
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein ubiquitination via dehydroalanine: development and insights into the diastereoselective 1,4-addition step†
Roman
Meledin
,
Sachitanand M.
Mali
,
Sumeet K.
Singh
and
Ashraf
Brik
*
Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa, 3200008 Israel. E-mail: abrik@technion.ac.il
First published on 4th May 2016
Abstract
We report a strategy for site-specific protein ubiquitination using dehydroalanine (Dha) chemistry for the preparation of ubiquitin conjugates bearing a very close mimic of the native isopeptide bond. Our approach relies on the selective formation of Dha followed by conjugation with hexapeptide bearing a thiol handle derived from the C-terminal of ubiquitin. Subsequently, the resulting synthetic intermediate undergoes native chemical ligation with the complementary part of the ubiquitin polypeptide. It has been proposed that the Michael addition step could result in the formation of a diastereomeric mixture as a result of unselective protonation of the enolate intermediate. It has also been proposed that the chiral protein environment may influence such an addition step. In the protein context these questions remain open and no experimental evidence was provided as to how such a protein environment affects the diastereoselectivity of the addition step. As was previously proposed for the conjugation step on protein bearing Dha, the isopeptide bond formation step in our study resulted in the construction of two protein diastereomers. To assign the ratio of these diastereomers, trypsinization coupled with high-pressure liquid chromatography analysis were performed. Moreover, the obtained peptide diastereomers were compared with identical synthetic peptides having defined stereogenic centers, which enabled the determination of the configuration of the isopeptide mimic in each diastereomer. Our study, which offers a new method for isopeptide bond formation and protein ubiquitination, gives insights into the parameters that affect the stereoselectivity of the addition step to Dha for chemical protein modifications.
Introduction
Protein ubiquitination is highly important and ubiquitous posttranslational modification (PTM) is utilized for controlling various cellular events.1 This process involves the enzymatic machinery consisting of E1, E2 and E3, which collaborate with each other to attach a ubiquitin monomer or polyubiquitin chain to a protein substrate.2 In the polyubiquitin chain, the monomers are linked through isopeptide bonds, which are formed between any of the seven-Lys side chains or the N-terminus in ubiquitin and the C-terminus of the subsequent ubiquitin.3 This leads to a wide landscape of ubiquitin bioconjugates, encompassing different ubiquitin chains of various lengths and linkage types that can be linked to a substrate at different sites.4 In order to terminate/edit the signal, ubiquitin or ubiquitin chains can be removed fully or partially by a family of enzymes known as deubiquitinases (DUBs).5In order to study ubiquitination and deubiquitination at the molecular level one has to obtain highly pure and homogeneous ubiquitin conjugates in workable quantities. Like many other posttranslationally modified proteins, the preparation of such complex conjugates using the enzymatic machinery is a formidable task. Chemical approaches offer many advantages for the preparation of free ubiquitin chains and ubiquitinated proteins as evident by several recent studies.6–11 The main privilege of the synthetic methods is the avoidance of using the E1–E3 enzymatic machinery and the opportunity to basically make any variation in these bioconjugates. These efforts include for example the preparation of ubiquitinated proteins having ubiquitin chains with the desired length and linkage type.12,13 These conjugates can also be stabilized to prevent cleavage by deubiquitinases14–16 or modified by an electrophile to trap ubiquitin interacting partners.17–19 Despite these advantages, there are still several limitations in the synthetic and semisynthetic approaches for the preparation of ubiquitinated proteins of large size.
Site selective modification of expressed proteins is another powerful approach to prepare protein conjugates including those that bear PTMs.20,21 The obvious advantage here is the ability to use protein substrates derived by recombinant technology or isolated from natural sources. The development of approaches for the site selective chemical ubiquitination of expressed proteins is still in its infancy. Only a few methods are available to non-enzymatically ubiquitinate proteins using unnatural linkages such as disulfide22–24 or applying click chemistry on a protein having an unnatural alkyne or azide incorporated via expression of proteins using the amber codon suppression technology.15,16,25 Notably, the latter technology was also used to incorporate thiolysine26 which has enabled the preparation of native ubiquitin chains.27 In addition, the GOPAL based strategy28 which also relies on the amber codon suppression approach was also developed and used to prepare ubiquitin chains.29
A step forward toward facilitating ubiquitination of expressed native proteins was made recently by our group where we succeeded in modifying the isolated α-globin with natively linked polyubiquitin chains of up to four monomers in length through the disulfide linkage.24 Since the disulfide bond is sensitive to the reducing environment these conjugates cannot be used for certain biochemical studies. To overcome this limitation, we also attempted alternative strategies linking the ubiquitin chains via thioether linkages, however this is still limited mainly to mono and di-ubiquitination.24 Moreover despite these successes, the obtained conjugates bear unnatural linkages between the chain and the substrate, which cannot be hydrolyzed by DUBs. Here we report on the development of a novel strategy for site selective protein ubiquitination based on dehydroalanine (Dha) chemistry. Subsequently, we examined the recognition of the isopeptide bond mimic in these conjugates by DUBs. For the first time, we also provide experimental information about the stereoselective addition of a nucleophile on the Dha moiety in the protein context.
Results and discussion
Dha based chemistry is a promising strategy for the preparation of protein conjugates and proteins bearing PTM mimics.30–32 Being the Michael acceptor for the 1,4-addition reaction with various thiols, Dha has become a useful synthetic precursor to obtain a variety of modified proteins. For example, mimics of acetylated histone H3 were prepared and shown to behave as substrates for histone deacetylases.30,32,33 One of the key aspect of this method is the ability of site selective incorporation of the Dha moiety into a desired protein. During the last decade, several research groups reported different strategies of Dha formation in such a way that it would be applicable for peptides34–36 and proteins.33,37–39 Davis and co-workers developed a highly efficient approach for site selective bis-alkylation–elimination of Cys to Dha.32 The bisamide of 1,4-dibromobutane was found to be the most efficient reagent for such a transformation. Importantly, this strategy is compatible for reactions in aqueous buffers with mild pH and temperature, making it very suitable for proteins. In addition, Dha formation can be selective even in the presence of more than one cysteine residue in the protein as long as the rest are participating in disulfide bonds.32Inspired by the recent approaches to prepare posttranslationally modified proteins via Dha, we envisioned a strategy for site selective ubiquitination based on the incorporation of a Cys residue at the desired position of substrate 1, followed by selective thiol elimination by reagent 2 to afford the Dha containing protein 3 (Scheme 1). This Dha will then serve as the electrophilic center for the conjugation with N-terminally thiazolidine (Thz) protected hexapeptide–thiol 4, derived from the C-terminal of ubiquitin. In this strategy, a single point mutation of Leu to Cys is introduced in ubiquitin to enable native chemical ligation (NCL).40 Later, the resulting conjugated product 6 will undergo Thz deprotection, followed by NCL with a complementary part of the ubiquitin(1–70)–thioester 7.
