
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studies in organic and physical photochemistry – an interdisciplinary approach
Michael
Oelgemöller
*a and
Norbert
Hoffmann
*b
aJames Cook University, College of Science and Engineering, Townsville, QLD 4811, Australia. E-mail: michael.oelgemoeller@jcu.edu.au
bCNRS, Université de Reims Champagne-Ardenne, ICMR, Equipe de Photochimie, UFR Sciences, B.P. 1039, 51687 Reims, France. E-mail: norbert.hoffmann@univ-reims.fr
First published on 22nd June 2016
Abstract
Traditionally, organic photochemistry when applied to synthesis strongly interacts with physical chemistry. The aim of this review is to illustrate this very fruitful interdisciplinary approach and cooperation. A profound understanding of the photochemical reactivity and reaction mechanisms is particularly helpful for optimization and application of these reactions. Some typical reactions and particular aspects are reported such as the Norrish-Type II reaction and the Yang cyclization and related transformations, the [2 + 2] photocycloadditions, particularly the Paternò–Büchi reaction, photochemical electron transfer induced transformations, different kinds of catalytic reactions such as photoredox catalysis for organic synthesis and photooxygenation are discussed. Particular aspects such as the structure and reactivity of aryl cations, photochemical reactions in the crystalline state, chiral memory, different mechanisms of hydrogen transfer in photochemical reactions or fundamental aspects of stereoselectivity are discussed. Photochemical reactions are also investigated in the context of chemical engineering. Particularly, continuous flow reactors are of interest. Novel reactor systems are developed and modeling of photochemical transformations and different reactors play a key role in such studies. This research domain builds a bridge between fundamental studies of organic photochemical reactions and their industrial application.
 Michael Oelgemöller | Associate Professor Michael Oelgemöller received his Diploma from the University of Münster in 1995 and his Ph.D. from the University of Cologne in 1999. He was a researcher at the ERATO-JST Photochirogenesis project in Osaka (1999–2001) and at Bayer CropScience Japan in Yuki (2001–2004). From 2004–2008 he held the position of Lecturer in Organic and Medicinal Chemistry at Dublin City University. In February 2009 he joined James Cook University in Townsville as an Associate Professor in Organic Chemistry. His research activities include continuous-flow photochemistry, solar manufacturing of chemicals, photochemical synthesis of bioactive compounds and photochemical degradation of pollutants.229 |
 Norbert Hoffmann | Norbert Hoffmann studied chemistry at the RWTH Aachen University, Germany and received his PhD degree in 1992 under the supervision of Hans-Dieter Scharf. In 1993, he obtained a permanent position (Chargé de Recherche) at the French National Center for Scientific Research CNRS in Reims, France. In 2004, he was appointed Research Director at the CNRS. His main research interests are in the field of organic photochemistry: electron transfer, photoinduced radical reactions, stereoselective reactions, cycloadditions of aromatic compounds, reactions in photochemical continuous flow reactors and application of these reactions to organic synthesis. Further research interests concern the production of fine chemicals from biomass and the synthesis of new organic semiconductor materials for microelectronics (chemistry of perylene derivatives). |
1 Introduction
When compared to reactions at the ground state, photochemical transformations are characterized by the fact that they involve electronic excitation.1 Thus, by light absorption the electron configuration of a molecule changes. Since the reactivity of a chemical compound is defined by its electron configuration, it is obvious that the photochemical reaction conditions considerably augment the number of transformations of a compound family. Often, photochemical reactivity is even complementary to ground state chemistry. More basically, this phenomenon may be described by potential-energy surface topology.2,3 Applying photochemical methods to organic synthesis, products can be synthesized which are difficultly or not available with conventional methods.4,5 Many catalytic reactions with organometallic compounds or enzymes are improved under these conditions. Since photochemical reactions can be performed under mild conditions, they further enable studying transformations in supramolecular structures or investigating particular phenomena of absolute chiral induction or chiral memory.6 Photochemical transformations are also applied to the synthesis of unusual or strained hydrocarbons.7 Such investigations contribute to a profound understanding of fundamental aspects of chemical structures and bonds. Electronic excitation, often avoids chemical activation with acids, bases, metals, and so on. Side reactions and waste products are thus limited. The photon is considered as a “traceless reagent”.8 Photochemical transformations provide a lot of perspectives for sustainable chemistry. They even belong to the standard methods of “green chemistry”.9 They can readily be optimized when performed in a microreactor or continuous flow systems. In many fields such as pharmaceutical or agricultural chemistry or in materials science photochemical reactions are therefore highly appreciated.Traditionally, the study of organic photochemical reactions is strongly linked to physical chemistry. This intense cooperation between two research domains enables a profound and comprehensive understanding of reaction mechanisms and consequently a facile optimization of the reactions and application to industrial production. Also more general topics of chemistry are often elucidated by this research. It further facilitates interaction with other scientific domains such as materials science, medicinal and biochemistry, supramolecular chemistry, environmental science, etc. Although photochemical transformations are investigated in many different domains, we would like to focus the present contribution on some reactions possessing high interest in the context of organic synthesis. After a period of stagnation, activities in this field are increasing once again in academic and industrial research. The aim of this article is therefore to indicate and stimulate this fruitful interaction with some examples related to synthetic organic photochemistry.
2 Norrish Type II reaction and cyclizations
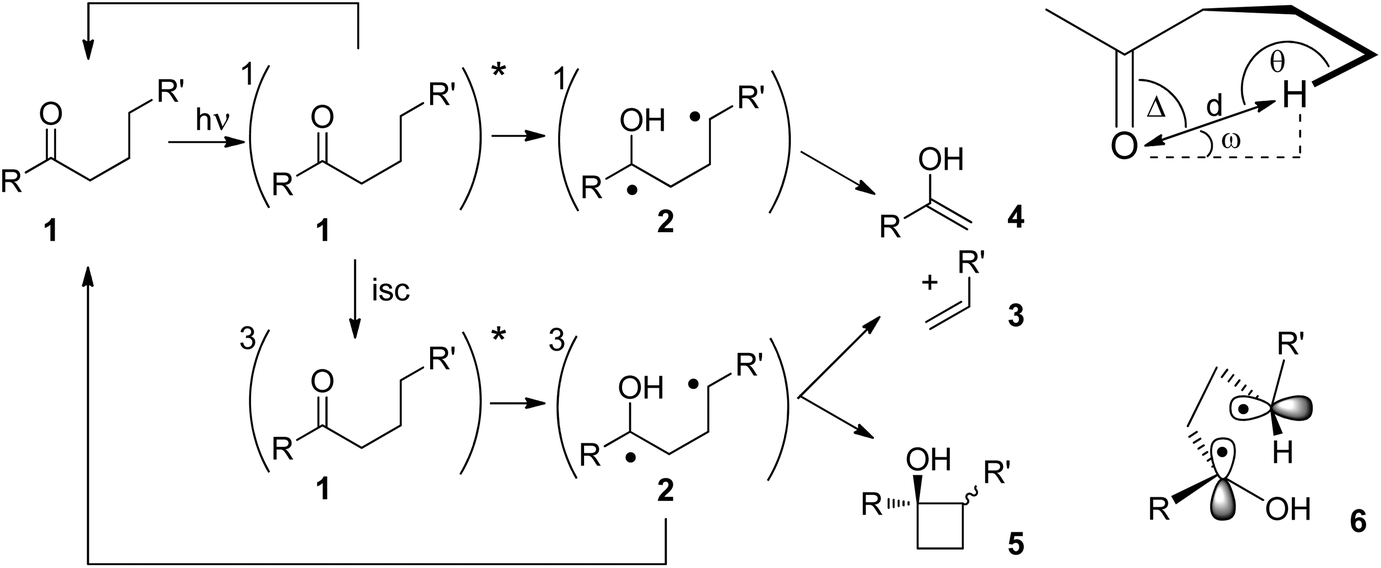
The Norrish Type II reaction and the competing Yang cyclization are classical photochemical reactions of carbonyl compounds.10 After photochemical nπ*-excitation of the carbonyl function in 1, intramolecular hydrogen abstraction takes place (Scheme 1). This reaction step occurs at both the singlet state and the triplet state. It requires specific orientations of the carbonyl group and the hydrogen atom which will be abstracted. The reactions were also performed in the solid state.11,12 Under these conditions the structure is rigid and the orientation of the participating atoms is determined by X-ray structure analysis. Thus, it was found that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–C dihedral angles Δ, ω and θ in 1 may vary greatly and that the O–H distance d is most relevant for the efficiency of this reaction step. Radical intermediates such as the resulting 1,4-diradical 2 play an important role in the outcome of many photochemical reactions.13 Depending on the intersystem crossing (isc) rate, the 1,4-diradical 2 may either be formed at its singlet or triplet state. At the singlet state, the intermediate undergoes cleavage easily leading to the alkene 3 and the enol 4. The latter product undergoes immediate tautomerization to the corresponding ketone. This overall reaction corresponds to the Norrish Type II reaction. This cleavage step is favored by entropy and it is not inhibited by intersystem crossing. At the triplet state, three reactions are in competition. Hydrogen back transfer may occur thus leading to substrate 1. The Norrish Type II reaction may also be in competition with the Yang cyclization by intramolecular radical combination. The latter reaction leads to cyclobutanols 5. In all these cases, intersystem crossing must occur since the final products possess singlet multiplicity. This step is therefore accelerated by efficient spin orbit coupling.14 Such an efficient interaction is linked to a particular transition state in which the two radical carrying orbitals are orthogonally orientated (6).15,16 As already mentioned, hydrogen abstraction can also occur at the singlet state. The quenching of the singlet state may even be faster than the corresponding triplet state. However, the hydrogen transfer is less efficient.17
O⋯H–C dihedral angles Δ, ω and θ in 1 may vary greatly and that the O–H distance d is most relevant for the efficiency of this reaction step. Radical intermediates such as the resulting 1,4-diradical 2 play an important role in the outcome of many photochemical reactions.13 Depending on the intersystem crossing (isc) rate, the 1,4-diradical 2 may either be formed at its singlet or triplet state. At the singlet state, the intermediate undergoes cleavage easily leading to the alkene 3 and the enol 4. The latter product undergoes immediate tautomerization to the corresponding ketone. This overall reaction corresponds to the Norrish Type II reaction. This cleavage step is favored by entropy and it is not inhibited by intersystem crossing. At the triplet state, three reactions are in competition. Hydrogen back transfer may occur thus leading to substrate 1. The Norrish Type II reaction may also be in competition with the Yang cyclization by intramolecular radical combination. The latter reaction leads to cyclobutanols 5. In all these cases, intersystem crossing must occur since the final products possess singlet multiplicity. This step is therefore accelerated by efficient spin orbit coupling.14 Such an efficient interaction is linked to a particular transition state in which the two radical carrying orbitals are orthogonally orientated (6).15,16 As already mentioned, hydrogen abstraction can also occur at the singlet state. The quenching of the singlet state may even be faster than the corresponding triplet state. However, the hydrogen transfer is less efficient.17
 | ||
| Scheme 1 Mechanism of the Norrish Type II reaction and the competing Yang cyclization. | ||
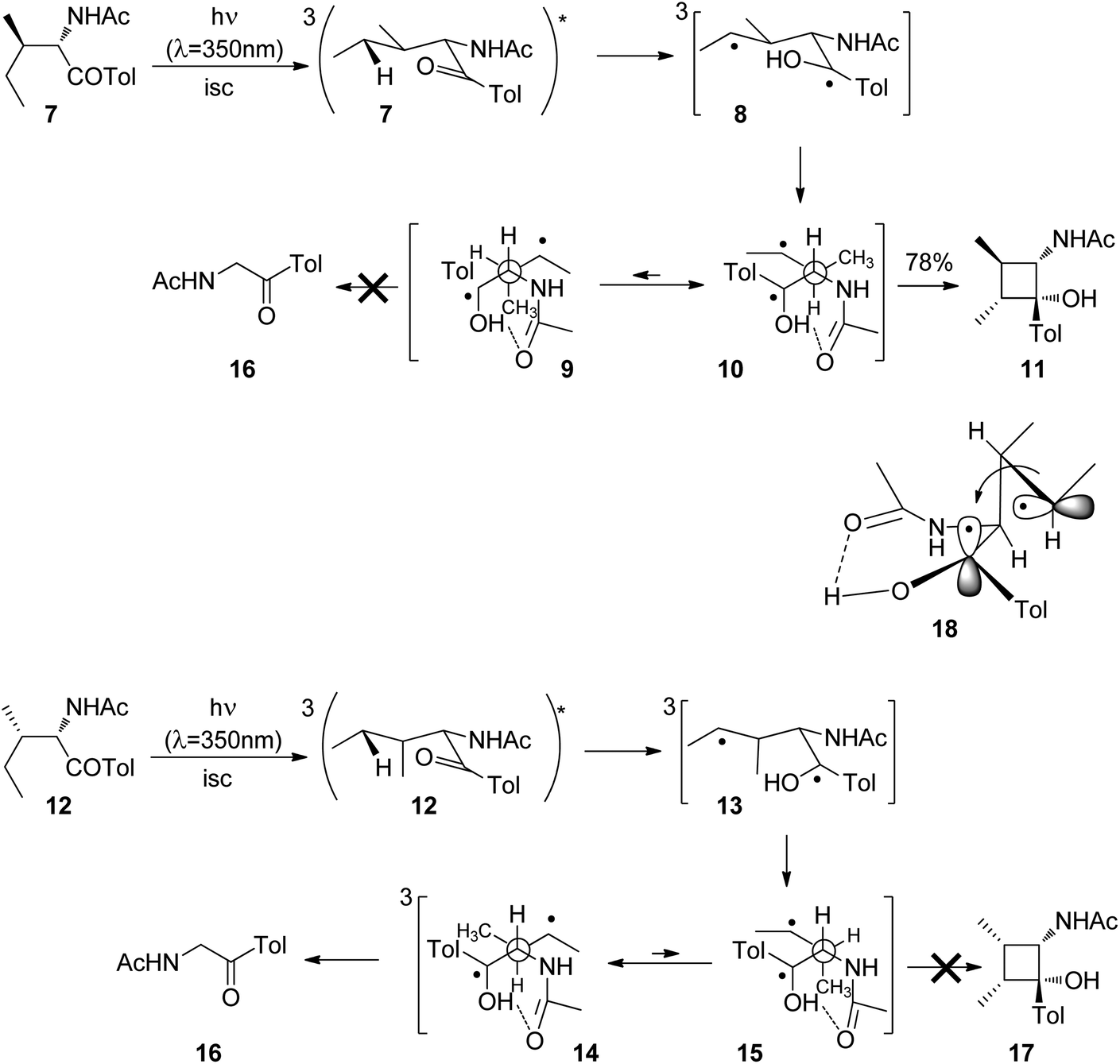
Cyclizations of photochemically generated diradicals are of particular interest since they may be applied to organic synthesis. Therefore, details of the stereochemical influence on such reactions have been intensively investigated. Thus, the influence of the relative configuration on the competition between the Norrish Type II fragmentation and the Yang cyclization was studied. When the aromatic ketone 7 derived from isoleucine was irradiated, hydrogen abstraction in the γ position at the ethyl group took place leading to the 1,4-diradical 8 (Scheme 2).18 Generally, in such aromatic ketones, intersystem crossing is fast and the nπ* triplet state is rapidly populated. Two main conformers 9 and 10 of the diradical are in equilibrium. Conformation 10 in which the two radical positions are orientated syn with respect to each other dominates in this equilibrium since it is 1.1 kcal mol−1 more stable than 9. For this reason, cyclization leading to the cyclobutanol derivative 11 is the only observed step. It must further be pointed out that a hydrogen bond between the newly formed hydroxyl function and the carbonyl group stabilizes these conformers. The corresponding diastereoisomer 12 after photochemical excitation, intersystem crossing and γ hydrogen abstraction is transformed into the 1,4-diradicals 13 with the main conformations 14 and 15. The anti configuration of the methyl groups in the cyclobutanol derivative 11 is explained by the structure of the transition state 18 of the cyclization step. In this structure, the radical carrying orbitals are orthogonally orientated as a means to minimize steric hindrance and spin orbit coupling. In contrast to the previous case, the conformer 14 with anti orientation of the radical centers is more stable than the corresponding syn conformer 15. The energy difference is 1.7 kcal mol−1. In this case, exclusively, the fragmentation product 16 is formed. No corresponding cyclobutane product 17 was observed.
 | ||
| Scheme 2 Stereoselectivity of the Norrish Type II reaction and the competing Yang cyclization. | ||
As already indicated, Yang cyclization has also been studied by irradiation of crystalline solids. In solution, conformational equilibria have a major influence on the competition between Norrish Type II fragmentation and Yang cyclization. The crystal lattice may also select and fix a conformation favoring the Yang cyclization thus inhibiting the fragmentation. High ee values are generally observed in photochemical reactions when they are performed with solids crystallizing in chiral space groups. In cases such as the present one, the crystal lattice selects conformations favorable for Yang cyclization in one enantiomeric form of the aromatic carboxylate. Photochemical reactions in chiral solids thus are an efficient method of asymmetric synthesis.11,19
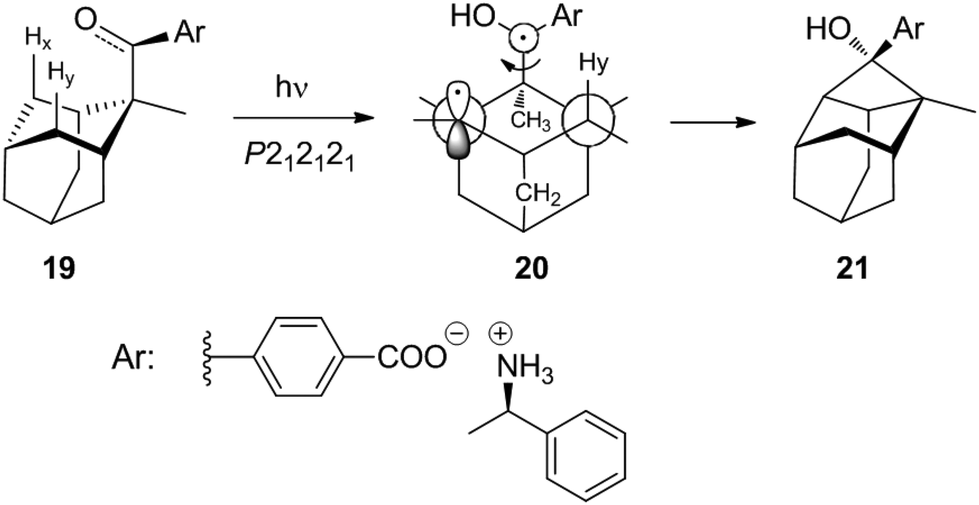
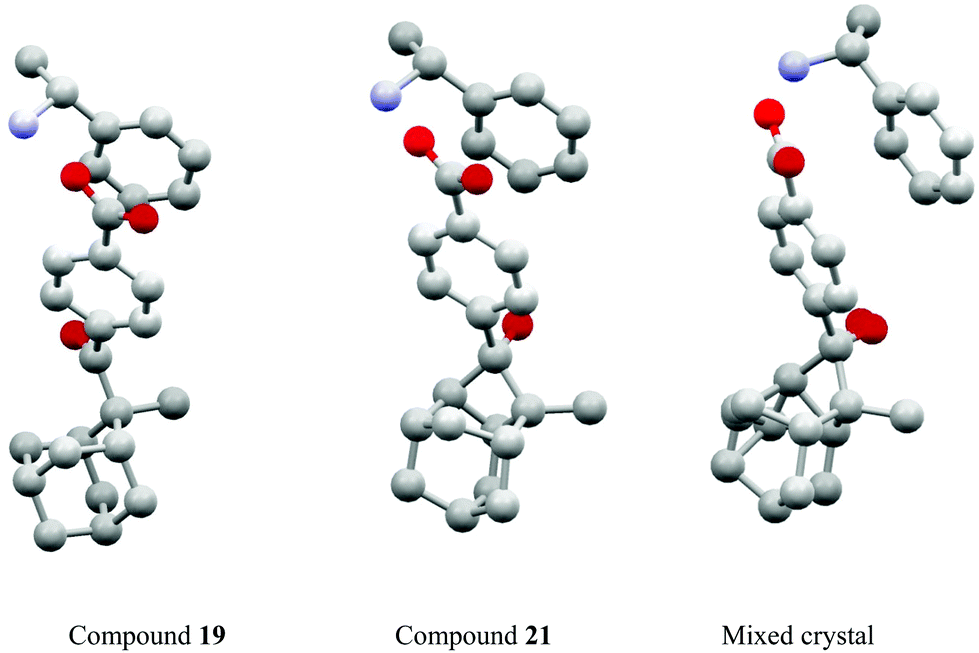
Studies of such transformations in crystals also provide general and fundamental information on crystallography. The chiral salt of the aromatic keto acid 19 crystallized in space group P212121. In these crystals, the oxygen of the carbonyl group is closer to Hx than to the enantiotopic Hy of the adamantane substituent (Scheme 3).12,20 Upon irradiation, therefore, only the hydrogen atom Hx is abstracted and the diradical intermediate 20 is formed. Cyclization leads to the cyclobutanol 21. Only this endo isomer is generated. Furthermore, this compound is obtained in high enantiomeric excess. It is particularly interesting to note that both the substrate 19 and the product 21 possess almost the same crystal structure (Fig. 1) in terms of space group, crystal system and cell constants. Thus, an X-ray crystal structure analysis of a mixed crystal containing 40% of the substrate 19 and 60% of the photoproduct 21 could be carried out.
 | ||
| Scheme 3 Yang cyclization with an adamantane derivative in the solid state using a chiral salt for asymmetric induction. | ||
 | ||
| Fig. 1 Crystal structures during a Yang cyclization in enantiomorphous crystals. | ||
The investigations of photochemical reactions in the solid state also enable a deeper understanding of how organic compounds crystallize depending on their structure. In a more detailed study, a large number of X-ray structure analyses were performed on single crystals containing substrate 19 and photoproduct 21 in different proportions (Fig. 2).21 Although, the space group does not change when the conversion advances, the dimensions, in particular two cell constants of the orthorhombic crystal system vary (Fig. 3). During the first 20 min of the irradiation, the conversion rapidly advances and the percentage P of photoproduct increased until it reaches 80%. During this period, the value of cell constant a slightly increases (Fig. 3(a)) while the value of constant b decreases (Fig. 3(b)). Cell constant c barely changes (Fig. 3(c)). During this period, the volume of the unit cell rapidly increases to a maximum of 2313 Å3 (Fig. 3(d)). An additional 70 min of irradiation was necessary to complete the conversion. During this period, the changes of the cell constants a and b are much larger and the constant c decreases slightly. The cell volume drastically diminishes to reach a final value of about 2300 Å3. All these changes are the result of the changes inside the cell which are monitored by atom distances as well as bond and dihedral angles. These parameters evolve in a very discontinuous manner. Some of them strongly change during the first period and remain constant during the final period of the conversion. Other parameters evolve in the opposite manner. Some parameters also change rapidly during the first period. They remain constant for the following period of conversion in order to evolve rapidly again at the end to attain their final values.
