
Synthesis and amylin receptor activity of glycomimetics of pramlintide using click chemistry†
Lauren R.
Yule
abc,
Rebekah L.
Bower
a,
Harveen
Kaur
bc,
Renata
Kowalczyk
abc,
Debbie L.
Hay
ac and
Margaret. A.
Brimble
*abc
aThe School of Biological Sciences, University of Auckland, 3A Symonds St, Auckland 1010, New Zealand. E-mail: dl.hay@auckland.ac.nz
bThe School of Chemical Sciences, University of Auckland, 23 Symonds St, Auckland 1010, New Zealand. E-mail: m.brimble@auckland.ac.nz
cMaurice Wilkins Centre for Molecular Biodiscovery, University of Auckland, Private Bag 92019, Auckland 1010, New Zealand
First published on 26th April 2016
Abstract
Pramlintide (Symlin®), a synthetic analogue of the neuroendocrine hormone amylin, is devoid of the tendency to form cytotoxic amyloid fibrils and is currently used in patients with type I and type II diabetes mellitus as an adjunctive therapy with insulin or insulin analogues. As part of an on-going search for a pramlintide analogue with improved pharmacokinetic properties, we herein report the synthesis of mono- and di-glycosylated analogues of pramlintide and their activity at the AMY1(a) receptor. Introduction of N-glycosylated amino acids into the pramlintide sequence afforded the native N-linked glycomimetics whilst use of Cu(I)-catalysed azide–alkyne 1,3-dipolar cycloaddition (click) chemistry delivered 1,2,3-triazole linked glycomimetics. AMY1(a) receptor activity was retained by incorporation of single or multiple GlcNAc moieties at positions 21 and 35 of native pramlintide. Importantly, no difference in AMY1(a) activity was observed between native N-linked glycomimetics and 1,2,3-triazole linked glycomimetics demonstrating that the click variants can act as surrogates for the native N-glycosides in a biological setting.
Introduction
Diabetes mellitus (DM) is a disorder characterised by hyperglycemia, as a result of defects in the secretion and/or action of insulin. This leads to disturbances in carbohydrate, fat and protein metabolism.1 In 2010, it was estimated that the worldwide prevalence of DM was 2.8%, and this has been projected to increase to 7.7% by the year 2030.2 Current DM therapies address high blood glucose levels by subcutaneous injections of either biosynthetic human insulin, insulin analogues,3 glucagon-like peptide-1 (GLP-1) receptor agonists4 and amylin receptor agonists. Amylin is a 37-amino acid neuroendocrine hormone involved in glucose homeostasis. Like insulin, it is secreted from pancreatic β-cells following an increase in blood glucose.5 Consequently, type I and late stage type II DM patients are deficient in amylin due to the destruction of β-cells.6 Functions of amylin include the suppression of glucagon release, reduction in food intake and gastric emptying, all of which result in long term reductions in body weight.7 As a result, amylin offers significant potential for the treatment of both diabetes and obesity. Recognition of the role of amylin led to the discovery of pramlintide 1, an amylin mimetic, which differs from amylin in that amino acids 25, 28 and 29 were replaced with prolines.8 This replacement strategy overcame the problematic physiochemical property of forming cytotoxic amyloid fibrils, which renders human amylin unsuitable as a drug (Fig. 1).9 | ||
| Fig. 1 Primary sequence of human amylin and amylin analogue pramlintide 1. | ||
Pramlintide 1 lowers post-prandial glucose levels in both type I and type II DM. For type I DM, it is used as an adjunctive therapy along with mealtime insulin alone. In late stage type II DM, it is used as an adjunctive treatment to mealtime insulin in patients who have failed to obtain glucose control despite optimal insulin therapy.10 Amylin and therefore pramlintide 1, demonstrate the ability to regulate nutrient intake by inducing satiety. Pramlintide 1 has been tested in several clinical trials and consistently reduces bodyweight.11 However, despite its numerous beneficial pharmacodynamic properties, problematic pharmacokinetic properties of the peptide still exist. For instance, pramlintide 1 has a circulatory half-life of only 48 minutes, and is cleared from the body within two to three hours after intravenous input is discontinued.10,12 Moreover, insulin and pramlintide 1 possess differences in both the isoelectric point and pH,13 thus co-formulation with insulin is currently not possible.10,12
The use of peptide hormones as therapeutic treatments is limited by their short half-lives and rapid proteolysis. Chemical techniques including the incorporation of unnatural amino acids such as β-amino acids,14 mutation of the enzymatic cleavage site15 and the covalent attachment of fatty acids (lipidation), sugars (glycosylation), or polyethylene glycol units (PEGylation)16,17 can be used to develop peptide hormone analogues with enhanced activity, half-life or physiochemical properties. This has led to the development of several peptide hormone analogues used to treat type II DM, such as Semaglutide,18 a GLP-1 receptor agonist with two amino acid substitutions (Aib-8, Arg-34) and a lipidated Lys-26, with increased resistance to proteolysis.19 A number of these procedures have been previously investigated on pramlintide 1, including lipidation using fatty acids incorporated via lysine or cysteine residue side chains,20 and glycosylation.21,22 Glycosylation is a technique that has been used successfully to improve the proteolytic stability of peptide hormones related to diabetes, including GLP-1,23 insulin24 and exendin-4.25 Our group have investigated the effects of the incorporation of three carbohydrates GlcNAc, Man3(GlcNAc)2 and (NeuAcGalGlcNAcMan)2Man(GlcNAc)2 at each of the six asparagine residues (Asn-3, Asn-14, Asn-21, Asn-22, Asn-31 and Asn-35) of pramlintide 1via N-glycosylation and subsequent enzymatic glycosylation.21 Nevertheless, the synthesis of the glycosylated pramlintides required the tedious preparation of the Fmoc-Asn(GlcNAc(OAc)3)-OH building block,26 maximally protected for 9-fluorenylmethoxycarbonyl solid phase peptide synthesis (Fmoc-SPPS). An alternative method to install the GlcNAc moiety without the need for extensive synthesis of glycosylated amino acid building blocks and using the unprotected sugar under aqueous conditions is desirable. Moreover, it is advantageous to develop a method for simultaneous introduction of several GlcNAc moieties. With the aim of preparing a pramlintide analogue with improved pharmacokinetic properties, taking into account the desire to use an improved method to introduce the GlcNAc moiety, we herein describe the synthesis and receptor activity of pramlintide analogues 2–13 (Fig. 2). Our earlier work focused on pramlintide analogues containing a single sugar substitution, using native N-glycosylation, of which pramlintide analogues with native sugars incorporated at positions 21 and 35 were found to exhibit the best activity.21 Thus, as a logical extension to our previously reported structure–activity relationship study,21 in the work described herein, single or multiple GlcNAc moieties were selectively incorporated at positions 21 and/or 35 of pramlintide 1 to further improve the solubility and pharmacokinetics of the native peptide. This was carried out using a combination of N-glycosylation to deliver a native N-GlcNAc linkage and/or the Cu(I)-catalysed azide–alkyne 1,3-dipolar cycloaddition (click chemistry)27,28 to deliver a non-native 1,2,3-triazole glycosidic linkage, providing a library of analogues 2–13 of pramlintide 1 (Fig. 2). The click reaction offers an efficient method to effect the glycosylation of peptides and proteins, in which a non-native 1,2,3-triazole glycosidic bond is formed following peptide assembly.29 Peptides incorporating the commercially available propargylglycine moiety (Fmoc-L-Pra) are readily assembled using Fmoc-SPPS, thus providing a handle with which glycosylazides react with selectively. Click chemistry therefore enables control over the structure and the site of glycosylation,30 and enables facile introduction of unprotected glycans into the peptide sequence under aqueous conditions.29,31 Moreover, the 1,2,3-triazole glycosidic linkage is likely to be resistant to enzymatic or digestive hydrolysis,7 further increasing the stability of the pramlintide analogues.