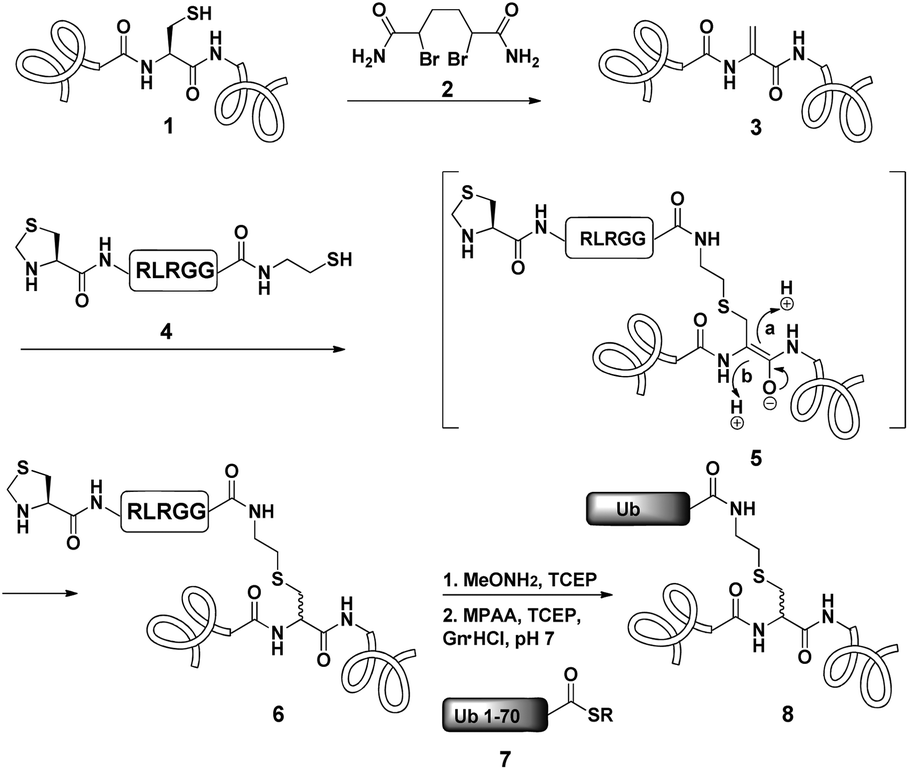 | ||
| Scheme 1 General strategy of Dha based protein ubiquitination. | ||
Notably, the final ubiquitinated product 8 would contain a close mimic of the isopeptide bond where the γ-carbon of the Lys side chain is replaced by sulfur. Despite the two-step approach, such a conjugation of a Dha containing substrate with a small size peptide is advantageous compared with that of a bigger molecular weight protein (such as ubiquitin–thiol), since it allows one to use a higher ratio of nucleophile/electrophile and hence to push the equilibrium of the reaction towards product formation. Remarkably, the Strieter group reported an elegant method based on the use of thiol–ene chemistry to prepare a thiol containing an isopeptide bond having an extra bond relative to the native isopeptide bond.41
As a starting point for developing this strategy we chose ubiquitin as a substrate to prepare Lys48-linked di-ubiquitin. The N-terminally Thz protected hexapeptide–thiol 4 was prepared with C-terminal N-acyl-benzimidazolinone (Nbz),42 followed by switching with cysteamine. The ubiquitin bearing Cys at position 48 was subjected to the elimination reaction by using reagent 2 to afford 9, followed by conjugation with 4 and methoxylamine treatment to afford 10 in 57% isolated yield. Ligation with ubiquitin (1–70)–thioester afforded di-ubiquitin 11 in 39% isolated yield (Scheme 2 and Fig. 1). At this stage, Cys71 was either kept intact to give 11, converted to Ala71 via desulfurization reaction43 to give 12, or alkylated with iodoacetamide to generate 13 (Scheme 2).
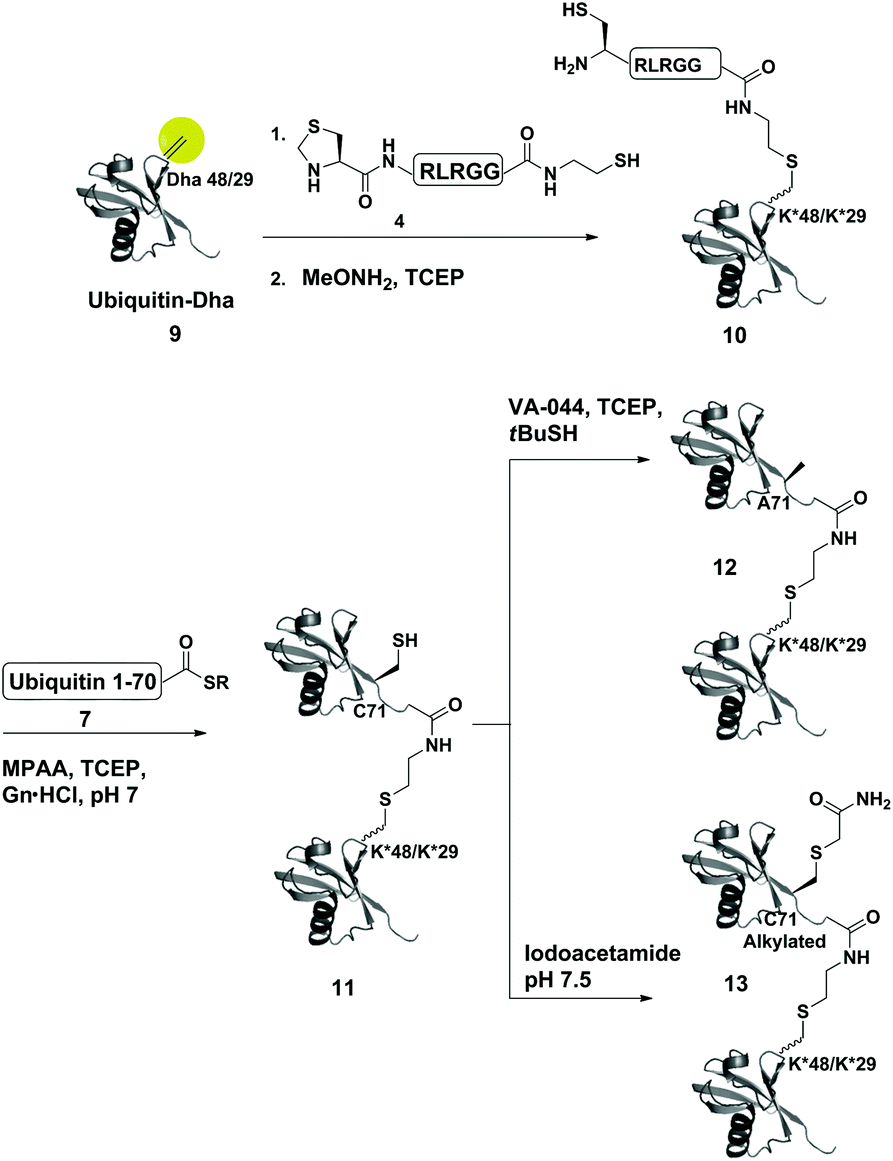 | ||
| Scheme 2 Synthesis of Lys48- and Lys29-linked di-ubiquitin analogues via the Dha strategy, (K* = lysine analogue). | ||
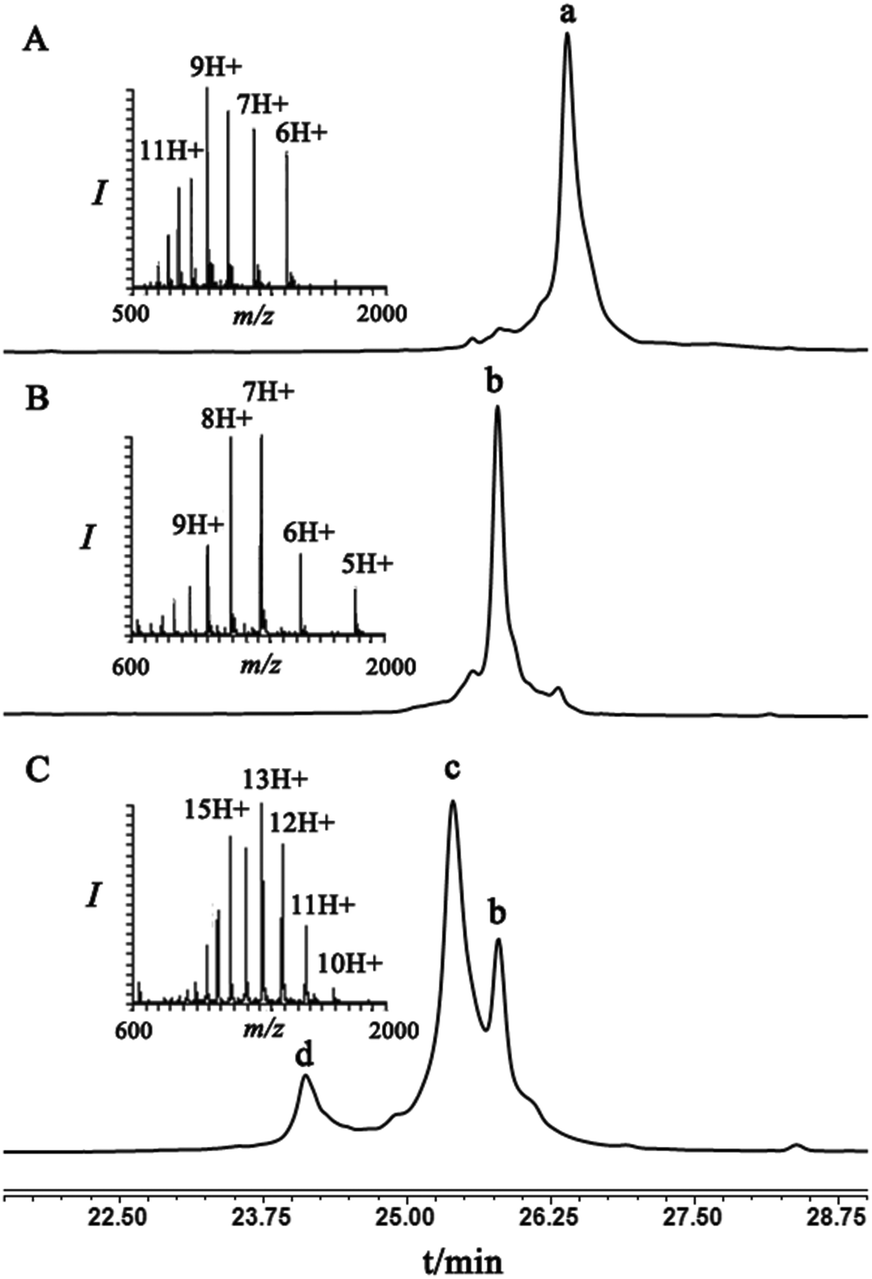 | ||
| Fig. 1 HPLC and MS analyses: (A) peak a corresponds to ubiquitin–Dha48, 9, with the observed mass 8485.9 ± 0.5 Da, calcd 8487.8; (B) peak b corresponds to 10, which is the result of conjugation of hexapeptide–thiol 4 to 9, with the observed mass 9206.3 ± 0.6 Da, calcd 9206.6 Da; (C) NCL of ubiquitin(1–70)-thioester 7 with 10, where peak c corresponds to the ligated product 11 with the observed mass 17082.1 ± 2 Da, calcd 17082.6 Da, peak b corresponds to unreacted 10, peak d corresponds to unreacted ubiquitin(1–70)-thioester 7 with the observed mass 8043.4 ± 0.7 Da, calcd 8045.0 Da. | ||
