
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkyl substituted [2.2]paracyclophane-1,9-dienes†
Benjamin J.
Lidster
,
Dharam R.
Kumar
,
Andrew M.
Spring‡
,
Chin-Yang
Yu§
,
Madeleine
Helliwell
,
James
Raftery
and
Michael L.
Turner
*
Organic Materials Innovation Centre, School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: Michael.Turner@Manchester.ac.uk; Fax: +44(0) 161-275-4273
First published on 1st June 2016
Abstract
[2.2]Paracyclophane-1,9-dienes substituted with n-octyl chains have been synthesised from the corresponding dithia[3.3]paracyclophanes using a benzyne induced Stevens rearrangement. The use of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate and tetra-n-butylammonium fluoride as the in situ benzyne source gave significantly improved yields over traditional sources of benzyne and enabled the preparation of n-octyl substituted [2.2]paracyclophane-1,9-dienes on a multi-gram scale.
Introduction
[2.2]Paracyclophanes have received considerable attention due to their confined molecular motions and the close proximity of the two stacked phenylene groups.1–3 This restricted geometry gives rise to unique electronic and optical properties, characteristic NMR spectra and applications in asymmetric catalysis for planar chiral derivatives.4–10 Linking of the phenylene groups by two cis-vinylene bonds in [2.2]paracyclophane-1,9-diene (1) results in extensive steric repulsion, with an intramolecular distance of just 2.85 Å between the phenylene group bridge head carbons.11 The high ring strain of this class of molecules, calculated to be 176 kJ mol−1 for cyclophanediene 1, makes them interesting monomers in polymer science.2,12,13 Specifically, alkoxy-substituted [2.2]paracyclophane-1,9-dienes have been used in ruthenium carbene initiated ring opening metathesis polymerisation (ROMP), forming soluble poly(phenylenevinylene)s (PPVs).14–18 These conjugated polymers have been extensively studied for application in electronic and optoelectronic devices, and recently in fluorescence imaging.19–22 Traditionally alkoxy groups have been appended to the polymer backbone to improve solubility, however, this results in a significant red-shift of the emission of these polymers over that of the parent unsubstituted PPV.23 By contrast alkyl substituted PPVs show minimal change in the energy of the band gap, in comparison to the parent PPV, whilst providing the required solubility.24The introduction of alkyl groups on the phenylene groups of 1 has not been reported. It is of interest as PPVs prepared by the ROMP of [2.2]paracyclophane-1,9-dienes show greater control of the polymer structure, molecular weight and enable the preparation of block copolymers.14
Synthetic routes towards [2.2]paracyclophane-1,9-dienes are extremely limited (Scheme 1).25–28 Boekelheide et al. reported a route from dithia[3.3]paracyclophane (2) that uses a benzyne induced Stevens rearrangement (BSR) to contract the dithiacyclophane ring (3), followed by oxidation of the phenyl sulfides (4) and a thermal syn-elimination to obtain 1.25 Recently Pasini et al. described a synthesis of 1 from 2 that involved a Pummerer rearrangement to give 5, followed by ring contraction through photochemical sulfur extrusion to give 6 and a final thermal elimination of the acetate groups to produce 1.26 The BSR route generally gives low yields of the desired paracyclophanedienes, whereas the Pummerer rearrangement gives improved yields but has been demonstrated only for synthesis of the parent [2.2]paracyclophane-1,9-diene and not for the substituted derivatives that are required to form soluble PPVs. The route of Boekelheide et al. utilises the diazotization of anthranilic acid with isoamyl nitrite as the in situ source of benzyne. A more convenient source of benzyne has been reported through the reaction of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (7) with tetra-n-butylammonium fluoride (TBAF).29,30 This system offers a wider substrate scope, less toxic reagents, milder reaction conditions and much greater yields.31,32
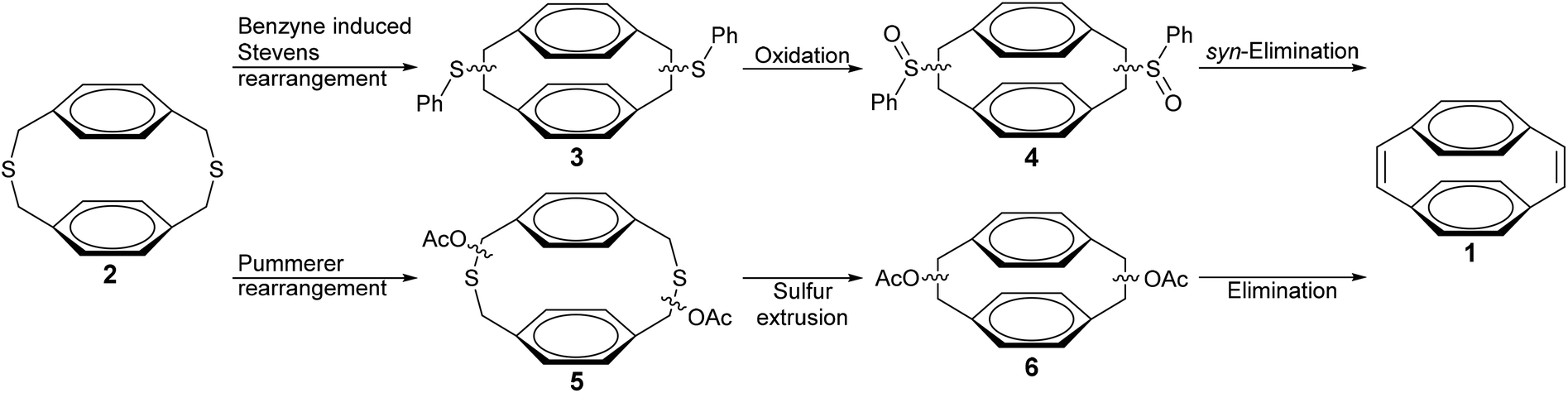 | ||
| Scheme 1 Synthetic routes to [2.2]paracyclophane-1,9-dienes.25,26 | ||
In our previous reports on the synthesis of tetraoctyloxy-substituted [2.2]paracyclophane-1,9-dienes we have used the method of Boekelheide et al., however, only low yields (<31%) were obtained.33 Herein we report an improved process to prepare substituted [2.2]paracyclophane-1,9-dienes using 7 and TBAF·3H2O as the benzyne source and the use of this process to prepare [2.2]paracyclophane-1,9-dienes with alkyl side chains, an important precursor to alkyl substituted PPVs by ROMP.