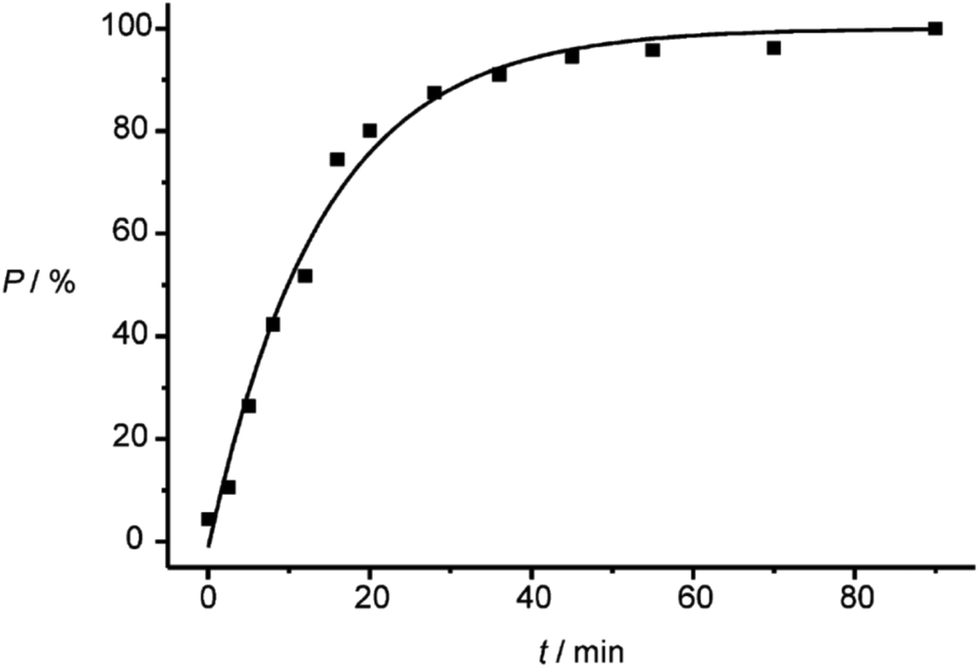 | ||
| Fig. 2 Transformation of compound 19 into 21. After 80% conversion, the reaction rate slows down. (Reprinted with permission from: I. Turowska-Tyrk, E. Trzop, J. R. Scheffer and S. Chen, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 128–134. DOI: 10.1107/S0108768105034014. Copyright (2006) IUCr.) | ||
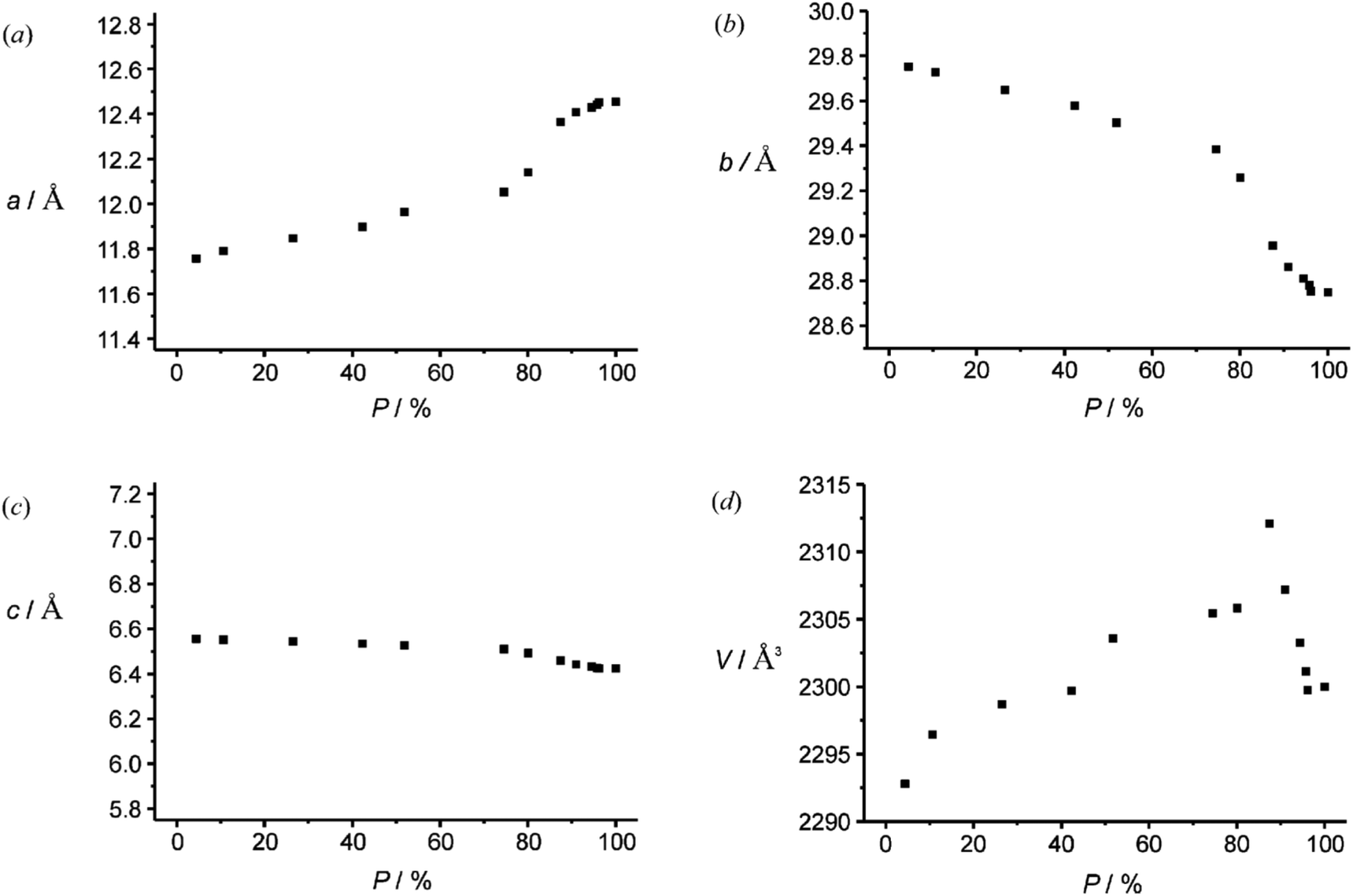 | ||
| Fig. 3 Variation of cell constants during the transformation of compound 19 into 21. (Reprinted with permission from: I. Turowska-Tyrk, E. Trzop, J. R. Scheffer and S. Chen, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 128–134. DOI: 10.1107/S0108768105034014. Copyright (2006) IUCr.) | ||
Globally, the volume of the crystal structure increases because it must host a large variety of different molecular structures. In the anionic part, the adamantane moiety moves more than the remaining part since it is less engaged in hydrogen bonding. When the conversion comes to its end, the structure seems to collapse because the number of different molecular structures diminishes. Only the molecules of the final product need to be packed in a crystal structure. The final structural parameters of the cyclobutanol moiety are established during this period. Although the changes at the molecular level are very discontinuous, the changes of the cell parameter are nevertheless very ordered. Cell constant a increases, constant b decreases and cell constant c remains almost unchanged.
Crystal-to-crystal [4 + 4] photocycloadditions with anthracene moieties were used to synthesize regular two dimensional polymers.22 In the present case, the crystal structures act as a template which orients three photoreactive centers of the monomers in the desired manner. These monomers possess D3h or C3h molecular symmetry. Thus layers are formed. Such polymers are of high interest, for example, in the domain of materials science as it has been frequently shown for the graphene prototype. Controlled two dimensional polymerization is particularly difficult.23 The presented method provides access to a larger variety of such compounds. These results are also good examples for the use of particularly mild photochemical reaction conditions for the construction of supramolecular structures.
Several electron active substitutions on the tether separating the two radical positions may affect the competition between Norrish Type II fragmentation and Yang cyclization.24 Such a competition has been observed in reactions of carbohydrate derivatives.25 The presence of an additional ketone function in α-diketones generally favors the formation of hydroxycyclobutanones via Yang cyclization with respect to the Norrish Type II cleavage. Such structures are also of high interest for organic synthesis. For example, the glucose derivative 22 was efficiently transformed into the corresponding hydroxycyclobutanone derivative 23 (Scheme 4).26 Only one anomer was obtained which was generated under retention of configuration. Furthermore, only one configuration was generated at the quaternary center carrying the hydroxyl function. The reaction was carried out with UV or sunlight with almost the same results. When the diketone moiety is placed in the former anomeric position of the glucose derivative 24, a Norrish Type II reaction takes place.27 After hydrogen abstraction in the γ position, in the resulting 1,4-diradical 25, a C–O bond is cleaved. An enol and a ketone function are thus formed in the intermediate 26. The final product 27 is then obtained by intramolecular aldol reaction followed by the formation of a half acetal.
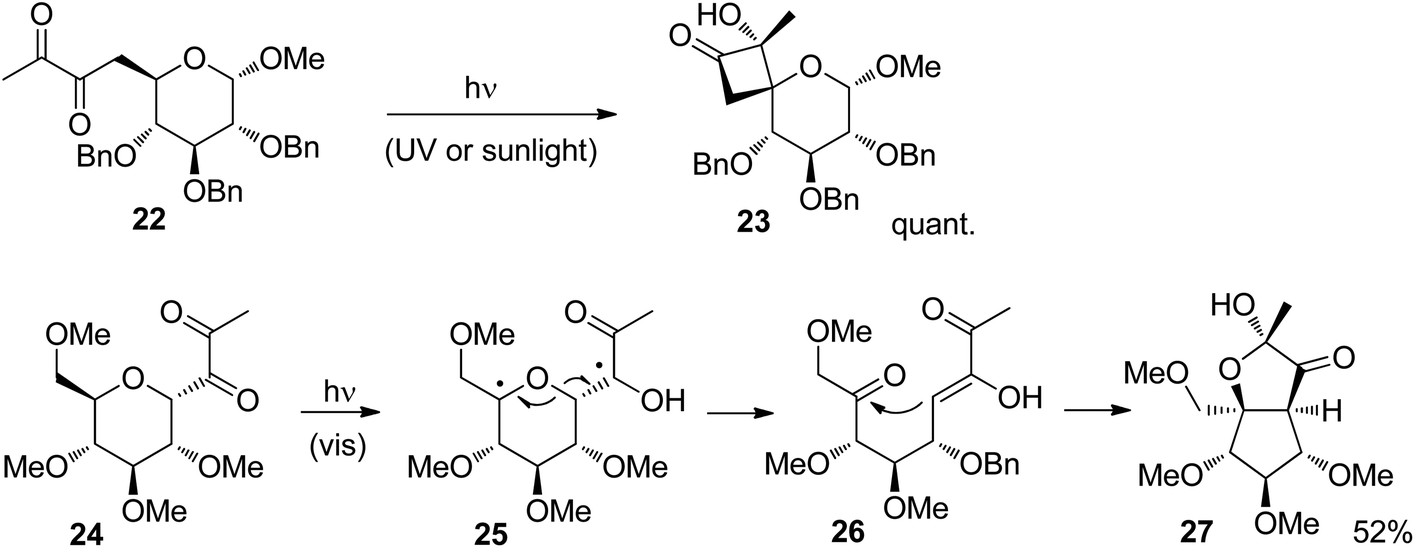 | ||
| Scheme 4 Reactions similar to the Norrish Type II reaction and the Yang cyclization. | ||
Photochemically induced intramolecular hydrogen abstraction followed by cyclization may be carried out with a variety of chromophores.24 Also macrocyclizations have been carried out.28 Such reactions are valuable tools in organic synthesis. The photochemistry of α,β-unsaturated carbonyl or carboxyl compounds resembles that one of the simple carbonyl compounds. Photochemical transformations of these compounds, particularly [2 + 2] photocycloadditions, were often applied to organic synthesis.29 Most frequently, such reactions start at the 3ππ* excited state which is the T1 state. Intramolecular hydrogen abstraction may also occur at this electronically excited state.30 Due to the high energy of the singlet state S1,31 such compounds are most frequently excited by sensitization, for instance by triplet energy transfer from acetone, when these reactions are applied to organic synthesis.
Such a reaction has been carried out with α,β-unsaturated lactones carrying a tetrahydropyranyl substituent on the site chain (Scheme 5).32 Compounds 28a,b are excited to their ππ* triplet state (29) by triplet energy transfer from the nπ* excited acetone. In this state, the π bond is suppressed and the spin density is particularly high in the β position (29).33–35 For this reason, hydrogen abstraction occurs in this position and the diradical 30 is formed. Hydrogen is exclusively transferred from the anomeric center of the tetrahydropyranyl moiety. Radical combination leads to the final products 31a and 31b in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. When an alkyl substituent is present on the site chain between the tetrahydropyranyl and the furanone moiety as in compounds 32a,b, only one spirocyclic product 33 was isolated possessing the lk relative configuration (S*,S*) at the anomeric center and the lactone ring. This results from hydrogen abstraction at the anomeric center (34) and radical combination. No stereoisomer possessing the ul relative configuration (S*,R*) was obtained. In the case of the substrate possessing the ul relative configuration (S*,R*), hydrogen abstraction also occurs in the 6′ position of the tetrahydropyranyl ring (36). Radical combination leads to the formation of the macrocyclic compound 35. Product 37 resulting from a radical combination in intermediate 38 with a lk configuration (S*,S*) was not obtained.
:1. When an alkyl substituent is present on the site chain between the tetrahydropyranyl and the furanone moiety as in compounds 32a,b, only one spirocyclic product 33 was isolated possessing the lk relative configuration (S*,S*) at the anomeric center and the lactone ring. This results from hydrogen abstraction at the anomeric center (34) and radical combination. No stereoisomer possessing the ul relative configuration (S*,R*) was obtained. In the case of the substrate possessing the ul relative configuration (S*,R*), hydrogen abstraction also occurs in the 6′ position of the tetrahydropyranyl ring (36). Radical combination leads to the formation of the macrocyclic compound 35. Product 37 resulting from a radical combination in intermediate 38 with a lk configuration (S*,S*) was not obtained.
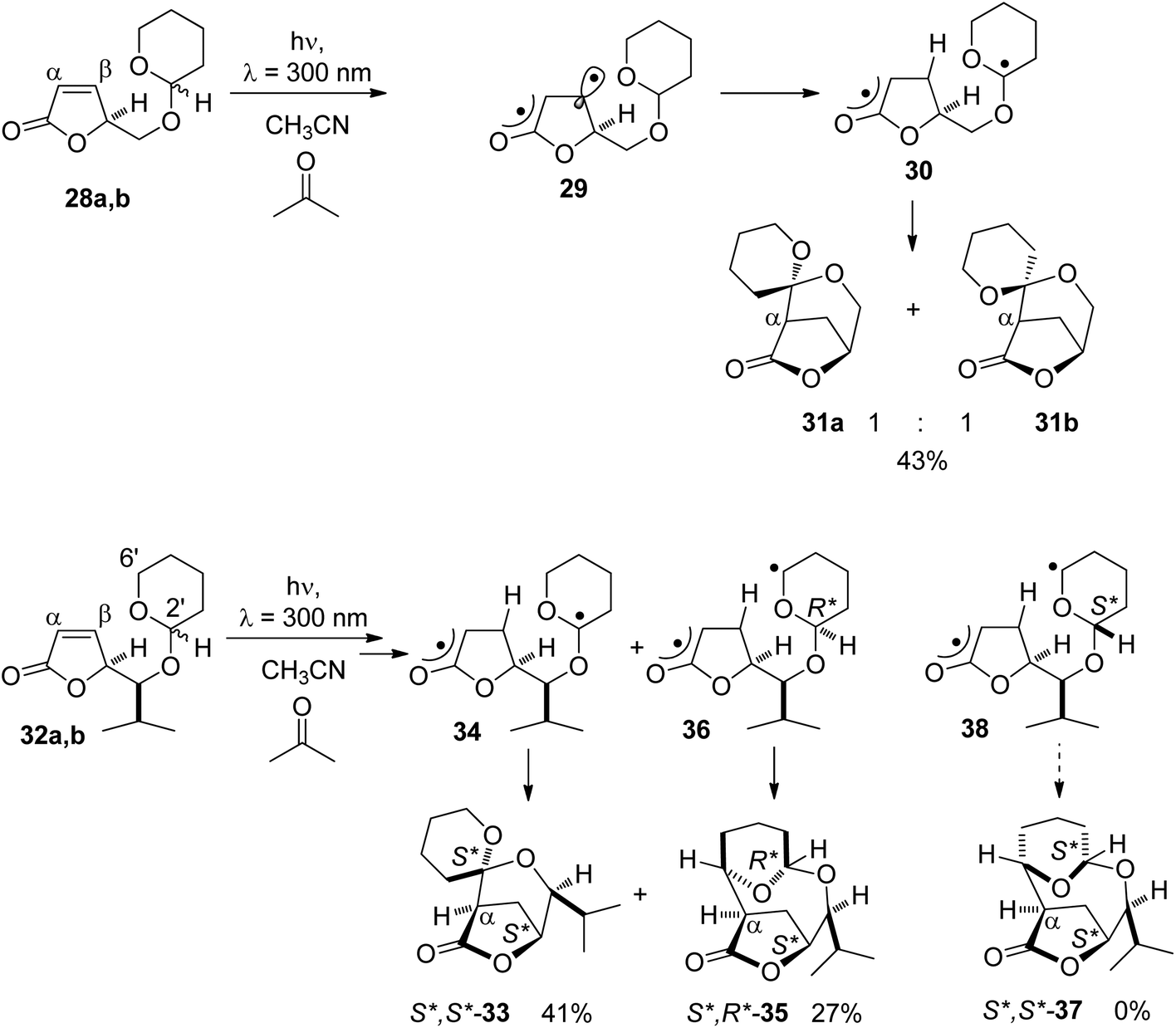 | ||
| Scheme 5 Regio- and stereoselective hydrogen abstraction followed by radical cyclization in α,β-unsaturated lactones. | ||
The relative configuration at the tetrahydropyranyl (39) and the furanone moiety and the equilibrium between equatorial and axial conformations (Scheme 6) are the two structural elements which mainly control the regioselectivity of hydrogen abstraction. In order to determine the influence of each of these elements on the outcome of the reaction, the transformation was carried out with the glucose derivatives 40, 41, 42 and 43. Due to the equatorial position of all further substituents in glucose derivatives, the equatorial (40 and 41) and axial orientations (42 and 43) at the anomeric center are locked.
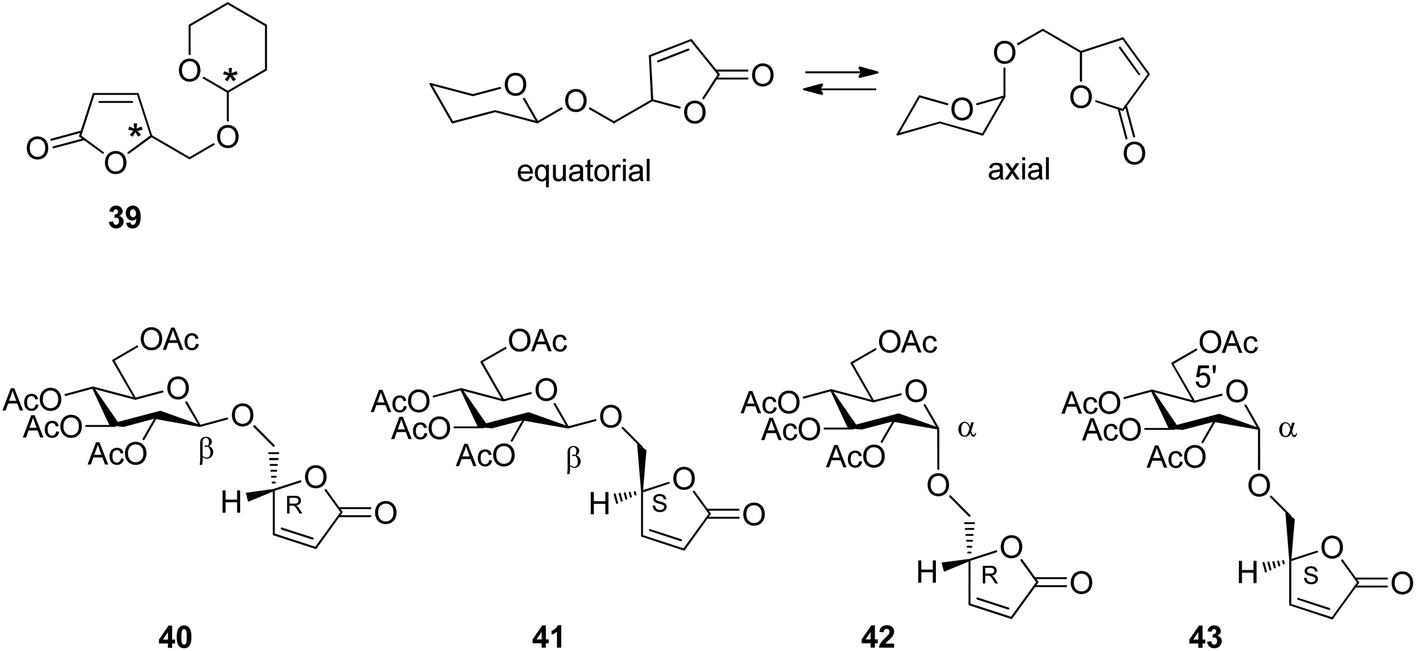 | ||
| Scheme 6 Stereochemical elements in α,β-unsaturated lactone derivatives and the corresponding glucosyl compounds. | ||
Upon irradiation under sensitization conditions, compounds 40 and 42 possessing R configurations at the furanone moiety yielded the same spirocyclic compounds 44 and 45 in almost the same diastereomeric ratio (Scheme 7). These products result from hydrogen abstraction at the 1′ position, the anomeric center of the glucose moiety. The higher yield in the case of the transformation of the β anomer 42 is explained by the fact that hydrogen abstraction at the anomeric center is easier than in 40.36 When compound 43 possessing an S configuration at the furanone moiety and α-configuration at the sugar part was transformed, only the macrocyclic compound 46 was isolated in good yield. This product results from hydrogen abstraction at the 5′ position of the glucosyl substituent. When the corresponding β-anomer 41 was transformed under the same conditions, again spirocyclic compounds 47 and 48 were isolated. Moreover, the macrocyclic compound 46 was also formed. The latter compound can only be formed when an epimerization at the anomeric center takes place during the transformation. Studies on reactions with partial conversion confirmed this hypothesis. This epimerization is also photochemically induced.
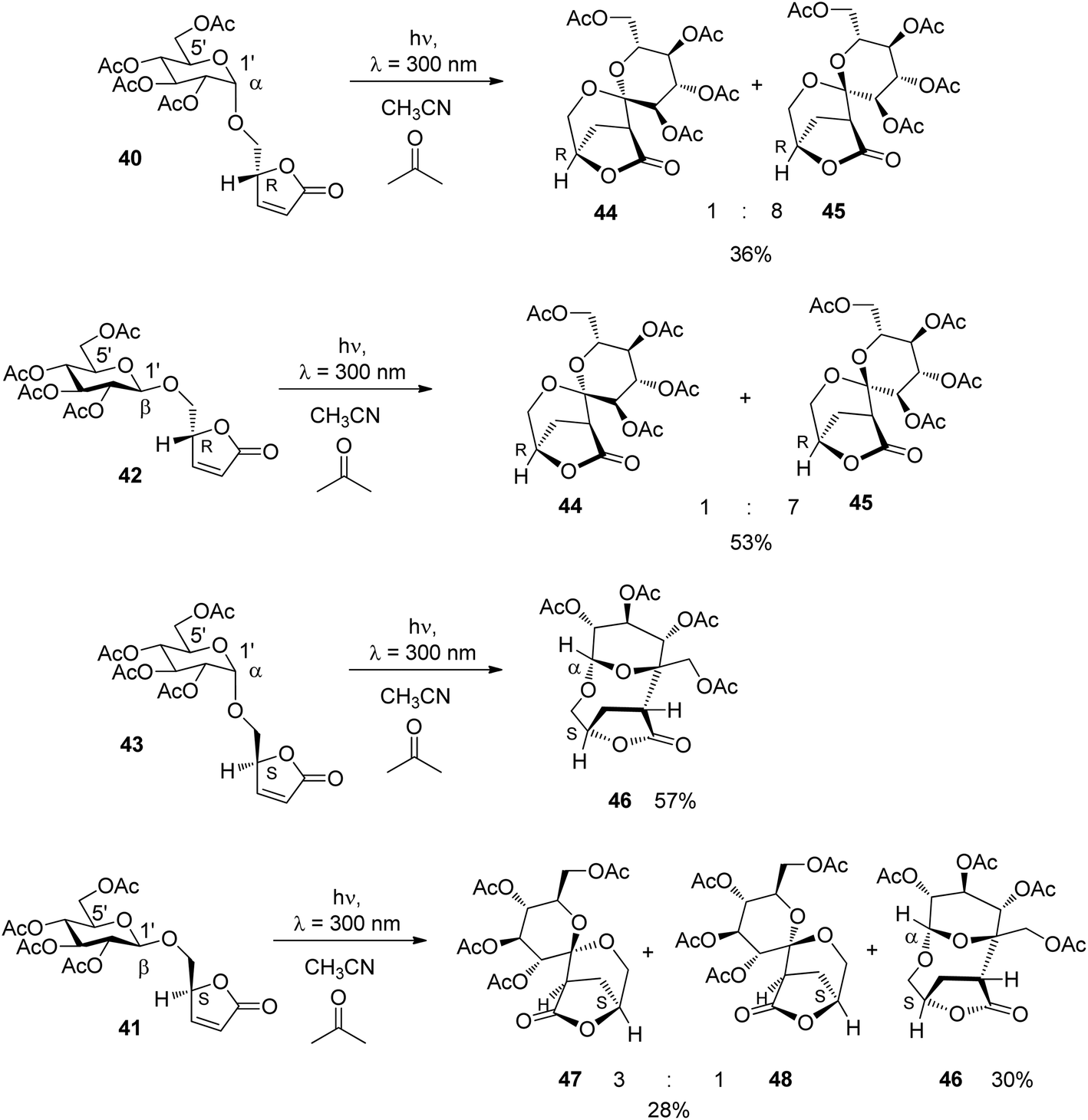 | ||
| Scheme 7 Stereoselective triplet sensitized transformation of α,β-unsaturated lactone derivatives. | ||
All detected mechanistic steps of these reactions are present in the transformation of compound 41 (Scheme 8). After excitation and hydrogen abstraction, the intermediates 49a and 49b are generated. They are in equilibrium. Radical combination in the intermediates leads to the formation of the spirocyclic compounds 47 and 48. In the case of the diradical intermediate 49b, hydrogen transfer from the lactone moiety back to the glucosyl moiety may occur leading to the formation of the α-anomeric compound 43. This compound was indeed detected in considerable amounts in the reaction mixture of 41 after 50% of conversion. As previously shown, under the same conditions via hydrogen abstraction in the 5′ position (50), compound 43 reacts selectively to yield the macrocyclic compound 46. It must be pointed out that significant parts of this mechanism have been evidenced by a stereochemical analysis.