To examine the influence of the changes on the isopeptide bond as well as the mutations of Leu71, the obtained analogues, 11, 12 and 13, were subjected to cleavage with USP2—a DUB that is known to cleave all Lys-linked di-ubiquitin chains.44 Interestingly, we observed similar cleavage patterns, compared with the WT di-ubiquitin in which within 30 min all analogues were hydrolyzed to near completion to the mono-ubiquitin (Fig. S17†). These results indicate that subtle changes in this site apparently do not harm USP2 activity although we cannot exclude a more noticeable effect on other DUBs. Nevertheless, one could overcome the Leu71Cys point mutation by introducing mercaptoleucine for NCL instead of N-terminal Cys in 4 followed by selective desulfurization to generate the native Leu at this site.45,46
Despite the wide usage of Dha based chemistry for protein modifications, it has been proposed that the Michael addition step could result in the formation of a diastereomeric mixture as a result of unselective protonation of the enolate intermediate in 5 (Scheme 1).30,32,33 It has also been proposed that the chiral protein environment may influence such an addition step.33 However, in the protein context this question remains open and no experimental evidence was provided as to how such a protein environment affects the diastereoselectivity of the addition step. Asymmetric induction was previously observed on a model system involving the addition of methanethiol to proline–dehydroalanine dipeptides in ethanol, which afforded the D- and L-isomers in a 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 ratio, respectively.47 Darses and coworkers have achieved even higher diastereoselectivity of up to 95% when conjugating potassium trifluoroorganoborates to Dha derivatives, catalyzed by a chiral rhodium catalyst, Binap as a chiral ligand and guaiacol as the proton source in toluene.48 In contrast, the group of van der Donk did not observe diastereoselectivity in the conjugation of farnesylthiolate and thioglycosides to dehydropeptides in DMF or methanol.35 Despite this, the two diastereomers were separable by reverse phase HPLC, which could be useful in the preparation of conjugates with defined diastereomeric forms. Proteins, however, are much more challenging to study when compared to these model peptides, yet they provide a unique chiral environment, which might affect the addition step.33 Moreover, since proteins prefer aqueous media for solubility, water is likely to act as a proton donor, which may limit the stereoselectivity.49 How these parameters as well as the protein sequence, folding and the nearby residues may affect the diastereoselectivity of the addition product remains unclear.
