
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron-mediated oxidative C–H coupling of arenes and alkenes directed by sulfur: an expedient route to dihydrobenzofurans†
Craig W.
Cavanagh
a,
Miles H.
Aukland
a,
Quentin
Laurent
a,
Alan
Hennessy
b and
David J.
Procter
*a
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.j.procter@manchester.ac.uk
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
First published on 11th May 2016
Abstract
A novel route to medicinally-relevant dihydrobenzofurans utilises a sulfur-directed C–H ortho-coupling of arenes and unactivated terminal alkenes mediated by iron, and a palladium-catalysed deallylation/heterocyclisation sequence. The iron-mediated coupling affords linear products of alkene chloroarylation in good yield and with complete regioselectivity. The coupling likely proceeds by redox-activation of the arene partner by iron(III) and alkene addition to the resultant radical cation.
Introduction
The search for more sustainable synthetic methods has led to increased interest in cross-couplings that unite two partners at the expense of a C–H bond in each, i.e. without the need for pre-functionalisation. Such processes would lead to lower costs, as fewer steps are required, and less waste results, when compared to standard metal-catalysed coupling reactions.1 The ubiquity of C–H bonds means that achieving regioselectivity in C–H functionalisation can often prove difficult, and directing groups are often necessary to facilitate functionalisation with directed ortho C–H bond activation on an aromatic or heteroaromatic ring being a common strategy.2 The oxidative ortho C–H coupling of arenes and heteroarenes with alkenes has been well investigated using various directing groups, reactive alkenes (e.g. acrylates and styrenes), and Pt group metals.3 In contrast, the use of a sulfur-based directing group to direct C–H coupling of an aromatic ring with alkenes is a recent advance and is largely unexplored (Scheme 1A).4 The replacement of Pt group metals with inexpensive first row transition metals for synthetic procedures, including those involving C–H functionalisation, has clear benefits.5 The low cost of iron, its high abundance, and its low toxicity, make iron-based reagents ideal for new transformations.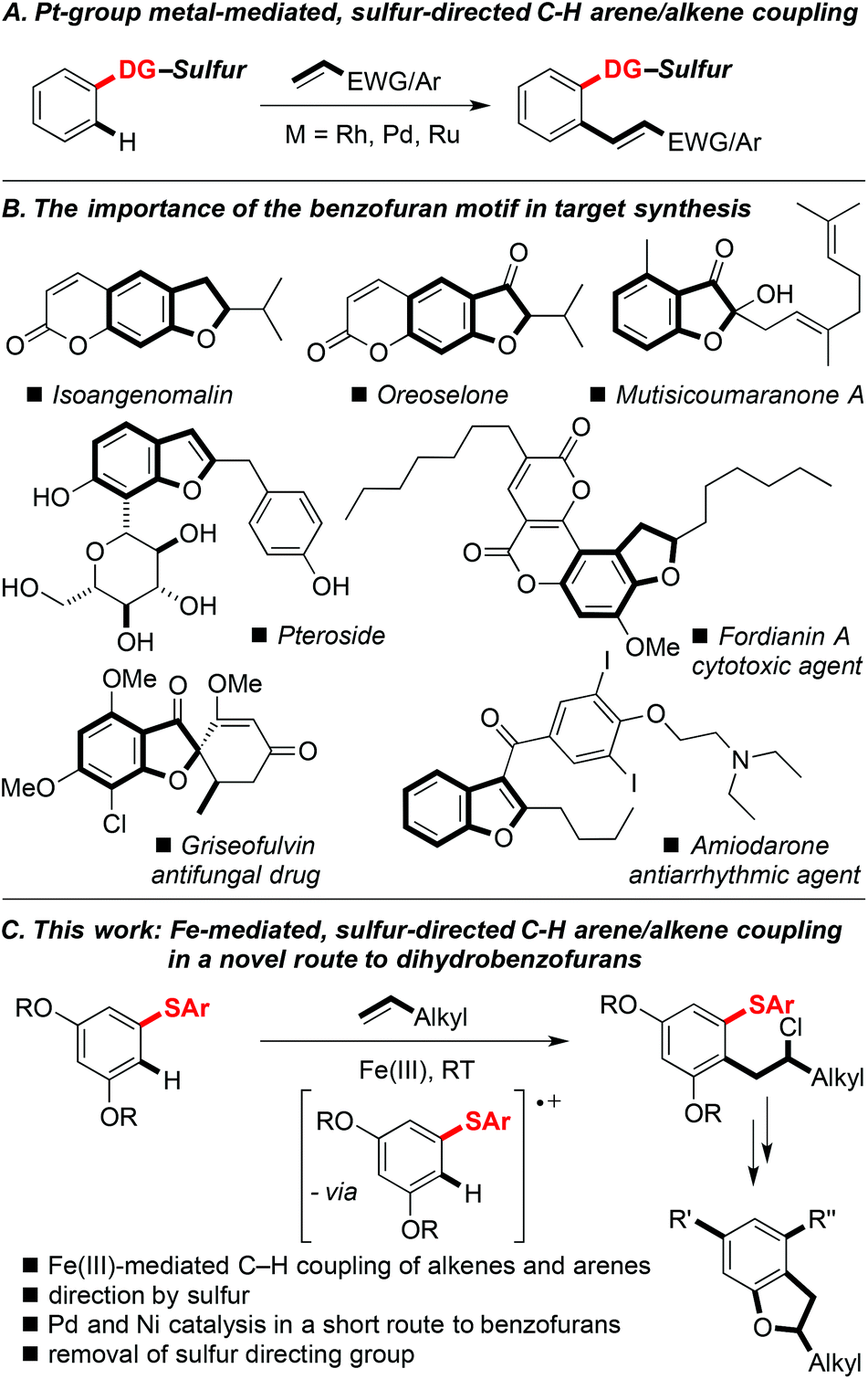 | ||
| Scheme 1 A. Pt-group metal-mediated, sulfur-directed C–H alkenylation. B. Benzofuran motifs in important synthetic targets. C. This work: Fe(III)-mediated, sulfur directed C–H coupling of arenes and alkenes exploited in a novel approach to dihydrobenzofurans. DG-Sulfur = Sulfur-containing directing group. | ||
Here we describe the Fe(III)-mediated oxidative C–H ortho coupling of aryl sulfides with simple, terminal alkenes to give linear β-chloroarenes.6 It is proposed that the two nucleophiles couple through oxidation of the arene partner by Fe(III), followed by addition of the alkene to quench the resulting radical cation.7,8 The sulfur-directed, iron-mediated coupling facilitates an expedient, novel route to decorated dihydrobenzofurans; a motif found in many natural products and drug molecules (Scheme 1B). The approach also features a Pd-catalysed deprotection/heterocyclisation and Ni-catalysed cross-couplings of sulfides (Scheme 1C).
Results and discussion
Scope and limitations
Previously reported optimisation studies found that FeCl3 was the most effective oxidant for the C–H coupling of diaryl sulfides and alkenes and that the reaction proceeded at ambient temperature under air in CH2Cl2.6 As the coupling is thought to involve reactive radical cation intermediates that are prone to decomposition, slow addition of the Fe(III) oxidant to a mixture of arene and alkene was employed to maintain a low concentration of radical cation. Slow addition of FeCl3 was found to give improved mass balance and yield. Regardless of how the oxidant was added, crude 1H NMR spectra showed clean product and mass balance was the only issue.We next assessed the scope of the process by varying the alkene partner in the coupling process (Scheme 2). Products were obtained in good isolated yields (37–65%) from couplings of alkene substrates bearing alkenyl (2b), iodide (2c), bromide (2d and 2e), chloride (2f), aryl (2g and 2h), and nitro (2j) functionality. Interestingly, 1,6-heptadiene underwent selective coupling to give the monoaddition product (formation of 2b): the corresponding double addition product was not observed (vide infra).