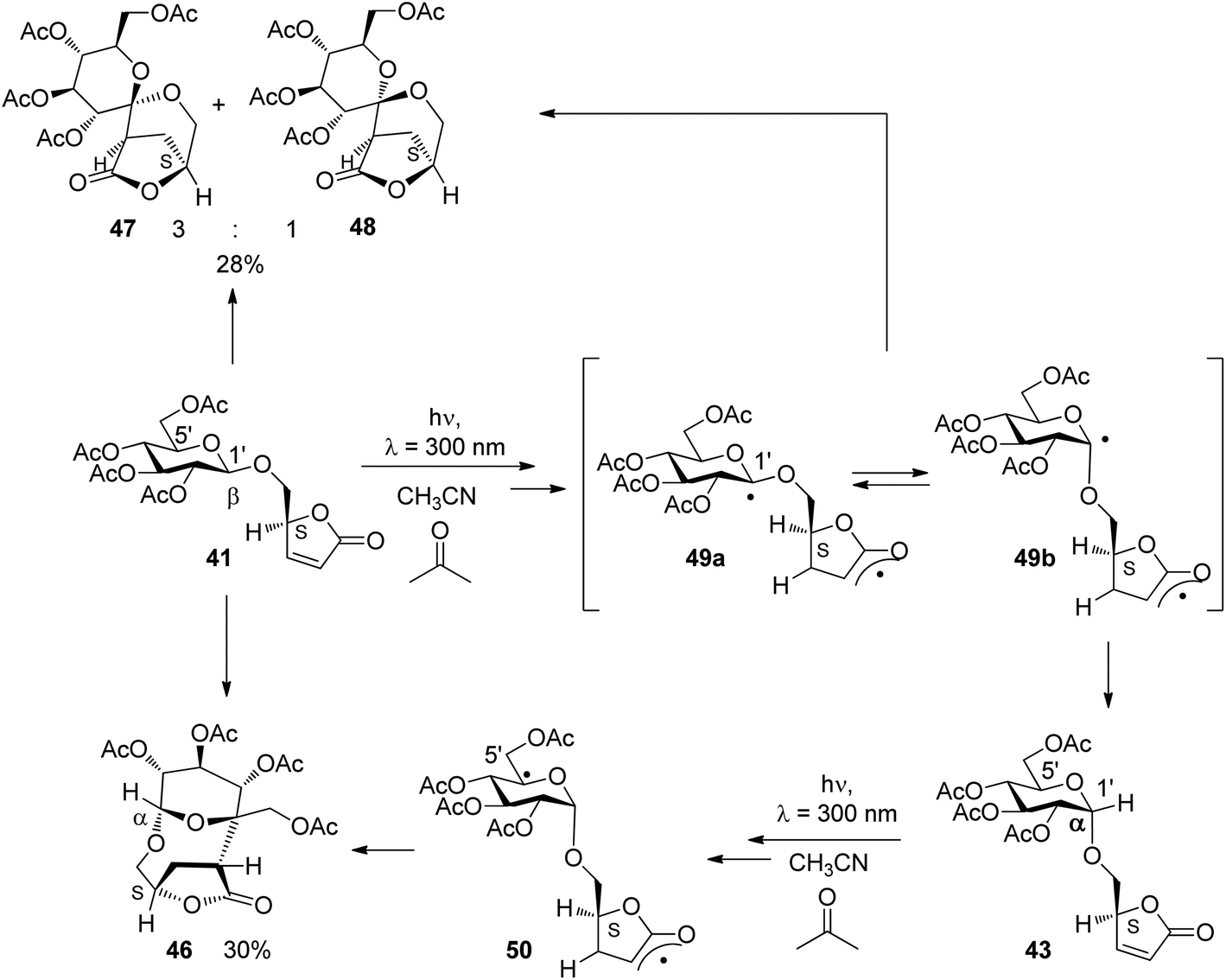 | ||
| Scheme 8 Radical steps in the stereoselective triplet sensitized reaction of α,β-unsaturated lactone derivatives. | ||
In order to get a deeper insight into the stereochemical requirements for hydrogen abstraction, DFT calculations have been carried out. The 3ππ* excited and vibrationally relaxed state has been modeled with structure 51 (Scheme 9). Transition states for hydrogen abstraction in positions 2′ (acetal center) and 6′ have been calculated. Due to different nomenclature systems, the positions 2′ and 6′ of these tetrahydropyranyl structures correspond in glucose derivatives to the positions 1′ and 5′ respectively. Table 1 reports the calculated differences in the free enthalpy of activation for hydrogen abstraction in positions 2′ and 6′ in model structure 51. These results confirm the experimental observation that in the case of 40 and 42 (R configuration at the furanone moiety, Scheme 9, model structures 51a,b, Table 1), hydrogen abstraction is favored at the anomeric center 52 (Scheme 9). In the case of the model structure 51c,d corresponding to the S configuration at the furanone moiety, only in the electronically excited α-anomer (51c), hydrogen abstraction in the 6′ position (53) is favored. Thus the computational results reproduce the experimental observations for the transformation of the glucosyl compounds 40, 41, 42 and 43 (Schemes 6 and 7). In particular the high spin density in the β-position of the 3ππ* excited and vibrationally relaxed state as modeled by structure 51 is essential for the observed regio- and stereoselectivity of the hydrogen abstraction step. Similar studies have been performed for the reaction of compounds 32a,b (Scheme 5). These reactions have also been discussed in the more general context of hydrogen transfer.32,37 The fact that the proton and the electron are transferred simultaneously leads to the formation of a C–C bond in the α position of the α,β-unsaturated lactone. A two-step mechanism (electron transfer followed by proton transfer) would have led to the formation of a C–C bond in the β position.
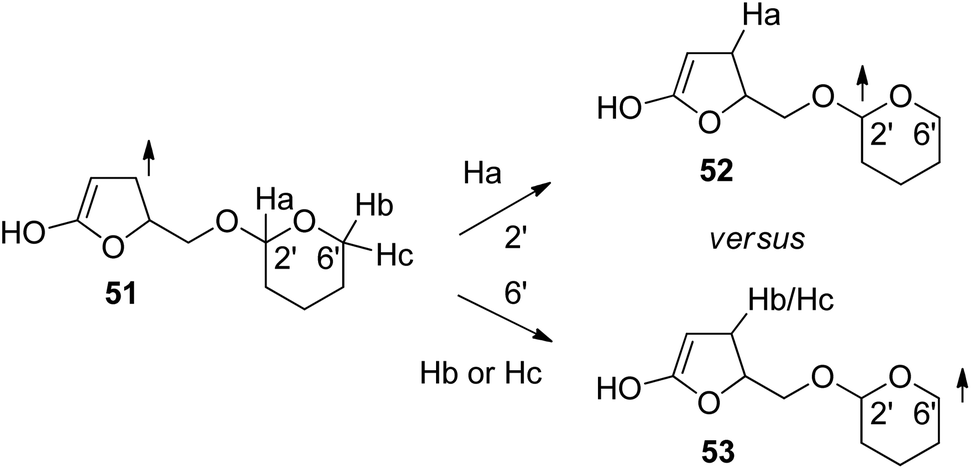 | ||
| Scheme 9 Model structures for the computational study of regio- and stereoselective hydrogen abstraction in the triplet sensitized reaction of α,β-unsaturated lactone derivatives. | ||
In such reactions, the phenomenon of the spin-center shift38 is also observed. After photochemical excitation of the acetophenone derivative 54 hydrogen abstraction occurs in one of the diastereotopic γ positions leading to the diradical intermediates R-55 and S-55 (Scheme 10).39 In contrast to the previously described reactions, in this case, the tosyl group in the α position is eliminated and the radical position is shifted in this position leading to the 1,3-diradical intermediates R-56 and S-56. Radical combination generates the bicyclic final products R-57 and S-57. The enantioselectivity of the overall reaction is mainly determined when the 1,4-diradical intermediates R-55 and S-55 are formed. The competition between hydrogen abstractions in the two diastereotopic γ positions depends on a ratio between conformers in an equilibrium. The enantioselectivity depends on the solvent and the reaction temperature. The latter is particularly interesting. When the reaction was carried out in methanol, the enantioselectivity decreases when the reaction temperature is diminished. When the reaction was carried out in methylene chloride, the ratio between R-57 and S-57 was 2.5:1 at −40 °C while it was 1:2.5 at −56 °C. Although often considered as unusual, such behavior is frequently observed.40 From Eyring diagrams such as the one depicted in Fig. 4, enthalpy and entropy effects are deduced. An extensive discussion in this context has been performed, for example, in the context of the diastereoselective [2 + 2] photocycloaddition.35 For an example of a photooxygenation see below.
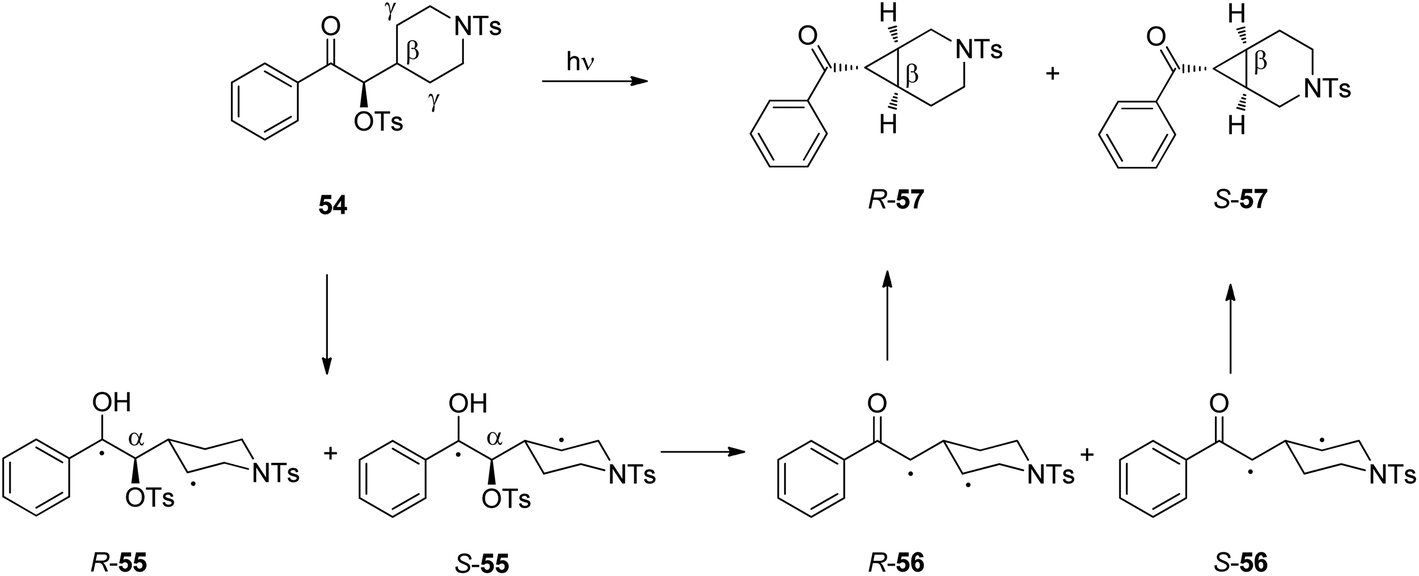 | ||
| Scheme 10 Intramolecular stereoselective hydrogen abstraction involving a spin-center shift followed by cyclization. | ||
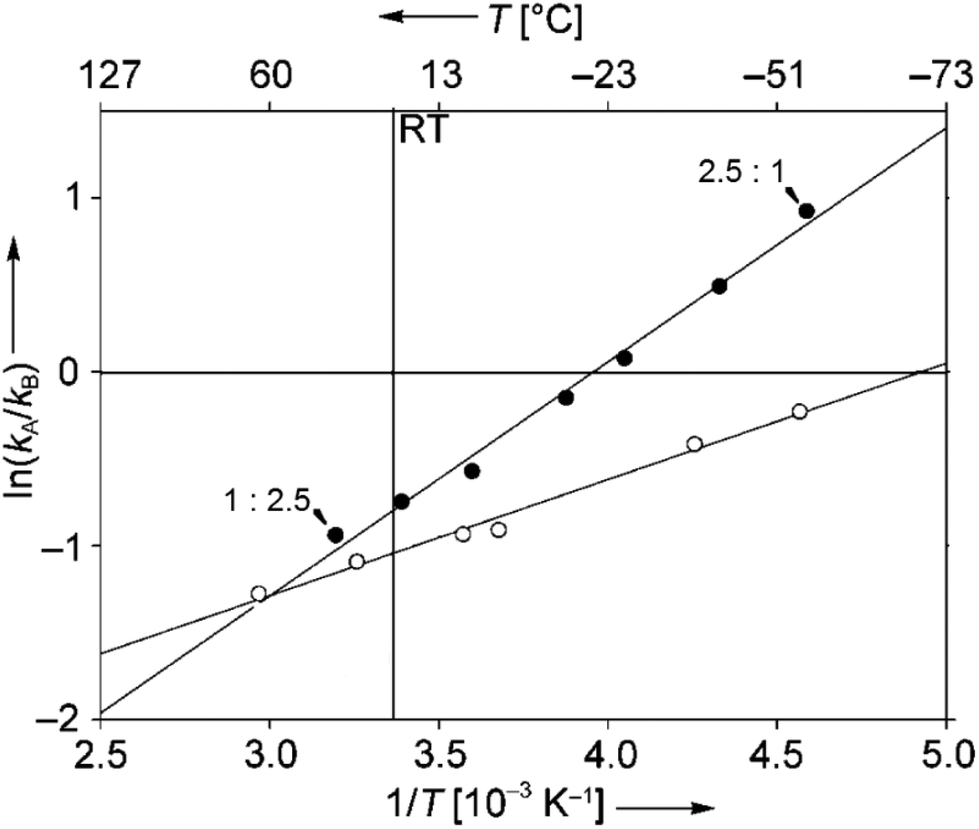 | ||
| Fig. 4 Stereoselective hydrogen abstraction involving a spin-center shift (Scheme 10). Enantioselective formation of bicyclic derivatives R-57 and S-57. kA: overall rate of the formation of R-57, kB: overall rate of the formation of S-57, ● CH2Cl2, ○ MeOH as the solvent. RT: room temperature. (Adapted with permission from: P. Wessig and O. Muehling, Chem. – Eur. J., 2008, 14, 7951–7960. Copyright (2008) John Wiley & Sons.) | ||
Similar reactions have been carried out with aromatic ketones or aldehydes. The reaction depicted in Scheme 1141,42 can be carried out with numerous derivatives. After photochemical excitation of the benzophenone derivative 58 and intersystem crossing (isc), the triplet diradical intermediate 59 is generated by intramolecular hydrogen abstraction. The dienol species 60 is then formed. Both intermediates 59 and 60 may be considered as the triplet and the singlet form, respectively, of one species, the photochemically generated enol tautomer. Intermediate 60 is placed on the singlet ground state potential energy surface. It easily undergoes [4 + 2] cycloaddition with alkenes such as 61. The final product 62 was obtained in high yields in a stereospecific manner which is typical for a Diels–Alder reaction at the ground state. This reaction was frequently applied to the synthesis of organic compounds such as natural and other biologically active compounds or organic materials.43 Similar reactions also play an important role in the photochemical removal of protecting groups.44 Intermediates such as 60 are also called phototautomers.42,45–47 Concerning the stability of tautomers, a complementary tendency between the ground state and electronic state is generally observed.48,49 Tautomers which are less stable at the ground state can be generated via photochemical excitation. In this context, photochemically generated achiral enols have been transformed into the corresponding chiral ketone forms using asymmetric catalysis.50 The determination of the enantioselectivity as a function of temperature was used to determine the influence of enthalpy and entropy on the stereoselectivity in such reactions.51
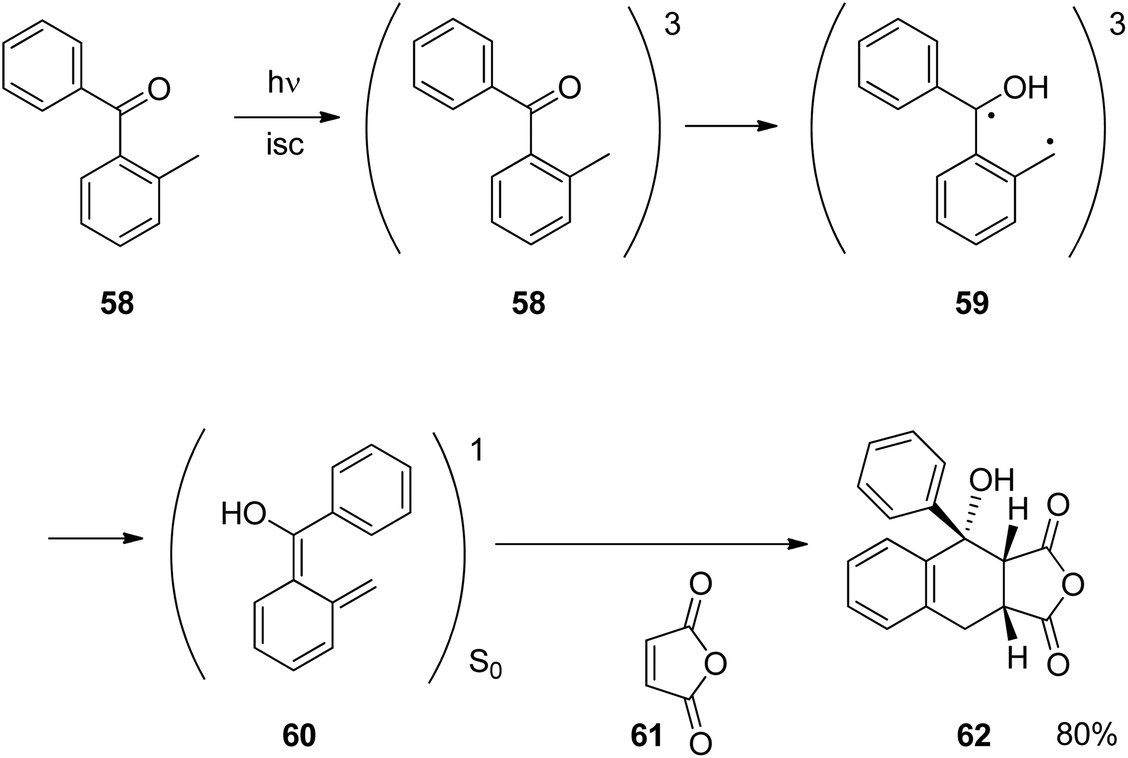 | ||
| Scheme 11 Trapping of a photodienol by a [4 + 2] cycloaddition. | ||
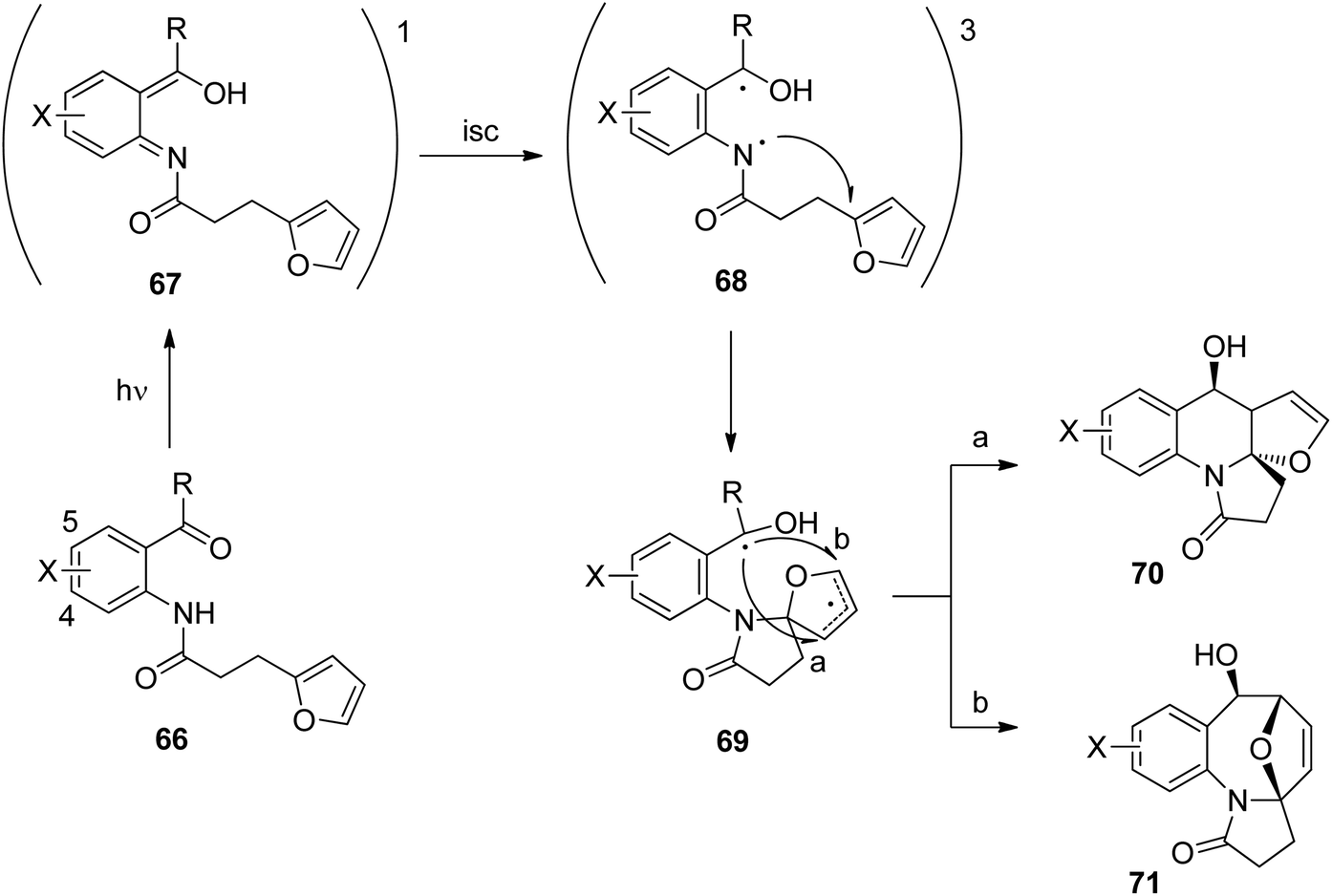
Anthranilic acid derivatives have been transformed in a similar manner. A typical example is depicted in Scheme 12.52 In the intramolecular photochemical reaction of compound 63, the cycloaddition product 64 resulting from the [4 + 2] photocycloaddition and product 65 resulting from a [4 + 4] cycloaddition are formed. The formation of the latter compound indicates that the reaction mechanism is different from the one suggested in Scheme 11 for the transformation of a similar benzophenone derivative. In contrast to the [4 + 2] cycloaddition, a concerted [4 + 4] cycloaddition at the singlet ground state is forbidden according to the Woodward–Hoffmann rules.53 In order to get a deeper insight into the mechanism, intensive photo-physical and theoretical studies have been carried out.54 Based on the results the following mechanism has been suggested (Scheme 13). After photochemical excitation of 66, a proton is transferred to the S1 state and the corresponding imine–enol 67 is formed. Intersystem crossing (isc) occurs and the triplet diradical 68 is formed. This intermediate possesses lifetimes in the range of τ ≈ 5.2–5.7 ns which is long enough to enable intramolecular trapping. The highly reactive nitrogen centered radical adds to the furan moiety which leads to the diradical intermediate 69. Radical combination occurs in two competitive ways. Pathway a generates the major compound 70 (formal [4 + 2] cycloaddition) while pathway b leads to the minor product 71 (formal [4 + 4] cycloaddition). Depending on structural variation in the substrates, the corresponding triplet diradical intermediates of type 68 undergo typical side reactions such as trapping with triplet oxygen or intramolecular hydrogen abstraction. The isc step plays an important role in the outcome of the reaction. The influence of the substitution pattern in 66 on the quantum yield of the photocyclization has been discussed in this manner (Table 2). When a bromine atom is added the quantum yield considerably increased (entries 2 and 3). Thus the introduction of bromine in position 5 (entry 2) leads to the highest value measured in this series of substrates. This effect was explained by a higher spin orbit coupling (SOC) due to a heavy atom effect. The significantly stronger effect in the case of a substitution in position 5 (entry 2) when compared to the substitution in position 4 (entry 3) was explained by the higher spin density on the bromine atom in position 5 of the corresponding intermediate 68. The spin densities have been determined by DFT calculations. As shown in the case of chlorine substitution (entry 4), electronegative atoms in the 5 position have also a favorable effect since they diminish the stability of the nitrogen centered radical in the intermediate 68. Concomitantly, its reactivity is increased. However, the effect of SOC on a more efficient isc is stronger in the case of chlorine. The SOC is also influenced by the rotation of the hydroxyl alkyl radical position in intermediate 68. It should further be noted that the quantum yield also depends on the R substituent. This should be small in the case of cyclic (tetralone) derivatives. A significant decrease of the quantum yield is also observed when hydrogen is replaced by a methyl group (entry 5).