:15 ratio, respectively.47 Darses and coworkers have achieved even higher diastereoselectivity of up to 95% when conjugating potassium trifluoroorganoborates to Dha derivatives, catalyzed by a chiral rhodium catalyst, Binap as a chiral ligand and guaiacol as the proton source in toluene.48 In contrast, the group of van der Donk did not observe diastereoselectivity in the conjugation of farnesylthiolate and thioglycosides to dehydropeptides in DMF or methanol.35 Despite this, the two diastereomers were separable by reverse phase HPLC, which could be useful in the preparation of conjugates with defined diastereomeric forms. Proteins, however, are much more challenging to study when compared to these model peptides, yet they provide a unique chiral environment, which might affect the addition step.33 Moreover, since proteins prefer aqueous media for solubility, water is likely to act as a proton donor, which may limit the stereoselectivity.49 How these parameters as well as the protein sequence, folding and the nearby residues may affect the diastereoselectivity of the addition product remains unclear.
To examine this we turned our attention towards the characterization of the diastereomeric mixture formed as a result of the conjugation of hexapeptide–thiol 4 to ubiquitin–Dha 9 at position 48 under folding and denaturing conditions. Since we did not observe two separable peaks in the RP-HPLC analysis having the same mass which correspond to the two isomers, assuming that these diastereomers indeed formed, we trypsinized the product(s) hoping that shorter peptides will likely separate in the HPLC. In the case of formation of two protein diastereomers, the trypsinization step will lead to the two peptides of interest, each containing the isopeptide bond mimic with L- or D-configuration in a ratio that likely reflects the diastereoselectivity of the addition step.
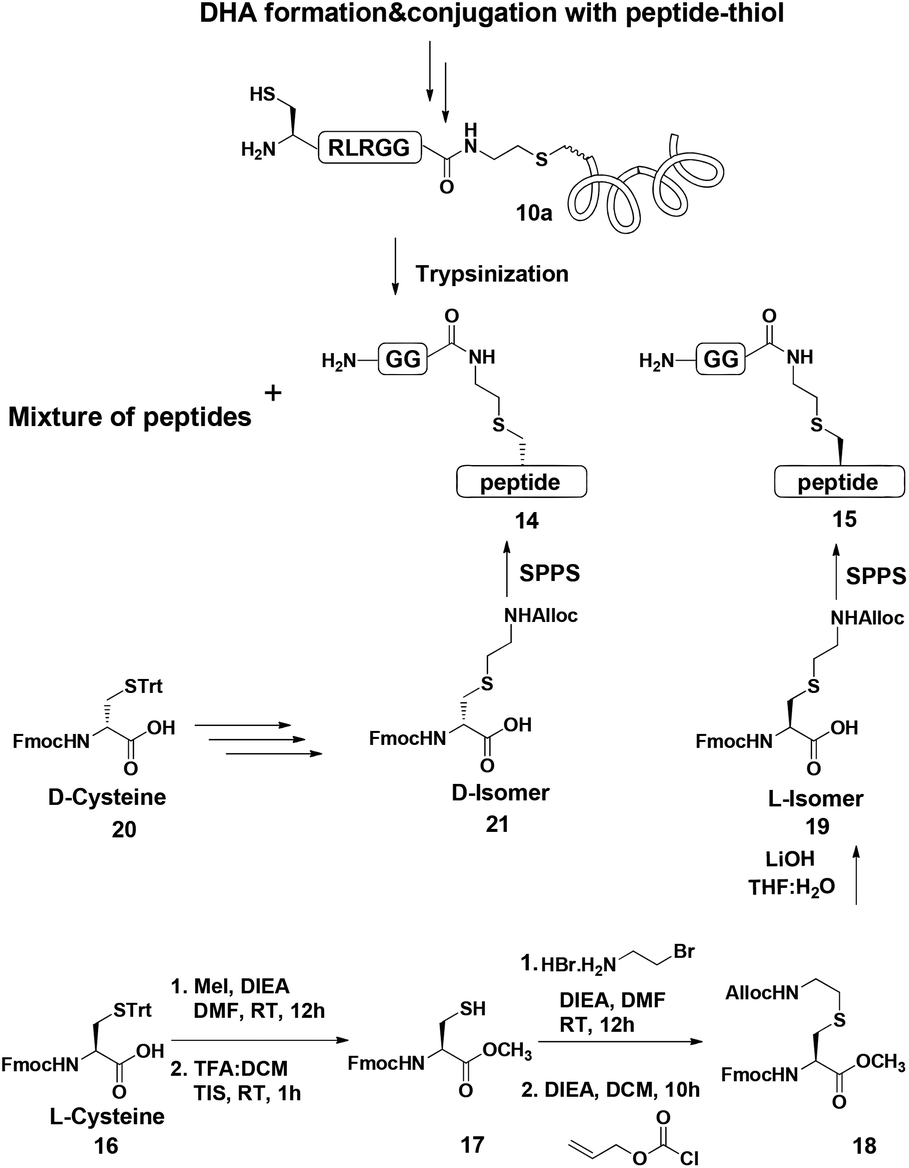
Indeed, when the peptide mixture of the digestion was analyzed by RP-HPLC, two peptides with the same mass were detected (Fig. 2C). These could correspond to the expected two peptide diastereomers bearing also the Gly–Gly from the C-terminal of ubiquitin linked to the peptide via the isopeptide mimic (Fig. 2A and Scheme 3). A similar procedure was repeated for the conjugation step, which was carried out under denaturing conditions followed by applying trypsin digestion and HPLC analysis (Fig. 2B). Under both conditions, we observed similar patterns in which the two peaks that corresponded to the peptides with the same mass (1477 Da) were obtained in a ratio of ∼60:40 based on the HPLC analysis.