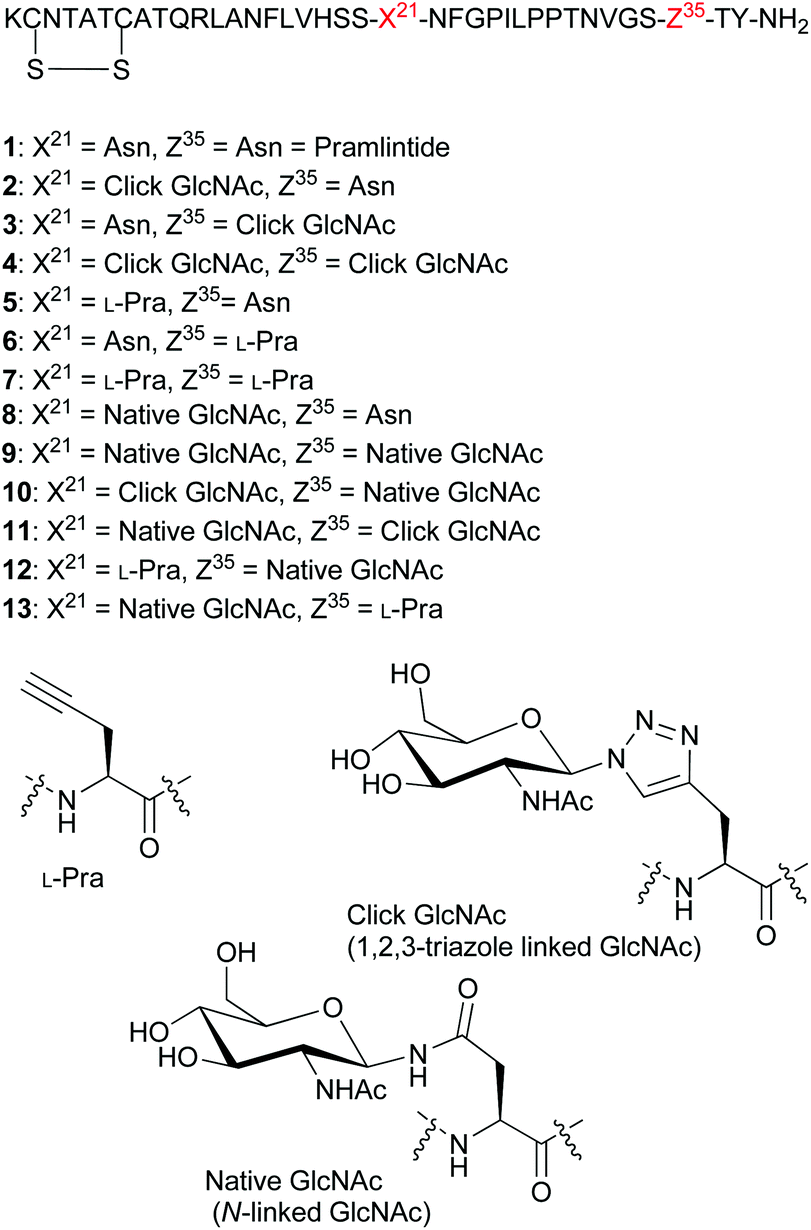 | ||
| Fig. 2 Sequences for pramlintide 1 and pramlintide analogues 2–13, and structures of L-Pra, click 1,2,3-triazole linked GlcNAc and the native N-linked GlcNAc. | ||
Our present study focused on the synthesis of click glycomimetics 2, 3 and 4 bearing a 1,2,3-triazole linked GlcNAc moiety at residues 21 or 35 and at both 21 and 35, respectively (Fig. 2). We also prepared pramlintide analogues 5, 6, and 7 containing an L-Pra moiety at residues 21, 35 and both 21 and 35, respectively. Glycopramlintide analogues 8 (used in previous studies)21 and 9 were prepared bearing a native N-linked GlcNAc at residue 21, and at both residues 21 and 35, respectively. In addition, glycopramlintide analogue 10, containing both a 1,2,3-triazole linked GlcNAc at residue 21 and a native N-linked GlcNAc at residue 35 was prepared together with glycopramlintide analogue 11, containing a 1,2,3-triazole linked GlcNAc at residue 35 and a native N-linked GlcNAc at residue 21. Lastly, we prepared analogues 12 and 13 containing both an L-Pra moiety and a native N-linked GlcNAc at residues 21 and 35 (analogue 12) and residues 35 and 21, respectively (analogue 13).