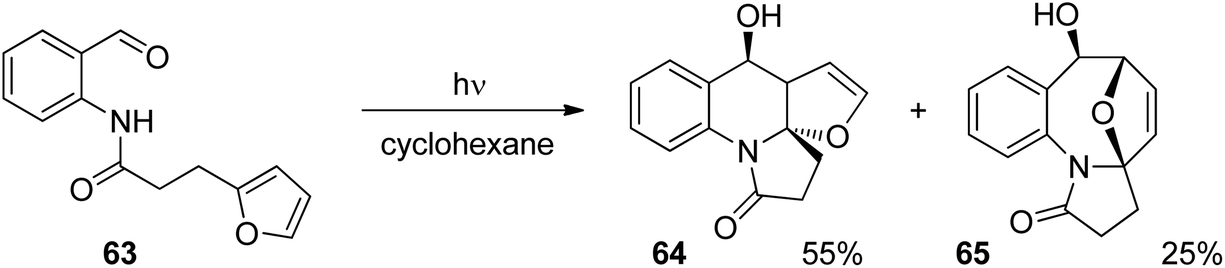 | ||
| Scheme 12 Intramolecular photocycloaddition with an anthranilic acid derivative. | ||
 | ||
| Scheme 13 Mechanism of the intramolecular photocycloaddition with an anthranilic acid derivative. | ||
| Entry | Compound 66 | Quantum yield (Φ) of photocyclization |
|---|---|---|
| 1 | X = H, R = H | 0.18 |
| 2 | X = 5-Br, R = H | 0.75 |
| 3 | X = 4-Br, R = H | 0.22 |
| 4 | X = 5-Cl, R = H | 0.32 |
| 5 | X = H, R = Me | 0.058 |
These photochemical reactions are now intensively investigated in the context of diversity oriented synthesis.55 They provide an access to compound families which are not available or difficult to achieve by more conventional ground state reactions. The previous examples also nicely illustrate how the change of the addition of functional groups on the side chain of an acetophenone moiety modifies the reaction pathways and thus yield products of new compound families. Recently, this has been demonstrated for the transformation of epoxides.56 The photochemical generation of radical species followed by hydrogen abstraction is also used for pharmaceutical applications such as cancer therapy. A typical example is the photo-Bergman reaction and related processes.57
3 Photochemical heterolytic cleavage – the structure and the reaction of aryl cations
Phenyl cations possess low stability. Only recently, the parent compound was generated and its reactivity was studied in cryogenic argon matrices.58 When substituted with electron donor groups such compounds become more stable. They are generated from the corresponding halogen derivatives (72) by photolysis and can act as intermediates in photochemical reactions (Scheme 14).59,60 The phenyl cation species possess singlet (73) or triplet spin multiplicity (74). Upon direct excitation by light absorption, the singlet state 73 is generated after release of the Cl− ion. For phenyl cations possessing strong electron donor substituents such as amino functions, the triplet energy is generally lower. Thus the triplet state 74 is generated by intersystem crossing. In the case of the presence of weaker electron donors such as 75 the singlet state is less energetic (Fig. 5).61 Nevertheless, some typical structural parameters for each spin multiplicity remain comparable. Thus the angle ∠(C3–C4–C5) for the singlet state (75) is larger than for the triplet state (76). In the first case, the partial structure is ketene-like while in the second case, it resembles a carbene.62 Various computational studies have been carried out on these structures.63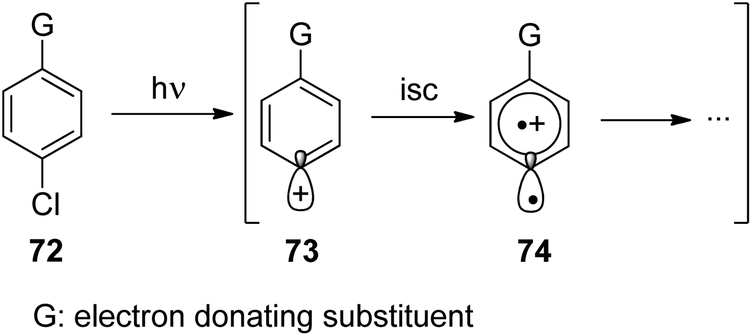 | ||
| Scheme 14 Generation and structure of phenyl cation derivatives. | ||
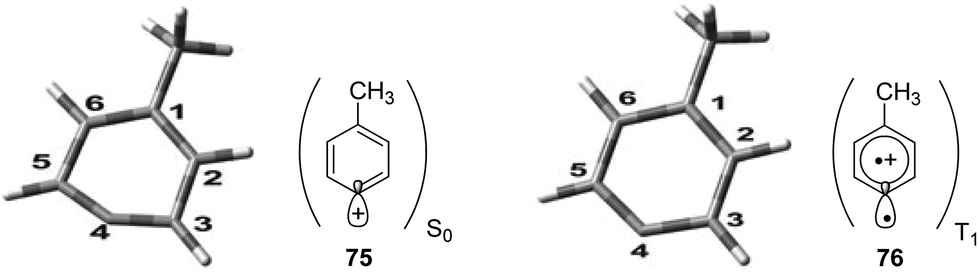 | ||
| Fig. 5 Calculated structures for the singlet and the triplet state of the phenyl cation 75, 76. E(T1)–E(S0) = 13.99 kcal mol−1. Angle ∠(C3–C4–C5) = 147° for the S0 and ∠(C3–C4–C5) = 127° for the T1 state. (Adapted with permission from: H. Qrareya, C. Raviola, S. Protti, M. Fagnoni and A. Albini, J. Org. Chem., 2013, 78, 6016–6024. Copyright (2013) American Chemical Society.) | ||
The phenyl cations easily react with a variety of nucleophilic species. In the case of a p-(N,N-dimethylamino)phenyl cation at the singlet state and the triplet state, rates for the reaction with different nucleophiles were determined.64 The results have been discussed with the aid of Mayr's parameters for nucleophilicity and electrophilicity.65
The addition of such phenyl cations to nucleophilic species is of great interest in organic chemistry. The reaction of compound 77 with the pyrrole derivative 78 leading to the coupling product 79 resembles a Suzuki Miyaura reaction (Scheme 15).66 However no palladium catalysis is needed. Furthermore, no borate substrates are involved which considerably facilitates the transformation. Only minor amounts of side products 80 and 81 are formed. The transformation of 82 with 83 resembles a Sonogashira reaction. Again the corresponding product 84 is formed avoiding any metal catalysis.59 Various other nucleophiles have been successfully transformed under similar conditions. Thus the allyl silane compound 85 reacts with chloroanisole 86 (R = Me) yielding 87.67 Interestingly, the unprotected chlorophenol 86 (R = H) was also efficiently transformed and the corresponding products 87 were obtained in high yields. Enol ethers such as 88 were transformed into 89 without any Lewis acid catalysis. The resulting ketone 90 was isolated in high yields.68 Reactions with alkenes possessing further functional groups such as 4-pentenoic acid 91 were successfully transformed, for example with chlorophenol 92.69 In an intramolecular trapping reaction, a butyrolactone derivative 93 is formed. It should be pointed out that this reaction was also carried out with phenol or aniline derivatives possessing other leaving groups such as chlorine. Thus triflates, phosphates or mesylates were transformed.
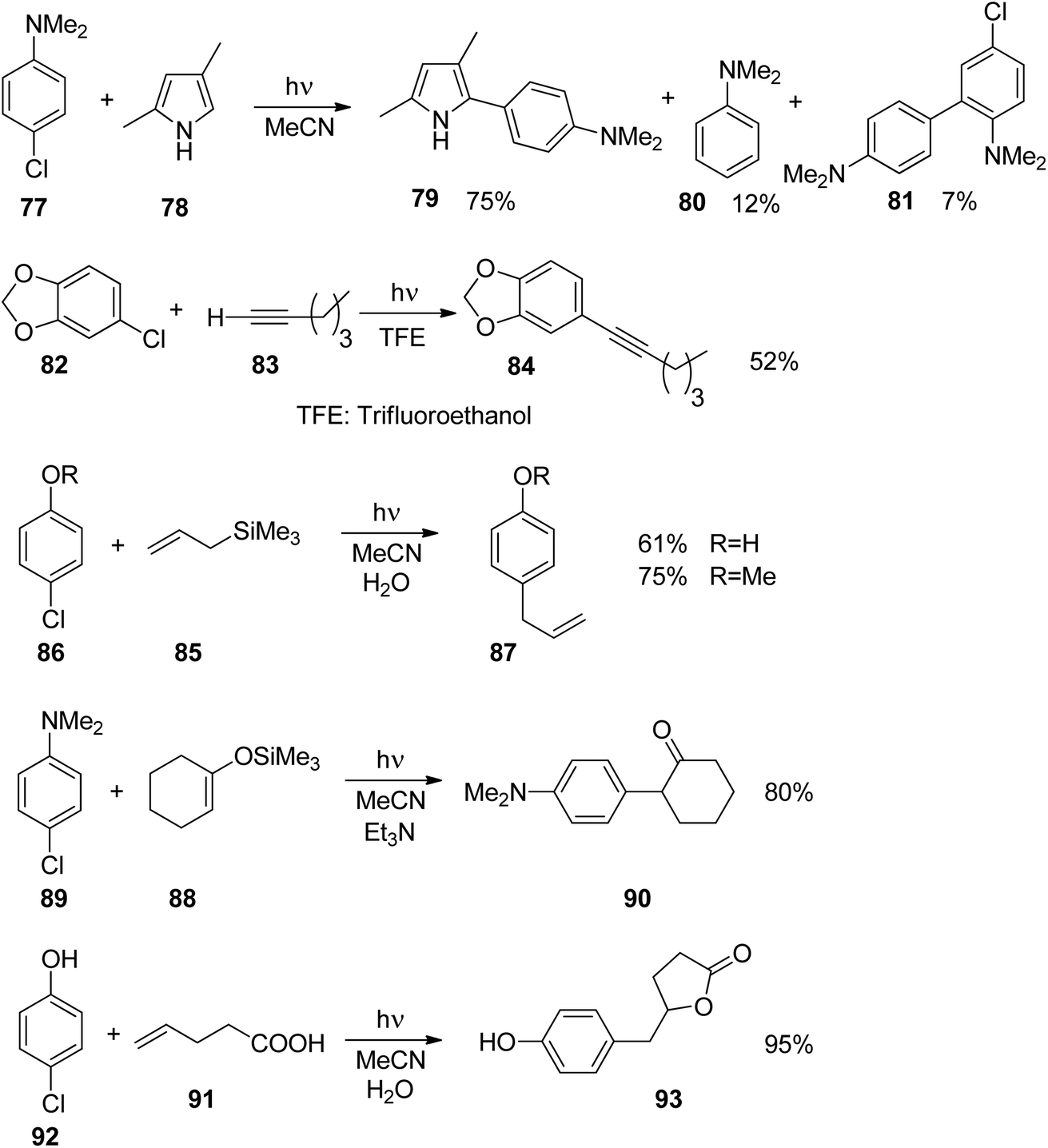 | ||
| Scheme 15 Reactions involving phenyl cations with various nucleophilic species. | ||
4 [2 + 2] Photocycloadditions
The photochemical reaction between carbonyl compounds and alkenes is certainly the most versatile method for the synthesis of oxetanes.70–72 These small heterocycles are encountered in some natural products. But they are also interesting synthesis intermediates. The generally accepted mechanism in most of these reactions is depicted in Scheme 16.72,73 The reactions start with the excitation of the carbonyl compound 94 to the nπ* state. The Paternò–Büchi reaction may take place at the singlet state or the triplet state.74 In the first case, the excited carbonyl function is added to the alkene leading to the formation of the biradical intermediate 95. In the case of a triplet reaction, intersystem crossing (isc) takes place. This step is particularly fast in the case of aromatic compounds such as benzaldehyde or benzophenone (kisc ∼ 1011 s−1). Therefore, many of the chemical reactions start at the triplet state. The reaction with an alkene generates a 1,4-diradical intermediate 95 by the formation of a C–O bond. In order to get the final product at its singlet ground state, intersystem crossing must take place. Radical combination leads to the oxetanes 96. Alternatively, the diradical 95 may generate the starting products. It must be pointed out that the second isc and the formation of the oxetane 96 or the formation of the starting products by cleavage of the newly formed C–O bond are strongly coupled (see below). Depending on the redox potentials of the substrates, photochemical electron transfer may also occur which leads to a radical ion pair 97. In this case, the oxetane 96 is formed preferentially via the singlet diradical intermediates 95 or is directly generated. In such reactions the intermediate possesses zwitterionic character.75,76 Unsaturated alcohols 98 are typical competitive products of this mechanism involving electron transfer. In this case, a C–C bond may also be formed. The two reaction pathways, involving electron transfer or not, have a different impact on the regioselectivity of the reaction.77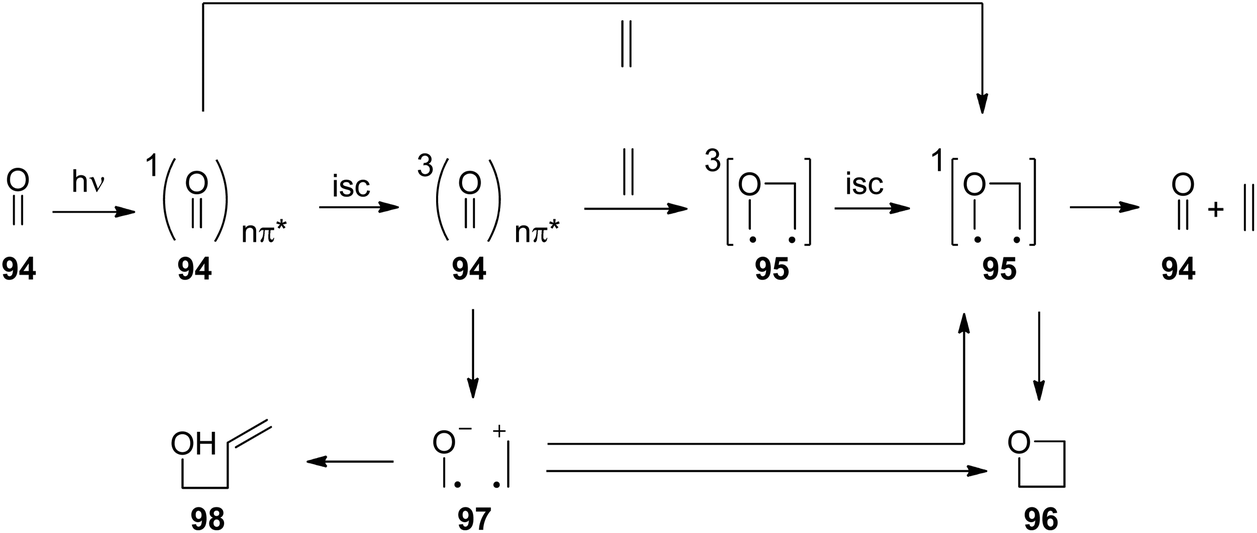 | ||
| Scheme 16 Mechanism of the Paternò–Büchi reaction. | ||
When electronically excited, the polarity of the carbonyl group is inversed with respect to the ground state. Thus the carbonyl oxygen becomes electrophilic and attacks an alkene at its most nucleophilic side. Therefore, the oxetanes 99a,b are the only products isolated from the reaction of dihydrofuran 100 with benzaldehyde 101 (Scheme 17).78 No electron transfer is involved in the formation of this isomer.79 The formation of a solvent separated ion pair is endothermic (ΔGet = +0.62 eV). The regioselectivity is again inversed when electron transfer is involved76 as in the case of the transformation of benzaldehyde 101 with the more electron rich dihydrofuran derivative 102 possessing a ketenacetal moiety (Scheme 17).80 A radical ion pair 103 and 104 is formed. This reaction step is exothermic by ΔGet ∼ −0.3 eV (compare ref. 77). In the radical cation 103, the positive charge is essentially localized on the carbon atom carrying the two oxygen atoms. This polarity determines the regioselective formation of product 105. This orthoester is easily transformed into the butyrolactone derivative 106 by hydrolysis. It must further be pointed out that in the reaction of dihydrofuran 100, the formation of the endo isomer 99a is favored while in the case of the transformation of compound 102, the thermodynamically favored exo diastereomer 105 is mainly formed. Concerning the understanding of the diastereoselection in the Paternò–Büchi reaction a lot of investigations have been carried out. Thus it has been evidenced that the 1,4-diradical intermediate 95 (Scheme 16) plays a key role in the diastereoselectivity of the reaction of α-ketoesters carrying a chiral auxiliary. The competition between the cyclization on the one hand and the formation of the starting materials (ketone and alkene) on the other hand contributes to the diastereoselectivity.51,81 By measuring the temperature dependence of the stereoselectivity the contribution of the enthalpy and the entropy to the activation free enthalpy of each of the two steps have been determined and discussed.
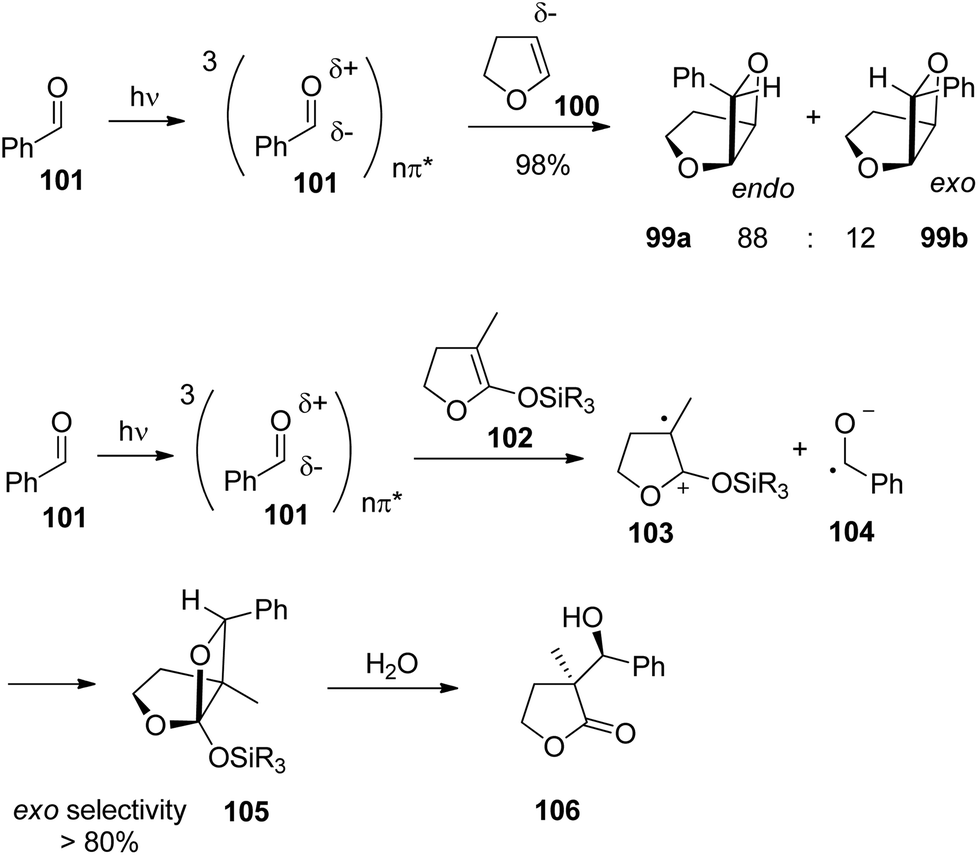 | ||
| Scheme 17 Regioselectivity of the Paternò–Büchi reaction. | ||
The herein reported Paternò–Büchi reactions have been carried out with electron rich alkenes. The reaction was also carried out with carbonyl compounds at the S1 state and electron deficient alkenes. In this case, an interaction between the alkene π system and the π* orbital of the carbonyl compound was discussed.82 In such an interaction, the nπ* excited carbonyl function acts a nucleophile. In this context, an amphoteric nature was attributed to the excited carbonyl group.