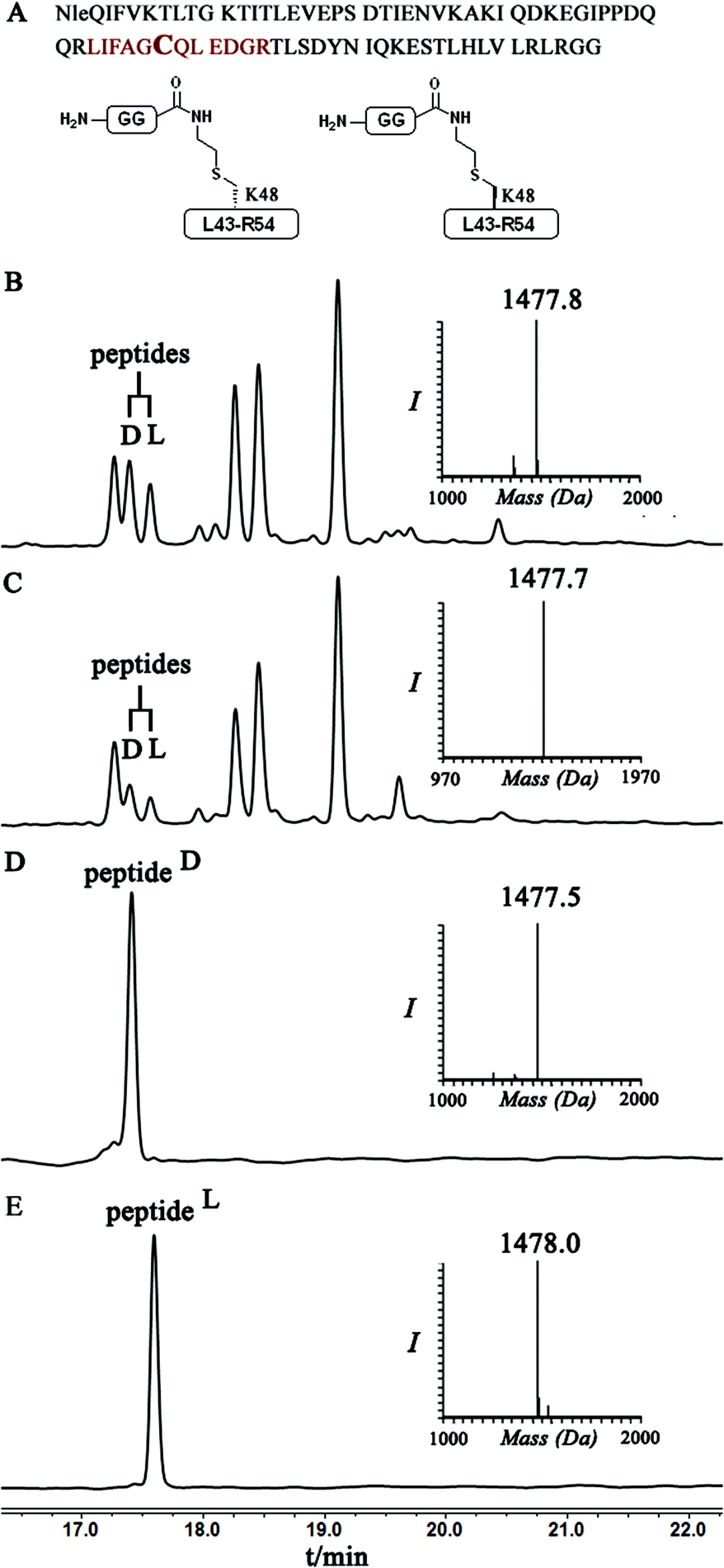 | ||
| Fig. 2 (A) Sequence of full ubiquitin, highlighting the products of trypsinization in D- and L-forms. Asterisk indicates Cys bearing an isopeptide bond mimic with the GG from ubiquitin (shown in bold). HPLC and mass analysis for: (B) trypsinization of the hexapeptide-conjugated product under denaturing conditions with the observed mass 1477.8 ± 0.2 Da, calcd 1479.5 Da. (C) Trypsinization of the hexapeptide conjugated product under folding conditions with the observed mass 1477.7 ± 0.2 Da, calcd 1479.5. (D & E) Synthetically prepared peptides corresponded to the D and L peptide diastereomers obtained from trypsinization, respectively. | ||
 | ||
| Scheme 3 General scheme of trypsinization of conjugated products (10a = hexapeptide–thiol conjugated to either ubiquitin48, ubiquitin29 or α-globin). Synthesis of L- and D-Lys mimics from L or D-Cys, followed by their incorporation into the expected peptide sequence for the comparison study with the actual peptide mixture obtained from trypsinization. | ||
Next, we aimed to confirm the structure of the two peptides and assign the HPLC peaks to the specific diastereomer. To achieve this, we prepared chemically two peptides bearing the defined D- and L-forms of an isopeptide bond mimic (Scheme 3). Briefly, the syntheses started with methyl esterification of L- or D-Cys(Trt)-OH (16, 20) followed by trityl group removal using 50% TFA in DCM. The obtained free thiol 17 was subjected to substitution with ethylamine bromide in the presence of DIEA to give the S-substituted ethylamine Cys. The side chain amine of this analogue was further protected with alloc to give 18 and subsequent hydrolysis of the ester gave the Lys mimic 19 or 21.
Having these analogues in hand, we incorporated each one in the expected peptide sequence 14 or 15 from the digestion mixture. After purification, these peptides were injected into RP-HPLC under similar conditions that we have used for analyzing the trypsinization mixture. Comparison of the retention times of these peptides confirmed the identity of each diastereomer, where the D-form was eluted first and found to be the slightly preferred isomer in the conjugation reaction under both conditions (Fig. 2).
Next, we were motivated to investigate if the nearby residues and the location in the same protein could have an influence on the diastereoselectivity of the conjugation step. Therefore, we chose to locate the Dha at position 29 in ubiquitin to prepare Lys29-linked di-ubiquitin. In this case, the conjugation products were separable and the two diastereomeric products, under folding conditions, were obtained in an ∼60:40 ratio. Interestingly, this ratio under denaturing conditions was reversed, highlighting the effect of folding on the stereoselectivity of the conjugation step (Fig. 3).
