSM 700). Differences in cell binding were assessed by comparing the number of cells between the test and control samples using the one-sided, unequal variance Student's t-test with a significance threshold of 0.05. The use of parametric statistics was justified because cell-binding data is normally distributed according to the Lilliefors test (Fig. 8d).Acknowledgements
The research was supported by the Alberta Glycomics Centre, SENTINEL Bioactive Paper Network and NSERC Discovery Grant # 401511. Canada Foundation for Innovation (CFI) provided infrastructure support. F. D. thanks the Government of Canada for a Grand Challenge Canada award, and Y. Y. is supported by special funds from the Alberta Glycomics Centre and SENTINEL Bioactive Paper Network.Notes and references
- S. P. Fodor, J. L. Read, M. C. Pirrung, L. Stryer, A. T. Lu and D. Solas, Science, 1991, 251, 767–773 CAS.
- A. C. Pease, D. Solas, E. J. Sullivan, M. T. Cronin, C. P. Holmes and S. P. Fodor, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5022–5026 CrossRef CAS.
- D. J. Lockhart, H. Dong, M. C. Byrne, M. T. Follettie, M. V. Gallo, M. S. Chee, M. Mittmann, C. Wang, M. Kobayashi, H. Norton and E. L. Brown, Nat. Biotechnol., 1996, 14, 1675–1680 CrossRef CAS PubMed.
- M. Amiram, F. G. Quiroz, D. J. Callahan and A. Chilkoti, Nat. Mater., 2011, 10, 141–148 CAS.
- E. S. Lander, Nat. Genet., 1999, 21(Suppl), 3–4 CrossRef CAS PubMed.
- U. Reineke, R. Sabat, A. Kramer, R. Stigler, M. Seifert, T. Michel, H. Volk and J. Schneider-Mergener, Mol. Diversity, 1996, 1, 141–148 CrossRef CAS PubMed.
- U. Reineke, C. Ivascu, M. n. Schlief, C. Landgraf, S. Gericke, G. Zahn, H. Herzel, R. Volkmer-Engert and J. Schneider-Mergener, J. Immunol. Methods, 2002, 267, 37–51 CrossRef CAS PubMed.
- R. Kato, C. Kaga, M. Kunimatsu, T. Kobayashi and H. Honda, J. Biosci. Bioeng., 2006, 101, 485–495 CrossRef CAS PubMed.
- M. Okochi, S. Nomura, C. Kaga and H. Honda, Biochem. Biophys. Res. Commun., 2008, 371, 85–89 CrossRef CAS PubMed.
- S. Ahmed, A. S. Mathews, N. Byeon, A. Lavasanifar and K. Kaur, Anal. Chem., 2010, 82, 7533–7541 CrossRef CAS PubMed.
- F. Deiss, W. L. Matochko, N. Govindasamy, E. Y. Lin and R. Derda, Angew. Chem., Int. Ed., 2014, 53, 6374–6377 CrossRef CAS PubMed.
- K. Kaur, S. Ahmed, R. Soudy and S. Azmi, in Peptide Libraries: Methods and Protocols, 2015, vol. 1248, pp. 239–247 Search PubMed.
- T. Praneenararat, G. D. Geske and H. E. Blackwell, Org. Lett., 2009, 11, 4600–4603 CrossRef CAS PubMed.
- C. W. Diehnelt, Front. Microbiol., 2013, 4, 402 Search PubMed.
- K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2007, 2, 1652–1660 CrossRef CAS PubMed.
- K. Hilpert, Mini-Rev. Org. Chem., 2011, 8, 157–163 CrossRef CAS.
- K. Hilpert, M. Elliott, H. Jenssen, J. Kindrachuk, C. D. Fjell, J. Koerner, D. F. H. Winkler, L. L. Weaver, P. Henklein, A. S. Ulrich, S. H. Y. Chiang, S. W. Farmer, N. Pante, R. Volkmer and R. E. W. Hancock, Chem. Biol., 2009, 16, 58–69 CrossRef CAS PubMed.
- S. Akita, N. Umezawa, N. Kato and T. Higuchi, Bioorg. Med. Chem., 2008, 16, 7788–7794 CrossRef CAS PubMed.
- A. Thiele, J. Zerweck and M. Schutkowski, in Peptide Microarrays: Methods and Protocols, 2009, vol. 570, pp. 19–65 Search PubMed.
- A. Thiele, G. I. Stangl and M. Schutkowski, Mol. Biotechnol., 2011, 49, 283–305 CrossRef CAS PubMed.
- A. Thiele, S. Poesel, M. Spinka, J. Zerweck, U. Reimer, U. Reineke and M. Schutkowski, Mini-Rev. Org. Chem., 2011, 8, 147–156 CrossRef CAS.
- S. Kudithipudi, D. Kusevic, S. Weirich and A. Jeltsch, J. Visualized Exp., 2014, 29, e52203 Search PubMed.
- F. Toepert, T. Knaute, S. Guffler, J. R. Pires, T. Matzdorf, H. Oschkinat and J. Schneider-Mergener, Angew. Chem., Int. Ed., 2003, 42, 1136–1140 CrossRef CAS PubMed.
- U. Hoffmuller, M. Russwurm, F. Kleinjung, J. Ashurst, H. Oschkinat, R. Volkmer-Engert, D. Koesling and J. Schneider-Mergener, Angew. Chem., Int. Ed., 1999, 38, 2000–2004 CrossRef CAS.