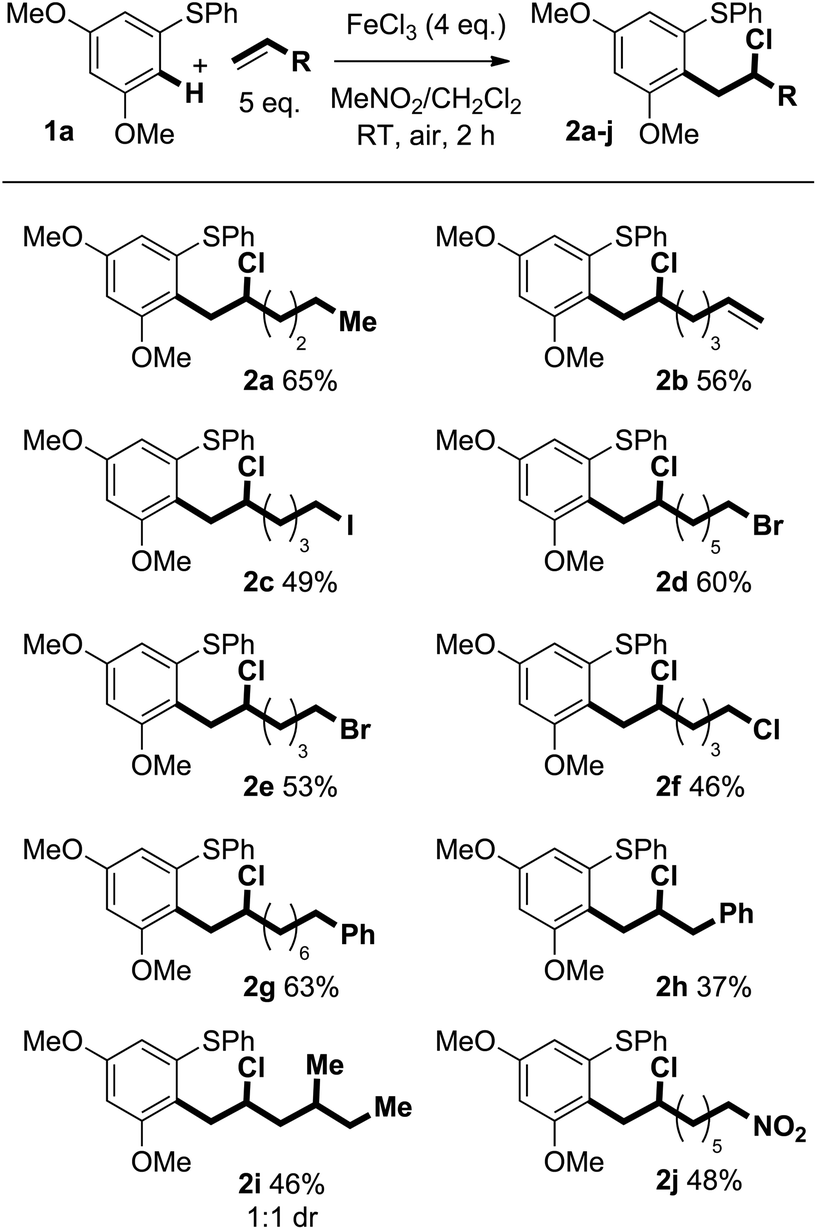 | ||
| Scheme 2 Variation of alkene in the Fe(III)-mediated C–H coupling. | ||
The scope with regard to the aryl sulfide partner was next assessed (Scheme 3). Although the coupling is sensitive to substitution in the aryl ring undergoing carbon–carbon bond formation (vide infra), the non-reacting, right-hand aryl ring can be varied: methyl (2k), bromide (2l), fluoride (2m and 2n), nitro (2o), trifluoromethyl (2p), and methoxy (2q and 2r) groups were compatible with the coupling and products were isolated in good yield.
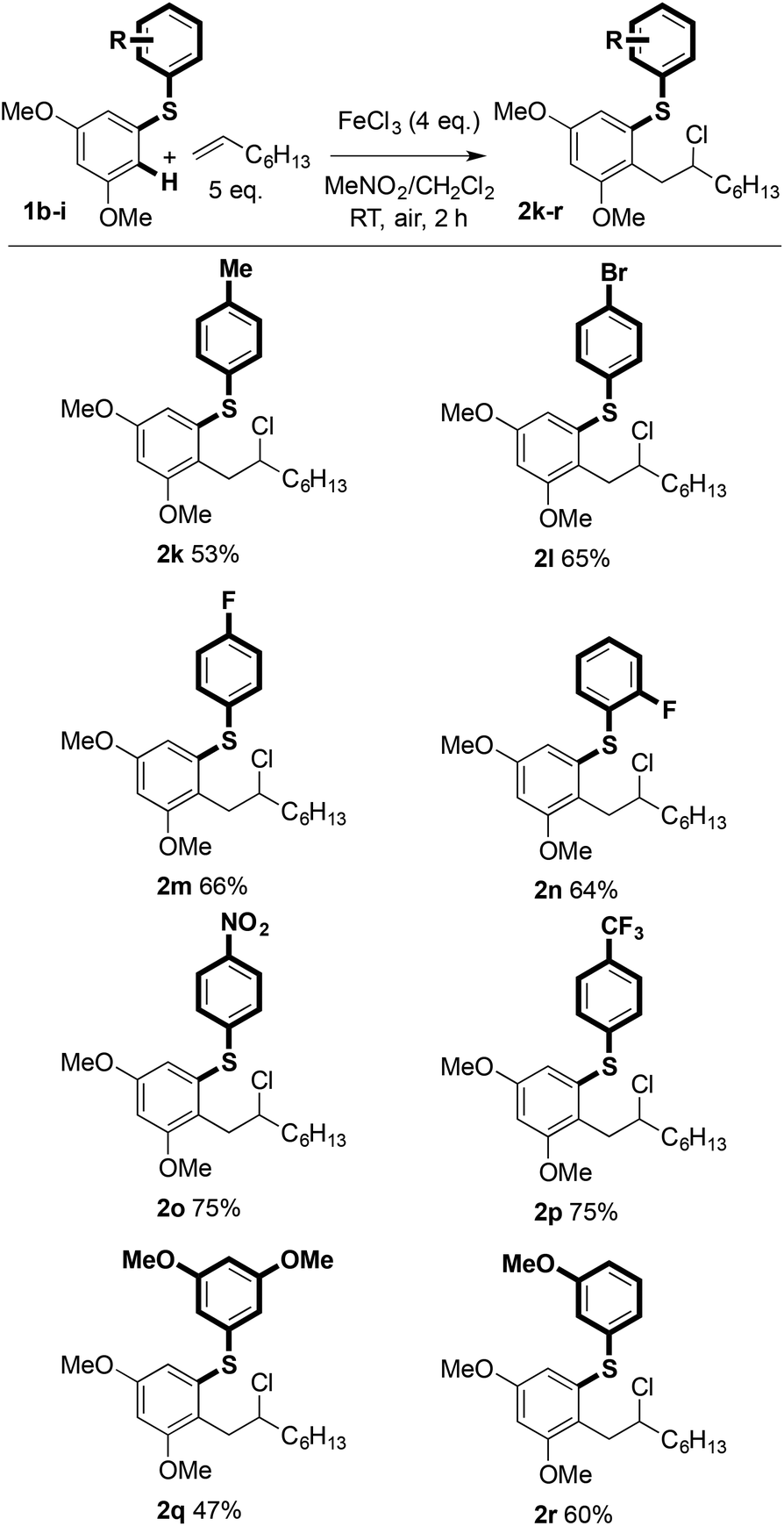 | ||
| Scheme 3 Variation of aryl sulfide in the Fe(III)-mediated C–H coupling. | ||
Whilst 3,5-dioxygenation in the left-hand aryl ring of the diaryl sulfides has to date proved indispensible, allyl- and isopropoxyethers can also be used in the C–H coupling and products were isolated in moderate yield. The use of an allyl ether-containing substrate allowed interesting avenues for product manipulation to be explored (vide infra, Scheme 4).