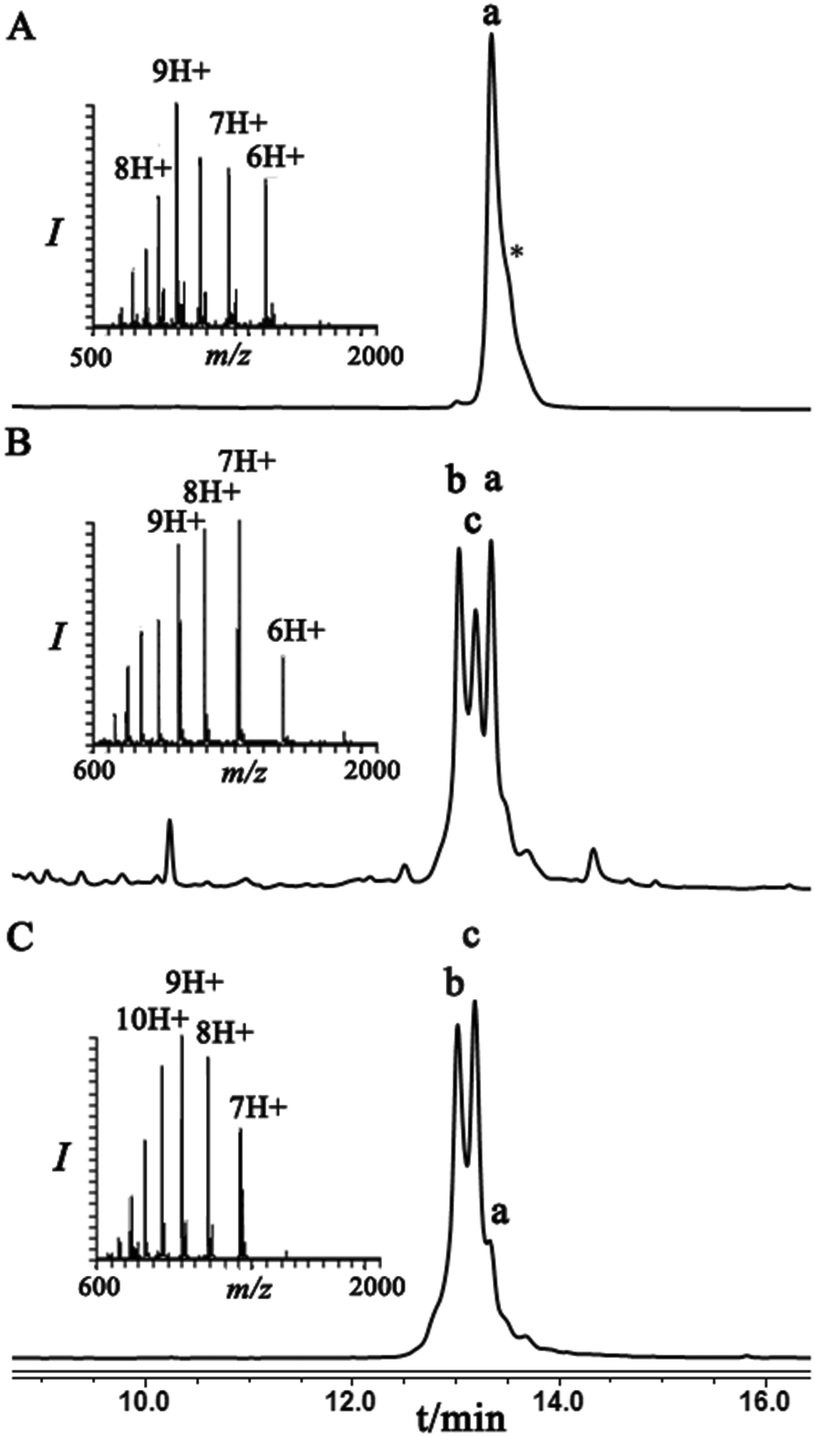 | ||
| Fig. 3 HPLC and mass analysis for (A) Ubiquitin–Dha29, 9 with the observed mass 8485.9 ± 0.6 Da, calcd 8487.8 Da. (B) and (C) Conjugation of hexapeptide–thiol 4 to ubiquitin–Dha29 9 under folding and denaturing conditions, respectively. Peak b and c correspond to the conjugation products with the observed mass 9206.1 ± 0.5 Da, calcd 9206.6. Peak a corresponds to unreacted ubiquitin–Dha29 9. Asterisk corresponds to an unidentified compound. | ||
Similar to the previous case with the ubiquitin–Dha at Lys48, we applied trypsinization on the whole conjugation mixture as well as on each individual peak and for their assignment we compared the retention times of the two peptides having the same mass with their respective synthetic peptides with the L- or D-isopeptide bond. Our analyses indicated that the diastereomer that eluted first is the one that has the L-configuration while the second has the D-configuration of the isopeptide mimic (Fig. S18†).
Finally, we examined the stereoselectivity of conjugation to α-globin–Dha as a different protein system with a single naturally occurring Cys at position 104. Here, also we observed separation of the two diastereomer proteins in which the two products were obtained in a ∼70:30 ratio under denaturing conditions. Notably, the conjugation of hexapeptide–thiol, 4, to folded α-globin–Dha did not proceed at all under different reaction conditions (Fig. 4), apparently due to this cysteine being in a less exposed region. The trypsinization-based analysis as described in the case of ubiquitin was performed and the retention time of each peak was compared with synthetic peptides having the D- and L-form of the isopeptide bond mimic. Importantly, the obtained ratio of the D- and L-peptides (∼70:30), derived from trypsin digestion was very similar to the observed ratio of the conjugation products in the full protein, highlighting the high efficiency of the trypsinization. In this system we found a preference for the product with the D-configuration, which eluted first in the HPLC (Fig. S19†).