- W. Haehnel, Mol. Diversity, 2004, 8, 219–229 CrossRef CAS PubMed.
- H. K. Rau, N. DeJonge and W. Haehnel, Angew. Chem., Int. Ed., 2000, 39, 250–253 CrossRef CAS.
- M. D. Bowman, M. M. Jacobson and H. E. Blackwell, Org. Lett., 2006, 8, 1645–1648 CrossRef CAS PubMed.
- G. M. Eldridge and G. A. Weiss, Bioconjugate Chem., 2011, 22, 2143–2153 CrossRef CAS PubMed.
- R. Volkmer, ChemBioChem, 2009, 10, 1431–1442 CrossRef CAS PubMed.
- C. Katz, L. Levy-Beladev, S. Rotem-Bamberger, T. Rito, S. G. D. Rudiger and A. Friedler, Chem. Soc. Rev., 2011, 40, 2131–2145 RSC.
- R. Volkmer and V. Tapia, Mini-Rev. Org. Chem., 2011, 8, 164–170 CrossRef CAS.
- R. Volkmer, I. Kretzschmar and V. Tapia, Eur. J. Cell Biol., 2012, 91, 349–356 CrossRef CAS PubMed.
- H. E. Blackwell, Curr. Opin. Chem. Biol., 2006, 10, 203–212 CrossRef CAS PubMed.
- G. MacBeath, A. N. Koehler and S. L. Schreiber, J. Am. Chem. Soc., 1999, 121, 7967–7968 CrossRef CAS.
- R. Frei and H. E. Blackwell, Chem. – Eur. J., 2010, 16, 2692–2695 CrossRef CAS PubMed.
- G. MacBeath and S. L. Schreiber, Science, 2000, 289, 1760–1763 CAS.
- O. Blixt, S. Head, T. Mondala, C. Scanlan, M. E. Huflejt, R. Alvarez, M. C. Bryan, F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Skehel, I. van Die, D. R. Burton, I. A. Wilson, R. Cummings, N. Bovin, C.-H. Wong and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17033–17038 CrossRef CAS PubMed.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- B. Bacsa, K. Horvati, S. Bosze, F. Andreae and C. O. Kappe, J. Org. Chem., 2008, 73, 7532–7542 CrossRef CAS PubMed.
- R. Biswas, N. Maillard, J. Kofoed and J.-L. Reymond, Chem. Commun., 2010, 46, 8746–8748 RSC.
- K. Hilpert, D. F. H. Winkler and R. E. W. Hancock, Nat. Protoc., 2007, 2, 1333–1349 CrossRef CAS PubMed.
- R. Frank, Tetrahedron, 1992, 48, 9217–9232 CrossRef CAS.
- M. D. Bowman, R. C. Jeske and H. E. Blackwell, Org. Lett., 2004, 6, 2019–2022 CrossRef CAS PubMed.
- J. P. Tam, Int. J. Pept. Protein Res., 1987, 29, 421–431 CrossRef CAS PubMed.
- K. Barlos, D. Papaioannou, S. Patrianakou and T. Tsegenidis, Liebigs Ann. Chem., 1986, 1986, 1950–1955 CrossRef.
- H. M. Yu, S. T. Chen and K. T. Wang, J. Org. Chem., 1992, 57, 4781–4784 CrossRef CAS.
- J. K. Murray and S. H. Gellman, Nat. Protoc., 2007, 2, 624–631 CrossRef PubMed.
- C. A. Hood, G. Fuentes, H. Patel, K. Page, M. Menakuru and J. H. Park, J. Pept. Sci., 2008, 14, 97–101 CrossRef CAS PubMed.
- M. D. Simon, P. L. Heider, A. Adamo, A. A. Vinogradov, S. K. Mong, X. Li, T. Berger, R. L. Policarpo, C. Zhang, Y. Zou, X. Liao, A. M. Spokoyny, K. F. Jensen and B. L. Pentelute, ChemBioChem, 2014, 15, 713–720 CrossRef CAS PubMed.
- M. D. Bowman, M. M. Jacobson, B. G. Pujanauski and H. E. Blackwell, Tetrahedron, 2006, 62, 4715–4727 CrossRef CAS.
- Q. Lin and H. E. Blackwell, Chem. Commun., 2006, 2884–2886 RSC.
- Q. Lin, J. C. O'Neill and H. E. Blackwell, Org. Lett., 2005, 7, 4455–4458 CrossRef CAS PubMed.
- R. Frei, A. S. Breitbach and H. E. Blackwell, Chem. Sci., 2012, 3, 1555–1561 RSC.
- D. F. H. Winkler, Mini-Rev. Org. Chem., 2011, 8, 114–120 CrossRef CAS.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods; heating set-up and distribution of temperature on arrays; Fmoc quantification and calibration curve; synthesis on paper of different porosities; synthesis of homohexapeptides. See DOI: 10.1039/c6ob00898d |
| ‡ Present address: Department of Chemistry and Chemical Biology, Indiana University-Purdue University, Indianapolis, 402 N Blackford Street, Indianapolis, IN, 46202, USA. |
| § These authors contributed equally to this work. |
| ¶ Present address: Department of Antibody Engineering, Genentech Inc., 1DNA Way, MS 433, South San Francisco, CA, USA. |
| This journal is © The Royal Society of Chemistry 2016 |