Results and discussion
Synthesis of native pramlintide 1 and pramlintide analogues 2–7 containing an L-Pra moiety or a 1,2,3-triazole linked GlcNAc
Based on our previous work,21,22 Fmoc-SPPS was chosen for the preparation of native pramlintide 1, and pramlintide analogues 2–7 (Scheme 1), which incorporated an L-Pra or the 1,2,3-triazole linked GlcNAc at either or both positions 21 and 35 of pramlintide.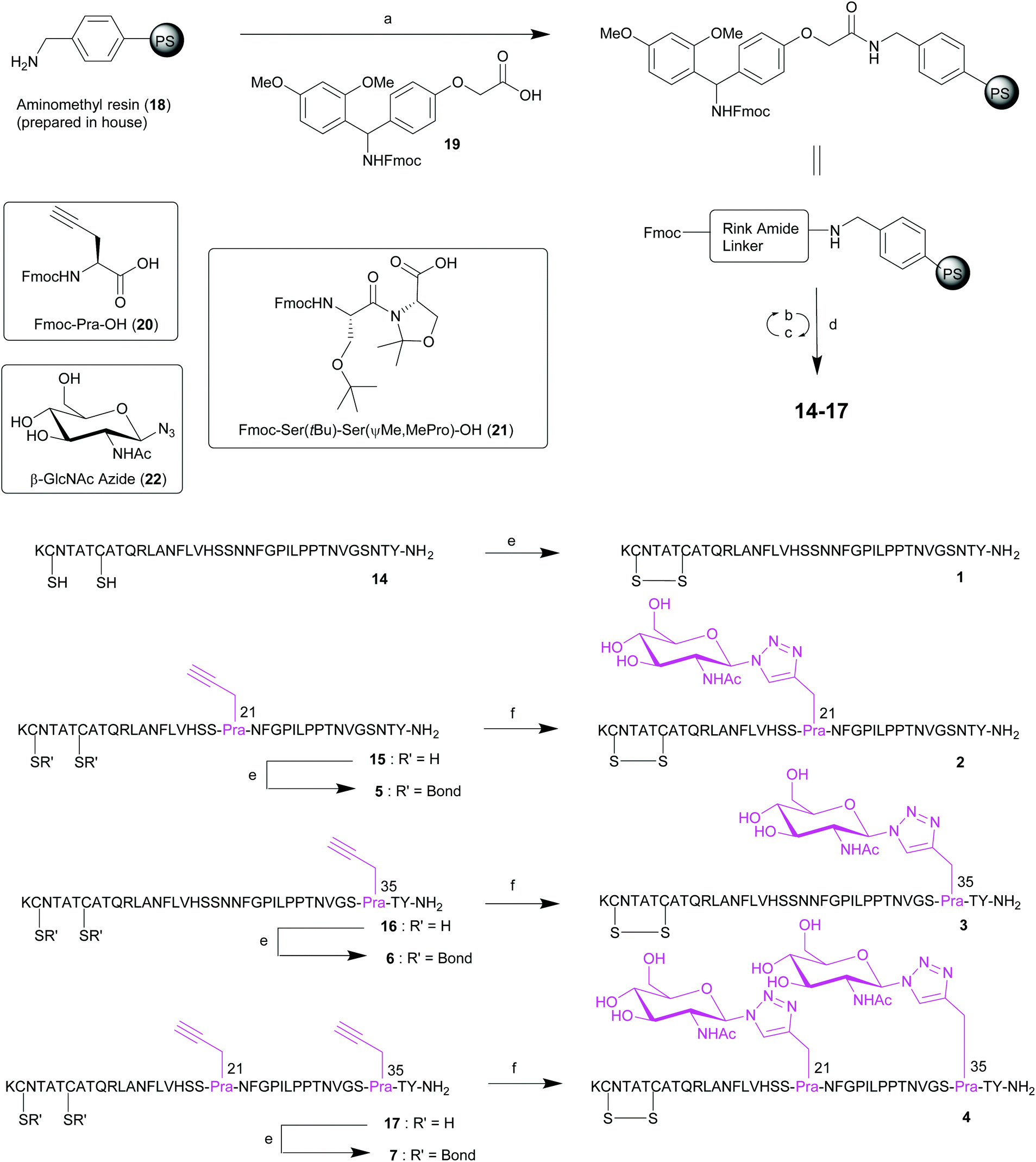 | ||
| Scheme 1 Synthesis of pramlintide 1 and pramlintide analogues 2–7 containing an L-Pra or click GlcNAc moiety at either position 21, 35 or both. Reagents and conditions: (a) 19, DIC, 6-Cl HOBt, DMF, rt, 2 h; (b) 20% piperidine in DMF, rt, 2 × 5 min; (c) Fmoc-AA-OH, HCTU, iPr2NEt, DMF, rt, 45 min; or for 20 or 21: HATU, collidine, DMF, rt, 90 min; (d) TFA/iPr3SiH/DODT/H2O (94/1/2.5/2.5), 2.5 h, rt; (e) Na2HPO4, Gu·HCl, rt, 2 h; (f) β-GlcNAcN322, Gu·HCl, Na2HPO4, TCEP·HCl, CuSO4·5 H2O, 60 °C, 20 W, 2 h. | ||
A disulfide bond (Cys-2/Cys-7) and a C-terminal amide are required for receptor affinity of pramlintide 1, and this was synthetically accommodated for. The corresponding linear precursors 14–17 to peptides 1–7 were prepared by initial functionalization of aminomethyl polystyrene resin 1832 with a 4-[(R,S)-α-[1-(9H-fluoren-9-yl)]-methoxycarbonylamino]-2,4-dimethoxyphenoxyacetic acid (Rink amide linker) 19 (Scheme 1). Nα-Fmoc-deprotection, using 20% piperidine in DMF, and subsequent peptide elongation was performed using an automated Tribute™ peptide synthesiser at room temperature (rt). Serine- and threonine-derived oxazolidines (pseudoprolines, ΨPro) serve as reversible protecting groups for serine or threonine during SPPS.33,34 The use of these building blocks results in the introduction of a “kink” in the backbone of the peptide, as well as removing hydrogen bond donors, which helps to prevent peptide aggregation by disrupting secondary structures such as β-sheets during chain assembly. Amylin, and a number of other long or hydrophobic peptides such as exenatide,35,36 have been successfully synthesised using a pseudoproline dipeptide.34,37–39 As a result, the Fmoc-Ser(tBu)-Ser(ΨMe,MePro)-OH 21 building block was incorporated into the synthesis of the crude pramlintide analogues 15–17 (containing a hydrophobic L-Pra) in place of the two native serine residues at positions 19 and 20 of the pramlintide sequence. For standard amino acid couplings, a mixture of O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU) and N,N-diisopropylethylamine (iPr2NEt) were employed. Alternatively, for the coupling of Fmoc-L-Pra-OH 20 and Fmoc-Ser(tBu)-Ser(ΨMe,MePro)-OH 21, O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) and 2,4,6-trimethylpyridine (collidine) were used for 90 min at rt. The linear precursors were then released from the resin with simultaneous side-chain deprotection, using a mixture of trifluoroacetic acid/triisopropylsilane/water/3,6-dioxa-1,8-octanedithiol (TFA/TIS/H2O/DODT) (v/v/v/v; 94/1/2.5/2.5), affording the corresponding crude peptides 14–17 (Scheme 1).