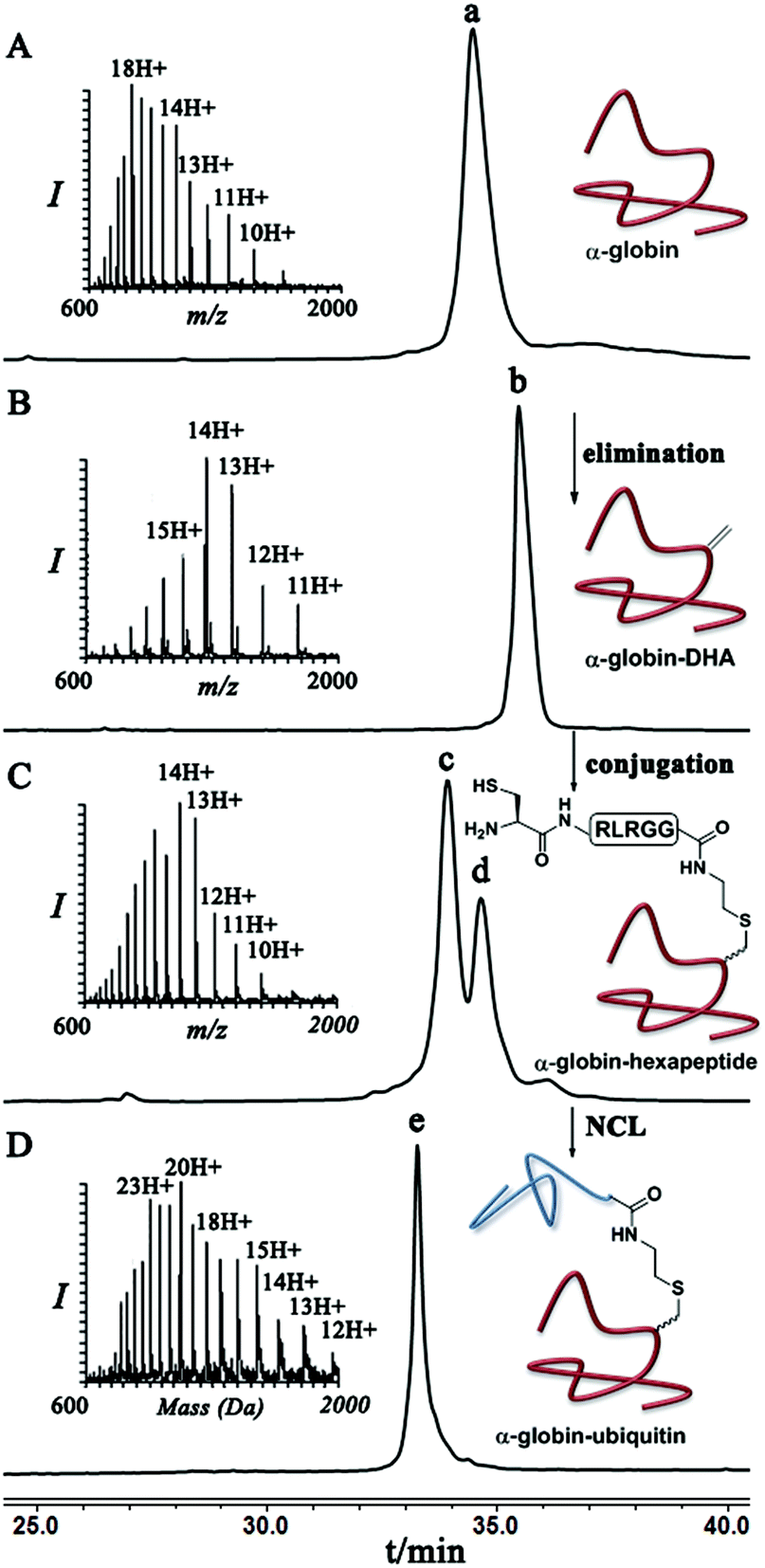 | ||
| Fig. 4 HPLC and mass analysis for (A) pure α-globin with the observed mass 15123.5 ± 1.4 Da, (calcd 15125.3 Da). (B) α-globin–Dha with the observed mass of 15090.5 ± 1.6 Da, (calcd 15093 Da). (C) Conjugation of hexapeptide–thiol to α-globin–Dha under denaturing conditions. Peak c and d correspond to conjugation products with the observed mass 15811.9 ± 2.0 Da and 15812.2 ± 2.1 Da, (calcd 15813.1 Da). (D) Ligation product of hexapeptide conjugated α-globin and ubiquitin (1–70)–thioester with the observed mass of 23686.3 ± 2.2 Da, (calcd 23689.1 Da). | ||
Conclusions
We have developed a new methodology, which can be used for ubiquitination of a variety of protein targets having a single Cys residue. Moreover, this approach could enable the preparation of bioconjugates having the isopeptide bond such as in the case of ubiquitin-like modifiers (e.g. neddylation). In principle, one could also evolve our approach to enable sortase mediated protein ubiquitination. This can be achieved by conjugating Gly–Gly–thiol to a protein–Dha and utilizing the product as a substrate for sortase mediated ligation with ubiquitin(1–75) bearing the sortase recognition sequence.50 In such a case this would lead to two point mutations in the flexible C-terminal region (LPXTG in sortase vs. LRLRG in ubiquitin).For the first time we provided experimental information on the stereoselective 1,4-addition of thiols to Dha in a protein environment. We showed that the location of the Dha within the same protein and in different ones as well as the folding conditions affect the diastereoselectivity of the addition step. One noticeable aspect is the possibility of having the D-configuration as the preferred one in some systems and conditions, which in return may have influence on the interactions with protein partners.30 Our results raise new questions and bring new opportunities in protein chemistry, such as how to improve the stereoselectivity of the conjugation step in the protein context under aqueous conditions. One could examine for example different proton sources to influence the protonation step.48 The current study also provides a platform for future studies with Dha based conjugations where the exact behavior of the different isomers could be characterized. For example, although in our study the USP2 seems to cleave both isomers rapidly one could obtain more detailed information about the enzymatic activity of the obtained conjugates with different DUBs. These and other directions are currently under investigation in our group.
Acknowledgements
We thank the Israel Science Foundation for financial support. A. Brik is a Neubauer Professor and a Taub Fellow-Supported by the Taub Foundations. SMM is thankful to the Schulich foundation for the postdoctoral fellowship.Notes and references
- M. H. Glickman and A. Ciechanover, Physiol. Rev., 2002, 82, 373–428 CrossRef CAS PubMed.
- C. M. Pickart, Annu. Rev. Biochem., 2001, 70, 503–533 CrossRef CAS PubMed.
- D. Komander, M. J. Clague and S. Urbe, Nat. Rev. Mol. Cell Biol., 2009, 10, 550–563 CrossRef CAS PubMed.
- Y. Kravtsova-Ivantsiv, T. Sommer and A. Ciechanover, Angew. Chem., Int. Ed., 2013, 52, 192–198 CrossRef CAS PubMed.
- F. E. Reyes-Turcu and K. D. Wilkinson, Chem. Rev., 2009, 109, 1495–1508 CrossRef CAS PubMed.
- R. Meledin, S. M. Mali and A. Brik, Chem. Rec., 2016, 16, 509–519 CrossRef CAS PubMed.
- G. H. Pham and E. R. Strieter, Curr. Opin. Chem. Biol., 2015, 28, 57–65 CrossRef CAS PubMed.
- L. Spasser and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 6840–6862 CrossRef CAS PubMed.
- C. E. Weller, W. Huang and C. Chatterjee, ChemBioChem, 2014, 15, 1263–1267 CrossRef CAS PubMed.
- T. Abeywardana and M. R. Pratt, ChemBioChem, 2014, 15, 1547–1554 CrossRef CAS PubMed.
- C. E. Weller, M. E. Pilkerton and C. Chatterjee, Biopolymers, 2014, 101, 144–155 CrossRef CAS PubMed.