Results and discussion
Synthesis of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate
Benzyne precursor 7 offers major advantages over traditional sources of benzyne, but it is exceptionally expensive. Consequently, a method to prepare compound 7 on a large scale was developed and this was adapted from a procedure reported by Brimble et al.34 The procedure involved the protection of the hydroxy group of 2-chlorophenol with TMSCl, followed by a retro-Brook rearrangement in the presence of molten sodium.35 Reaction of 2-(trimethylsilyl)phenol with trifluoromethanesulfonic anhydride gave compound 7 in a good yield (overall 78%). The procedure could be scaled to provide multigram quantities (experimental procedure in ESI†).Synthesis of 4,7-dioctyl-[2.2]paracyclophane-1,9-diene
The initial step in the synthesis of 4,7-dioctyl-[2.2]paracyclophane-1,9-diene (15) and the two isomeric cyclophanedienes; 4,7,12,15-tetraoctyl-[2.2]paracyclophane-1,9-diene (20) and 5,8,12,15-tetraoctyl-[2.2]paracyclophane-1,9-diene (21) is the synthesis of 1,4-bis(bromomethyl)-2,5-bis(octyl)benzene (9). Preparation of compound 9 required the incorporation of two bromomethyl groups on the 2,5-positions of dioctylbenzene (8) and this was achieved by adapting the procedure of Tanaka et al. (Scheme 2).36 Compound 8 was heated at reflux in acetic acid with HBr and paraformaldehyde, over 5 days to give 9 in a single step (see ESI†).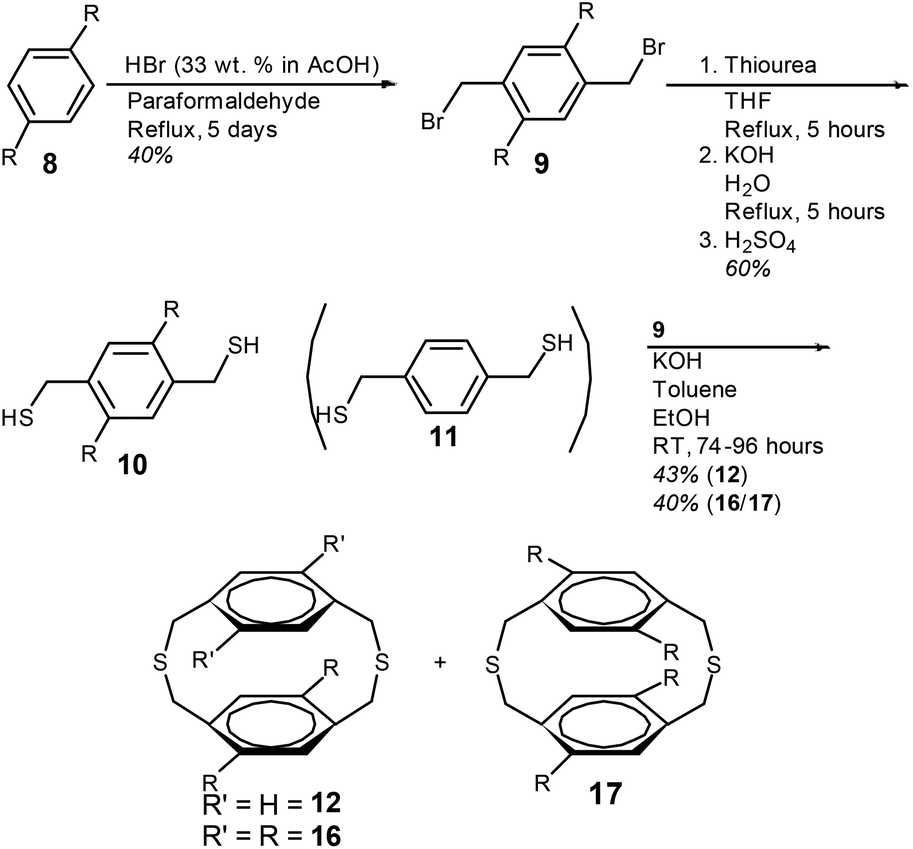 | ||
| Scheme 2 Synthesis of dithia[3.3]paracyclophanes; 12, 16 and 17, R = n-octyl. | ||
5,8-Dioctyl-2,11-dithia[3.3]paracyclophane (12) was prepared by the intramolecular cyclisation between an equimolar amount of 1,4-benzenedimethanethiol (11) and 9 (Scheme 2). A toluene solution of the two coupling components was added dropwise to a large volume of ethanol containing KOH. A slow addition is required to prevent oligomerisation. After column chromatography dithiacyclophane 12 was obtained as a colourless viscous oil in a yield of 43%.
The BSR of dithiacyclophane 12 was performed by the slow addition of TBAF·3H2O to a THF solution of 7 and 12 (Scheme 3). Isolation of the mixture of phenyl sulfides (13) was achieved using simple filtration through silica to remove biphenylene (by-product from the dimerisation of benzyne). Compound 13 was obtained as a highly viscous yellow oil in a yield of 67%. The analogous reaction with isoamyl nitrite and anthranilic acid in 1,2-dichloroethane gave 13 in a yield of only 25% (ESI).