Subsequent disulfide bond formation (Cys-2/Cys-7) for analogues 1, 5, 6 and 7 was performed directly on crude peptides 14–17 using 6 M guanidinium hydrochloride (Gu·HCl) and disodium hydrogen phosphate (Na2HPO4) as buffer. After 2 h, the desired disulfide-containing pramlintide 1 (from 14) and disulfide-containing pramlintide analogues 5 (from 15), 6 (from 16) and 7 (from 17) were formed quantitatively as evidenced by liquid chromatography-mass spectrometry (LCMS) (see ESI†). Alternatively, crude peptides 14–17 underwent one-pot click chemistry and simultaneous disulfide bond formation (Cys-2/Cys-7) in solution using β-GlcNAcN322 (synthesised in one step from unprotected GlcNAc40) using previously optimised conditions31 to afford the 1,2,3-triazole linked GlcNAc at position 21 (analogue 2), position 35 (analogue 3) and at both positions 21 and 35 (analogue 4). The reaction was quantitative, and purification using reverse phase high-performance liquid chromatography (RP-HPLC) enabled the isolation of pure samples of the desired native pramlintide 1, 1,2,3-triazole linked GlcNAc pramlintide analogues 2, 3 and 4 and L-Pra pramlintide analogues 5, 6 and 7 in excellent purities ranging from 94–99% (see ESI† for details).
Synthesis of pramlintide analogues 8–13 containing a native N-GlcNAc, L-Pra and/or 1,2,3-triazole linked GlcNAc
With an established procedure for the synthesis of native pramlintide 1 and pramlintide analogues 2–7 in hand, attention focused on the preparation of analogues 8–13 that contained a GlcNAc moiety linked via a native N-bond by incorporation of Fmoc-Asn(GlcNAc(OAc)3)-OH 23.26 This included analogues containing a single native N-GlcNAc at position 21 (analogue 8), or at both positions 21 and 35 (analogue 9) as well as analogues containing both a native N-GlcNAc and a 1,2,3-triazole linked GlcNAc (analogues 10 and 11), along with their acetylene-containing precursor peptides 12 and 13, respectively (Fig. 2). Linear pramlintide analogues 24 and 26 containing a native N-linked GlcNAc were synthesised via Fmoc-SPPS conducted at rt using analogous conditions to the synthesis of analogues 14–17. In addition, for the synthesis of linear pramlintide analogues 25 and 27, microwave (mw) irradiation was also employed (Scheme 2). Fmoc-Asn(GlcNAc(OAc)3)-OH 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 was coupled using HATU and collidine activation for either 90 min (24 and 26) or 15 min (25 and 27) sequence (Scheme 2). LCMS analysis of a sample of crude pramlintide analogues 25 and 27 (synthesised under mw conditions) revealed partial deprotection of the sugar hydroxyls had occurred; however, this was not a concern as the next synthetic step was the removal of the hydroxyl protecting groups to obtain pramlintide analogues 9, 11 and 13. Deprotection of the acetate protecting groups on the sugar hydroxyls with simultaneous disulfide bond formation (Cys-2/Cys-7) was carried out on crude linear peptides 24–27 using 5% hydrazine hydrate (NH2NH2·5 H2O) in DMSO for 3 h at rt to afford, with full conversion, the desired crude mono- and di-glycosylated analogues 8 (from 24), 9 (from 25), 12 (from 26), 13 (from 27). Subsequently, the crude monoglycopeptides 12 and 13, containing one native N-GlcNAc and an L-Pra moiety, were subjected to the click chemistry conditions used for the preparation of click glycopeptides 2, 3 and 4. This resulted in the attachment of a second GlcNAc moiety to the L-Pra residue of 12 and 13via a non-native 1,2,3-triazole linkage to afford crude doubly glycosylated pramlintide analogues 10 and 11, containing both a native N-GlcNAc and a non-native 1,2,3-triazole glycosidic linkage. Again, RP-HPLC purification enabled the isolation of pure samples of the desired glycopeptides 8–13 in excellent purities, ranging from 96% to 99% (see ESI† for details).
26 was coupled using HATU and collidine activation for either 90 min (24 and 26) or 15 min (25 and 27) sequence (Scheme 2). LCMS analysis of a sample of crude pramlintide analogues 25 and 27 (synthesised under mw conditions) revealed partial deprotection of the sugar hydroxyls had occurred; however, this was not a concern as the next synthetic step was the removal of the hydroxyl protecting groups to obtain pramlintide analogues 9, 11 and 13. Deprotection of the acetate protecting groups on the sugar hydroxyls with simultaneous disulfide bond formation (Cys-2/Cys-7) was carried out on crude linear peptides 24–27 using 5% hydrazine hydrate (NH2NH2·5 H2O) in DMSO for 3 h at rt to afford, with full conversion, the desired crude mono- and di-glycosylated analogues 8 (from 24), 9 (from 25), 12 (from 26), 13 (from 27). Subsequently, the crude monoglycopeptides 12 and 13, containing one native N-GlcNAc and an L-Pra moiety, were subjected to the click chemistry conditions used for the preparation of click glycopeptides 2, 3 and 4. This resulted in the attachment of a second GlcNAc moiety to the L-Pra residue of 12 and 13via a non-native 1,2,3-triazole linkage to afford crude doubly glycosylated pramlintide analogues 10 and 11, containing both a native N-GlcNAc and a non-native 1,2,3-triazole glycosidic linkage. Again, RP-HPLC purification enabled the isolation of pure samples of the desired glycopeptides 8–13 in excellent purities, ranging from 96% to 99% (see ESI† for details).