The exo/endo selectivity was also investigated in detail. When derivatives of benzaldehyde 107 were transformed with dihydrofuran 100 the endo isomers of the oxetanes were formed in excess (Table 3 and Scheme 18) and high yields.15,78,83 The endo isomers 108 are less stable than the corresponding exo isomers 109 due to their higher steric hindrance. By increasing the steric encumbrance by increasing the size of R, one should expect that endo/exo ratio is reduced. However, the opposite effect is observed. By increasing the size of this substituent (going from entry 1 to entry 3 in Table 3), the endo/exo selectivity is even improved while the product yield remains high. Increased steric hindrance on the olefinic reaction partner only slightly affects this selectivity.84 In contrast, the corresponding naphthalene derivatives 113 yielded the exo isomers 114 selectively (entries 4 and 5). Unlike the benzaldehyde derivatives, the naphthylaldehydes react at the singlet state yielding the thermodynamically more stable exo products. In the triplet reaction, 1,4-diradicals such as 110, 111 or 112 are formed themselves possessing triplet multiplicity also. In order to obtain the final products 108 or 109, an intersystem crossing must take place (Scheme 18). As discussed in the previous section on the Norrish–Yang reaction this process is favored by a strong spin orbit coupling.14 Spin orbit coupling in such intermediates is increased when the two p orbitals carrying the unpaired electrons are orthogonally orientated (115). Among numerous possibilities, the three conformations 110, 111 and 112 are significant. Due to low steric hindrance, conformer 110 is dominant in the conformational equilibrium. Cyclization leads to the formation of the thermodynamically disfavored endo product 108. Cyclization of conformers 111 and 112 in which the two p orbitals carrying the unpaired electrons are also orthogonally orientated would lead to the exo products 109. By increasing the steric hindrance by modification of the phenyl substituent R, the dominance of conformer 110 in the equilibrium is enhanced and therefore the formation of the endo portion of product 108 is increased. In the case of the transformation of the naphthylaldehydes 113 which react at the singlet excited state, the corresponding diradical intermediate 116 can react immediately. No change of the spin multiplicity is required. The cyclization step occurs at the conformation 116 in which the naphthyl substituent is placed in the sterically favored exo position. It should further be pointed out that the product yields are lower. The triplet multiplicity is advantageous for the Paternò–Büchi reaction. A detailed analysis of conformational equilibria and spin orbit coupling in these Paternò–Büchi 1,4-diradicals using high level ab initio methods has been recently performed.85
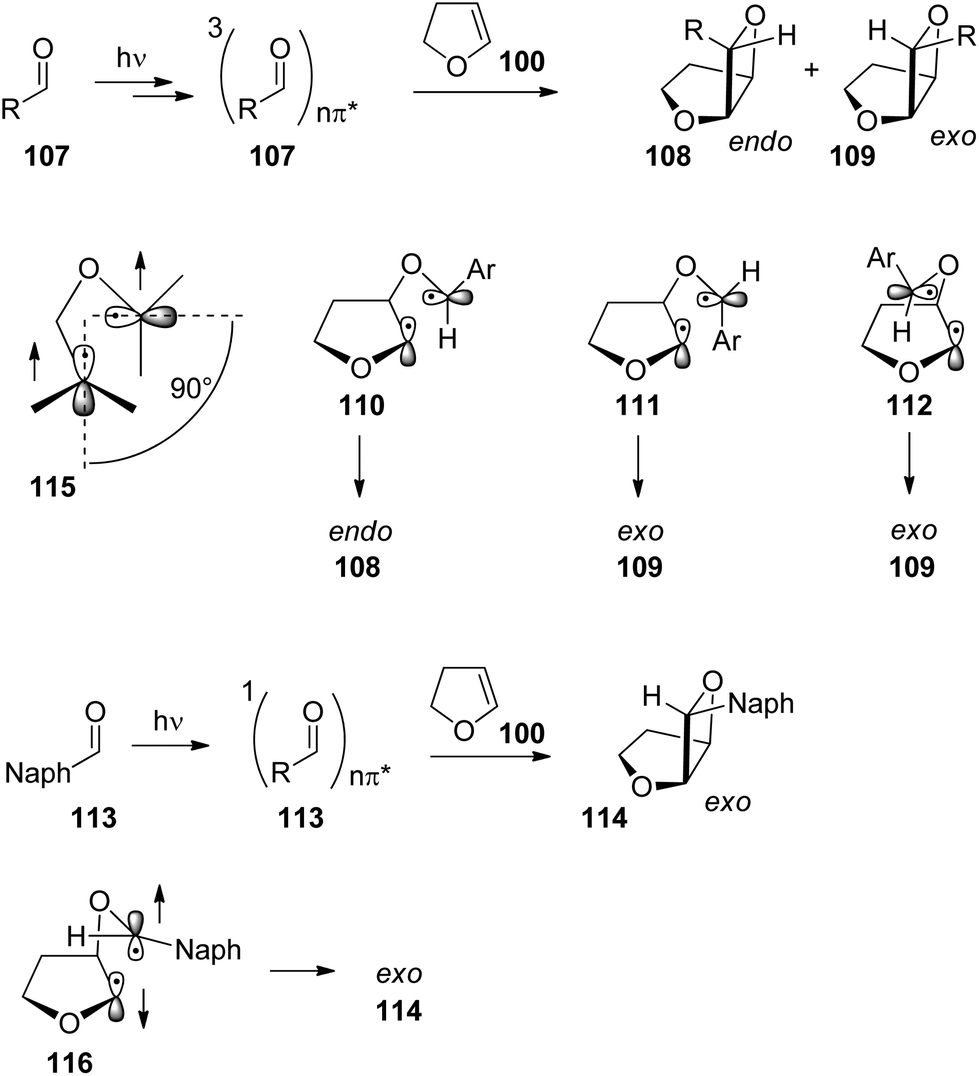 | ||
| Scheme 18 Mechanisms of the exo/endo stereoselection in the Paternò–Büchi reaction. | ||
| Entry | R |
endo:exo |
Yield (%) |
|---|---|---|---|
| 1 | Phenyl | 88:12 |
98 |
| 2 | o-Tolyl | 93:7 |
97 |
| 3 | Mesityl | >98:2 |
90 |
| 4 | 1-Naphthyl | <2:98 |
55 |
| 5 | 2-Naphthyl | <2:98 |
87 |
In the case of aliphatic aldehydes such as propionaldehyde 117 the endo/exo selectivity depends on the concentration of the alkene (dihydrofuran 100) in the reaction mixture (Scheme 19 and Fig. 6).16,86 At low concentrations, the endo product 118 is mainly formed while at higher concentrations both products are obtained in almost equal amounts. Obviously, singlet and triplet diradical intermediates are generated. The triplet diradicals are formed in a unimolecular process with kisc ranging from 109 to 1012 s−1. Thus diffusion has an influence on the competitive bimolecular formation of singlet or triplet diradicals. The fluorescence quenching rate of propionaldehyde 117 by dihydrofuran 100 is kq = 2.2 × 109 M−1 s−1 with a corresponding lifetime τ(S1) = 1.8 × 10−9 s.87 In the case of high concentrations, singlet diradicals are generated leading to low endo/exo selectivity while at low alkene concentration, unimolecular inter system crossing is faster and 117 is transformed only at its triplet state in a bimolecular reaction with 100. In the latter case, the thermodynamically disfavored product 118 was formed in large excess. A more detailed kinetic evaluation of the plot in Fig. 6 revealed that at a concentration of 0.05 M of alkene 100, almost equal amounts of singlet and triplet excited propionaldehyde molecules are trapped in the bimolecular reaction leading to an endo/exo ratio of about 68 to 32. It must be pointed out that the transformation of benzaldehyde 101 with 100 under the same conditions (compare Scheme 18 and Table 3) yielded the corresponding endo/exo isomers with ratios around 88:12 regardless of the concentration of the alkene 100. Aromatic aldehydes generally possess low singlet lifetimes and particularly high values of kisc so that only triplet species of the carbonyl compound are trapped in the bimolecular reaction. The viscosity of the reaction mixture also has an influence on the endo/exo selectivity. At higher viscosity, diffusion and consequently the bimolecular trapping of the excited carbonyl species are slowed down. The formation of endo isomers resulting from triplet diradicals is thus favored.87
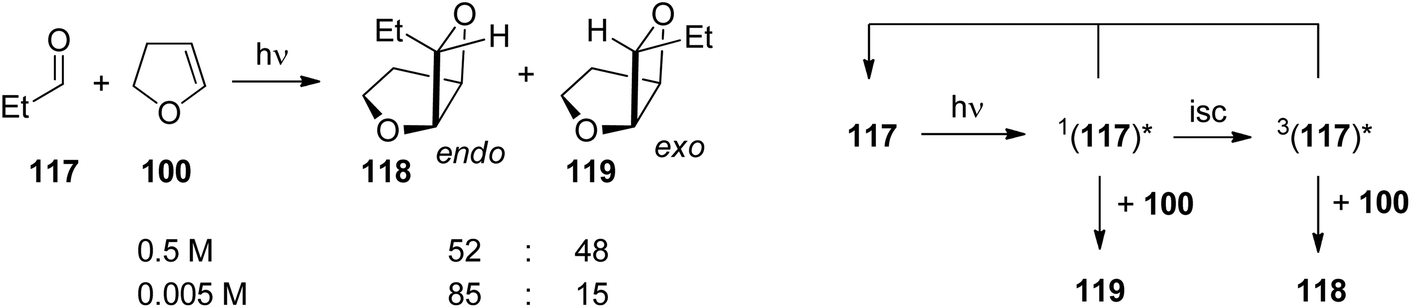 | ||
| Scheme 19 Concentration dependence of the exo/endo-selectivity of the Paternò–Büchi reaction on aliphatic ketones. | ||
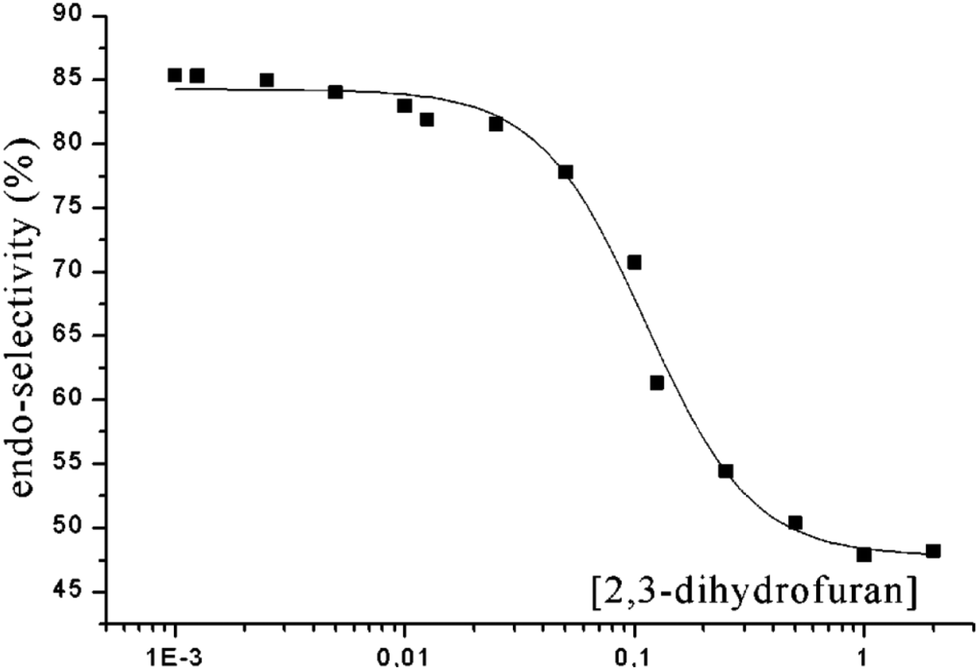 | ||
| Fig. 6 Dependence of the endo/exo selectivity (ratio of endo to exo isomers) in the reaction of 117 with 2,3-dihydrofuran 100 on the concentration of 100 (compare Scheme 19). (Adapted with permission from: A. G. Griesbeck, M. Fiege, S. Bondock and M. S. Gudipati, Org. Lett., 2000, 2, 3623–3625. Copyright (2000) American Chemical Society.) | ||
The viscosity is also influenced by the temperature. Thus it was possible to change from a singlet reaction in a reaction medium with low viscosity and favored formation of the exo isomer to a triplet reaction in the same reaction medium with high viscosity and favored formation of the endo isomer by decreasing the temperature. Therefore, at a particular temperature, a sudden change of the temperature dependence of the endo/exo selectivity occurs. Nonlinear Eyring plots are observed. Such a phenomenon has frequently been observed in stereoselective reactions.16,51 In the present case, it is related to a change in mechanism induced by the reaction medium.88,89 Often such changes of selectivities in chemical reactions depending on the temperature are linked to enthalpy–entropy compensation.89,90
Nonlinear Eyring plots are also observed for the regioselectivity of benzophenone with methylfuran.91 Two cases are discussed. In the first one, the regioselectivity is determined by the approach of the triplet excited carbonyl compound to the furan derivative. In the second one, the regioselectivity also depends on the conformation equilibrium in the 1,4-diradical intermediates. Conformations leading to oxetanes via cyclization are in equilibrium with conformers leading to the starting products via fragmentation of the newly formed C–O bond. Although there is no photochemical electron transfer involved and in contrast to the reaction with dihydrofuran, the carbonyl oxygen of benzaldehyde is added in position 2 to furan derivatives such as in the case of 2-methylfuran 120 (Scheme 20).72,91 This regioselectivity is explained by the higher HOMO coefficient in position 2. Furthermore, and also in contrast to the same reaction with dihydrofuran, the thermodynamically more stable exo isomers 121 and 122 are formed in excess.16,72,91 This particular exo/endo selectivity is explained by the fact that the conformational equilibrium of the 1,4-diradicals is shifted to the structures 123 and 124. The benzyl radical is located above the furan moiety. This sterically less favored arrangement is induced by an anomeric effect92 which is caused by the interaction of an n-orbital of the initial oxygen atom with the σ*-orbital of the C–O bond of the furan moiety as indicated in structures 123 and 124 (compare also ref. 85). The endo isomers 125 and 126 are formed from the corresponding diradical intermediates 127 and 128 respectively.
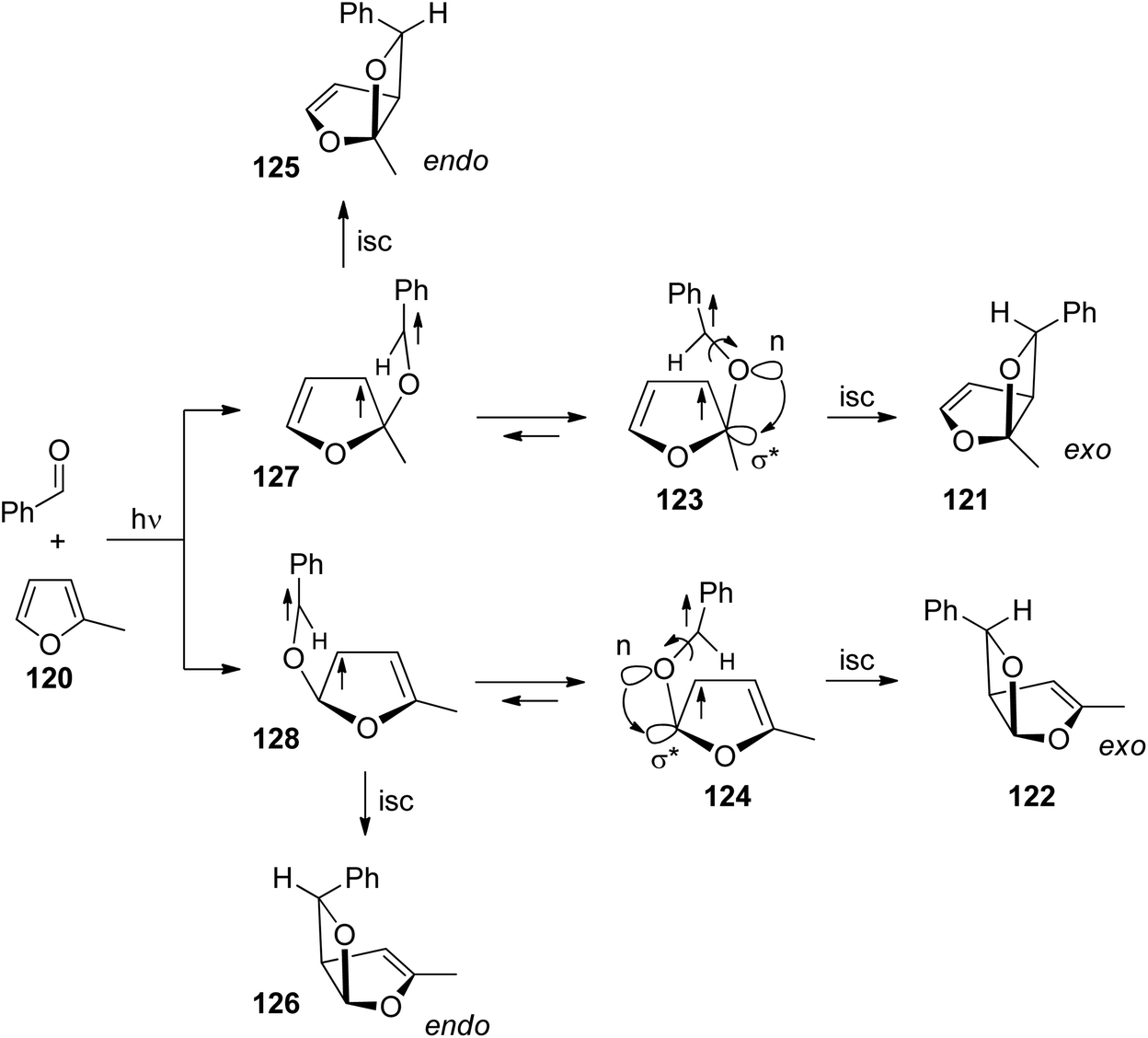 | ||
| Scheme 20 Electronic effects influencing the exo/endo-selectivity of the Paternò–Büchi reaction. | ||
Hydrogen bonding is a powerful directing element also in photochemical reactions. Hydrogen bonding of the carbonyl group has also an impact on the regio- and stereoselectivity of the Paternò–Büchi reactions.93 It orients the substrates with respect to each other. The effect was successfully applied, for example, to the asymmetric Paternò–Büchi reaction.94 For further examples of photocycloadditions see ref. 95. Concerning hydrogen bonding at the 3nπ* excited carbonyl function, it must be pointed out that such an interaction should be significantly weaker than a hydrogen bond at the corresponding ground state carbonyl function.96 Due to the inversion of polarization at the excited state, the carbonyl oxygen is electrophilic, the energy of the hydrogen bond and its influence on the regio- and stereoselectivity in the Paternò–Büchi reaction should therefore be low.3,97,98 A recent and very detailed investigation provides deep insight into the different influences of hydrogen bonding of an electronically excited carbonyl function in the Paternò–Büchi reaction. When the bicyclic furan derivative 129 was transformed with benzophenone 130, only trans adducts 131 and 132 were formed (Scheme 21).99 These products result from the addition of 3nπ* excited benzophenone 130. The reaction was carried out in toluene as a non-polar and aprotic solvent. The regioselectivity slightly depends on the temperature. At low temperature, the sterically less hindered regioisomer 131 is formed in somewhat higher quantities. Nevertheless, it remains the minor product. The privileged formation of compound 132 may be explained by the higher electron density at the higher substituted CC bond of the furan moiety. No major influence of the alkene (129) concentration on the product ratio was detected. The stereoselectivity is essentially controlled by the steric hindrance of the methoxy substituent.
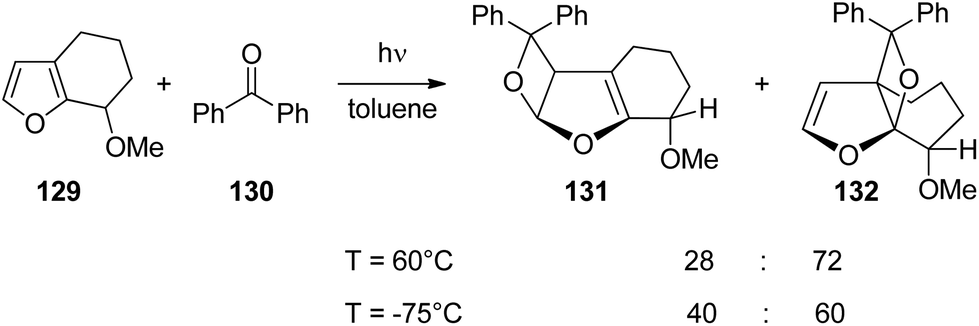 | ||
| Scheme 21 Temperature dependence of the regioselectivity of the Paternò–Büchi reaction. | ||
When the same reaction was carried out with the unprotected alcohol 133 in toluene as the non-polar and aprotic solvent, both cis (134a,b) and trans products (135a,b) were formed (Scheme 22 and Table 4).99 These products were obtained in yields between 63% and 91%. Benzpinacol was a major side product. The formation of considerable amounts of the cis products 134a,b was attributed to a directing effect caused by hydrogen bonds. The stereoselectivity and the regioselectivity are influenced in a characteristic manner by the temperature and the concentration of the alkene 133. Since the hydrogen bond between the electronically excited benzophenone 130 and the hydroxyl function of 133 is of low energy, its effect on the regio- and stereoselectivity is reduced and hydrogen bond formation with the reaction medium also becomes relevant. This competition is nicely shown by the present results. When the reaction is carried out in DMSO as the solvent (Table 4, entry 7), only trans products 135a,b were isolated as in the case of the reaction of the methyl ether 129 (Scheme 21). The hydroxyl function in 133 is thus “protected” from complexation by the electronically excited benzophenone. Furthermore, it has been shown by NMR studies that the complexation of benzophenone at the ground state with compound 133 by hydrogen bonds depends on the temperature and the concentration of 133.99 Significant complexation in dry d8-toluene was observed at −60 °C and an alkene concentration of 100 mM. At higher temperatures or at lower alkene concentrations, hydrogen bond formation was considerably reduced. In light of these additional studies, the results reported in Table 4 may be interpreted as follows. At high temperature (entries 1–3), no significant influence of the alkene concentration on the stereo- and the regioselectivity is detected. Formation of hydrogen bonds between molecules of the alkene 133 is low. Therefore hydrogen bond formation between the substrates, the electronically excited benzophenone and the hydroxyfuran 133 becomes competitive at the transition state of the reaction. In the case of the regioisomers 134b and 135b, the formation of the cis isomer 135a dominates. In the case of the regioisomers 134a and 135a, this effect is weaker and the formation of the trans isomer is favored 135a. Hydrogen bonding also strongly directs the ketone into a sterically disfavored position leading to the formation of the regioisomers 134b and 135b in large excess. At low temperature (entries 4–6), hydrogen bond formation between the hydroxyl functions of molecules of alkene 133 becomes competitive to the complexation of electronically excited benzophenone. At low alkene concentration (entry 6) this effect is still weak and the product ratio still resembles those at high temperature. However, at a high alkene concentration (entry 4), the influence of hydrogen bonding between alkene 133 and the excited benzophenone is low. Due to the presence of larger amounts of the hydroxyl compound 133, the reaction medium can't be considered anymore as an aprotic one. In all cases, the formation of the trans isomer is dominant (entries 4–6). This result may be compared to the results obtained from the transformation of the methylether derivative 129 (Scheme 21). Also, the formation of the sterically hindered regioisomers 134b and 135b becomes less favorable. Although, its influence has been reduced, hydrogen bonding between the electronically excited benzophenone and the hydroxyfuran derivative 133 remains important in the control of the stereo- and the regioselectivity of this Paternò–Büchi reaction.
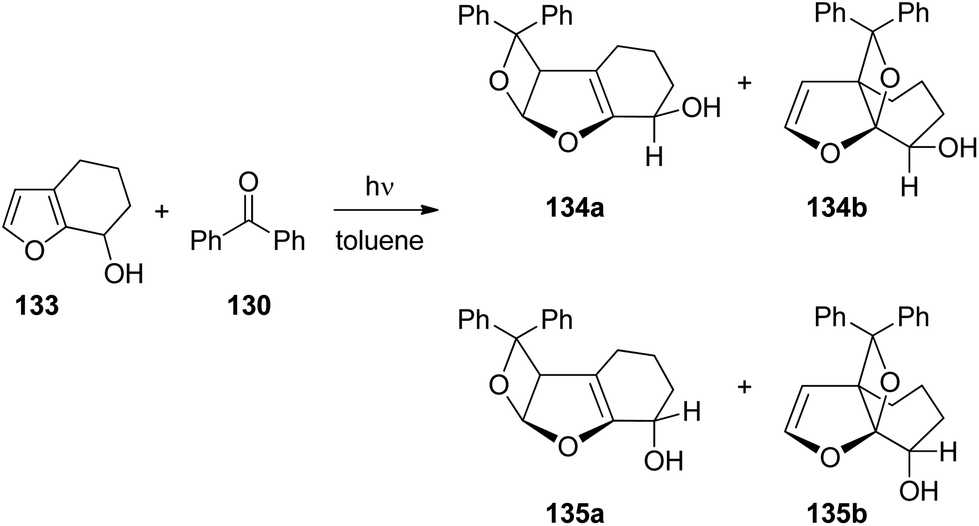 | ||
| Scheme 22 Regio- and diastereoselective Paternò–Büchi reaction by a hydroxyl function. | ||
| Entry | Temp (°C) | [133] (mM) | Stereoselectivitya | Regioselectivitya | ||
|---|---|---|---|---|---|---|
| trans/cis (135a/134a) | trans/cis (135b/134b) | trans (135a/135b) | cis (134a/134b) | |||
| a The selectivities are normalized to 100%. b The reaction was performed in a dry and degassed DMSO solution. | ||||||
| 1 | 60 | 1000 | 70/30 | 41/59 | 22/78 | 8/92 |
| 2 | 60 | 100 | 67/33 | 41/59 | 21/79 | 7/93 |
| 3 | 60 | 10 | 67/33 | 42/58 | 21/79 | 8/92 |
| 4 | −75 | 1000 | 85/15 | 81/19 | 53/47 | 47/53 |
| 5 | −75 | 100 | 81/19 | 63/37 | 47/53 | 26/74 |
| 6 | −75 | 10 | 79/21 | 51/49 | 45/55 | 19/81 |
| 7b | 20 | 340 | >97/3 | >97/3 | 31/69 | — |
In order to better characterize the unexpectedly strong hydrogen bonding at the 3nπ* excited benzophenone, comprehensive computational studies were carried out. Early studies estimated the stabilization energy of hydrogen bonded 3nπ* excited formaldehyde with water to be 0.2–0.4 kcal mol−1 while the corresponding interaction at the ground state was estimated to be 3.5 kcal mol−1.96 Recent studies of the stabilization energy of hydrogen bonded 3nπ* excited formaldehyde with methanol was calculated to range between 0.5 and 0.7 kcal mol−1 and the corresponding interaction at the ground state was found to be 4.68 kcal mol−1.99 Somewhat higher stabilization energies were calculated for the hydrogen bonds with benzophenone. Values between 2.1–3.2 kcal mol−1 for the 3nπ* excited state and values between 4.5–6.0 kcal mol−1 for the ground state were found. To get further information on the interaction between 3nπ* excited carbonyl compounds and furan derivatives, the Mulliken charge at the carbonyl oxygen atom was determined during the addition of the photochemical excited formaldehyde or benzophenone 130 to the furan 136. The benzophenone case is depicted in Fig. 7. The approach occurs via transition state TS1 leading to the formation of an exciplex. The diradical intermediate 137 is then formed via transition state TS2. At the beginning of this reaction step, the Mulliken charge on the oxygen is around −0.13. This increases until reaching a maximum of −0.26 near the transition state TS2. In the diradical intermediate 137 the Mulliken charge of the initial carbonyl oxygen was calculated to be −0.13. These results clearly show that this oxygen is particularly basic at TS2 and at this stage, hydrogen bonding is most efficient in controlling the stereoselectivity. The effect is less pronounced before TS1 and, more importantly, in the 1,4-diradical 137.