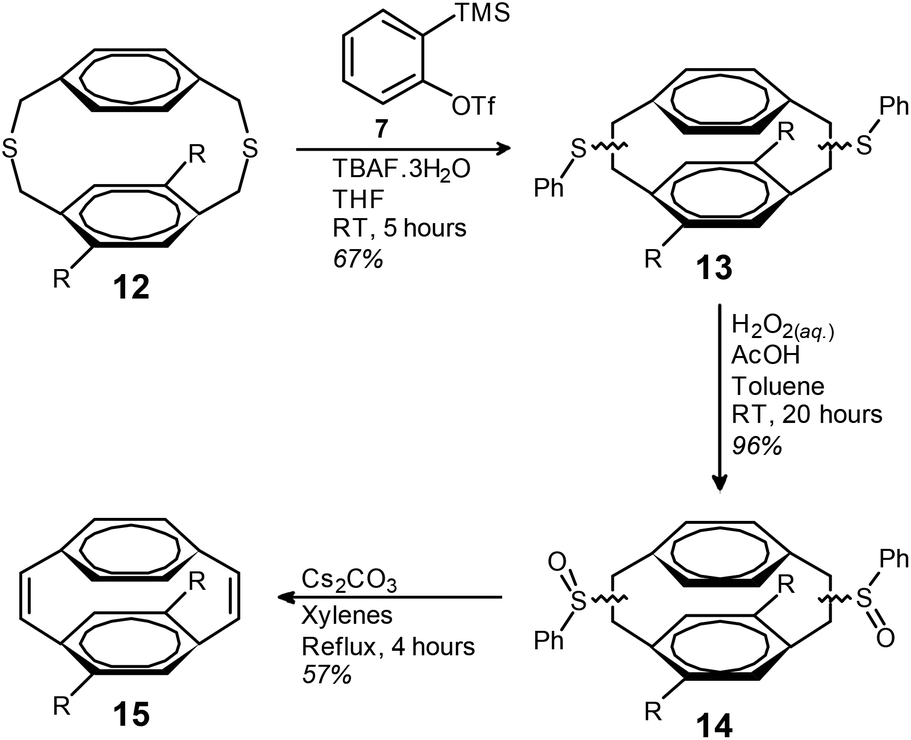 | ||
| Scheme 3 Synthesis of [2.2]paracyclophane-1,9-diene 15, R = n-octyl. | ||
Oxidation of the phenyl sulfides to the corresponding phenyl sulfoxides (14) was performed using aqueous H2O2 in acetic acid and toluene at room temperature. Compound 14 was isolated as a highly viscous oil in a yield of 96% after work-up, with no further purification performed. The thermal syn-elimination of the phenyl sulfoxide groups of compound 14 to obtain the corresponding cis-vinylene bonds of 15 was performed by refluxing in a mixture of xylenes in the presence of Cs2CO3. The addition of Cs2CO3 gave slightly improved yields, presumably due to reaction of the acidic by-products of the elimination reaction. Purification by column chromatography gave cyclophanediene 15 as a clear oil in a yield of 57%.
Synthesis of 4,7,12,15-tetraoctyl-[2.2]paracyclophane-1,9-diene and 5,8,12,15-tetraoctyl-[2.2]paracyclophane-1,9-diene
The synthesis of tetraoctyl-substituted [2.2]paracyclophane-1,9-dienes; 20 and 21 required the conversion of compound 9 to dithiol compound 10, to introduce n-octyl groups on to both phenylene groups. This was achieved by the reaction of 9 with thiourea followed by hydrolysis of the intermediate isothiouronium salt and acidification. Dithiacyclophanes 16 and 17 were prepared by the same procedure as described for dithiacyclophane 12 by the slow addition of a toluene solution containing an equimolar amount of 9 and 10, to a large volume of ethanol containing KOH (Scheme 2). Purification by column chromatography gave a mixture of dithiacyclophanes 16 and 17 as a colourless solid in a combined yield of 40%. Integration of the ArCH2S groups in the 1H NMR spectrum gave a ratio of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 of 6:94. In relation to the synthesis of tetraoctyloxy-substituted [2.2]paracyclophane-1,9-dienes, performing the BSR on a single dithiacyclophane resulted in the formation of both cyclophanediene isomers, after the final syn-elimination, as a consequence of isomerisation in this reaction.33 Consequently, separation of 16 and 17 was not conducted due to this expected isomerisation.33
:17 of 6:94. In relation to the synthesis of tetraoctyloxy-substituted [2.2]paracyclophane-1,9-dienes, performing the BSR on a single dithiacyclophane resulted in the formation of both cyclophanediene isomers, after the final syn-elimination, as a consequence of isomerisation in this reaction.33 Consequently, separation of 16 and 17 was not conducted due to this expected isomerisation.33
The mixture of dithiacyclophanes 16 and 17 was subjected to the BSR with compound 7 and TBAF·3H2O (Scheme 4). After column chromatography the mixture of phenyl sulfides (18) was isolated as a viscous yellow oil in a yield of 83%. The analogous reaction with isoamyl nitrite and anthranilic acid in 1,2-dichloroethane gave 18 in a yield of only 32% (ESI). Oxidation of the phenyl sulfides of 18 to give 19 was performed with aqueous H2O2 in toluene and acetic acid in an excellent yield (95%). Heating compound 19 at reflux in a mixture of xylenes with Cs2CO3 resulted in the thermal syn-elimination of the phenyl sulfoxide groups and the formation of cyclophanedienes 20 and 21. Purification by column chromatography gave cyclophanedienes 20 and 21 as a colourless oil in a combined yield of 45%. Integration of the phenylene group hydrogens in the 1H NMR spectrum gave the ratio of 20:21 to 17:83. Preparative separation of the two isomers was not possible through column chromatography, HPLC or recrystallisation (see ESI†). However, a single crystal of 20 was obtained for X-ray crystallography through multiple fractional crystallisations from hexane, at −10 °C.