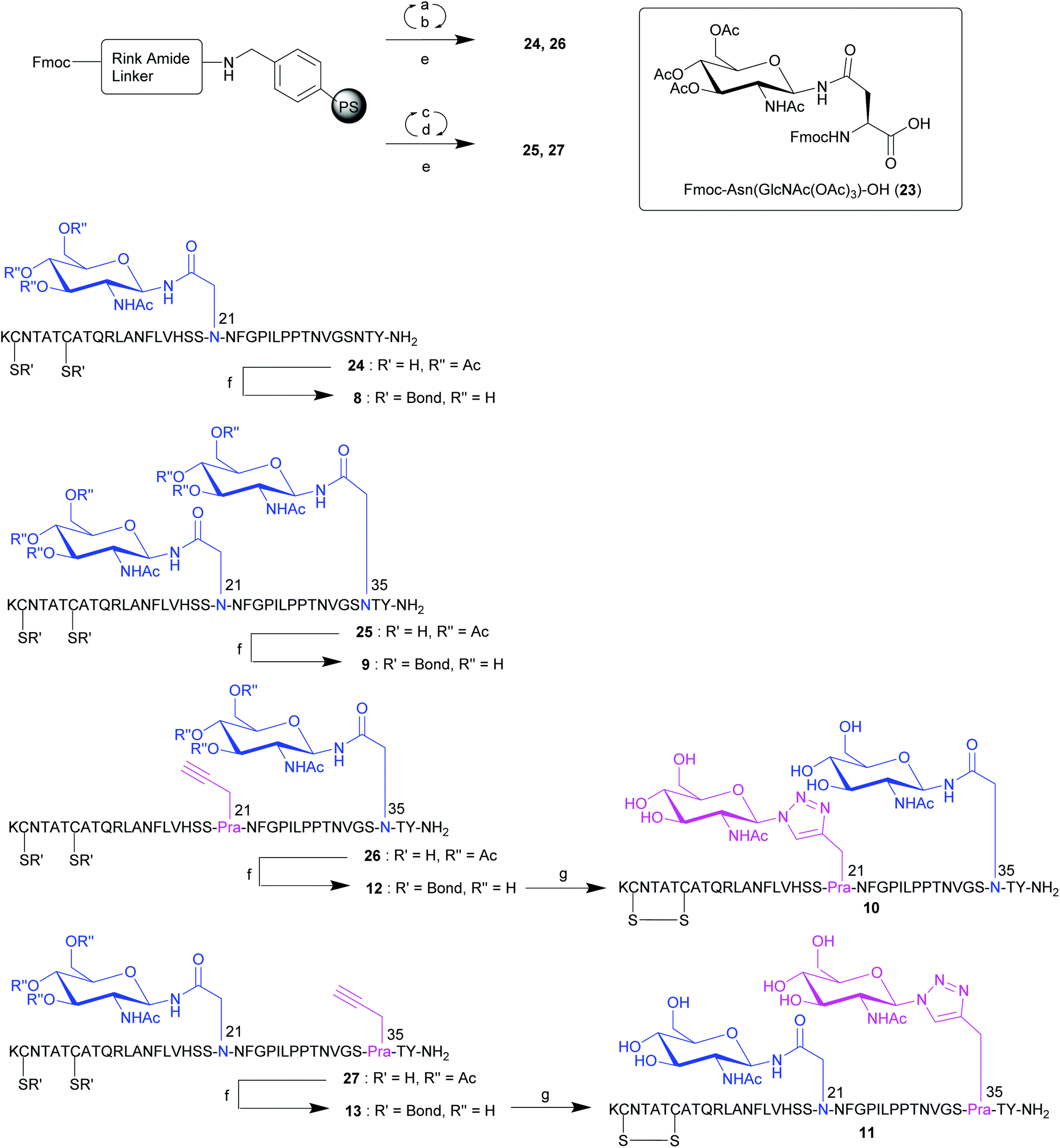 | ||
| Scheme 2 Synthesis of pramlintide analogues 8–13 containing a native N-GlcNAc, L-Pra and/or click GlcNAc moiety at positions 21 or 35 of pramlintide. Reagents and conditions: (a) 20% piperidine in DMF, rt, 2 × 5 min; (b) Fmoc-AA-OH, HCTU, iPr2NEt, DMF, rt, 45 min; or for 20, 21 or 23 HATU, collidine, DMF, rt, 90 min; (c) 20% piperidine in DMF, 62 W, 70 °C, 2 × 3 min; (d) Fmoc-AA-OH, HCTU, iPr2NEt, DMF, 25 W, 75 °C, 5 min; 25 min at rt then 5 min at 25 W, 72 °C, (Fmoc-Arg(Pbf)-OH); or 10 min at rt then 5 min at 25 W, 47 °C (Fmoc-His(Trt)-OH and Fmoc-Cys(Trt)-OH); or for 20, 21 or 23: HATU, collidine, DMF, 25 W, 70 °C, 15 min; (e) TFA/iPr3SiH/DODT/H2O (94/1/2.5/2.5), 2.5 h, rt; (f) 5% N2H4·1.5 H2O in DMSO, rt, 3 h; (g) β-GlcNAcN322, Gu·HCl, Na2HPO4, TCEP·HCl, CuSO4·5H2O, 60 °C, 20 W, 2 h. | ||
Amylin receptor testing of native pramlintide 1 (control) and pramlintide analogues 2–13
Native pramlintide 1 and pramlintide analogues 2–13 were then tested at an amylin and pramlintide receptor (the AMY1(a) receptor),5,7 and were directly compared to native pramlintide 1 (control) in each experiment (Table 1, Fig. 3). Biological activity was tested in accordance with our previous studies on pramlintide 1.21 Briefly, Cos 7 cells were transfected with the necessary receptor DNA components, and cAMP production was measured according to our published methods (see ESI† for further details).41,42 This revealed that glycosylated pramlintide analogues 2–13 exhibited only a mild loss of activity when compared to native pramlintide 1 at AMY1(a), and these results were consistent with activities observed by Kowalczyk et al.,21 for the same residues of the pramlintide sequence (Table 1). Although substitution of the native Asn residue at either position 21 or 35 with an L-Pra residue (analogues 5, 6, 7, 12 and 13) resulted in a mild reduction of AMY1(a) activity, when the GlcNAc moiety was then introduced to the peptide using click chemistry (analogues 2, 3, 4, 10 and 11), no significant further loss of activity was observed. The change in the backbone structure of pramlintide to incorporate the L-Pra in place of the Asn residue may have altered the conformation of pramlintide 1, leading to the slight decrease in activity. More importantly, no greater loss of AMY1(a) activity was observed with the incorporation of the GlcNAc using click chemistry when compared to the incorporation of a native N-linked GlcNAc residue (i.e. similar activities for analogue 4, containing two non-native 1,2,3-triazole GlcNAc linkages, and analogue 9, containing two native N-GlcNAc linkages). Analogue 2, containing a single non-native glycosidic linkage at position 21, performed slightly better at AMY1(a) than analogue 8, containing