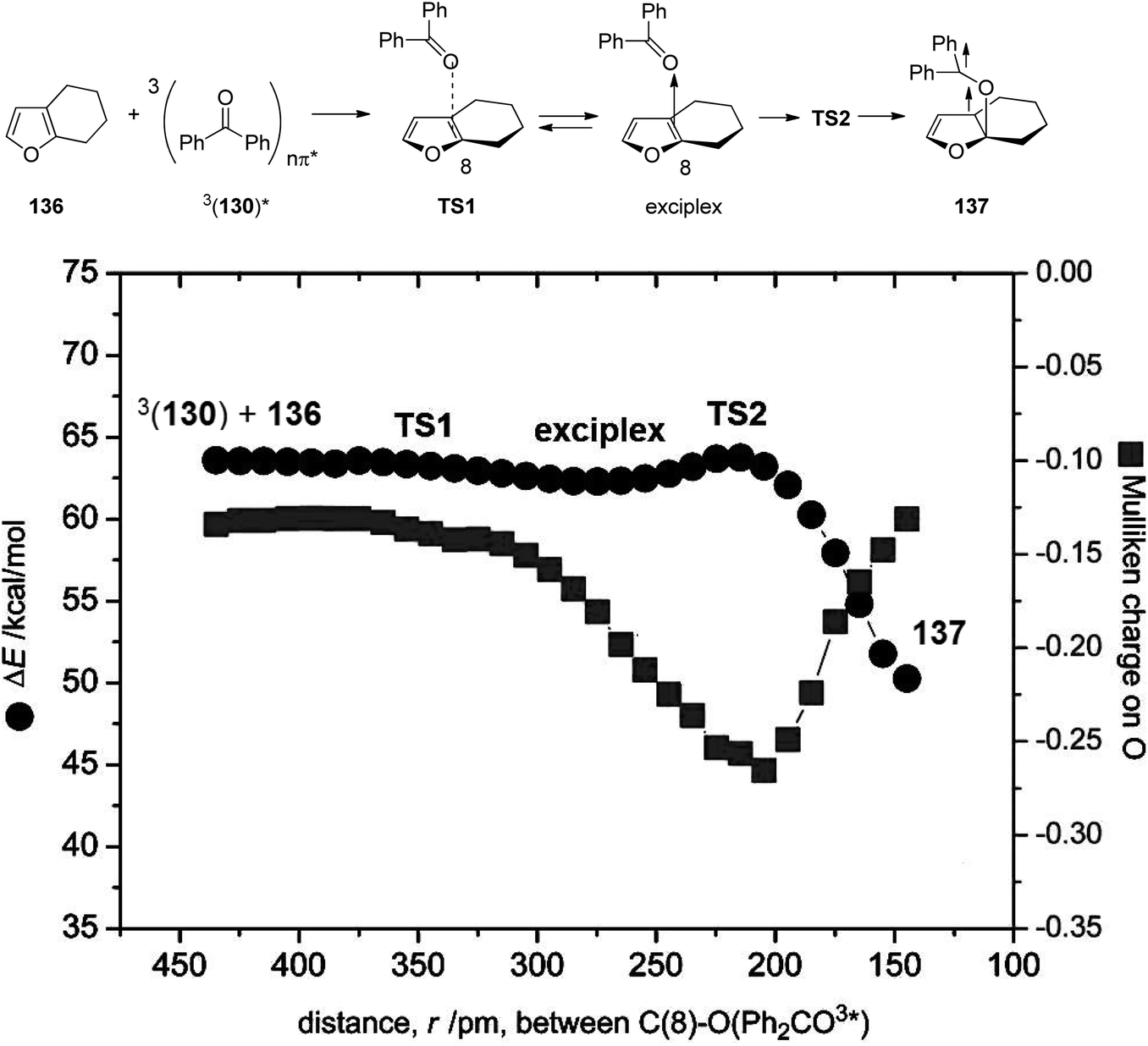 | ||
| Fig. 7 UB3LYP/6-31+G(d) analysis of the energy profile and the distance dependent Mulliken charge of the carbonyl oxygen during the approach of 3nπ* excited benzophenone to 136 and the C–O bond formation between the two species. (Adapted with permission from: Y. Yabuno, Y. Hiraga, R. Takagi and M. Abe, J. Am. Chem. Soc., 2011, 133, 2592–2604. Copyright (2011) American Chemical Society.) | ||
The hydrogen bonding influence on the cis/trans selectivity in the reaction is described in Scheme 23 and Table 4. It is particularly strong at the exciplex and at its irreversible transformation into the 1,4-diradical intermediate. A computational study was carried out on the reaction of 3nπ* excited benzophenone with the bicyclic hydroxyl furan derivative 133. The results for the formation of the diastereoisomers 134b and 135b are depicted in Scheme 23.99 A considerable energy difference of 1.4 kcal mol−1 due to hydrogen bonding has been calculated for the cis-TS and the trans-TS. For the corresponding diradical intermediates cis-DR and trans-DR the energy difference is only 0.2 kcal mol−1 in favor of the cis isomer. These two intermediates undergo either cyclization leading to the final products or they undergo fragmentation leading to the starting products 130 and 133. The intermediates are also in equilibrium with the anti conformers cis-anti-DR and trans-anti-DR. The energy difference between the cis-anti and trans-anti conformer is large (2.1 kcal mol−1) in favor of the trans isomer. This high energy difference is essentially caused by steric hindrance in the case of cis-anti-DR. No influence of hydrogen bonding is detected for the anti conformers. Apart from the equilibrium with the syn conformers, the anti conformers may only undergo fragmentation. Thus the tendency to form oxetanes from the cis-approach of the electronically excited benzophenone to 133 is enhanced by hydrogen bonding at the stage of the exciplex and its transformation to the diradical intermediate. This tendency is also increased by the relatively high energy of the corresponding cis-anti-DR which reduces fragmentation of the diradical intermediate to the starting material.
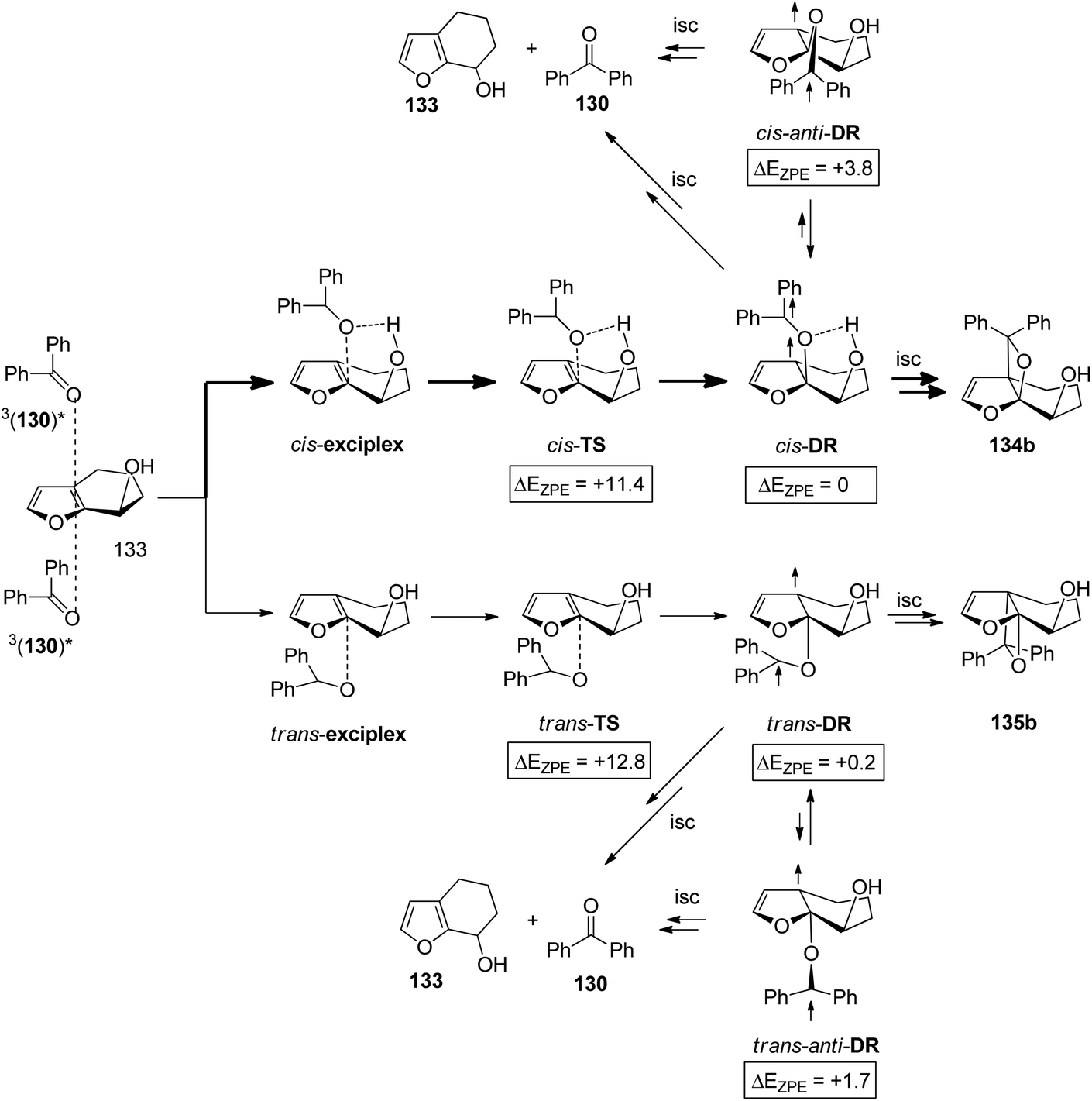 | ||
| Scheme 23 Energies (ΔEZPE values are given in kcal mol−1) for the reaction of 3nπ* excited benzophenone with the furan derivative 133 leading to oxetanes 134b and 135b calculated at the UB3LYP/6-31+G(d) level. | ||
The [2 + 2] photocycloaddition of α,β-unsaturated carbonyl compounds is frequently applied to organic synthesis.100 For this reason, the stereoselectivity is particularly studied. More recently, particular asymmetric catalysis was applied to these reactions.101 Chirality may be induced by complexation of the substrate to a template via hydrogen bonds or Lewis acid complexation.
Also UV absorption of such nitrogen containing heterocycles can also be shifted towards the visible (bathochromic) when efficient Lewis acid complexation takes place. In Fig. 8, UV spectra of compound 136 (Scheme 24) are depicted.102 In the absence of a Lewis acid, an absorption maximum is observed around λ = 290 nm. This maximum is shifted to λ ≈ 345–350 nm. The photochemical reactivity of compounds such as 136 is generally low when they are electronically excited by direct light absorption. This is due to the fast internal conversion from the first excited singlet state S1 to the ground state S0. Lewis acid complexation increases the life time of the S1 state.103 Intersystem crossing to the T1 state becomes competitive which favors [2 + 2] photocycloaddition. Complexation with the chiral Lewis acid 137 was used in the intramolecular asymmetric [2 + 2] photocycloaddition of the cyclic enone 136 (Scheme 24). Chiral induction was efficient when 0.5 equivalent of the Lewis acid was used. The formation of the enantiomer 138 is explained by structure 139. Recently, a computational study on the effect of Lewis acids on the mechanism of such [2 + 2] photocycloadditions has been published.104
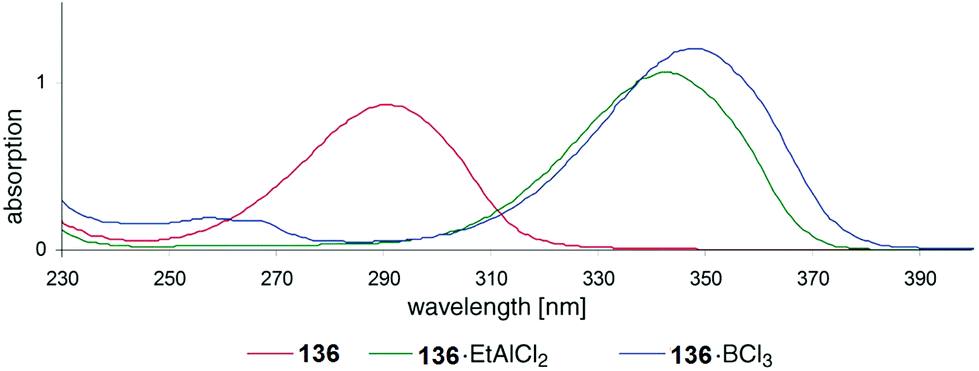 | ||
| Fig. 8 Bathochromic shift of absorption maxima by Lewis acid complexation. (Reproduced with permission from: R. Brimioulle and T. Bach, Science, 2013, 342, 840–843. Copyright (2013) The American Association for the Advancement of Science.) | ||
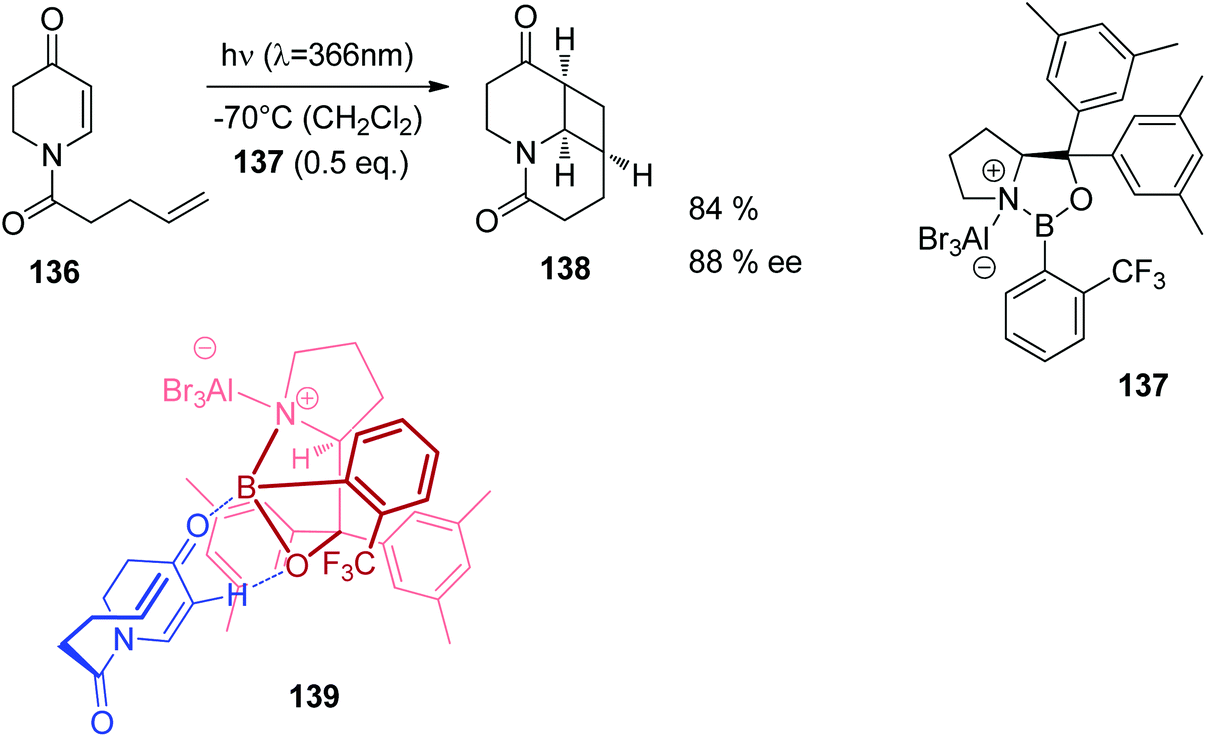 | ||
| Scheme 24 Asymmetric Lewis acid catalyzed [2 + 2] photocycloaddition of the dihydropyridone derivative 136. | ||
Enantioselective [2 + 2] photocycloaddition with visible light and Lewis acid activation was also carried out using photoredox catalysis with [Ru (bpy)3]2+ complexes.105 An intramolecular reaction with two different α,β-unsaturated carbonyl compounds was studied. Eu3+ complexes with peptide ligands were used as Lewis acids. Dimerization of cinnamates via [2 + 2] photocycloaddition is difficult when it is carried out in solution. However, when such compounds are complexed by thiourea derivatives, this reaction becomes efficient, especially when it is carried out in a continuous flow reactor.106 In this case, the reactivity is increased by two hydrogen bonds between the thiourea compound and the carboxyl group of the cinnamate.
5 Photochemical electron transfer induced reactions
Photoinduced electron transfer reactions play a dominant role in the chemistry of life.107 Despite this, their application to organic synthesis started only a few decades ago.108 The understanding of electron transfer in general and of photoinduced electron transfer (PET) processes in particular developed relatively late in the middle of the twentieth century.109,110 Before that time, many PET reactions were originally thought to proceed via homolytic steps.Upon electronic excitation of a compound, its redox properties may become enhanced, either as an electron donor (D) or an acceptor (A). The possibility of a subsequent electron transfer process can be estimated from the Rehm–Weller approach (eqn (1)).111 Only for exergonic processes (ΔG < 0), a PET process is considered thermodynamically favorable.
| ΔG = [EOx1/2(D) − ERed1/2(A)] − ΔEexcit + ΔEcoul | (1) |
In intermolecular PET reactions, radical ions are formed either as close pairs or as free species from neutral molecules (Scheme 25).110 These species can undergo a variety of subsequent follow-up processes, for example, an addition (or recombination) reaction of a radical or radical ion or a nucleophilic capture of a cation or radical cation, respectively.
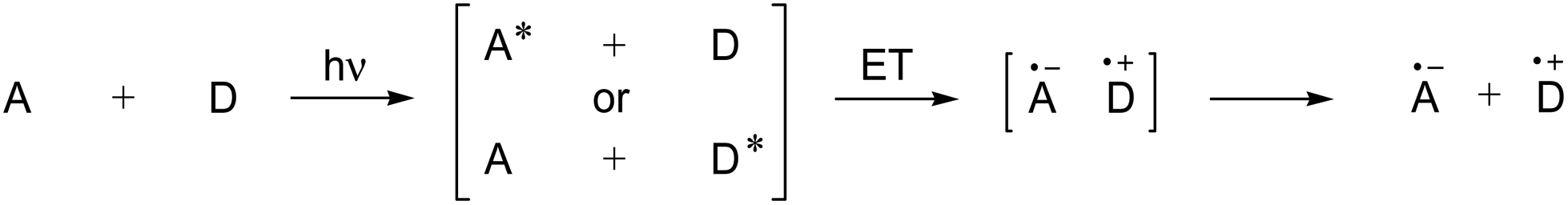 | ||
| Scheme 25 Photoinduced electron transfer between neutral donor and acceptor molecules. | ||
In intramolecular PET processes, a donor and an acceptor are connected via a linker (donor–bridge–acceptor molecules). The photochemical behavior of these has been described as “a bug that opens its mouth when struck by light, can hold it open for only a short time, and during that time can swallow anything within striking range of its open mouth including its own tail”.112
Rapid back electron transfer (BET, also called return electron transfer RET) commonly limits the efficiency of the PET process, resulting in low yields and prolonged irradiation times.113 Consequently, various strategies have been developed to circumvent this energy-inefficient process, among these PET-sensitization, co-sensitization and salt effects.110,114,115
5.1 Cyclization and addition reactions
Due to its favorable photophysical and electrochemical properties,116,117 the phthalimide chromophore has been intensively utilized for PET studies.118 From this data, the limiting oxidizing power of phthalimides for an exergonic electron transfer is ca. 2.4 V (vs. SCE) for the first excited singlet state (E00 = 3.8 eV) and ca. 1.7 V (vs. SCE) for the first excited triplet state (E00 = 3.1 eV). In some cases, electron transfer from the spectroscopically non-detectable second triplet state (E00 = 3.6 eV) has been suggested, thus increasing the oxidizing power to ca. 2.3 V (vs. SCE).Thioethers (Me2S: EOx. = ca. 1.25 V vs. SCE119) are easily oxidized by excited phthalimides. For example, irradiation of the simple alkylthio-substituted phthalimide derivatives (140) in acetone furnished the corresponding unbranched azathiacyclols (141) as major products in moderate to good yields (Scheme 26).120 The branched products 142 were obtained in smaller amounts, if at all, thus demonstrating the higher kinetic acidity of the primary vs. secondary α-CH group. Solely the butylene-linked substrate exclusively gave the branched product 142 and this was explained by a geometrically favored ε-CH2-activation.
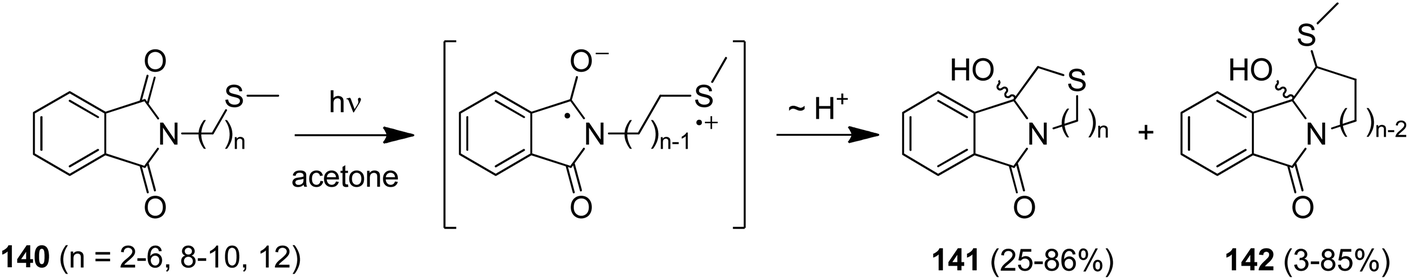 | ||
| Scheme 26 PET-cyclizations of simple alkylthio-substituted phthalimide derivatives. | ||
The phthalimide/methylthioether system was successfully utilized for the synthesis of medium to macrocyclic products with a ring size of up to 38 atoms, among these sulfur containing amines, lactams, lactones, and crown ether analogues (Scheme 27).120,121
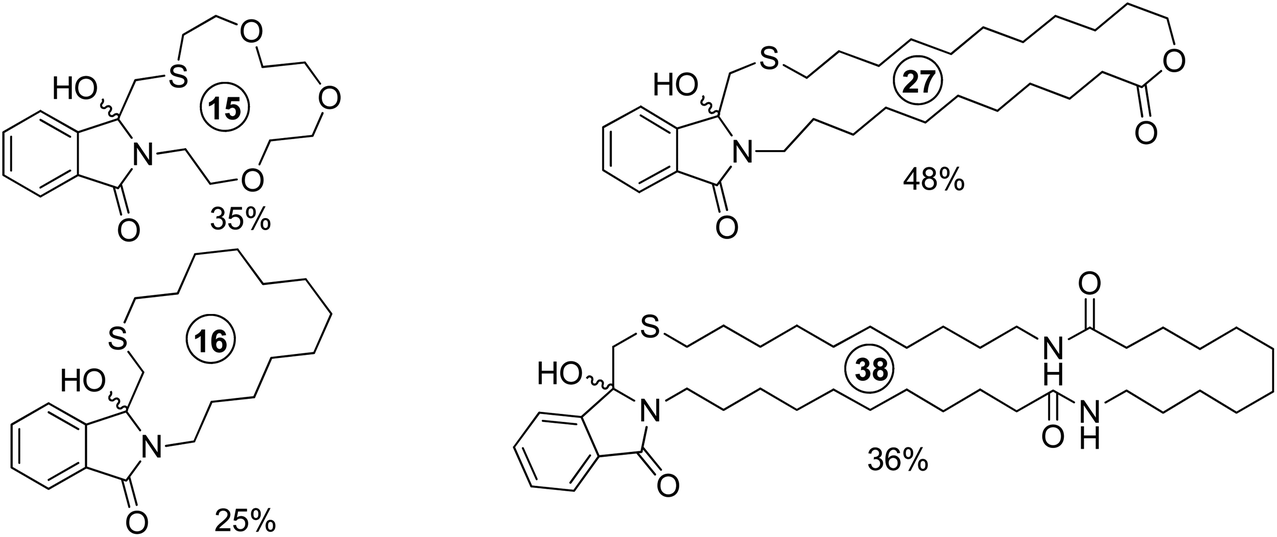 | ||
| Scheme 27 Examples of sulfur containing medium to macrocyclic products. | ||
Due to the very low oxidation potential of tertiary amines (R3N: EOx. = 0.7–1.3 V vs. SCE119), photoinduced electron transfer reactions of aminoalkyl-substituted phthalimides are highly exergonic. However, since amines are potent hydrogen donors, photoreductions and photopinacolizations are commonly observed side reactions. As a result, yields and selectivities are generally low.122 For dibenzylated aminoalkyl-substituted derivatives, higher conversions and yields of up to 39% were obtained.122c
Efficiencies and selectivities of the above mentioned cyclization reactions could be significantly improved by incorporating a suitable leaving group in the α-position to the electron donor. The trimethylsilyl, trialkylstannyl or carboxylate function has been successfully utilized for this strategy.123
Intramolecular PET reactions involving alkenyl phthalimides show a remarkable solvent dependence.124 When irradiated in MeCN, [π2 + σ2]-addition to the C(O)–N bond takes place and benzazepinediones are isolated. In contrast, irradiation in alcohol furnished alcohol-trapping products. Their formations were explained by an anti-Markovnikov trapping of the intermediary generated radical cations by the nucleophilic solvent.125 In an extension of this reaction, the tetrachlorophthalimide (143) with a remote hydroxyalkyl substituent furnished the macrocyclic lactone 144 in 50% yield when irradiated in the presence of α-methylstyrene (Scheme 28).126
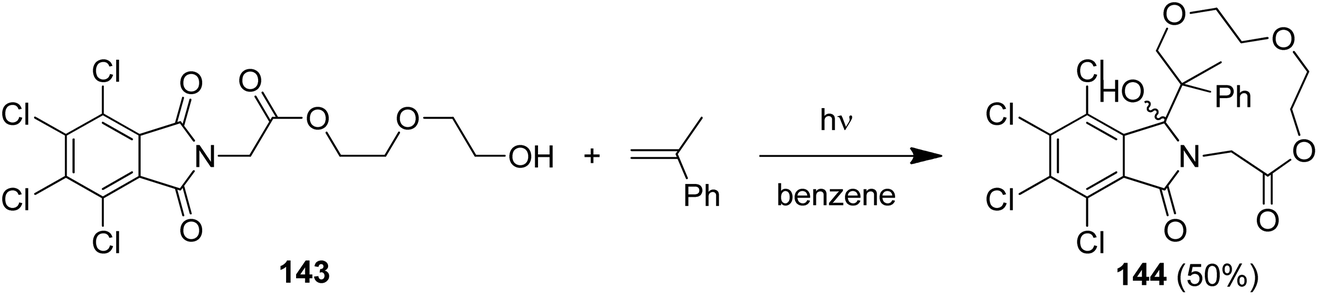 | ||
| Scheme 28 α-Methylstyrene trapping and macrocyclization of tetrachlorophthalimide 143. | ||
Despite their relatively high oxidation potentials (e.g. acetate:127EOx. = 1.54 V in MeCN, 2.65 V in H2O vs. SCE), carboxylates can be efficiently decarboxylated under PET conditions. Due to the irreversible exclusion of carbon dioxide, these decarboxylation reactions operate with high quantum yields of up to 0.6 and enable short irradiation times.116 Using this mechanistic feature, a range of phthalimido ω-alkylcarboxylates (145) have been successfully converted to ring systems (146), among these medium and macrocyclic amines, polyethers, lactams, lactones, as well as cycloalkynes and arenes (Scheme 29).128,129 Examples of photodecarboxylations were also realized in macro- and micro-formats.130,131
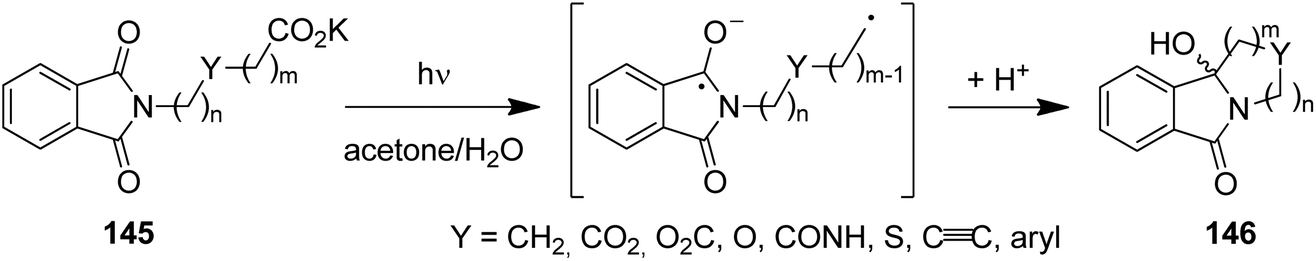 | ||
| Scheme 29 Photodecarboxylative cyclizations of phthalimido ω-alkylcarboxylates. | ||
The photodecarboxylation was furthermore applied to the diastereoselective synthesis of [1,4]benzodiazepines and pyrrolo[1,4]benzodiazepines (Scheme 30).132 Irradiation of the chiral, α-amino acid-derived substrates 147 showed high diastereoselectivities and the trans-diastereoisomers 148 were solely formed. Using proline and the proline-analogue, a remarkably high memory of chirality effects was furthermore determined with ee values of 86% and >98%, respectively. The memory-of-chirality concept has been recently expanded to adamantane-type dipeptides.133
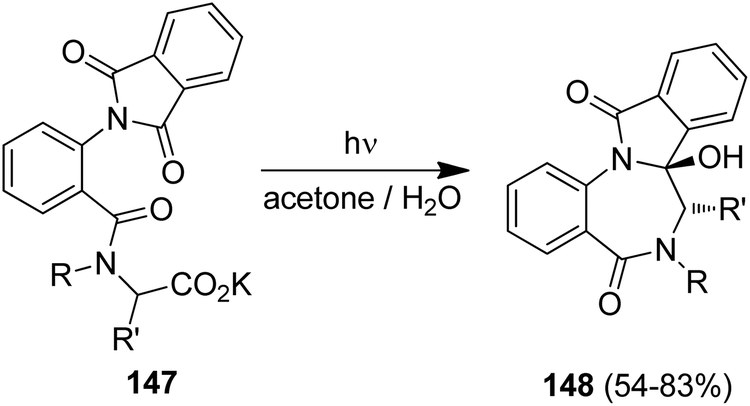 | ||
| Scheme 30 Memory-of-chirality in photodecarboxylative cyclizations. | ||
The intermolecular photodecarboxylative addition protocol has been used for the synthesis of known and potentially bioactive 3-(alkyl and aryl)methylene-1H-isoindolin-1-ones as target compounds.134 As an example, the phthalimide derivative 149 was converted in two simple steps and in overall yields of 19–65% into the corresponding products 150 (Scheme 31). Some representatives of 150 have documented cardiovascular activity.134b This transformation was furthermore realized in series in an advanced continuous-flow reactor.134c
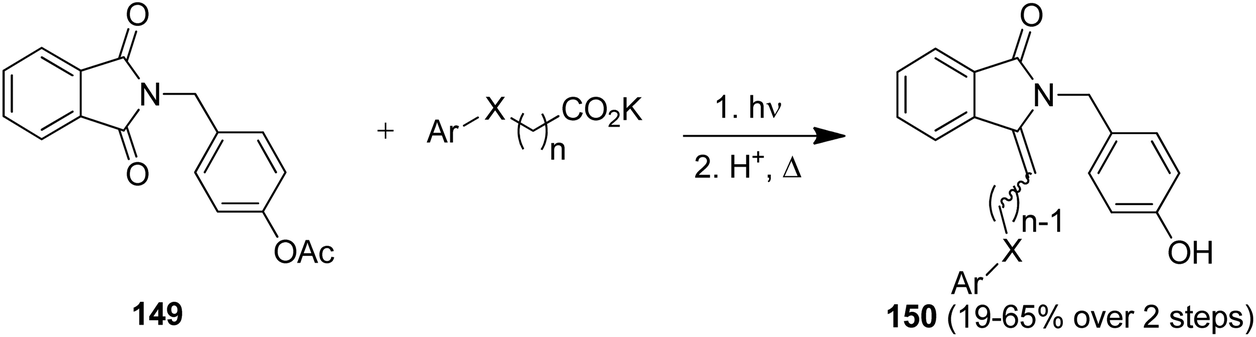 | ||
| Scheme 31 Photodecarboxylative alkylations and dehydrations cascade. | ||
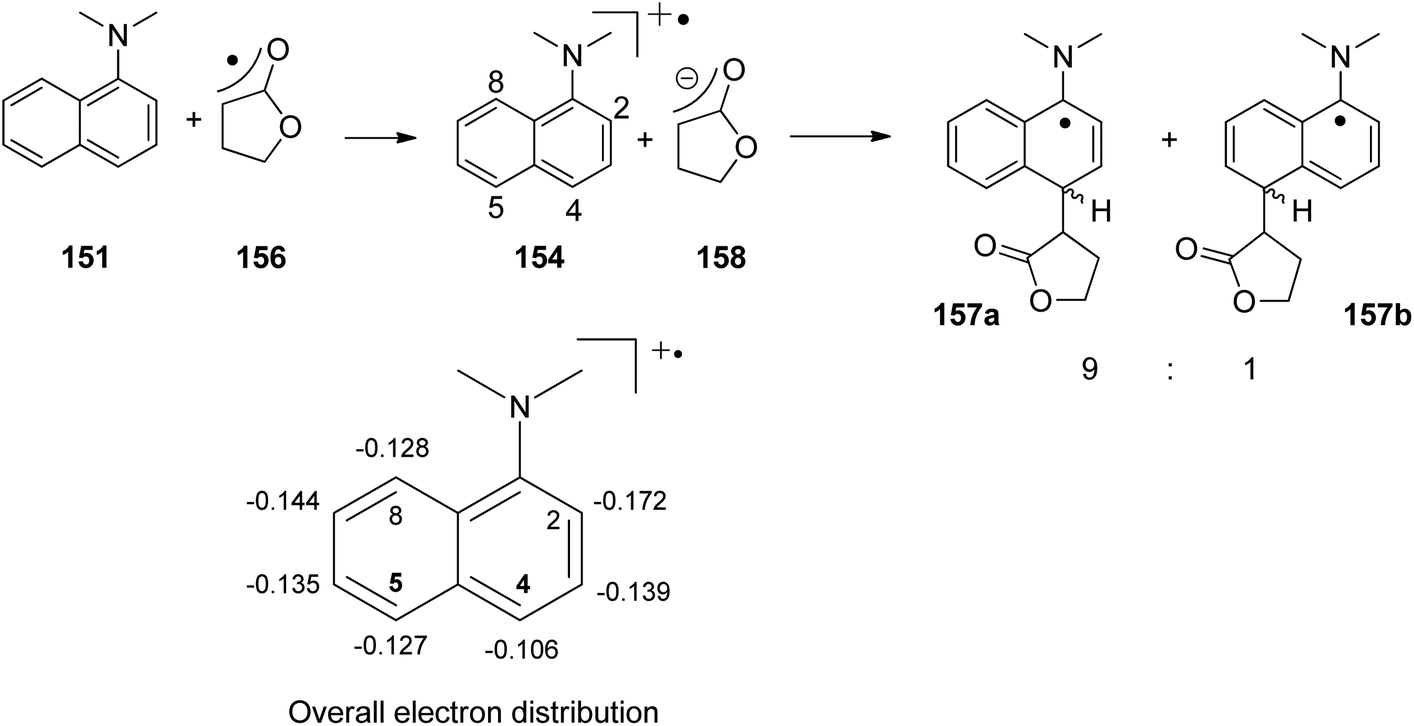