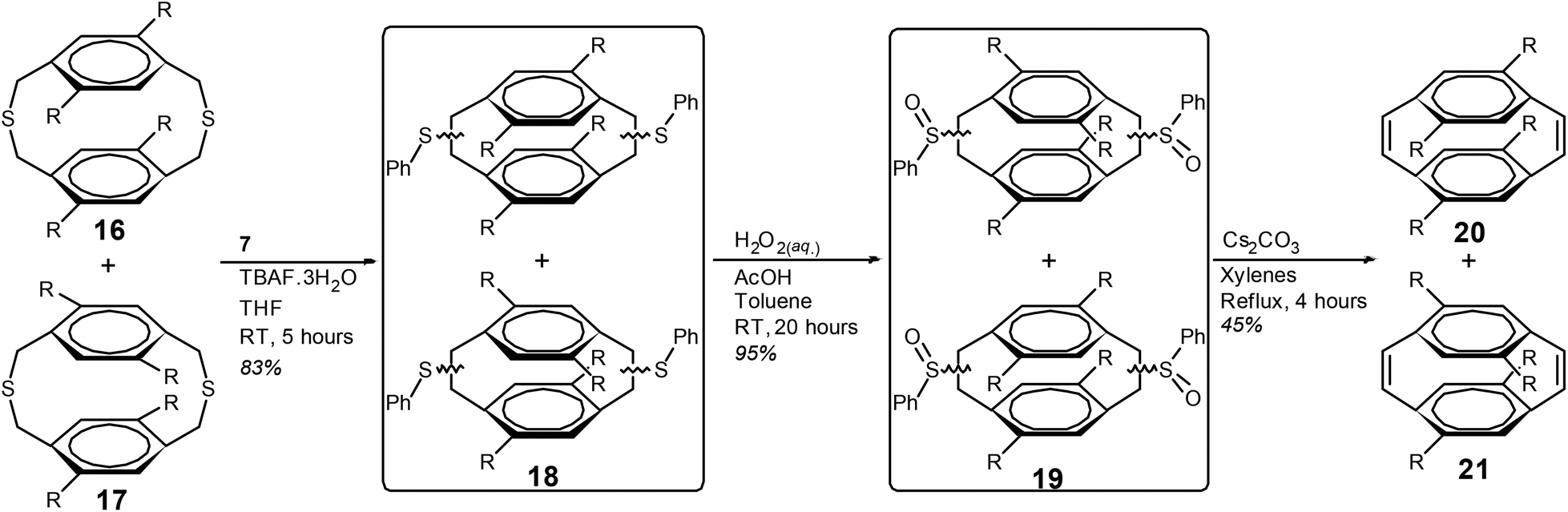 | ||
| Scheme 4 Synthesis of [2.2]paracyclophane-1,9-dienes 20 and 21, R = n-octyl. | ||
Characterisation of intermediates 12, 13, 14 and 4,7-dioctyl-[2.2]paracyclophane-1,9-diene (15)
Dithia[3.3]paracyclophanes and [2.2]paracyclophane-1,9-dienes, where at least one phenylene group is substituted, are planar chiral as a consequence of restricted rotation of the phenylene group through the dithiacyclophane ring. Substitution with n-octyl chains causes both the methylene hydrogens of the alkyl chain and of the thioether bonds to become diastereotopic. The 1H NMR spectrum of dithiacyclophane 12 (Fig. 1a) showed two hydrogen environments for the unsubstituted phenylene group, which were observed as AA′XX′ spin systems at 6.96 and 6.86 ppm. The hydrogens of the substituted phenylene group appear as a singlet at 6.64 ppm. Due to the planar chirality of these molecules the methylene hydrogens of the thioether bridges are diastereotopic and four doublets are observed, between 3.83–3.62 ppm, for the 8 hydrogens. A large difference in chemical shift is observed between the two diastereotopic methylene hydrogens bonded to the substituted phenylene group. This is a consequence of the close proximity of one of these hydrogens to the n-octyl chain. The methylene hydrogens adjacent to the unsubstituted phenylene group have a similar chemical shift and appear close to the equivalent hydrogens in the unsubsistutued dithiacyclophane 2.26 The hydrogens of the methylene group of the n-octyl chain adjacent to the phenylene group are also diastereotopic and appear as doublet of doublet of doublets at 2.71 and 2.44 ppm.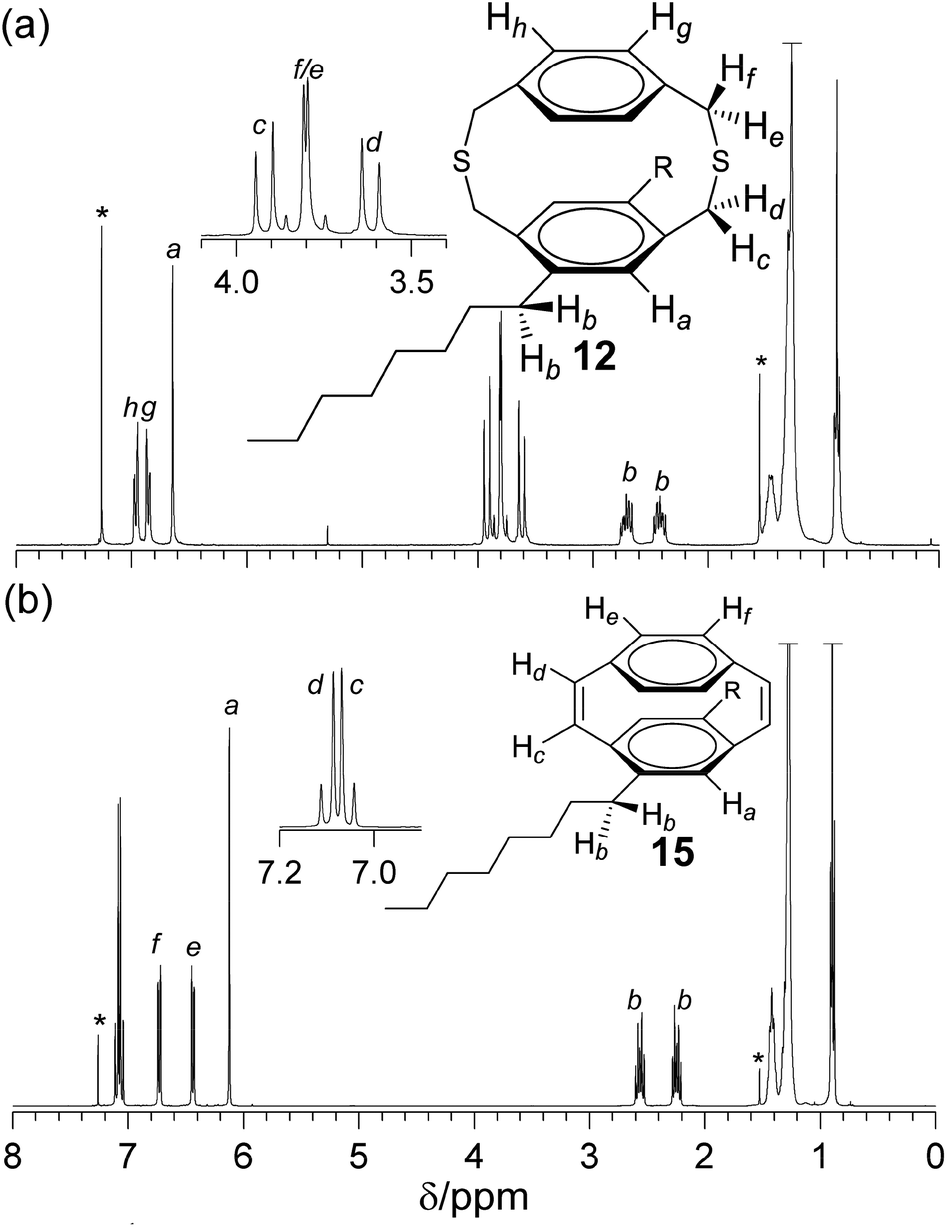 | ||
| Fig. 1 1H NMR spectra (CDCl3) of (a) dithia[3.3]paracyclophane 12 and (b) [2.2]paracyclophane-1,9-diene 15. | ||
The 1H NMR spectrum of compound 13 exhibited a complex range of signals and complete characterisation was not possible. This is a consequence of the BSR not being regioselective and migration of the phenyl sulfide groups occurring to either carbon of the thioether bond. Consequently each of these carbon centres becomes a stereogenic centre and a large number of regio and stereoisomers are possible. The success of the BSR was confirmed by mass spectrometry (MS) with the molecular ion observed at 650 Da (m/z) and through the fragmentation species by loss of a phenyl group and a phenyl sulfide group. Characterisation of phenyl-sulfoxide compound 14 by 1H NMR spectroscopy again proved difficult due to the large number of possible stereoisomers, and this was further complicated as each sulfoxide group is also a stereocentre. FT-IR spectroscopy confirmed the oxidation of the sulfide group through an intense absorbance observed at 1041 cm−1, which corresponded to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch. Over oxidation to the sulfone did not occur to any significant degree, as minimal absorbances are observed between 1160–1120 cm−1, that are characteristic for this group.37 MS measurements showed major molecular ions at 681 and 556 Da (m/z) which corresponded to the expected species and the fragmentation species by loss of a phenyl sulfoxide group, respectively.