a single native N-GlcNAc at position 21, demonstrating the power of click chemistry for the easy sugar attachment of non-protected glycans in aqueous conditions while retaining bioactivity. Moreover, “one-pot” click and disulfide bond formation conveniently afforded bio-active pramlintide analogues, and therefore can be used for future modifications using more complex unprotected glycans to further improve the solubility and stability of pramlintide peptidomimetics. Finally, AMY1(a) activity was maintained or only slightly reduced when two GlcNAc moieties were introduced to the pramlintide sequence at positions 21 and 35 using N-glycosylation (analogue 9), click chemistry (analogue 4) or a combination of both techniques (analogues 10 and 11), further emphasising the ability of click chemistry to effect the facile introduction of multiple GlcNAc or other more complex sugar moieties into the pramlintide 1 sequence.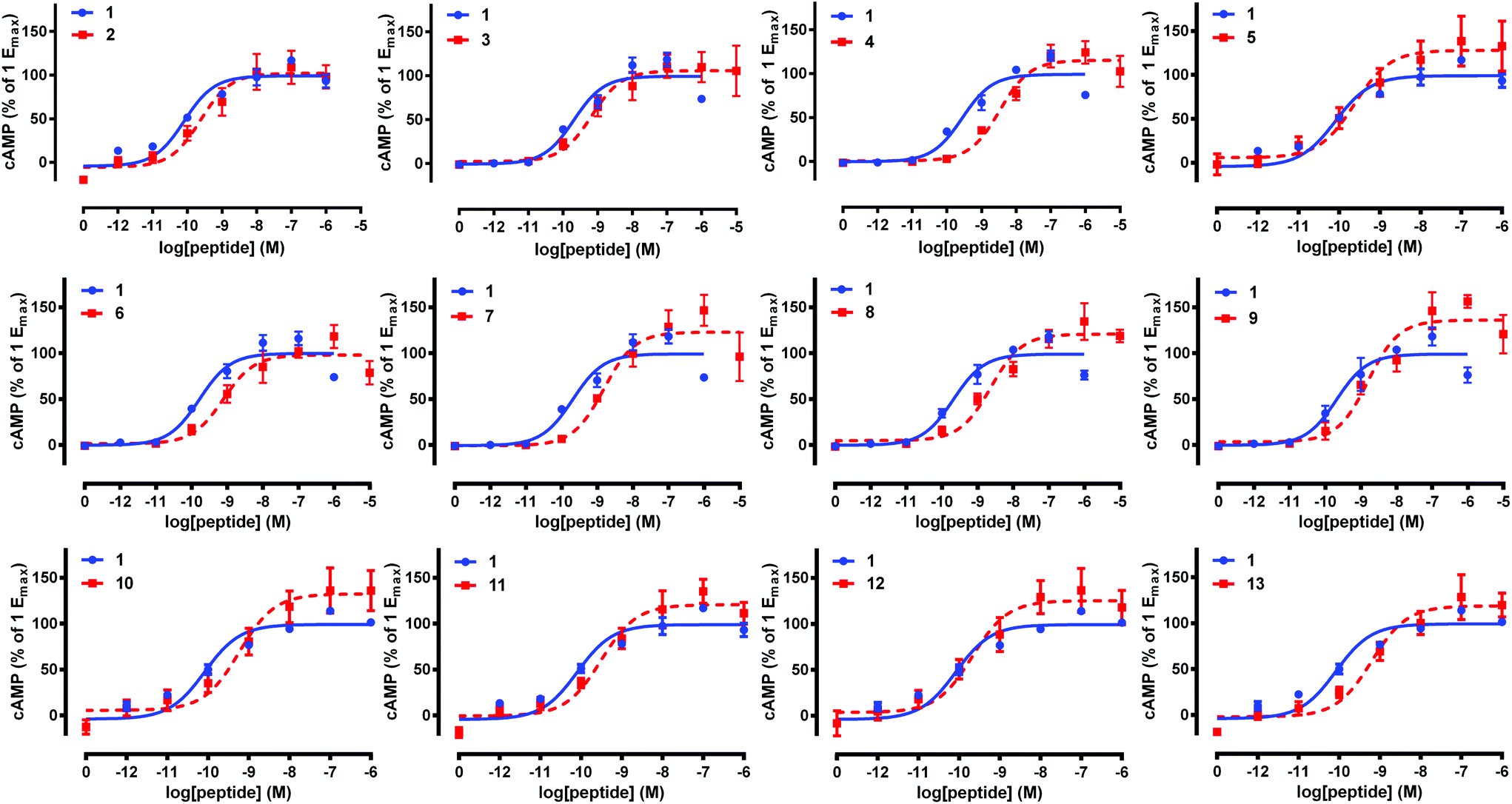 | ||
| Fig. 3 Activity of pramlintide analogues 2–13 at the AMY1(a) receptor. Data are combined from at least three independent experiments and show the mean ± S.E.M. | ||
| Analogue | X21 |
Z35 |
hAMY1(a) | |||
|---|---|---|---|---|---|---|
| Analogue pEC50 ([nM]) | Control (1) pEC50, ([nM]) | Fold-change | E max | |||
| Values are mean ± SEM. Numbers in parentheses represent the number of independent experiments analysed. Unpaired t-tests were performed to compare 1 (control), *P < 0.05; one-way ANOVA followed by Tukey's multiple comparisons test was used to compare analogues 2–13 with one-another, aP < 0.05 vs.2, bP < 0.05 vs.4, cP < 0.05 vs.5, dP < 0.05 vs.7, eP < 0.05 vs.8, fP < 0.05 vs.9; other analogues were not significantly different from one another. | ||||||
| 2 | Click GlcNAc | Asn | 9.56 ± 0.09 (4)* (0.28) | 10.0 ± 0.11 (4) (0.10) | −2.9 | 102.5 ± 15.8 |
| 3 | Asn | Click GlcNAc | 9.17 ± 0.02 (3)* (0.68) | 9.64 ± 0.14 (3) (0.23) | −2.9 | 105.7 ± 18.7 |
| 4 | Click GlcNAc | Click GlcNAc | 8.51 ± 0.11 (3)*,a (3.09) | 9.50 ± 0.15 (3) (0.32) | −9.7 | 115.1 ± 14.6 |
| 5 | L-Pra | Asn | 9.59 ± 0.22 (4)b (0.26) | 10.0 ± 0.11 (4) (0.10) | −2.7 | 129.8 ± 26.1 |
| 6 | Asn | L-Pra | 9.08 ± 0.10 (3)* (0.83) | 9.77 ± 0.09 (3) (0.17) | −4.8 | 98.3 ± 10.6 |
| 7 | L-Pra | L-Pra | 8.84 ± 0.11 (3)*,a,c (1.45) | 9.64 ± 0.14 (3) (0.23) | −6.3 | 123.2 ± 20.1 |
| 8 | Native GlcNAc | Asn | 8.58 ± 0.15 (3)*,a,c (2.63) | 9.63 ± 0.17 (3) (0.23) | −11.2 | 122.2 ± 7.57 |
| 9 | Native GlcNAc | Native GlcNAc | 8.71 ± 0.21 (3)*,a,c (1.95) | 9.63 ± 0.17 (3) (0.23) | −8.3 | 137.8 ± 1.13 |
| 10 | Click GlcNAc | Native GlcNAc | 9.25 ± 0.08 (4)*,b (0.56) | 9.98 ± 0.16 (4) (0.10) | −5.4 | 132.6 ± 21.7 |
| 11 | Native GlcNAc | Click GlcNAc | 9.45 ± 0.12 (4)*,b,e,f (0.35) | 10.0 ± 0.11 (4) (0.10) | −3.7 | 121.5 ± 13.6 |