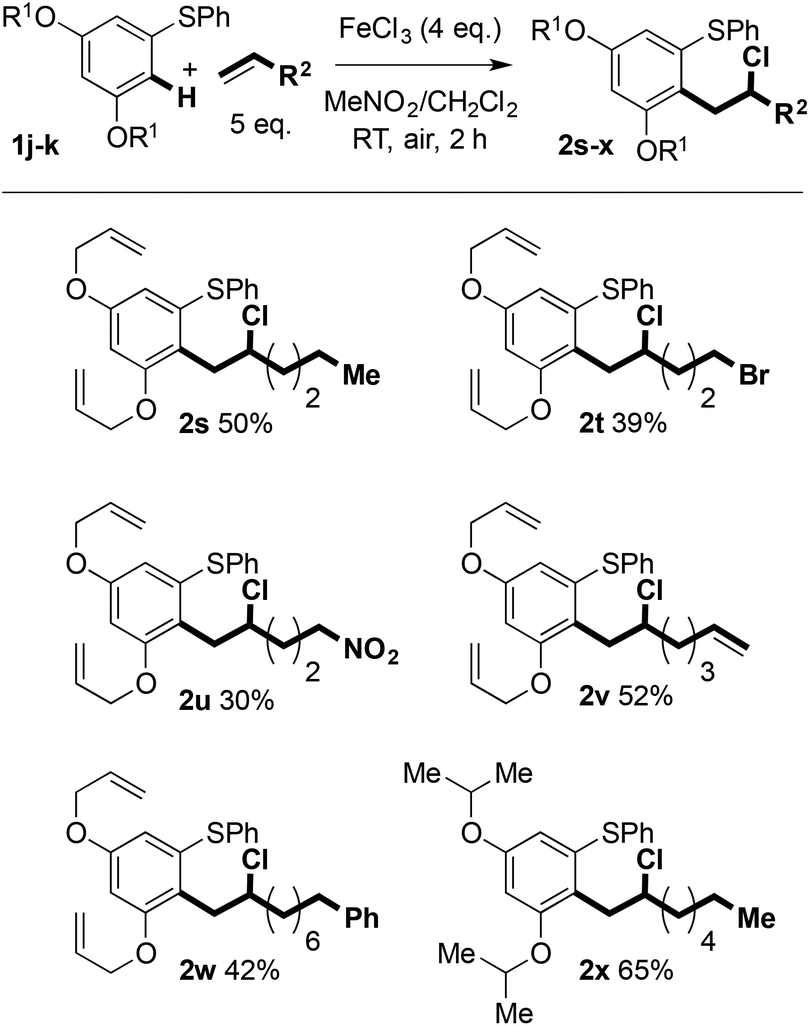 | ||
| Scheme 4 Variation of ether protection in the aryl sulfides undergoing Fe(III)-mediated C–H coupling. | ||
Fig. 1 shows a selection of substrate combinations that did not result in successful coupling. Styrene was an ineffective partner potentially due to it being easily oxidised and thus reacting with FeCl3. Alkenes bearing oxygen-containing functionality, such as ketones and alcohols, also proved ineffective coupling partners. This is likely due to competitive coordination of Fe(III) to the Lewis basic sites in the alkene partner. Finally, internal alkenes, such as cyclohexene, did not undergo coupling. In terms of the arene partner, 3-methoxyphenyl phenyl sulfide did not undergo coupling with 1-octene (vide infra). Also, in contrast to substrates bearing nitro and trifluoromethyl groups in the right-hand aryl ring, the presence of an electron-donating substituent in the para-position of that ring adversely affected coupling. This may be due to formation of a thioquinone-type species. To date, alkyl aryl sulfides have also proved to be incompatible with the coupling reaction. Additional substrates that failed to undergo coupling can be found in the ESI.†
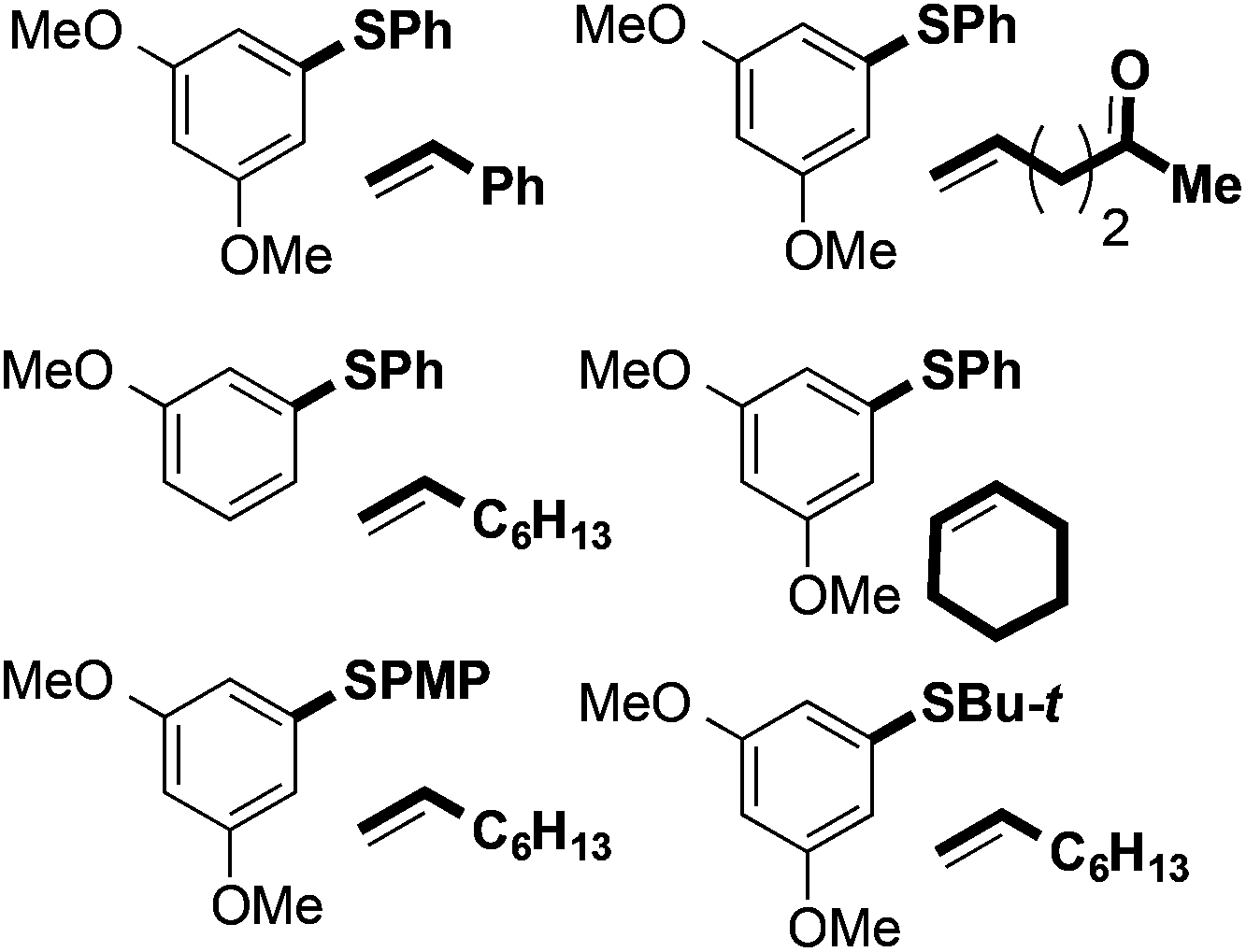 | ||
| Fig. 1 Unsuccessful arene and alkene combinations for C–H coupling. PMP = 4-MeOC6H4. | ||
Mechanistic insights
Carbon–carbon bond formation at the terminal position of the alkene, and the formation of linear products, rather than coupling at the internal position of the alkene, and the formation of branched products, suggests that an SEAr process is not operational.9 In addition, control experiments using non-redox-active Lewis acids did not result in product formation (Scheme 5).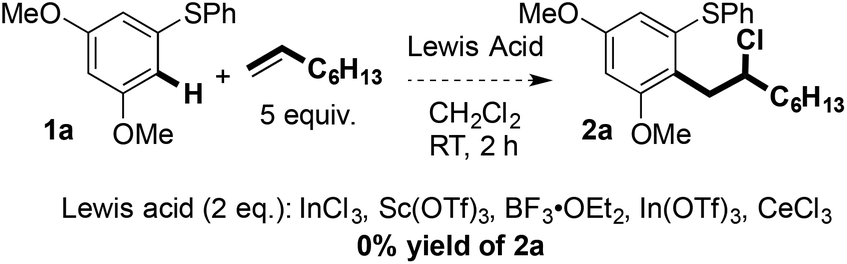 | ||
| Scheme 5 Control reactions employing a variety of Lewis acids. | ||
Scheme 6 shows a plausible mechanism for the Fe(III)-mediated arene/alkene coupling.10 Oxidation of the aryl sulfide 1 by the anhydrous oxidant FeCl3 [approx. +2.00 V (vs. SHE) in MeCN]11 gives radical cation A. Addition of the terminal alkene partner then gives intermediate radical cation B. The preference for bond formation ortho to sulfur may indicate that chelation with the metal centre in D directs reaction towards this position.12 Substrates in which the arylsulfanyl substituent was replaced by an alkoxy or alkylamino substituent failed to undergo coupling, thus highlighting the important role played by sulfur (see the ESI†).