O stretch. Over oxidation to the sulfone did not occur to any significant degree, as minimal absorbances are observed between 1160–1120 cm−1, that are characteristic for this group.37 MS measurements showed major molecular ions at 681 and 556 Da (m/z) which corresponded to the expected species and the fragmentation species by loss of a phenyl sulfoxide group, respectively.
The 1H NMR spectrum of cyclophanediene 15 (Fig. 1b) showed two hydrogen environments for the cis-vinylenes at 7.08 and 7.04 ppm. The two hydrogen environments of the unsubstituted phenylene group appear as two AA′XX′ spin systems at 6.71 and 6.42 ppm, and the hydrogens of the substituted phenylene group appear as a upfield singlet at 6.11 ppm. As with dithiacyclophane 12 the hydrogens of the methylene group of the n-octyl chain bonded to the phenylene group are diastereotopic and appear as two sets of doublet of triplets at 2.55 and 2.23 ppm. The loss of the sulfoxide group was confirmed by FT-IR spectroscopy with no intense absorbance at 1041 cm−1. Further, an absorbance at 690 cm−1 was associated with the out-of-plane stretch of the C–H bond of the cis-vinylene bond. High resolution MS (HR-MS) observed the molecular ion corresponding to 15 at 429.3510 Da (m/z) (calc. 429.3516).
Characterisation of intermediates 16, 17, 18, 19 and 4,7,12,15-tetraoctyl-[2.2]paracyclophane-1,9-diene (20) and 5,8,12,15-tetraoctyl-[2.2]paracyclophane-1,9-diene (21)
The tetra-octyl substituted dithiacyclophane 17 is planar chiral as previously described for the dithiacyclophane 12. However, as dithiacyclophane 16 exhibits a plane of symmetry it is not chiral. The 1H NMR spectrum (Fig. 2a) of mixture of dithiacyclophanes 16 and 17 shows two singlets at 6.76 and 6.78 ppm, respectively, which correspond to the four equivalent hydrogens of the phenylene groups of each isomer. The hydrogens of methylene groups of the thioether bond are diastereotopic and two doublets are observed at 4.07 and 3.57 ppm for 16 and at 3.82 and 3.63 ppm for 17. The chemical shift difference between the two diastereotopic hydrogens of 16 is 0.50 ppm, whereas, for 17 a difference of just 0.19 ppm is observed. The multiplets between 2.82–2.69 and 2.48–2.35 ppm are assigned to the hydrogens of the methylene groups of the n-octyl chains adjacent to the phenylene group in both 16 and 17. As expected the product of the BSR, compound 18, exhibits a complex 1H NMR spectrum and as with 13 complete characterisation was not possible. MS confirmed the BSR with the molecular ion of the expected product observed at 874 Da (m/z). Additional mass ions were observed that correspond to the fragmentation species by the loss of a phenyl group and a phenyl sulfide group. | ||
| Fig. 2 1H NMR spectra (CDCl3) of (a) dithia[3.3]paracyclophanes 16 and 17, and (b) [2.2]paracyclophane-1,9-dienes 20 and 21, R = n-octyl. | ||
After oxidation characterisation of the phenyl-sulfoxide compound 19 by NMR spectroscopy again proved difficult, however, a higher chemical shift of the phenyl sulfoxide groups was observed, in comparison to the phenyl sulfide group of 18. Oxidation was confirmed by FT-IR spectroscopy by the absorbance of the SO stretch at 1044 cm−1. Over oxidation to the sulfone again was not detected. MS showed the presence of the oxidised product at 906 Da (m/z) corresponding to the expected molecular species and the fragmentation species by loss of a phenyl sulfoxide group at 780 Da (m/z).
The 1H NMR spectrum of the mixture of cyclophanedienes 20 and 21 (Fig. 2b) showed the four hydrogens of the phenylene groups of each isomer to appear as singlets at 6.10 and 6.34 ppm, respectively. The four equivalent cis-vinylene hydrogens are observed as singlets at 7.07 ppm for 20 and at 6.92 ppm for 21. The hydrogens of the methylene group of the octyl chain bonded to the phenylene group are diastereotopic and are observed in cyclophanediene 20 as two doublets of triplets at 2.59 and 2.31 ppm. In cyclophanediene 21 the equivalent hydrogens appear as two doublet of triplets at 2.52 and 2.15 ppm. HR-MS of the mixture of isomers 20 and 21 observed the molecular ion at 652.5925 Da (m/z) (calc. 652.5942).