| 12 | L-Pra | Native GlcNAc | 9.64 ± 0.14 (4)b,d,e,f (0.23) | 9.98 ± 0.16 (4) (0.10) | −2.2 | 126.0 ± 19.4 |
| 13 | Native GlcNAc | L-Pra | 9.23 ± 0.10 (4)*,b (0.59) | 9.98 ± 0.16 (3) (0.10) | −5.6 | 118.9 ± 16.6 |
Conclusions
In conclusion, we have prepared pramlintide analogues 2–13 using glycosylation to improve the physicochemical and pharmacokinetic properties of pramlintide 1. The GlcNAc moiety was selectively incorporated at positions 21 and 35 of the pramlintide sequence, using an N-glycosylated building block to deliver a native N-linkage and click chemistry to afford a non-native 1,2,3-triazole linkage, or both. We have demonstrated that receptor activity at AMY1(a) is largely maintained with the incorporation of GlcNAc at positions 21 and 35 using a non-native 1,2,3-triazole linkage. In addition, AMY1(a) activity is also maintained when two GlcNAc moieties were introduced to the pramlintide sequence at positions 21 and 35, using either N-glycosylation, click chemistry, or a combination of both. Click chemistry thus provides an effective tool for the glycosylation of pramlintide 1 while retaining receptor bioactivity. Click chemistry delivers additional synthetic advantages over native N-glycosylation, including facile incorporation of an unprotected glycan into the peptide sequence under aqueous conditions whilst eliminating the requirement for total synthesis of a glycosylated amino acid building block. Moreover, glycosylation using click chemistry may provide additional improvements to the physicochemical and pharmacokinetic properties of pramlintide 1. To the best of our knowledge, we believe this work demonstrates the first application of click chemistry to prepare glycomimetics of peptide hormones as a strategy to improve the physicochemical and pharmacokinetic properties of the native peptide sequence.Acknowledgements
We thank the Maurice Wilkins Centre for Molecular Biodiscovery, (PhD Scholarship, LY) and the Royal Society of New Zealand Rutherford Foundation (Postdoctoral Fellowship, RK) for financial support of this work.References
- K. G. M. M. Alberti and P. Z. Zimmet, Diabetic Med., 1998, 15, 539–553 CrossRef CAS PubMed.
- J. E. Shaw, R. A. Sicree and P. Z. Zimmet, Diabetes Res. Clin. Pract., 2010, 87, 4–14 CrossRef CAS PubMed.
- G. Grunberger, J. Diabetes, 2013, 5, 110–117 CrossRef CAS PubMed.
- A. J. Garber, Diabetes Care, 2011, 34, S279–S284 CrossRef CAS PubMed.
- J. J. Gingell, E. R. Burns and D. L. Hay, Endocrinology, 2014, 155, 21–26 CrossRef PubMed.
- S. Mäkimattila, M. S. Fineman and H. Yki-Järvinen, J. Clin. Endocrinol. Metab., 2000, 85, 2822–2827 Search PubMed.
- D. L. Hay, S. Chen, T. A. Lutz, D. G. Parkes and J. D. Roth, Pharmacol. Rev., 2015, 67, 564–600 CrossRef CAS PubMed.