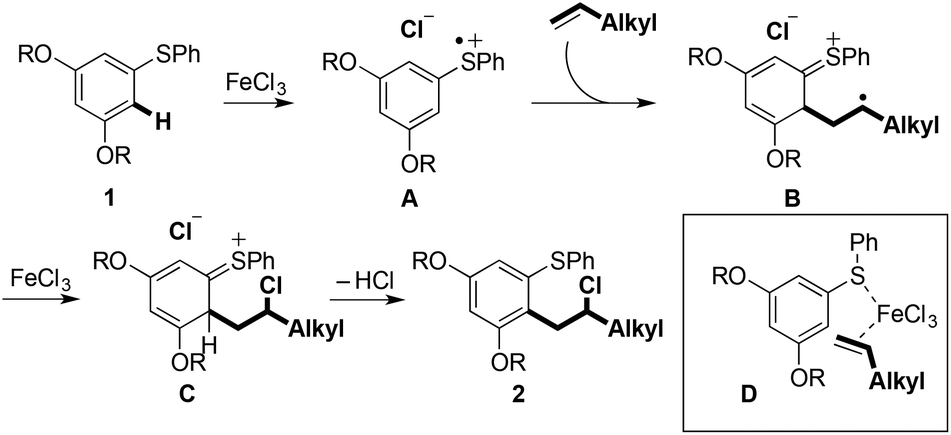 | ||
| Scheme 6 Proposed mechanism of Fe(III)-mediated C–H arene/alkene coupling. | ||
Our studies show that the oxidation potential of 1a [+1.71 V (vs. SHE) in MeCN] is compatible with the initial step of the proposed mechanism (Fig. 2). A carbocation is then formed by the oxidation of radical cation B with FeCl3 that is then quenched by chloride to deliver the products 2. Indirect evidence for this mechanism comes from the observation that well-known oxidant CAN mediates a similar transformation: coupling of 1a with 1-octene gave the nitrate analogous to 2a in 56% yield (Scheme 7).13,14 Furthermore, the oxidation of diaryl sulfides to the corresponding sulfoxides using CAN is proposed to proceed through radical cations analogous to A.15 The terminal alkene partners are known to have higher oxidation potentials than the aryl sulfides.16
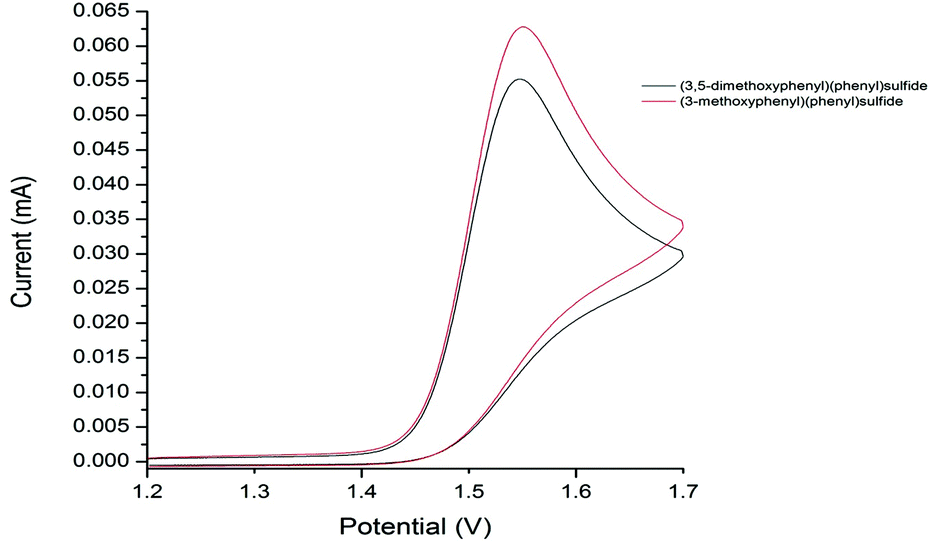 | ||
| Fig. 2 Voltammograms for 3-methoxyphenyl phenyl sulfide and (3,5-dimethoxyphenyl)phenyl sulfide versus reference electrode. | ||
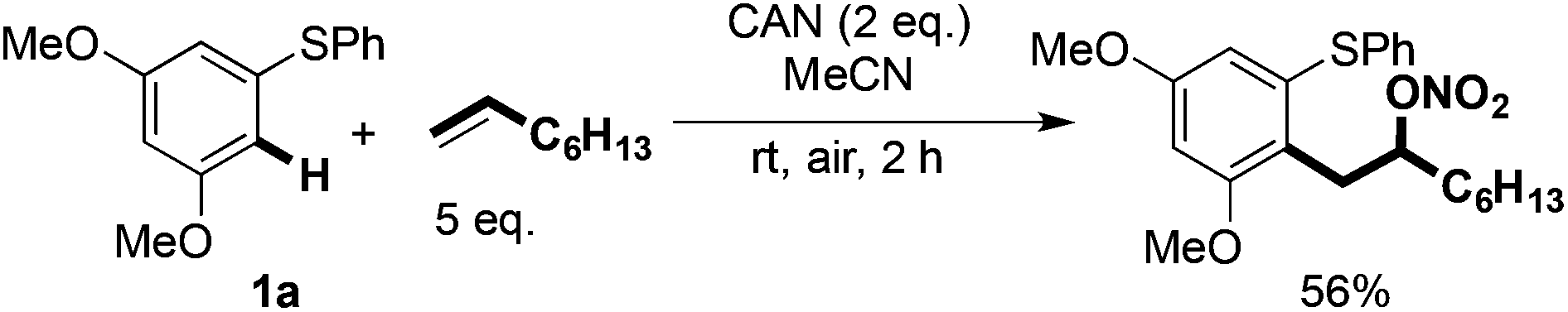 | ||
| Scheme 7 CAN-mediated C–H coupling to give an analogous product. | ||
5-hexenyl ‘radical clock’ cyclisation was used in an unsuccessful attempt to intercept radical B (Scheme 2: 2b was the only coupled product observed). Thus, under the experimental conditions, radical oxidation appears to be faster than cyclisation (i.e. >1 × 105 s−1) (Scheme 8).17,18 Interestingly, attempts to promote cyclative radical trapping using dienes bearing geminal dialkyl substitution led to alternative coupling products 2y and 2z in low yield (Scheme 8). This may be due to the increased steric bulk impeding trapping of the carbocation intermediate with chloride.