Solid-state structure of 4,7,12,15-tetraoctyl-[2.2]paracyclo-phane-1,9-diene
The solid-state structure of cyclophanediene 20 was determined by X-ray crystallography (Fig. 3). The intramolecular distance between the two carbon atoms of the phenylene groups bonded to the n-octyl group is 3.21 Å, whilst the equivalent distance between the two unsubstituted carbons is 3.13 Å. The intramolecular distance between the two carbons of the phenylene group bonded to the vinylene bond is shorter at 2.80 Å. | ||
| Fig. 3 Solid-state structure of 20 as ORTEP plot. Hydrogen atoms are omitted for clarity. | ||
The highly strained nature of the molecule is shown by the large distortion in the phenylene groups as the carbon atoms bonded to the vinylene bond lie between 0.17 and 0.21 Å out of the plane of the ring. The vinylene bond length is 1.34 Å, which is in agreement with that of a standard cis-vinylene bond 1.32 Å).38 In comparison to the unsubstituted cyclophanediene 1, no significant differences in the intramolecular distances of the phenylene groups are present and a comparable vinylene bond length of 1.33 Å is observed.11
Conclusion
[2.2]Paracyclophane-1,9-dienes substituted with two and four n-octyl chains have been prepared from the corresponding dithia[3.3]paracyclophanes. The use of benzyne precursor 7 and TBAF·3H2O as the in situ source of benzyne in the BSR gave significantly improved yields over the traditional source of anthranilic acid and isoamyl nitrite. A yield of 67% was obtained for 13 and 83% for 18, in comparison to 25 and 32% from the traditional reaction. The tetraoctyl substituted [2.2]paracyclophane-1,9-dienes were isolated as a mixture of the structural isomers 20 and 21 in ratio of 17:83. A single crystal of 20 was obtained by fractional crystallisation and the highly strained nature of this molecule was shown from the solid state structure by the large distortion of the bridging atoms of the phenylene group from the plane of the ring. The ability to prepare these alkyl substituted cyclophanedienes on a multi-gram scale offers great potential in polymer science where they can be subjected to ROMP to obtain PPVs with semiconducting and fluorescent properties.
Experimental
Methods
Nuclear magnetic resonance (NMR) spectra were obtained on Bruker spectrometers operating at either 300, 400 or 500 MHz, for 1H nuclei and 75, 101 or 126 MHz for 13C nuclei. Chemical shifts are reported in ppm relative to the indicated residual solvent (1H NMR spectroscopy: CDCl3: δ = 7.26 ppm and 13C NMR spectroscopy: CDCl3: δ = 77.00 ppm). All coupling constants (J values) are reported in hertz (Hz) and the following abbreviations are used to indicate multiplicity: s = singlet, d = doublet, t = triplet, dt = doublet of triplets, ddd = doublet of doublet of doublets, q = quartet, AA′XX′ = AA′XX′ spin system, and m = multiplet. HR-APCI mass spectra were obtained using a Thermo Scientific Exactive plus EMR. APCI-MS analysis was performed on Agilent technologies 6120 quadrupole LC/MS. HR-EI-MS were recorded on a Thermo Finningham MAT95XP. GC-MS (EI/CI) were performed on an Agilent 5975C Triple Axis GC-MS. Fourier transform-infrared spectroscopy (FT-IR) was conducted using a Nicolet iS5 (Thermo Scientific) with an iD5 attenuated total reflection accessory. The abbreviations: w = weak, m = medium, s = strong and vs = very strong are used to denote relative absorbance intensities. Elemental compositions of carbon, hydrogen and sulfur atoms were measured using a Flash 2000 Organic Elemental Analyser (Thermo Scientific). Slow additions were performed using a Watsons-Marlow 205S peristaltic pump.Materials
All reactions were carried out under an argon or nitrogen atmosphere using standard Schlenk techniques. Anhydrous THF was freshly distilled over sodium/benzophenone. All other anhydrous solvents and reagents were purchased from Apollo Scientific, Sigma-Aldrich, Fisher Scientific, Alfa Aesar or Acros and used as received. Deoxygenated solvents were prepared either by purging with argon or nitrogen. Column chromatography was performed using silica gel (60 Å, 230–400 mesh). Petroleum ether refers to the fraction obtained at 40–60 °C.:petroleum ether, 10:90 (v/v)) yielding a colourless oil (10.9 g, 43%). 1H NMR, (CDCl3, 500 MHz): δ 6.96 (2H, AA′XX′, Japp = 7.9, 1.7 Hz, ArH), 6.86 (2H, AA′XX′, Japp = 7.9, 1.7 Hz, ArH), 6.64 (2H, s, ArH), 3.92 (2H, d, J = 14.7 Hz, ArCH2S), 3.83 (2H, d, J = 15.2 Hz, ArCH2S), 3.77 (2H, d, J = 15.2 Hz, ArCH2S), 3.62 (2H, d, J = 14.7 Hz, ArCH2S), 2.71 (2H, ddd, J = 14.7, 8.8, 6.0 Hz, ArCH2CH2), 2.42 (2H, ddd, J = 14.7, 8.8, 6.0 Hz, ArCH2CH2), 1.55–1.22 (24H, m, CH2(CH2)6CH3), 0.90 (6H, t, J = 6.7 Hz, CH2CH3). 13C NMR, (CDCl3, 101 MHz): δ 137.64, 134.96, 132.94, 131.65, 129.28, 127.72, 37.72, 34.68, 32.78, 31.86, 30.56, 29.78, 29.52, 29.27, 22.65, 14.10. MS (APCI, M+): calc. for [C32H48S2 + 1H]+: m/z 497, found m/z 497. HR-MS (EI, M+): calc. for [C32H48S2]+: m/z 496.3197 found; m/z 496.3208. Elemental analysis: found: C, 77.23; H, 10.20; S, 12.50%; calc. for C32H48S2: C, 77.36; H, 9.74; S, 12.91%. FT-IR (thin film, cm−1) 2954m, 2922s, 2852m, 1510m, 1464m, 1422m, 907s, 731s.
:petroleum ether, 20:80 (v/v)) yielding a pale yellow oil (9.58 g, 67%). MS (APCI, M+): calc. for [C44H56S2 + 1H]+: m/z 650, found m/z 650; calc. for [C44H56S2 − 1SPh]+: m/z 540, found; m/z 540; calc. for [C44H56S2 − 1Ph]+: m/z 572, found; m/z 572. HR-MS (EI, M+): calc. for [C44H56S2]+: m/z 648.3823 found; m/z 648.3825. Elemental analysis: found: C, 81.79; H, 9.09; S, 9.81, calc. for C44H56S2: C, 81.42; H, 8.70; S, 9.88%. FT-IR (thin film, cm−1) 2953m, 2923s, 2853m, 1480m, 1466m, 1438m, 1215m, 755s, 734s, 689s.