- A. A. Young, W. Vine, B. R. Gedulin, R. Pittner, S. Janes, L. S. L. Gaeta, A. Percy, C. X. Moore, J. E. Koda, T. J. Rink and K. Beaumont, Drug Dev. Res., 1996, 37, 231–248 CrossRef CAS.
- P. Westermark, A. Andersson and G. T. Westermark, Physiol. Rev., 2011, 91, 795–826 CrossRef CAS PubMed.
- D. Singh-Franco, G. Robles and D. Gazze, Clin. Ther., 2007, 29, 535–562 CrossRef CAS PubMed.
- K. C. Dunican, N. M. Adams and A. R. Desilets, Ann. Pharmacother., 2010, 44, 538–545 CrossRef CAS PubMed.
- O. Schmitz, B. Brock and J. Rungby, Diabetes, 2004, 53, S233–S238 CrossRef CAS PubMed.
- C. Weyer, M. S. Fineman, S. Strobel, L. Shen, J. Data, O. G. Kolterman and M. F. Sylvestri, Am. J. Health-Syst. Pharm., 2005, 62, 816–822 CAS.
- C. Cabrele, T. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed.
- M. Prothiwa, I. Syed, M. O. Huising, T. van der Meulen, C. J. Donaldson, S. A. Trauger, B. B. Kahn and A. Saghatelian, J. Am. Chem. Soc., 2014, 136, 17710–17713 CrossRef CAS PubMed.
- V. Mäde, S. Els-Heindl and A. G. Beck-Sickinger, Beilstein J. Org. Chem., 2014, 10, 1197–1212 CrossRef PubMed.
- A. Trabocchi and A. Guarna, Peptidomimetics in Organic and Medicinal Chemistry: The Art of Transforming Peptides in Drugs, John Wiley & Sons, Ltd., Chichester, UK, 2014 Search PubMed.
- J. Lau, P. Bloch, L. Schäffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire, D. B. Steensgaard, H. M. Strauss, D. X. Gram, S. M. Knudsen, F. S. Nielsen, P. Thygesen, S. Reedtz-Runge and T. Kruse, J. Med. Chem., 2015, 58, 7370–7380 CrossRef CAS PubMed.
- B. Ahrén and O. Schmitz, Horm. Metab. Res., 2003, 36, 867–876 CrossRef PubMed.
- T. K. Hansen, J. Lau and L. Schaeffer, Amylin derivatives, WO Pat, 2007104789A2, 2008 Search PubMed.
- R. Kowalczyk, M. A. Brimble, Y. Tomabechi, A. J. Fairbanks, M. Fletcher and D. L. Hay, Org. Biomol. Chem., 2014, 12, 8142–8151 CAS.
- Y. Tomabechi, G. Krippner, P. M. Rendle, M. A. Squire and A. J. Fairbanks, Chem. – Eur. J., 2013, 19, 15084–15088 CrossRef CAS PubMed.
- T. Ueda, K. Tomita, Y. Notsu, T. Ito, M. Fumoto, T. Takakura, H. Nagatome, A. Takimoto, S.-I. Mihara and H. Togame, J. Am. Chem. Soc., 2009, 131, 6237–6245 CrossRef CAS PubMed.
- M. Sato, T. Furuike, R. Sadamoto, N. Fujitani, T. Nakahara, K. Niikura, K. Monde, H. Kondo and S.-I. Nishimura, J. Am. Chem. Soc., 2004, 126, 14013–14022 CrossRef CAS PubMed.
- T. Ueda, T. Ito, K. Tomita, H. Togame, M. Fumoto, K. Asakura, T. Oshima, S.-I. Nishimura and K. Hanasaki, Bioorg. Med. Chem. Lett., 2010, 20, 4631–4634 CrossRef CAS PubMed.
- T. Inazu, Synlett, 1993, 869–870 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 114, 2708–2711 CrossRef.
- Z. J. Witczak and R. Bielski, Click chemistry in glycoscience: New developments and strategies, John Wiley & Sons, New Jersey, 2013 Search PubMed.
- L. Pollaro and C. Heinis, Med. Chem. Commun., 2010, 1, 319–324 RSC.
- D. J. Lee, P. W. Harris and M. A. Brimble, Org. Biomol. Chem., 2011, 9, 1621–1626 CAS.
- P. W. R. Harris, S. H. Yang and M. A. Brimble, Tetrahedron Lett., 2011, 52, 6024–6026 CrossRef CAS.
- K. Page, C. A. Hood, H. Patel, G. Fuentes, M. Menakuru and J. H. Park, J. Pept. Sci., 2007, 13, 833–838 CrossRef CAS PubMed.
- T. Wöhr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. Sun and M. Mutter, J. Am. Chem. Soc., 1996, 118, 9218–9227 CrossRef.
- B. T. King, P. A. Bury, R. A. Gabel, J. E. Crider, T. C. I. I. Robert and B. S. DeHoff, Insulinotropic peptide synthesis using solid and solution phase combination techniques, WO Pat., 2009053315A1, 2009 Search PubMed.
- M. B. Davidson, G. Bate and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2005, 4, 713–714 CrossRef CAS PubMed.
- A. Abedini and D. P. Raleigh, Org. Lett., 2005, 7, 693–696 CrossRef CAS PubMed.
- P. White, J. W. Keyte, K. Bailey and G. Bloomberg, J. Pept. Sci., 2004, 10, 18–26 CrossRef CAS PubMed.
- E. K. Tiburu, P. C. Dave, J. F. Vanlerberghe, T. B. Cardon, R. E. Minto and G. A. Lorigan, Anal. Biochem., 2003, 318, 146–151 CrossRef CAS PubMed.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S. Shoda, Chem. Commun., 2009, 23, 3378–3379 RSC.
- R. J. Bailey and D. L. Hay, Peptides, 2006, 27, 1367–1375 CrossRef CAS PubMed.
- J. J. Gingell, T. Qi, R. J. Bailey and D. L. Hay, Peptides, 2010, 31, 1400–1404 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data for the synthesis and receptor activity testing of compounds 1–13. See DOI: 10.1039/c6ob00850j |
| This journal is © The Royal Society of Chemistry 2016 |