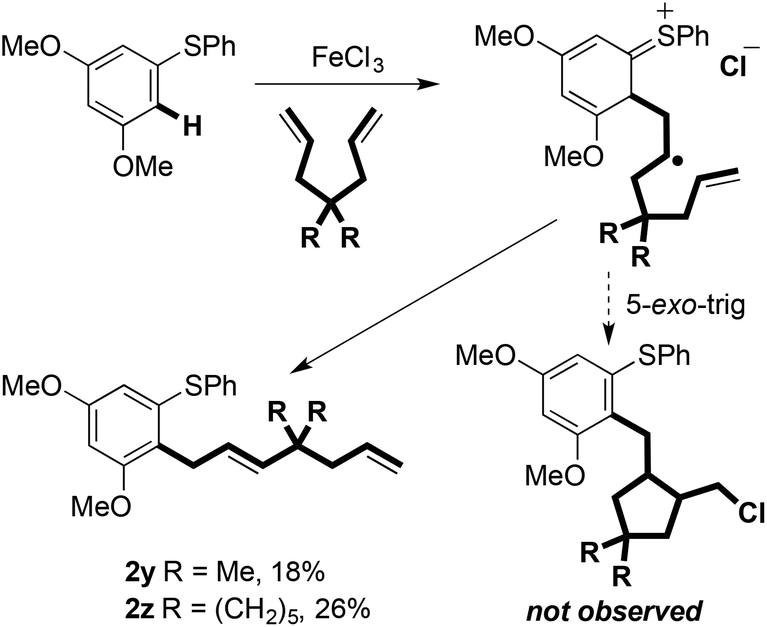 | ||
| Scheme 8 Attempts to trap radical intermediates formed during the Fe(III)-mediated C–H coupling. | ||
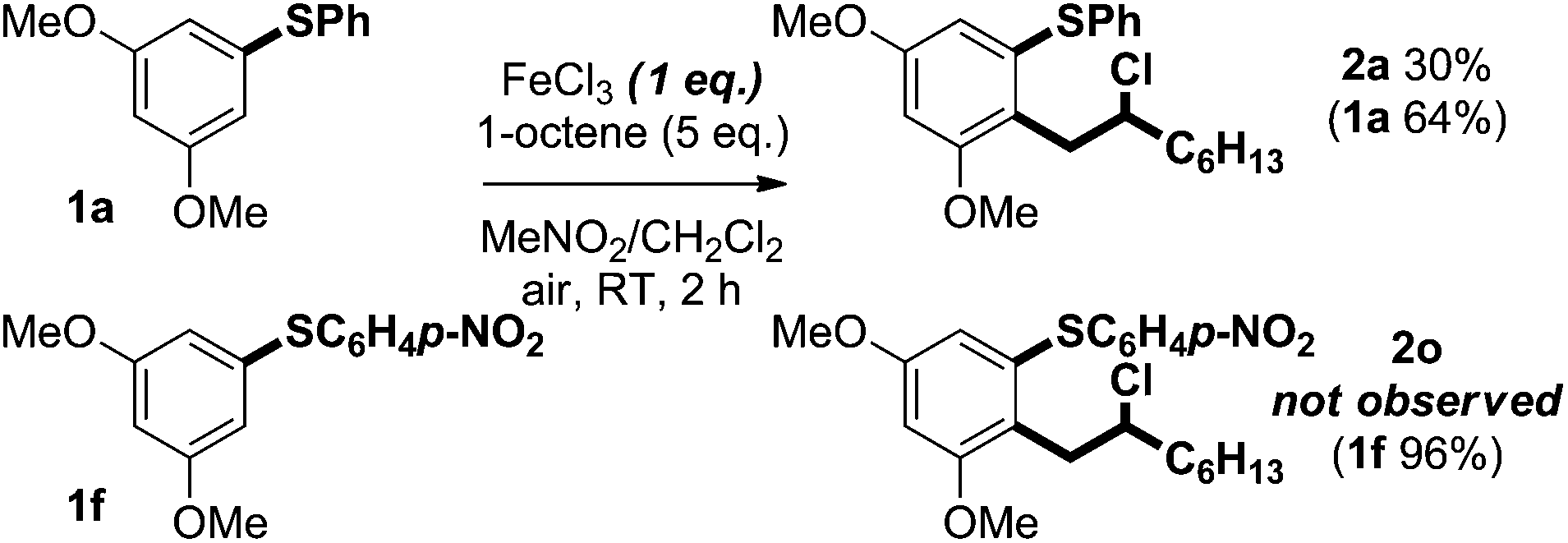
Two factors appear crucial to the success of the cross-coupling process; (i) the ease of oxidation of the aryl sulfide, and (ii) the stability of the resultant radical cation A. For example, attempts to use 3-methoxyphenyl phenyl sulfide as a coupling partner with 1-octene led to only a trace of product even though the aryl sulfide starting material was consumed. We have measured the oxidation potential of 3-methoxyphenyl phenyl sulfide and found it to be similar to that of 1a [+1.72 V (vs. SHE) in MeCN] (Fig. 2), therefore, we propose that deleterious side reactions of the radical cation derived from 3-methoxyphenyl phenyl sulfide are likely responsible for inefficient carbon–carbon bond formation. Steric stabilisation of the radical cation A may therefore be the most important role played by substituents in the ring undergoing coupling in the aryl sulfide.
Switching the electronic properties of the para substituent in the non-coupling, right-hand aryl ring of substrates affected the yield of the process (Scheme 3). Coupling reactions of aryl sulfide substrates bearing nitro and trifluoromethyl electron-withdrawing groups, to give sulfides 2o and 2p, gave products in higher yield than processes involving comparable substrates lacking these substituents. As expected, the competition experiment shown in Scheme 9 using limiting FeCl3 suggests that the rate of coupling is highest for electron-rich substrates: a mixture of 1-octene, 1a and 1f selectively delivered 2a with no trace of 2o. It appears likely that a decreased rate of aryl sulfide oxidation, thus avoiding large concentrations of radical cations that lead to decomposition,19 results in the improved yields obtained for the coupling of aryl sulfides to give 2o and 2p (Scheme 3).
 | ||
| Scheme 9 Competition experiment exploring the influence of the right-hand aryl ring. | ||
Product manipulation
The products of C–H arene/alkene coupling possess a range of functional handles for further manipulation (Fig. 3). For example, directed ortho-metalation of 2a followed by reaction with various electrophiles (to form 3a–c), oxidation of sulfur (to give sulfone 3d), reduction and elimination of chloride (to form 3e and 3f, respectively), and SN2 displacement of chloride (to give azide 3g) can be efficiently performed.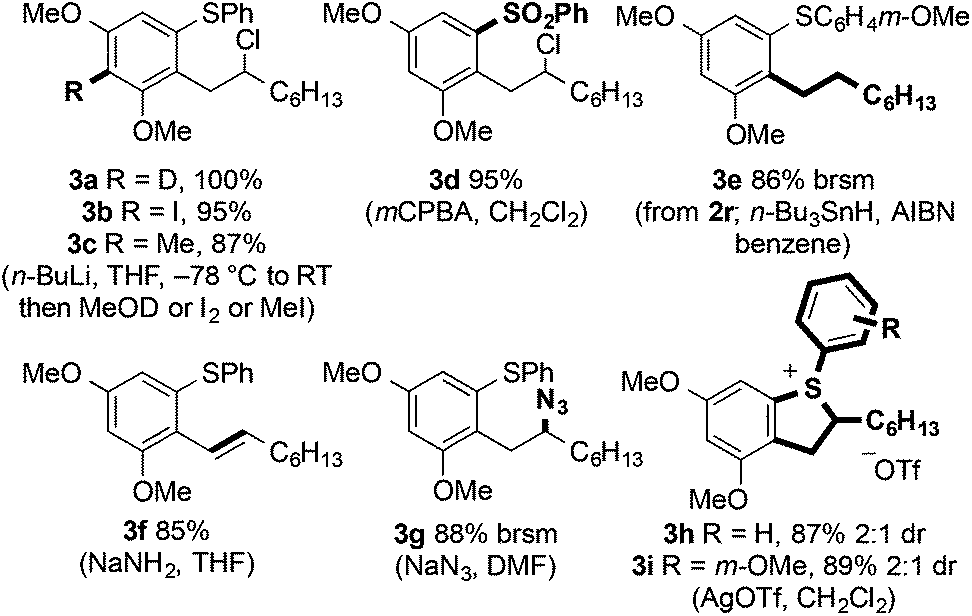 | ||
| Fig. 3 Synthetic manipulation of coupled product 2a. | ||
Notably, styrene 3f is formally the product of an oxidative Heck reaction involving aryl sulfide 1a and 1-octene.20 Coupling products can also undergo dehalogenation when treated with silver(I) salts to give the cyclic sulfonium salts (3h and 3i) as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers. Carrying out the Fe(III)-mediated C–H alkene/arene coupling in the presence of AgOTf allowed the sulfonium salt 3h to be prepared directly in moderate isolated yield (Scheme 10).