CH), 7.04 (2H, d, J = 10.5 Hz, ArCHCH), 6.71 (2H, AA′XX′, Japp = 8.0, 1.5 Hz, ArH), 6.42 (2H, AA′XX′, Japp = 8.0, 1.5 Hz, ArH), 6.11 (2H, s, ArH), 2.55 (2H, dt, J = 13.9, 7.8 Hz, ArCH2CH2), 2.23 (2H, dt, J = 13.9, 7.8 Hz, ArCH2CH2), 1.45–1.35 (4H, m, ArCH2CH2), 1.35–1.19 (20H, m, CH2(CH2)5CH3), 0.88 (6H, t, J = 7.0 Hz, CH2CH3). 13C NMR, (CDCl3, 126 MHz): δ 139.49, 138.95, 136.74, 136.64, 135.00, 133.03, 130.92, 126.80, 34.22, 31.89, 30.60, 29.60, 29.49, 29.26, 22.67, 14.11. MS (APCI, M+): calc. for [C32H44 + 1H]+: m/z 429, found m/z 429. HR-MS (APCI, M+): calc. for [C32H44 + 1H]+: m/z 429.3516 found; m/z 429.3510. Elemental analysis: Found: C, 89.31; H, 10.41, calc. for C32H44: C, 89.65; H, 10.35%. FT-IR (thin film, cm−1) 3000w, 2954w, 2922s, 2852m, 1585w, 1465m, 1435w, 903m, 690s.
:petroleum ether, 10:90 (v/v)) to reveal a white solid (11.7 g, 40%). The solid consisted of a mixture of 16 and 17 in ratio of 6:94. 1H NMR, (CDCl3, 500 MHz): δ 6.78 (3.75H, s, ArH (17)), 6.76 (0.25H, s, ArH (16)). 4.07 (0.25H, d, J = 15.1 Hz, ArCH2S(16)), 3.82 (3.75H, d, J = 14.9 Hz, ArCH2S(17)), 3.63 (3.75H, d, J = 14.9 Hz, ArCH2S(17)), 3.57 (0.25H, d, J = 15.1 Hz, ArCH2S(16)), 2.82–2.69 (4H, m, ArCH2CH2), 2.48–2.35 (4H, m, ArCH2CH2), 1.51–1.19 (48H, m, CH2(CH2)6CH3), 0.92–0.82 (12H, m, CH2CH3). 13C NMR, (CDCl3, 126 MHz): δ dithia[3.3]paracyclophane 17; 137.88, 132.72, 129.95, 34.70, 32.82, 31.89, 30.36, 29.87, 29.57, 29.31, 22.68, 14.11. Signals for dithia[3.3]paracyclophane 16 were not observed due to low abundance. HR-MS (EI, M+): calc. for [C48H80S2]+: m/z 720.5696: found: m/z 720.5695. Elemental analysis: found: C, 80.05; H, 11.04; S, 8.77; calc. for C48H80S2: C, 79.93; H, 11.18; S, 8.89%. FT-IR (thin film, cm−1) 2949m, 2921s, 2846m, 1500w, 1466m, 1221w, 894m, 753w, 723m, 693w.
:petroleum ether, 10:90 (v/v)) yielding a pale yellow oil (11.6 g, 83%). MS (APCI, M+): calc. for [C60H88S2 + 1H]+: m/z 874, found m/z 874; calc. for [C60H88S2 − 1SPh]+: m/z 764, found; m/z 764, calc. for [C60H88S2 − 1Ph]+: m/z 796, found; m/z 796. HR-MS (APCI, M+): calc. for [C60H88S2 + H]+: m/z 873.6406: found: m/z 873.6389. Elemental analysis: found: C, 81.64; H, 10.19; S, 7.14, calc. for C60H88S2: C, 82.50; H, 10.15; S, 7.34%. FT-IR (thin film, cm−1) 2953w, 2922s, 2852m, 1582w, 1478m, 1465m, 1438m, 1090w, 736s, 689s.
:83. 1H NMR, (CDCl3, 500 MHz): δ 7.07 (0.68H, s, ArCHCH (20)), 6.92 (3.32H, s, ArCHCH (21)), 6.34 (3.32H, s, ArH (21)), 6.10 (0.68H, s, ArH(20)), 2.59 (0.68H, dt, J = 13.7, 7.6 Hz, ArCH2CH2(20)), 2.52 (3.32H, dt, J = 13.8, 7.7 Hz, ArCH2CH2 (21)), 2.31 (0.68H, dt, J = 13.7, 7.6 Hz, ArCH2CH2 (20)), 2.15 (3.32H, dt, J = 13.8, 7.7 Hz, ArCH2CH2 (21)), 1.45–1.14 (48H, m, CH2(CH2)6CH3), 0.92–0.84 (12H, m, CH2CH3). 13C NMR, (CDCl3, 126 MHz): δ cyclophanediene 20; 139.70, 135.50, 135.38, 131.03, 34.79, 32.08, 31.87, 29.44, 29.33, 29.26, 22.66, 14.11, cyclophanediene 21; 140.10, 137.00, 134.61, 128.78, 34.03, 31.90, 30.71, 29.62, 29.50, 29.26, 22.68, 14.11. MS (APCI, M+): calc. for [C48H76 + H]+: m/z 654, found m/z 654. HR-MS (EI, M+): calc. for [C48H76]+: m/z 652.5942 found; m/z 652.5925. Elemental analysis: found: C, 88.75; H, 11.26. calc.; C48H76: C, 88.27; H, 11.73%. FT-IR (thin film, cm−1) 2997w, 2954m, 2921s, 2852s, 1585w, 1465m, 904m, 687m.
Acknowledgements
The authors acknowledge the support of the EPSRC for award of studentships (BJL and AMS) and project (EP/F056397). The high commission of India Education Wing-London, Government of India for studentship (DK) and project (11015/16/2012-SCD-V).References
- D. J. Cram and N. L. Allinger, J. Am. Chem. Soc., 1955, 77, 6289–6294 CrossRef CAS.