At the ground state, electron transfer between naphthylamine derivatives such as 151 and α,β-unsaturated lactones or furanones such as 152 (Scheme 32) is not possible.135 However, when irradiated with UV light an efficient transformation was observed and products 153a,b were isolated in high yields. When the 1-naphthylamine derivative 151 is electronically excited by irradiation λ = 365 nm, fluorescence is efficiently quenched (diffusion control) by electron transfer and a radical ion pair 154 and 155 is formed. This process is endothermic by −0.37 eV. From product quantum yield determination of the overall reaction, it was concluded that about 90% of these radical ion pairs undergo back electron transfer leading to the substrates at their ground state. However, in the presence of a proton donor (water was added to the reaction mixture), the radial anion 155 is trapped by protonation in the β position of the furanone and the electrophilic neutral radical 156 is formed. This regioselectivity is unusual since the basic center of such a radical anion is generally located at the carbonyl oxygen as is also the case for enolates. Based on previous results, this observation was explained by different charge densities in the radical anion depending on whether it is free in solution or attached to another species. In the present case, such an arrangement in a contact ion pair would induce an unusual charge distribution on the anion part. The resulting neutral radical 156 is electrophilic and thus added easily to the naphthalene derivative 151 which is used in excess. Thus the intermediate 157 is formed and after oxidation, the final product 153a is obtained. The protonation of the radical anion in the β position of 155 induces the unusual addition of a nucleophile in the α position of an α,β-unsaturated carboxylic compound (153a,b). The unusual regioselectivity of the protonation was additionally proven by isotopic labeling experiments.
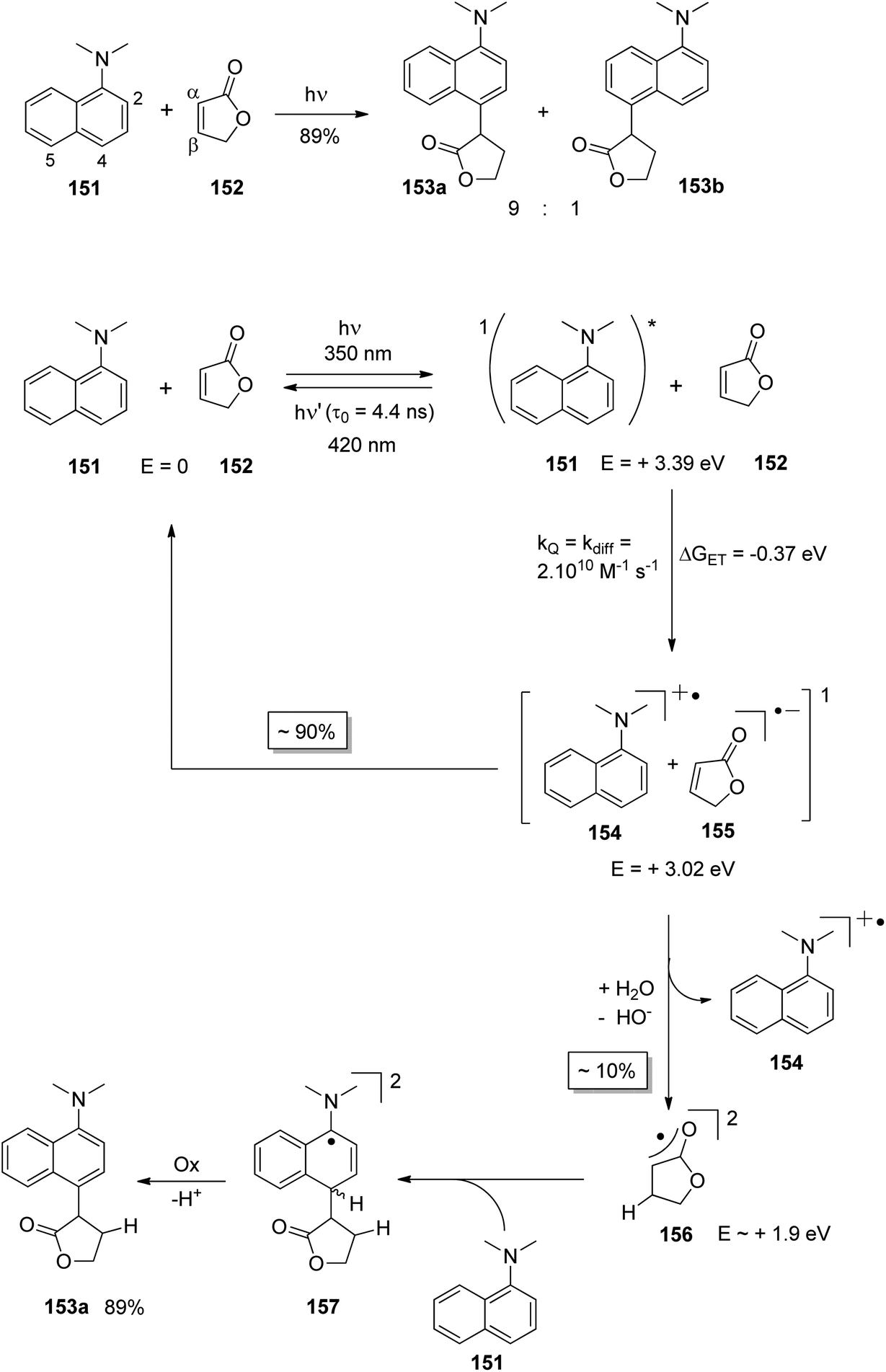 | ||
| Scheme 32 Photochemical electron transfer induced addition of a naphthylamine derivative to an α,β-unsaturated lactone. | ||
Electrophilic substitutions in 1-naphthylamine derivatives generally take place in positions 4 and 2 of the naphthalene system. It is therefore surprising that the addition of an electrophilic species was observed in position 5 as in the formation of compound 153b. In order to explain this observation, a two-step process for the addition of the radical intermediate 156 was proposed (Scheme 33). First an electron transfer occurs from the naphthylamine compound 151 to the electrophilic radical 156 leading to the radical cation 154 and the enolate 158. Charge combination then yields the radicals 157a,b. The overall electron distribution in the radical cation 154 has been calculated and the results indicate particularly low negative charge in position 4. They also show low densities in positions 5 and 8 while the negative charge density is high in position 2. Thus the addition of the enolate 158 by charge combination occurs preferentially in positions 4 and 5. The reaction in position 8 is sterically hindered. In this context, it must be mentioned that electrophilic neutral radicals may also add directly to 1-naphthylamine derivatives without previous electron transfer. Thus, the addition of electrophilic trifluoromethyl radicals to 1-naphthyl amine occurs in one step and only in positions 2 and 4.136 Both regioisomers were obtained in a 1:1 ratio. This latter result is in line with the regioselectivities observed in a large number of electrophilic aromatic substitutions. Compounds such as 153a possess a variety of pharmaceutical activities. Conventionally they are synthesized using methods of organometallic chemistry or organometallic catalysis.137
 | ||
| Scheme 33 Regioselectivity of the photochemical electron transfer induced addition of a naphthylamine derivative to an α,β-unsaturated lactone. | ||
In a similar reaction, perfluoroalkylations of 1-naphthylamine derivatives have been carried out (Scheme 34).138 After photochemical excitation, electron transfer from the excited naphthylamine derivative 151 to the perfluoroalkyliodide 159 becomes exothermic (ΔGET = −1.78 eV). Very fast release of iodide leads to the neutral perfluoroalkyl radical 161 which adds to the naphthalene derivative 151. The intermediate 162 is generated. Attempts to quench the reaction by trapping of the radical anion by addition of dinitrobenzene failed which indicates the formation of the neutral perfluoroalkyl radical 161 is fast. α-Aminoalkyl radicals such as 162 are easily oxidized. In the present case, the perfluoroalkyliodide 159 is reduced by a corresponding electron transfer from 162. This procedure also allows the formation of perfluoroalkyl radicals 161 after release of iodide. The resulting cation 163 is a typical Wheland intermediate of an electrophilic aromatic substitution which yields the final product 160 after deprotonation. The electrophilic radicals 161 add easily with the electron rich 1-naphthylamine derivative 151. Thus a radical chain process is established. The high product quantum yields of around 200 clearly indicate the efficiency of this process. The reaction was also carried out with aniline derivatives. Depending on the substrate structure and the precise reaction conditions, product yields up to 88% have been observed.
 | ||
| Scheme 34 Photochemical electron transfer induced addition of a fluoroalkyl radical to a naphthyl amine derivative involving a chain mechanism. | ||
5.2 Catalytic reactions
Catalytic reactions play an important role in synthetic chemistry. In particular, catalytic conditions accelerate transformations with high activation energies. This effect is based on the formation of energetically low reaction intermediates between the substrate and the catalyst. In many catalytic processes no activation with chemical reagents used in stoichiometric amounts is necessary. Thus the formation of side products is considerably reduced. As already pointed out, photochemical reaction conditions significantly alter the reactivity of organic compounds. They also have an impact on the elementary steps of catalytic reactions. It is therefore obvious that the combination of both domains, catalysis and photochemistry, should enable synergy effects.139,140 Many photochemical reactions are induced by sensitization in which an additive (sensitizer) transfers its excitation energy on a substrate.141,142 This process may be compared to catalysis and indeed definitions for a sensitizer and a photocatalyst are often very similar. The next procedure well illustrates the proximity of these terms. It deals with the radical addition of simple tertiary amines to electron deficient alkenes. Recently, an efficient methodology has been developed to perform this transformation.143 Thus the N-alkylpyrrolidines such as 165 were added efficiently to α,β-unsaturated lactones or furanones such as 164 (Scheme 35) when electron donor substituted aromatic ketones were used as sensitizers such as 4,4′-dimethoxybenzophenone 167.144 Product (166) yields up to 94% and quantum yields up to 4 have been observed. However, reactions with many acyclic tertiary amines such as triethylamine were less efficient. Similar reactions have also been carried out with unsubstituted benzophenone and this sensitizer has also been used in this case. When comparing these two types of sensitizers, it was observed that in the case of electron donor substituted aromatic ketones as sensitizers such as 167, the reactions were particularly fast. The sensitizer was used in catalytic amounts and was recovered up to 80% after the reaction. However, in the case of benzophenone for example, large amounts, often more than one equivalent of the sensitizer, were used and considerable decomposition via photoreduction and photopinacolization were observed.145 These are typical reactions of these ketones in the presence of amines.146 Electron donor substituted aromatic ketones undergo such transformations less efficiently. The difference in reactivity was attributed to different excited states. While benzophenone possesses an nπ* configuration for the T1 state, compounds such as 4,4′-dimethoxybenzophenone 167 possess a ππ* configuration at this triplet state. In order to check whether these different excited states have an influence on the reaction, among other parameters, the triplet quenching rates kq with the reactive amine 165 and the unreactive triethylamine (TEA) and the lifetime t1/2 of the corresponding ketyl radicals have been determined (Table 5).147 The luminescence quenching occurs via electron transfer. Concerning these photophysical steps, no significant difference between the two types of aromatic ketones on the one hand and between the reactive tertiary amine 165 and the unreactive TEA on the other hand have been detected. | ||
| Scheme 35 Photochemically sensitized addition of tertiary amines to electron poor double bonds using electron donor substituted aromatic ketones as sensitizers. | ||
| Ketone | Amine | k q (109 M−1 s−1) | t 1/2 (ms) |
|---|---|---|---|
| Benzophenone | 165 | 3.6 | 0.1–0.2 |
| TEA | 3.5 | 0.1–0.2 | |
| 4,4′-Dimethoxybenzophenone 167 | 165 | 1.5 | 0.1–0.2 |
| TEA | 0.8 | 0.1–0.2 | |
| Xanthone | 165 | 5 | 0.1–0.2 |
| TEA | 4.4 | 0.1–0.2 | |
| Acetophenone | 165 | 2 | 0.1 |
| TEA | 2.4 | 0.1 | |
| 4-Methoxyacetophenone | 165 | 1 | >0.05 |
| TEA | 0.28 | >0.05 |
Based on these observations the mechanism depicted in Scheme 36 is discussed. After light absorption of the sensitizer, electron transfer occurs from the tertiary amine 165 to the sensitizer 167 and the radical ion pair 168 and 169 is generated. Proton transfer between the ions leads to the neutral radicals 170 and 171 which enter into the radical chain reaction. The nucleophilic α-aminoalkyl radical 171 easily adds to the electron deficient double bond of the furanone 164 and the electrophilic oxoallyl radical 172 is thus formed.148 After hydrogen transfer from the tertiary amine 165 to 172 the final product 166 is formed and an α-aminoalkyl radical 171 is also generated. Such photochemically induced radical chain reactions often afford very high quantum yields. In the present case, the relatively low quantum yield Φ = 4 indicates that the termination step is also efficient. Thus the oxoallyl radical 172 may react with the ketyl radical 170 of the sensitizer leading to the final product 166 and the sensitizer 167 is regenerated. Due to the efficiency of this second radical cycle, the sensitizer becomes an efficient photocatalyst. As the difference in efficiency between the electron donor substituted aromatic ketones such as 167 and unsubstituted aromatic ketones such as benzophenone cannot be explained by the photophysical primary processes, the observed effects must be linked to the reactivity of the different ketyl radicals. Thus, the reaction of ketyl radicals such as 170 with electrophilic radicals such as 172 must be particularly efficient. Perhaps, an electron transfer is also involved in this step.
 | ||
| Scheme 36 Mechanism of the photochemically sensitized addition of tertiary amines to electron poor double bonds. | ||
The same reaction was carried out with inorganic semiconductors such as TiO2 and ZnS as photochemical electron transfer sensitizers.149 Thus heterogeneous photocatalysis was applied for this transformation.
In the context of asymmetric catalysis, the reaction has also been carried out in an intramolecular manner with a chiral sensitizer.150 In such reactions, the tertiary amine partner is present in stoichiometric amounts with respect to the olefinic partner and no radical chain process is possible. However due to the proximity of the oxoallyl radical and the ketyl radical, an efficient regeneration of the sensitizer is also possible and the productiveness of the overall reaction is thus guaranteed.
Due to the fact that no further optimization of the reaction seemed to be possible by modifying the structure of the aromatic ketone as a sensitizer or a photocatalyst, particular interest was paid to the radical mechanism. The complex interplay of different neutral radical intermediates may be affected by the addition of thiocarbonyl derivatives. The addition of these compounds to radical species is reversible151 and this behavior has been applied in polymer chemistry in order to control the radical polymerization.152 In the present case of the photochemically induced radical addition of amines to electron deficient alkenes, it was found that the addition of thiocarbonyl compounds such as the thiocarbamide 173 to the reaction mixture increases the reactivity of the tertiary amine (Scheme 37).153 Thus, the addition of triethylamine (TEA) to furanone 174 became possible and the adduct 175 was isolated in high yield. As previously discussed, α-aminoalkyl radical 176 generated by a two-step hydrogen transfer forms the tertiary amine to the photochemically excited sensitizer (compare also Scheme 36). The addition of such radicals to furanones is reversible. The equilibrium depends on the structure of the tertiary amine. In the case of TEA, either the formation of the intermediate 177 is less favorable or this intermediate is less efficiently trapped by the reaction with a TEA molecule. In the presence of thiocarbamide 173, the equilibrium is shifted to the product side by efficient addition of 173 to the oxoallyl radical 177 leading to the intermediate 178. The latter intermediate also forms the thiocarbamide adduct 179. In the presence of TEA and the corresponding α-aminoalkyl radicals 176, this adduct yields the final product 175 by release of the thiocarbamide group. Thus, a second reaction channel to the final product is opened. The thiocarbamide adduct 179 was detected by mass spectroscopy. The corresponding signal intensities and thus the stationary concentration of 179, depend on the excess of TEA in the reaction mixture. When the excess of TEA is diminished, the stationary concentration of 179 increases and vice versa. The transformation of 179 into the final product 175 is induced by such an addition. Under these modified reaction conditions, a variety of less reactive tertiary amines were successfully added to electron deficient double bonds.153,154 The addition of thiocarbamate 173 to nucleophilic radicals, α-aminoalkyl radicals, 176 is less efficient. Nevertheless, a corresponding adduct 180 was also detected by mass spectroscopy.