:1 mixture of diastereoisomers. Carrying out the Fe(III)-mediated C–H alkene/arene coupling in the presence of AgOTf allowed the sulfonium salt 3h to be prepared directly in moderate isolated yield (Scheme 10).
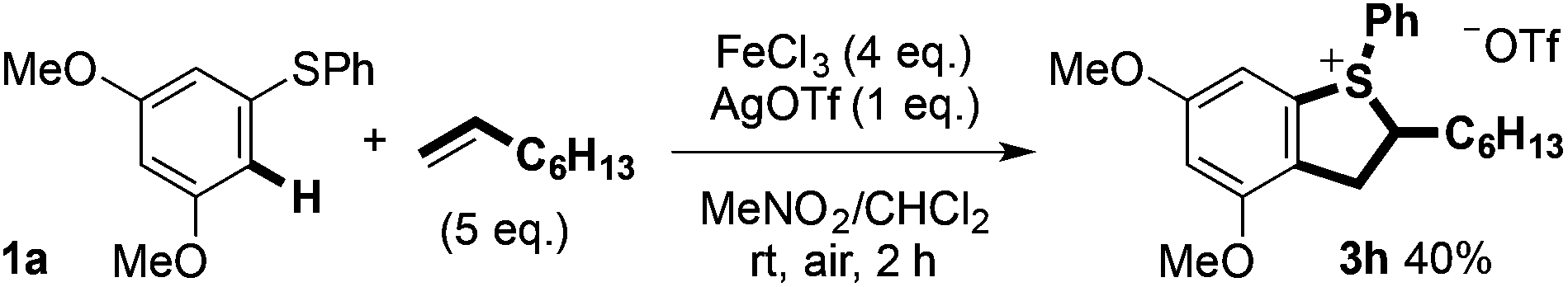 | ||
| Scheme 10 Direct Fe(III)-mediated C–H coupling to form cyclic sulfonium salt. | ||
Pd-catalysed deprotection of the allyl ethers in the products of coupling 2s–w triggered cyclisation to give important benzofuran scaffolds. The reaction proceeds efficiently under mild conditions employing NaBH4 and the allyl scavenger, morpholine. Interestingly, when NaH was used in place of NaBH4, only the mono-deprotected benzofuran product 4e was observed (Scheme 11).
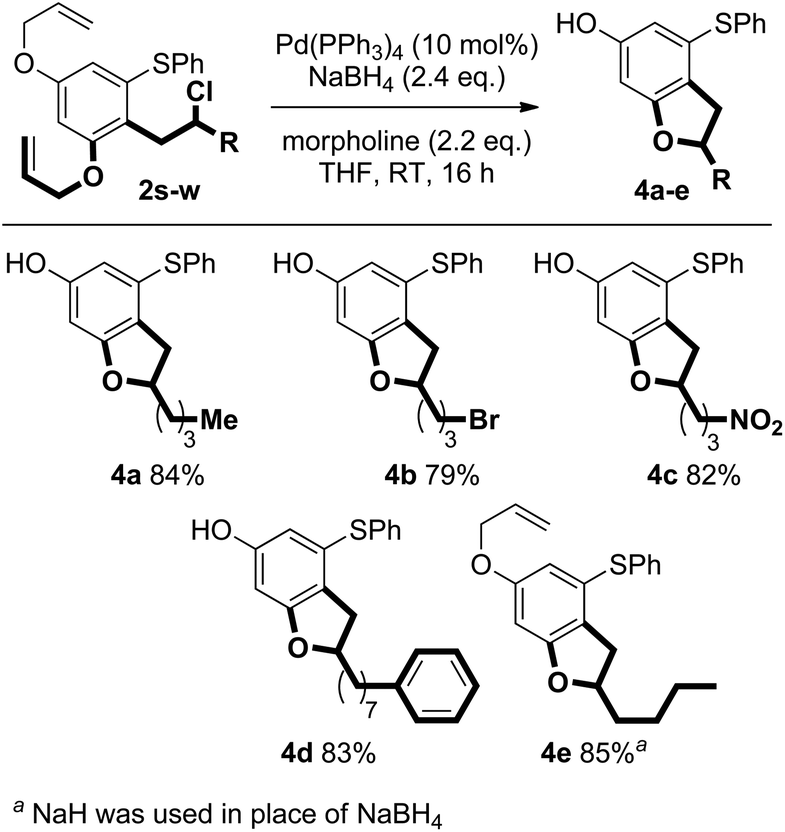 | ||
| Scheme 11 Pd-catalysed deallylation/cyclisation. | ||
Conversion of dihydrobenzofuran 4a to the corresponding triflate 5 (Tf2NPh, tBuONa, THF, 2 h; 87%) paved the way for further decoration of the heterocyclic scaffold. Pd-catalysed Suzuki-Miyaura cross-coupling21 gave 6a–c in high isolated yield. Alternatively, copper-free, Pd-catalysed Sonogashira cross-coupling gave 6d in excellent isolated yield (Scheme 12).22
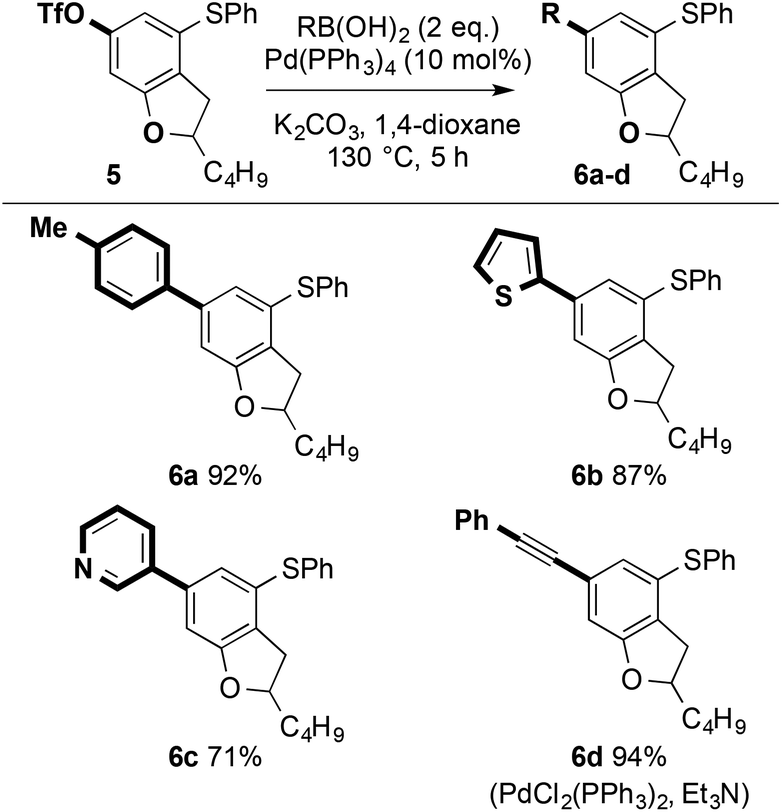 | ||
| Scheme 12 Pd-catalysed couplings for the decoration of dihydrobenzofurans. | ||
The arylsulfanyl directing group can be removed using RANEY® nickel in excellent yield (RANEY® Ni, EtOH, 1 h) (Fig. 4). Notably, reductive removal of the arylsulfanyl from the products of Fe(III)-mediated C–H coupling completes a concise approach to biologically important alkylresorcinols (e.g.7a).23
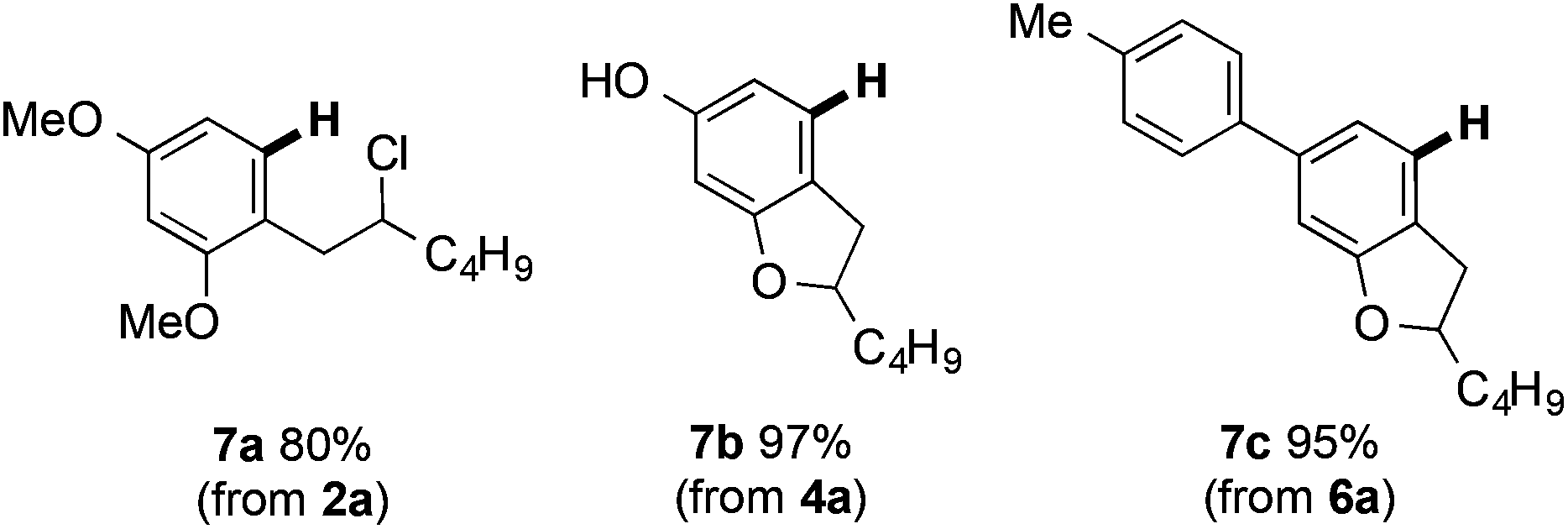 | ||
| Fig. 4 Reductive removal of the arylsulfanyl directing group using RANEY® Ni. | ||