- H. Hopf, Angew. Chem., Int. Ed., 2008, 47, 9808–9812 CrossRef CAS PubMed.
- P. G. Ghasemabadi, T. Yao and G. J. Bodwell, Chem. Soc. Rev., 2015, 44, 6494–6518 RSC.
- S. Mukhopadhyay, S. P. Jagtap, V. Coropceanu, J.-L. Brédas and D. M. Collard, Angew. Chem., Int. Ed., 2012, 51, 11629–11632 CrossRef CAS PubMed.
- M. Gon, Y. Morisaki and Y. Chujo, Eur. J. Org. Chem., 2015, 7756–7762 CrossRef CAS.
- Y. Morisaki, M. Gon, T. Sasamori, N. Tokitoh and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 3350–3353 CrossRef CAS PubMed.
- P. J. Pye, K. Rossen, R. A. Reamer, N. N. Tsou, R. P. Volante and P. J. Reider, J. Am. Chem. Soc., 1997, 119, 6207–6208 CrossRef CAS.
- S. Kitagaki, T. Ueda and C. Mukai, Chem. Commun., 2013, 49, 4030–4032 RSC.
- A. J. Roche, K. A. Abboud and W. R. Dolbier, J. Org. Chem., 2015, 80, 5355–5358 CrossRef CAS PubMed.
- L. Ernst, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 37, 47–190 CrossRef CAS.
- K. C. Dewhirst and D. J. Cram, J. Am. Chem. Soc., 1958, 80, 3115–3125 CrossRef CAS.
- A. de Meijere, S. I. Kozhushkov, K. Rauch, H. Schill, S. P. Verevkin, M. Kümmerlin, H.-D. Beckhaus, C. Rüchardt and D. S. Yufit, J. Am. Chem. Soc., 2003, 125, 15110–15113 CrossRef CAS PubMed.
- U. H. F. Bunz, D. Mäker and M. Porz, Macromol. Rapid Commun., 2012, 33, 886–910 CrossRef CAS PubMed.
- C.-Y. Yu and M. L. Turner, Angew. Chem., Int. Ed., 2006, 45, 7797–7800 CrossRef CAS PubMed.
- B. J. Lidster, J. M. Behrendt and M. L. Turner, Chem. Commun., 2014, 50, 11867–11870 RSC.
- E. Elacqua and M. Weck, Chem. – Eur. J., 2015, 21, 7151–7158 CrossRef CAS PubMed.
- F. Menk, M. Mondeshki, D. Dudenko, S. Y. Shin, D. Schollmeyer, O. Ceyhun, T. L. Choi and R. Zentel, Macromolecules, 2015, 48, 7435–7445 CrossRef CAS.
- C.-Y. Yu, J. W. Kingsley, D. G. Lidzey and M. L. Turner, Macromol. Rapid Commun., 2009, 30, 1889–1892 CrossRef CAS PubMed.
- Y. Yamada, T. Nakamura and K. Yano, J. Colloid Interface Sci., 2016, 468, 292–299 CrossRef CAS PubMed.
- S. Jeon, S. Park, J. Nam, Y. Kang and J.-M. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 1813–1818 CAS.
- S. Wang, J. W. Ryan, A. Singh, J. G. Beirne, E. Palomares and G. Redmond, Langmuir, 2016, 32, 329–337 CrossRef CAS PubMed.
- L. Xiong, F. Cao, X. Cao, Y. Guo, Y. Zhang and X. Cai, Bioconjugate Chem., 2015, 26, 817–821 CrossRef CAS PubMed.
- H. S. Woo, S. C. Graham, D. A. Halliday, D. D. C. Bradley, R. H. Friend, P. L. Burn and A. B. Holmes, Phys. Rev. B: Condens. Matter, 1992, 46, 7379–7389 CrossRef CAS.
- F. Hide, M. A. DiazGarcia, B. J. Schwartz, M. R. Andersson, Q. B. Pei and A. J. Heeger, Science, 1996, 273, 1833–1836 CrossRef CAS.
- T. Otsubo and V. Boekelheide, Tetrahedron Lett., 1975, 16, 3881–3884 CrossRef.
- M. Montanari, A. Bugana, A. K. Sharma and D. Pasini, Org. Biomol. Chem., 2011, 9, 5018–5020 CAS.
- M. Stöbbe, O. Reiser, R. Näder and A. de Meijere, Chem. Ber., 1987, 120, 1667–1674 CrossRef.
- C.-Y. Yu, C.-H. Sie and C.-Y. Yang, New J. Chem., 2014, 38, 5003–5008 RSC.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 8, 1211–1214 CrossRef.
- A. Cobas, E. Guitián and L. Castedo, J. Org. Chem., 1997, 62, 4896–4897 CrossRef CAS.
- A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 CAS.
- T. Kitamura, Aust. J. Chem., 2010, 63, 987–1001 CrossRef CAS.
- C.-Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Chem. – Eur. J., 2011, 17, 6991–6997 CrossRef CAS PubMed.
- D. J. Atkinson, J. Sperry and M. A. Brimble, Synthesis, 2010, 6, 911–913 Search PubMed.
- B. Michel and M. F. Greaney, Org. Lett., 2014, 16, 2684–2687 CrossRef CAS PubMed.
- K. Tanaka, T. Hori, T. Osaka, K. Noguchi and M. Hirano, Org. Lett., 2007, 9, 4881–4884 CrossRef CAS PubMed.
- K. C. Schreiber, Anal. Chem., 1949, 21, 1168–1172 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of 1,4-benzenedimethanethiol, 1,4-dioctylbenzene, compound 7, 1H and 13C NMR spectra and HPLC chromatogram of 20/21 and synthesis of 13 and 18 from isoamyl nitrite and anthranilic acid. Metrical parameters for the structure of 20. CCDC 1473670. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob00885b |
| ‡ Current address: Institute for Materials Chemistry and Engineering, Kyushu University, 6-1 Kasuga-koen Kasuga, Fukuoka 816-8580, Japan. |
| § Current address: Department of Materials Science and Engineering, National Taiwan University of Science and Technology, 43, Section 4, Keelung Road, Taipei, 10607, Taiwan. |
| This journal is © The Royal Society of Chemistry 2016 |