- M. Haj-Yahya, B. Fauvet, Y. Herman-Bachinsky, M. Hejjaoui, S. N. Bavikar, S. V. Karthikeyan, A. Ciechanover, H. A. Lashuel and A. Brik, Procl. Natl. Acad. Sci. U. S. A., 2013, 110, 17726–17731 CrossRef CAS PubMed.
- K. Yang, P. Gong, P. Gokhale and Z. Zhuang, ACS Chem. Biol., 2014, 9, 1685–1691 CrossRef CAS PubMed.
- N. Haj-Yahya, M. Haj-Yahya, C. A. Castaneda, L. Spasser, H. P. Hemantha, M. Jbara, M. Penner, A. Ciechanover, D. Fushman and A. Brik, Angew. Chem., Int. Ed., 2013, 52, 11149–11153 CrossRef CAS PubMed.
- S. Eger, B. Castrec, U. Hubscher, M. Scheffner, M. Rubini and A. Marx, ChemBioChem, 2011, 12, 2807–2812 CrossRef CAS PubMed.
- S. Sommer, N. D. Weikart, A. Brockmeyer, P. Janning and H. D. Mootz, Angew. Chem., Int. Ed., 2011, 50, 9888–9892 CrossRef CAS PubMed.
- A. Borodovsky, H. Ovaa, N. Kolli, T. Gan-Erdene, K. D. Wilkinson, H. L. Ploegh and B. M. Kessler, Chem. Biol., 2002, 9, 1149–1159 CrossRef CAS PubMed.
- N. Haj-Yahya, H. P. Hemantha, R. Meledin, S. Bondalapati, M. Seenaiah and A. Brik, Org. Lett., 2014, 16, 540–543 CrossRef CAS PubMed.
- A. de Jong, R. Merkx, I. Berlin, B. Rodenko, R. H. Wijdeven, D. El Atmioui, Z. Yalcin, C. N. Robson, J. J. Neefjes and H. Ovaa, ChemBioChem, 2012, 13, 2251–2258 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. Bernardes, Nat. Chem., 2016, 8, 103–113 CAS.
- C. Chatterjee, R. K. McGinty, B. Fierz and T. W. Muir, Nat. Chem. Biol., 2010, 6, 267–269 CrossRef CAS PubMed.
- J. Chen, Y. Ai, J. Wang, L. Haracska and Z. Zhuang, Nat. Chem. Biol., 2010, 6, 270–272 CrossRef CAS PubMed.
- H. P. Hemantha, S. N. Bavikar, Y. Herman-Bachinsky, N. Haj-Yahya, S. Bondalapati, A. Ciechanover and A. Brik, J. Am. Chem. Soc., 2014, 136, 2665–2673 CrossRef CAS PubMed.
- T. Schneider, D. Schneider, D. Rosner, S. Malhotra, F. Mortensen, T. U. Mayer, M. Scheffner and A. Marx, Angew. Chem., Int. Ed., 2014, 53, 12925–12929 CrossRef CAS PubMed.
- K. S. Ajish Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel and A. Brik, Angew. Chem., Int. Ed., 2009, 48, 8090–8094 CrossRef CAS PubMed.
- S. Virdee, P. B. Kapadnis, T. Elliott, K. Lang, J. Madrzak, D. P. Nguyen, L. Riechmann and J. W. Chin, J. Am. Chem. Soc., 2011, 133, 10708–10711 CrossRef CAS PubMed.
- S. Virdee, Y. Ye, D. P. Nguyen, D. Komander and J. W. Chin, Nat. Chem. Biol., 2010, 6, 750–757 CrossRef CAS PubMed.
- C. Castaneda, J. Liu, A. Chaturvedi, U. Nowicka, T. A. Cropp and D. Fushman, J. Am. Chem. Soc., 2011, 133, 17855–17868 CrossRef CAS PubMed.
- J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1835–1839 CrossRef CAS PubMed.
- M. R. Vallee, M. W. Schombs, Z. J. Balaban, J. Colyer and B. G. Davis, Chem. Commun., 2016, 52, 3014–3017 RSC.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernandez-Gonzalez, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- J. Guo, J. Wang, J. S. Lee and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 6399–6401 CrossRef CAS PubMed.
- K. Fukase, M. Kitazawa, A. Sano, K. Shimbo, H. Fujita, S. Horimoto, T. Wakamiya and T. Shiba, Tetrahedron Lett., 1988, 29, 795–798 CrossRef CAS.
- Y. Zhu and W. A. van der Donk, Org. Lett., 2001, 3, 1189–1192 CrossRef CAS PubMed.
- Y. O. You, M. R. Levengood, L. A. Ihnken, A. K. Knowlton and W. A. van der Donk, ACS Chem. Biol., 2009, 4, 379–385 CrossRef CAS PubMed.
- D. H. Strumeyer, W. N. White and D. E. Koshland Jr., Procl. Natl. Acad. Sci. U. S. A., 1963, 50, 931–935 CrossRef CAS.
- H. Weiner, W. N. White, D. G. Hoare and D. E. Koshland Jr., J. Am. Chem. Soc., 1966, 88, 3851–3859 CrossRef CAS PubMed.
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849–6851 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- E. M. Valkevich, R. G. Guenette, N. A. Sanchez, Y. C. Chen, Y. Ge and E. R. Strieter, J. Am. Chem. Soc., 2012, 134, 6916–6919 CrossRef CAS PubMed.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- S. M. Nijman, M. P. Luna-Vargas, A. Velds, T. R. Brummelkamp, A. M. Dirac, T. K. Sixma and R. Bernards, Cell, 2005, 123, 773–786 CrossRef CAS PubMed.
- Z. Harpaz, P. Siman, K. S. Kumar and A. Brik, ChemBioChem, 2010, 11, 1232–1235 CrossRef CAS PubMed.
- Z. Tan, S. Shang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2010, 49, 9500–9503 CrossRef CAS PubMed.
- U. Schmidt and E. Ohler, Angew. Chem., Int. Ed. Engl., 1976, 15, 42 CrossRef CAS PubMed.
- L. Navarre, R. Martinez, J. P. Genet and S. Darses, J. Am. Chem. Soc., 2008, 130, 6159–6169 CrossRef CAS PubMed.
- R. J. Moss, K. J. Wadsworth, C. J. Chapman and C. G. Frost, Chem. Commun., 2004, 1984–1985, 10.1039/b406905f.
- M. W. Popp and H. L. Ploegh, Angew. Chem., Int. Ed., 2011, 50, 5024–5032 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedures, analytical data for synthetic small molecules, peptides, proteins and enzymatic hydrolysis studies. See DOI: 10.1039/c6ob00882h |
| This journal is © The Royal Society of Chemistry 2016 |