More attractively, the arylsulfanyl directing group can also act as a synthetic handle for the introduction of further diversity. Nickel-catalysed Kumada couplings with aromatic, heteroaromatic, and aliphatic Grignard reagents allow efficient further decoration of the dihydrobenzofuran motifs (Scheme 13).24 Notably, nickel-catalysed coupling to give 8a and 8b, proceeded smoothly with no nickel insertion into thiophene C–S bonds.25
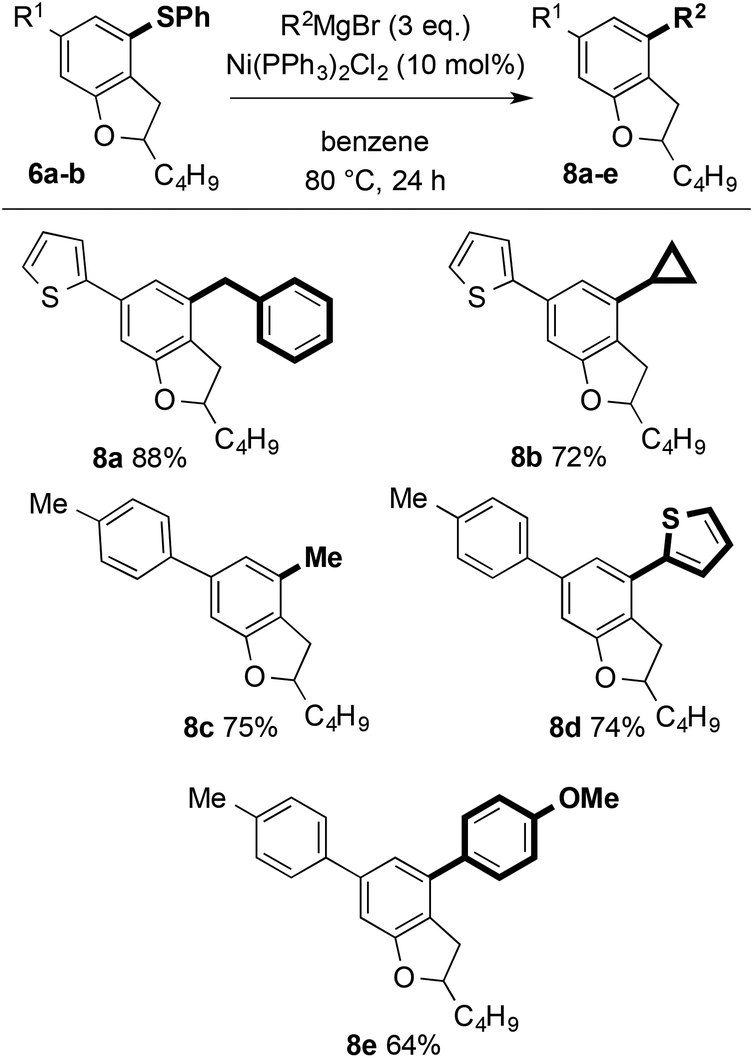 | ||
| Scheme 13 Ni-catalysed couplings for the decoration of dihydrobenzofurans. | ||
Conclusions
A novel route to medicinally-relevant dihydrobenzofurans has been developed that utilises a sulfur-directed C–H ortho-coupling of arenes and unactivated terminal alkenes mediated by iron, and a palladium-catalysed deallylation/heterocyclisation. The iron-mediated coupling affords linear products of alkene chloroarylation with complete regioselectivity. Redox-activation of the arene partner by iron(III) and coupling of the resultant radical-cation with the alkene partner is proposed. The dihydrobenzofuran scaffolds can be readily decorated using Pd and Ni-catalysed cross-couplings.Acknowledgements
EPSRC (Established Career Fellowship to DJP), Syngenta (CASE PhD Studentship to C. W. C), AstraZeneca (CASE PhD Studentship to M. H. A), and The University of Manchester.Notes and references
- For recent reviews see: (a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (b) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC; (c) L. Zhou and W. Lu, Chem. – Eur. J., 2014, 20, 634–642 CrossRef CAS PubMed.
- (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (c) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (d) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (e) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed.
- For seminal studies, see: (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (b) Y. Fujiwara, I. Moritani, S. Danno, R. Asano and S. Teranishi, J. Am. Chem. Soc., 1969, 91, 7166–7169 CrossRef CAS. For selected examples: using Pd, see: (c) Z. Xu, B. Xiang and P. Sun, Eur. J. Org. Chem., 2012, 3069–3073 CrossRef CAS; (d) L. Wang, S. Liu, Z. Li and Y. Yu, Org. Lett., 2011, 13, 6137–6139 CrossRef CAS PubMed; (e) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586–1587 CrossRef CAS PubMed; (f) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315–319 CrossRef CAS PubMed; (g) C. Jia, W. Lu, T. Kitamura and Y. Fujiwara, Org. Lett., 1999, 1, 2097–2100 CrossRef CAS; (h) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602–13605 CrossRef CAS PubMed. Using Ru, see: (i) K. Graczyk, W. Ma and L. Ackermann, Org. Lett., 2012, 14, 4110–4113 CrossRef CAS PubMed; (j) K. Padala and M. Jeganmohan, Org. Lett., 2012, 14, 1134–1137 CrossRef CAS PubMed; (k) B. Li, J. Ma, N. Wang, H. Feng, S. Xu and B. Wang, Org. Lett., 2012, 14, 736–739 CrossRef CAS PubMed; (l) L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728–731 CrossRef CAS PubMed; (m) K. Padala, S. Pimparkar, P. Madasamy and M. Jeganmohan, Chem. Commun., 2012, 48, 7140–7142 RSC; (n) K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144–6147 CrossRef CAS PubMed; (o) R. Manikandan, P. Madasamy and M. Jeganmohan, ACS Catal., 2016, 6, 230–234 CrossRef CAS. Using Rh, see: (p) P. Becker, D. L. Priebbenow, R. Pirwerdjan and C. Bolm, Angew. Chem., Int. Ed., 2014, 53, 269–271 CrossRef CAS PubMed; (q) X. Huang, J. Huang, C. Du, X. Zhang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12970–12974 CrossRef CAS PubMed; (r) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed; (s) C. Wang, H. Chen, Z. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2012, 51, 7242–7245 CrossRef CAS PubMed; (t) S. H. Park, J. Y. Kim and S. Chang, Org. Lett., 2011, 13, 2372–2375 CrossRef CAS PubMed; (u) T. K. Hyster, D. M. Dalton and T. Rovis, Chem. Sci., 2015, 6, 254–258 RSC; (v) X. Xue, J. Xu, L. Zhang, C. Xu, Y. Pan, L. Xu, H. Li and W. Zhang, Adv. Synth. Catal., 2016, 358, 573–583 CrossRef CAS.
- (a) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188–1191 CrossRef CAS PubMed; (b) B. Wang, C. Shen, J. Yao, H. Yin and Y. Zhang, Org. Lett., 2014, 16, 46–49 CrossRef CAS PubMed; (c) X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem. – Eur. J., 2013, 19, 11898–11903 CrossRef CAS PubMed; (d) M. Yu, Y. Xie, C. Xie and Y. Zhang, Org. Lett., 2012, 14, 2164–2167 CrossRef CAS PubMed; (e) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 704–707 CrossRef CAS PubMed; (f) T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef CAS; (g) P. Villuendas and E. P. Urriolabeitia, Org. Lett., 2015, 17, 3178–3181 CrossRef CAS PubMed; (h) X.-S. Zhang, Y.-F. Zhang, K. Chen and Z.-J. Shi, Org. Chem. Front., 2014, 1, 1096–1100 RSC.
- C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed.
- C. W. Cavanagh, M. H. Aukland, A. Hennessy and D. J. Procter, Chem. Commun., 2015, 51, 9272–9275 RSC.
- For reviews on metal-mediated oxidative coupling, see: (a) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed; (b) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459–3470 CrossRef CAS PubMed; (c) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed.
- For oxidative radical couplings of sulfides, see: (a) H. Cao, D. Liu, C. Liu, X. Hu and A. Lei, Org. Biomol. Chem., 2015, 13, 2264–2266 RSC; (b) A. Kamimura, T. Nokubi, K. Nasu, Y. Takechi, Y. Ishihara, K. Kato, S. Noguchi, M. Watanabe, M. Shirai, M. Sumimoto and H. Uno, Chem. Lett., 2012, 41, 950–951 CrossRef CAS. Similar pathways have been proposed for other transformations. See: (c) L. Menini and E. V. Gusevskaya, Chem. Commun., 2006, 209–211 RSC; (d) L. Yang, Z. Lu and S. S. Stahl, Chem. Commun., 2009, 6460–6462 RSC; (e) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed; (f) Z. Huang, L. Jin, Y. Feng, P. Peng, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7151–7155 CrossRef CAS PubMed; (g) Y. Ma, D. Zhang, Z. Yan, M. Wang, C. Bian, X. Gao, E. E. Bunel and A. Lei, Org. Lett., 2015, 17, 2174–2177 CrossRef CAS PubMed.
- J. Kischel, I. Jovel, K. Mertins, A. Zapf and M. Beller, Org. Lett., 2006, 8, 19–22 CrossRef CAS PubMed.
- An alternative process involving electrophilic metalation of the aryl sulfide followed by carbometalation of the alkene is less likely. For nucleophilic metalation, see: (a) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098–13101 CrossRef CAS PubMed; (b) Y. Hirata, A. Yada, E. Morita, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2010, 132, 10070–10077 CrossRef CAS PubMed; (c) Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 16170–16171 CrossRef CAS PubMed.
- Formal potential of Fe(III)/Fe(II) in anhydrous solution is +1.57 V vs. Ag/0.01 M Ag+. K. Izutsu, Electrochemistry in Nonaqueous Solutions, Wiley-VCH, Verlag GmbH & Co, KGaA, 2002 Search PubMed.
- (a) C. Krüger, B. L. Barnett and D. Brauer, Structure and Bonding in Organic Iron Compounds in The Organic Chemistry of Iron, ed. E. A. Koerner Von Gustorf, Elsevier, 1978, vol. 1 Search PubMed; (b) R. B. King, Monoolefin Iron Complexes in The Organic Chemistry of Iron, ed. E. A. Koerner Von Gustorf, Elsevier, 1978, vol 1 Search PubMed.
- It should also be noted that MeNO2 is used with CH2Cl2 only to aid solubility of FeCl3 in the Fe(III)-mediated cross-coupling. MeNO2 was therefore not used in the control reactions with other Lewis acids. See ref. 6.
- The CAN process may also be directed by sulfur as cerium is known to have a strong affinity for sulfur. For an example of a thiophenol-Ce(IV) complex, see: A. A. Soliman and W. Linert, Synth. React. Inorg. Met. –Org. Chem., 1999, 29, 1133–1151 CrossRef CAS.
- T.-L. Ho and C. M. Wong, Synthesis, 1972, 561–562 CrossRef.
- D. A. Nicewicz and D. S. Hamilton, Synlett, 2014, 1191–1196 CrossRef.
- A. L. J. Beckwith, Tetrahedron, 1981, 37, 3073–3100 CrossRef CAS.
- Alternatively, cyclisation of the radical may be prevented by its association with an iron chloride species. Attempts to use vinylcyclopropane as a coupling partner and radical probe were unsuccessful: no carbon–carbon bond formation product was observed.
- This is consistent with the observation that the slow addition of a solution of FeCl3 in MeNO2 to the aryl sulfide and alkene gave the highest yields of coupled product.
- (a) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef CAS PubMed; (b) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (b) L. Chen, F. Zhou, T.-D. Shi and J. Zhou, J. Org. Chem., 2012, 77, 4354–4362 CrossRef CAS PubMed.
- A. B. Ross, A. Kamal-Eldin and P. Aman, Nutr. Rev., 2004, 62, 81–95 CrossRef PubMed.
- (a) E. Wenkert, T. W. Ferreira and E. L. Michelotti, J. Chem. Soc., Chem. Commun., 1979, 637–638 RSC; (b) S. Kanemura, A. Kondoh, H. Yorimitsu and K. Oshima, Synthesis, 2008, 2659–2664 CAS; (c) J. Ma, L. Peng, X. Zhang, Z. Zhang, M. Campbell and J. Wang, Chem. – Asian J., 2010, 5, 2214–2219 CrossRef CAS PubMed; (d) A. J. Eberhart, J. E. Imbriglio and D. J. Procter, Org. Lett., 2011, 13, 5882–5885 CrossRef CAS PubMed; (e) K. Murakami, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 7510–7513 CrossRef CAS PubMed; (f) A. Baralle, S. Otsuka, V. Guérina, K. Murakami, H. Yorimitsu and A. Osuka, Synlett, 2015, 327–330 CAS.
- E. Wenkert, M. H. Leftin and E. L. Michelotti, J. Chem. Soc., Chem. Commun., 1984, 617–618 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experiments, full experimental details for the preparation of compounds, 1H and 13C NMR spectra for new compounds. See DOI: 10.1039/c6ob00883f |
| This journal is © The Royal Society of Chemistry 2016 |