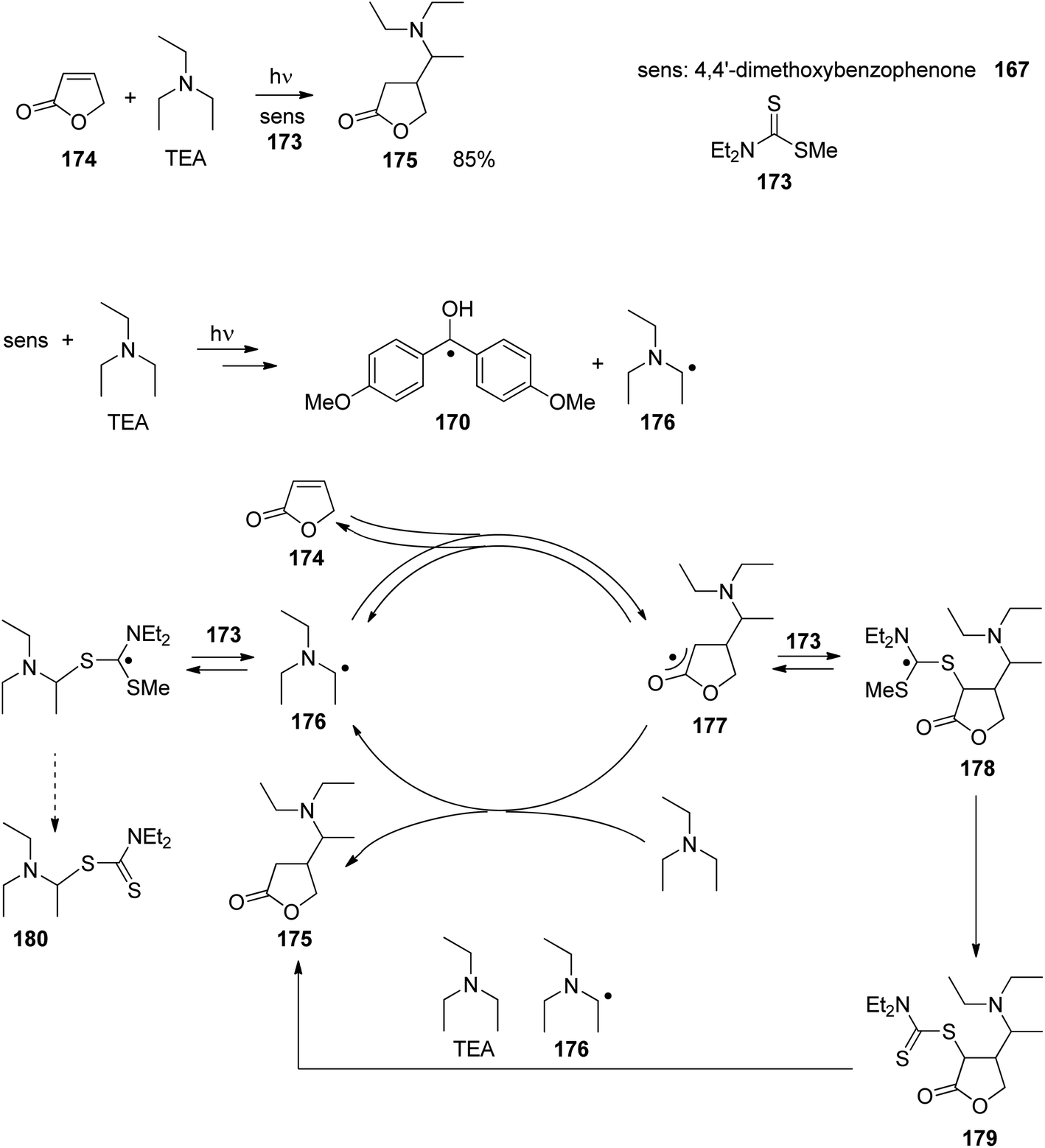 | ||
| Scheme 37 Photochemically induced radical addition of unreactive amines to electron deficient alkenes in the presence of thiocarbonyl compounds. | ||
In this context, it should be mentioned that photochemistry generally provides a large variety of methods to generate radical species and induce radical tandem or domino reactions as they are often observed in radical chemistry.155 The presented method of the photochemically induced radical addition of tertiary amines to alkenes was also applied to related tandem addition cyclization reactions. Particularly interesting transformations have been performed with aniline derivatives. When a solution of N,N-dimethylaniline 182 and the chiral furanone 181 was irradiated in the presence of the electron donor substituted aromatic ketone 184 as the sensitizer, the two diastereomers 183a,b of a tetrahydroquinoline were isolated (Scheme 38).156 However, considerable amounts of the side products 185 and 186 were also obtained. These products result from partial reduction of the furanone 181. Using isotopic labeling, it was shown that this side reaction is coupled to the formation of tetrahydroquinolines 183a,b. When the reaction was performed with the aniline derivative 187 carrying deuterium atoms in the ortho and para positions, deuterium was detected in the α-position of the lactone 188 and in one of the α-positions of the coupling product 189 (Scheme 39). When this reaction was carried out with the derivative 190 carrying deuterium atoms on the methyl groups, deuterium was detected in the β-position of the lactone 191 and again in one of the α-positions of the coupling product 189. The incorporation of one deuterium atom in the side products was between 45 and 60%. The results obtained from these experiments are explained by the mechanism depicted in Scheme 40. Photochemical electron transfer from the aniline derivatives to the excited sensitizer 184 leads to α-aminoalkyl radicals 192. They readily add to the electron deficient double bond of the α,β-unsaturated lactone 181 and oxoallyl radicals 193 are formed. After cyclization, the resulting tricyclic radical 194 needs to be oxidized in order to generate the final product 195. In this step, deuterium is transferred from the intermediate 194 to the furanone 181. Most probably, this reaction occurs in two steps. First, electron transfer occurs and in the second step, a proton follows. This sequence explains well the formation of the deuteroxyallyl radical intermediate 196. After hydrogen abstraction in the β-position from one of the methyl groups of the aniline derivative 187, and tautomerization the reduction side product 188 carrying a deuterium atom in the α-position is obtained. Intermediates such as 196 are nucleophilic and may therefore be added easily to the α,β-unsaturated carbonyl or carboxyl functions. Thus the oxoally radical 197 is formed and after hydrogen abstraction, the second side product 189, carrying a deuterium in one of the α-positions is obtained. In the case of the aniline derivative 190, the deuterium transfer occurs one step later in the mechanism. Deuterium is transferred from the CD3 group to the hydroxyallyl intermediate 198. After tautomerization, the lactone side product is obtained with deuterium in the β-position (191). This mechanistic investigation was necessary to optimize the process. In this reaction, the furanone substrate acts as an oxidant in the rearomatization step when the tricyclic radical intermediate (194) is transformed into the final product 195 or 183a,b. It was therefore concluded that the furanone 181 needs to be replaced by another oxidant in order to stop the consumption of this substrate in the side reaction. This was achieved when simple ketones such as acetone was added to the reaction mixture. Consequently, the yield of the desired tetrahydroquinoline compounds 183a,b was doubled and the formation of the undesired side products 185 and 186 was completely suppressed. Under the optimized conditions, the reaction was successfully performed with a variety of aniline derivatives.
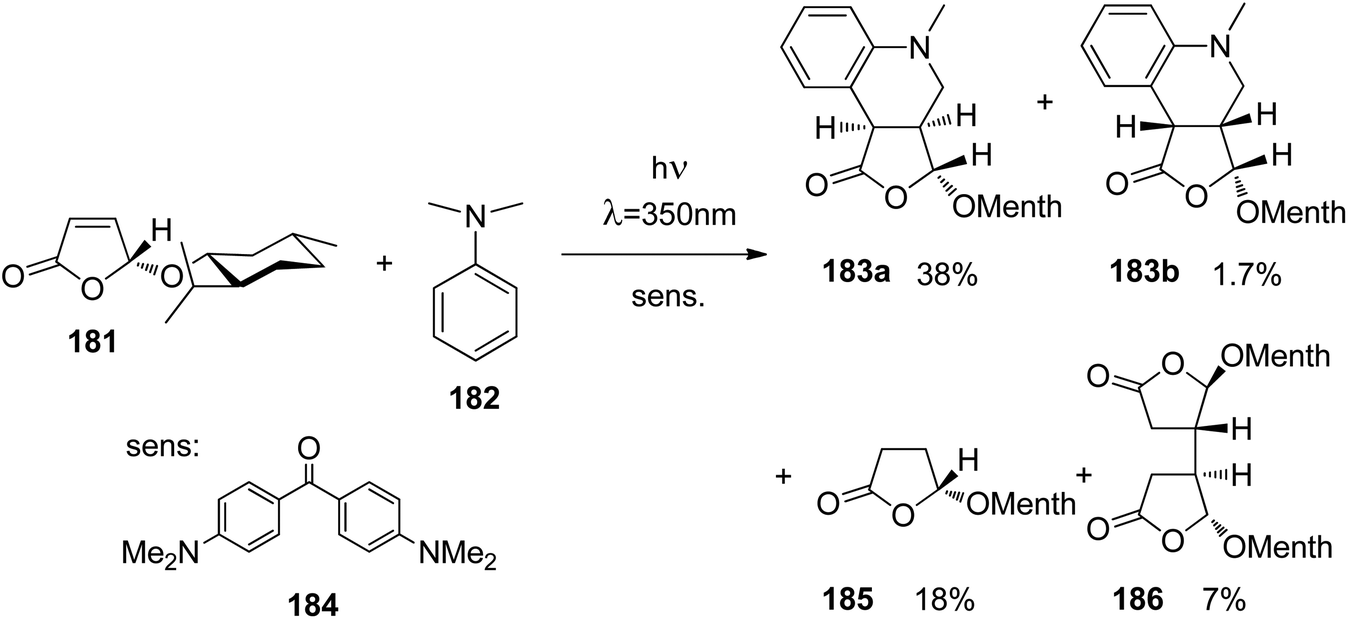 | ||
| Scheme 38 Radical addition cyclization reaction of aromatic tertiary amines with a furanone. | ||
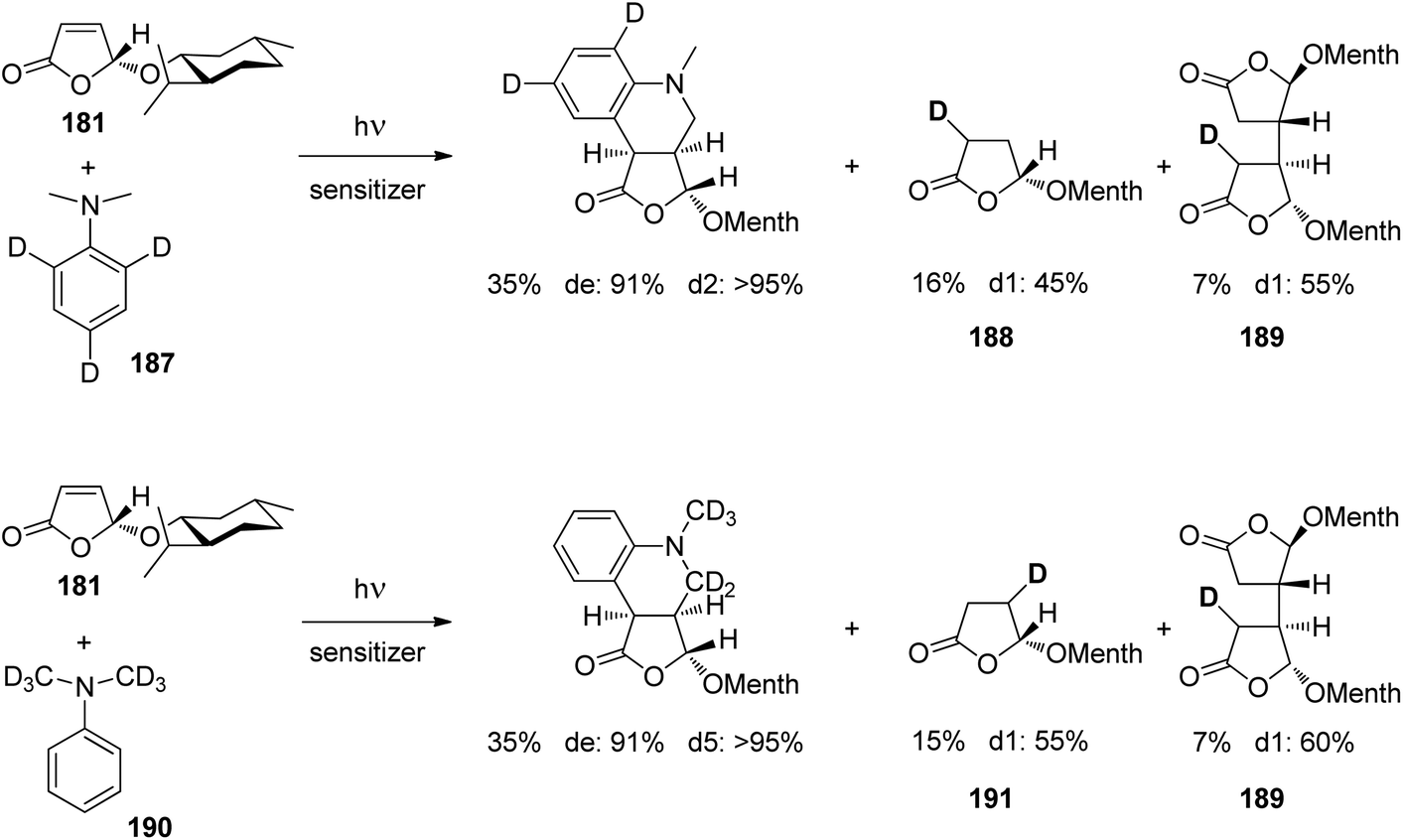 | ||
| Scheme 39 Isotopic labeling in the radical addition cyclization reaction of aromatic tertiary amines with a furanone. | ||
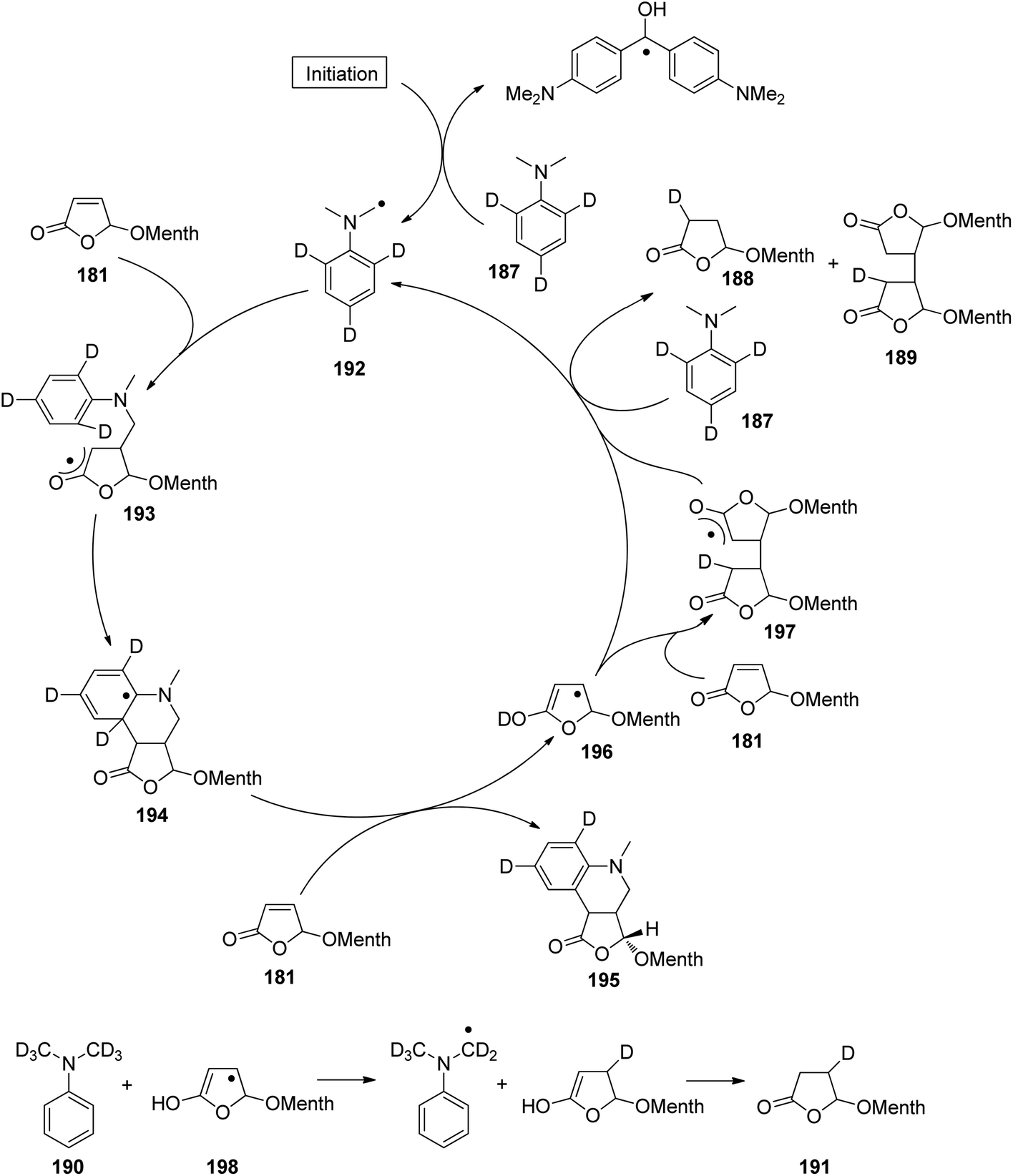 | ||
| Scheme 40 Proposed mechanism of the radical addition cyclization reaction of aromatic tertiary amines with a furanone based on isotopic labeling. | ||
The diastereoselectivity of addition reactions in the β-position of the chiral furanone 181 at its ground state is almost always complete. The presented tandem addition cyclization reaction with N,N-dimethylaniline 182 is somewhat less selective. Due to this fact, the transformation was used to investigate in detail the chiral induction in transformations with such alkoxyfuranones.157
The tandem addition cyclization reaction of aniline derivatives with furanones was also performed with inorganic semiconductors such as TiO2 or ZnS as the photochemical electron transfer sensitizer thus applying heterogeneous photocatalysis.158,159 As in the case of homogeneous photocatalysis, the corresponding isotopic labeling experiments gave a deep insight into some mechanistic steps of the reaction.
Most of the previously discussed photoredox catalytic reactions use UV light.142,160 These conditions were quite efficient since a lot of chemical reactions of interest for application to organic synthesis have been developed. For example, sophisticated systems of photochemical co-catalysis have been developed. Photoredox catalysis using visible light is now frequently applied to organic synthesis.140,161 Coordination compounds162 such as ruthenium trisbipyridine complexes or similar derivatives play a central role in this domain.163 Very often [Ru(bpy)3]2+ salts are used. The energetically lowest electronic excitation possesses a MLCT character. By light absorption the corresponding singlet state is populated. The values given in Scheme 41 have been determined for aqueous solutions. Intersystem crossing is very efficient and the triplet state is reached. The luminescence quantum yield of this lowest excited state is low and its lifetime is high. Thus many intermolecular quenching processes are possible. Among them reductive and oxidative electron transfers are efficient. Photochemical excitation facilitates both processes. Due to the MLCT character of the excited state, electron transfer from the ligand to the quencher takes place in the oxidative quenching cycle. Consequently, in the reductive quenching cycle, an electron is transferred from the quencher to the metal center. Similar properties are also reported for iridium complexes and many applications to organic synthesis of photoredox catalysis are also reported with such catalysts. The redox potentials also depend on the substitution pattern of the ligands. A lot of such compounds, in particular from ruthenium, are available and the redox properties of the catalyst can easily be adapted to the requirements of the organic reaction.
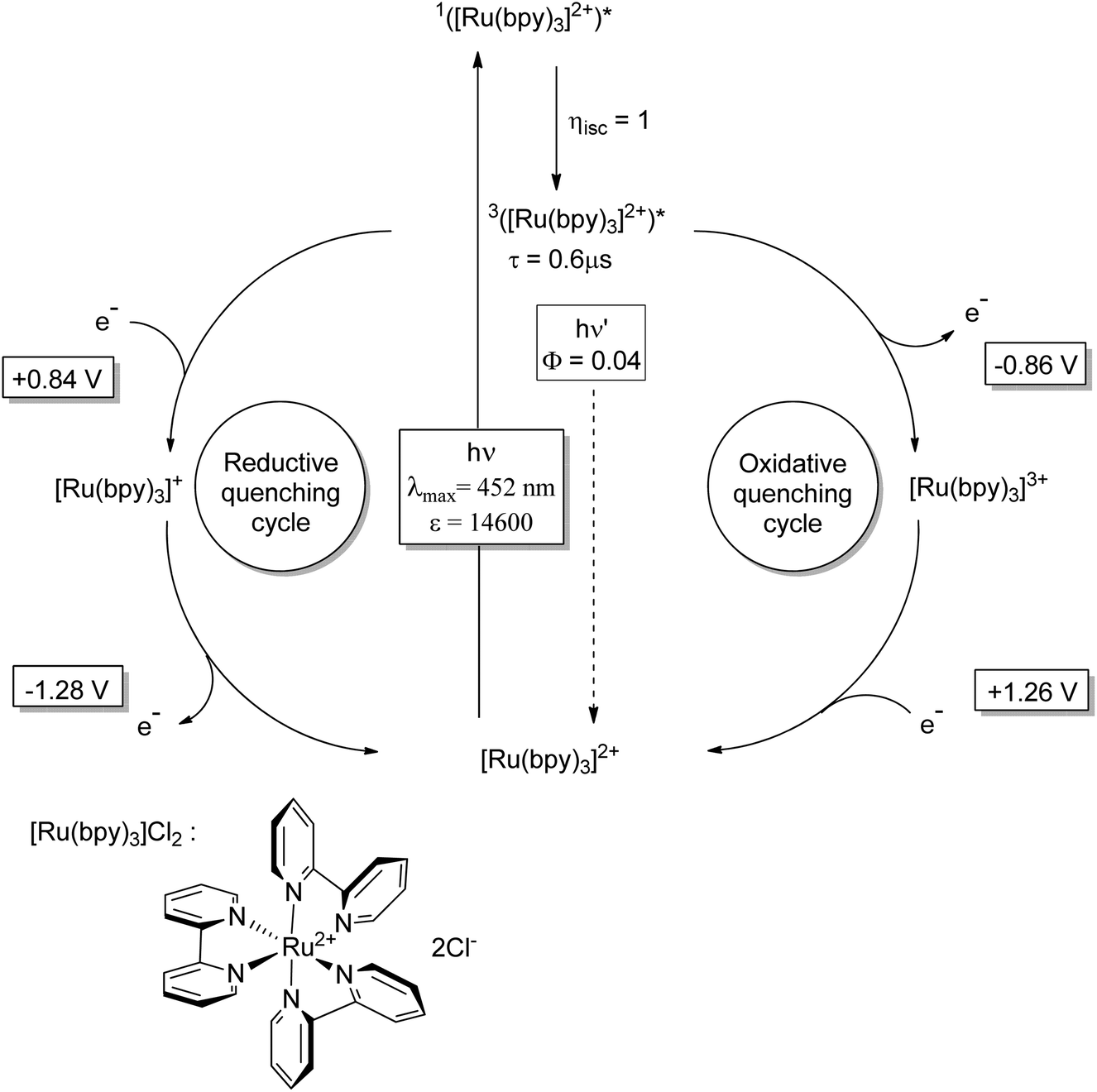 | ||
| Scheme 41 Redox potentials of [Ru(bpy)3]2+ salts at the ground and excited states. | ||
Although known for a long time,164 photoredox catalysis with organometallic and coordination compounds was only recently developed in a systematic manner by many research groups. In particular the photoredox systems involving ruthenium or iridium complexes are very robust and thus a lot of different reactions can successfully be performed. Furthermore, these systems can be combined with other types of catalysis such as organocatalysis. A prominent recent example is the asymmetric alkylation of aldehydes. Combining asymmetric organocatalysis involving enamine intermediates with [Ru(bpy)3]2+ based photoredox catalysis with visible light, phenacyl bromide 199 was added to aldehydes such as 200 (Scheme 42).165 The mechanism depicted in Scheme 43 has been proposed. The organocatalytic cycle starts with the addition of the organocatalyst 201 to octylaldehyde 200 leading to the enamine 202. Phenacyl bromide 199 is not electrophilic enough to be added to the enamine 202. However, the corresponding radical 203 possesses higher reactivity and its addition to 202 yields the α-aminoalkyl radical 204 with high enantiomeric excess. In order to join the normal organocatalytic cycle this intermediate needs to be oxidized to an iminium ion 205. The latter yields the final product 206 and the catalyst 201 by hydrolysis. This oxidation is performed by the photochemically excited [Ru(bpy)3]2+. The oxidation potentials of α-aminoalkyl radicals such as 204 range between −0.92 and −1.12 V (SCE in acetonitrile).166 The temporary participation of open shell intermediates in catalytic cycles which otherwise only involve closed shell intermediates is also called SOMO catalysis. During the oxidation of 204, a ruthenium(I) species is generated. This intermediate is a strong reductant (−1.33 V versus SCE in acetonitrile).167 It is therefore capable of reducing phenacyl bromide 199 which has an oxidation potential of −0.49 V (SCE in acetonitrile).168 The electrophilic phenacyl radical 203 is formed after release of bromide.
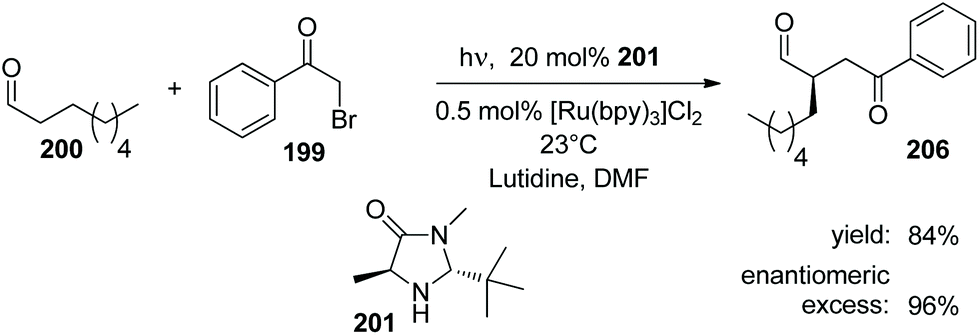 | ||
| Scheme 42 Photoredox catalysis combined with asymmetric organocatalysis. | ||
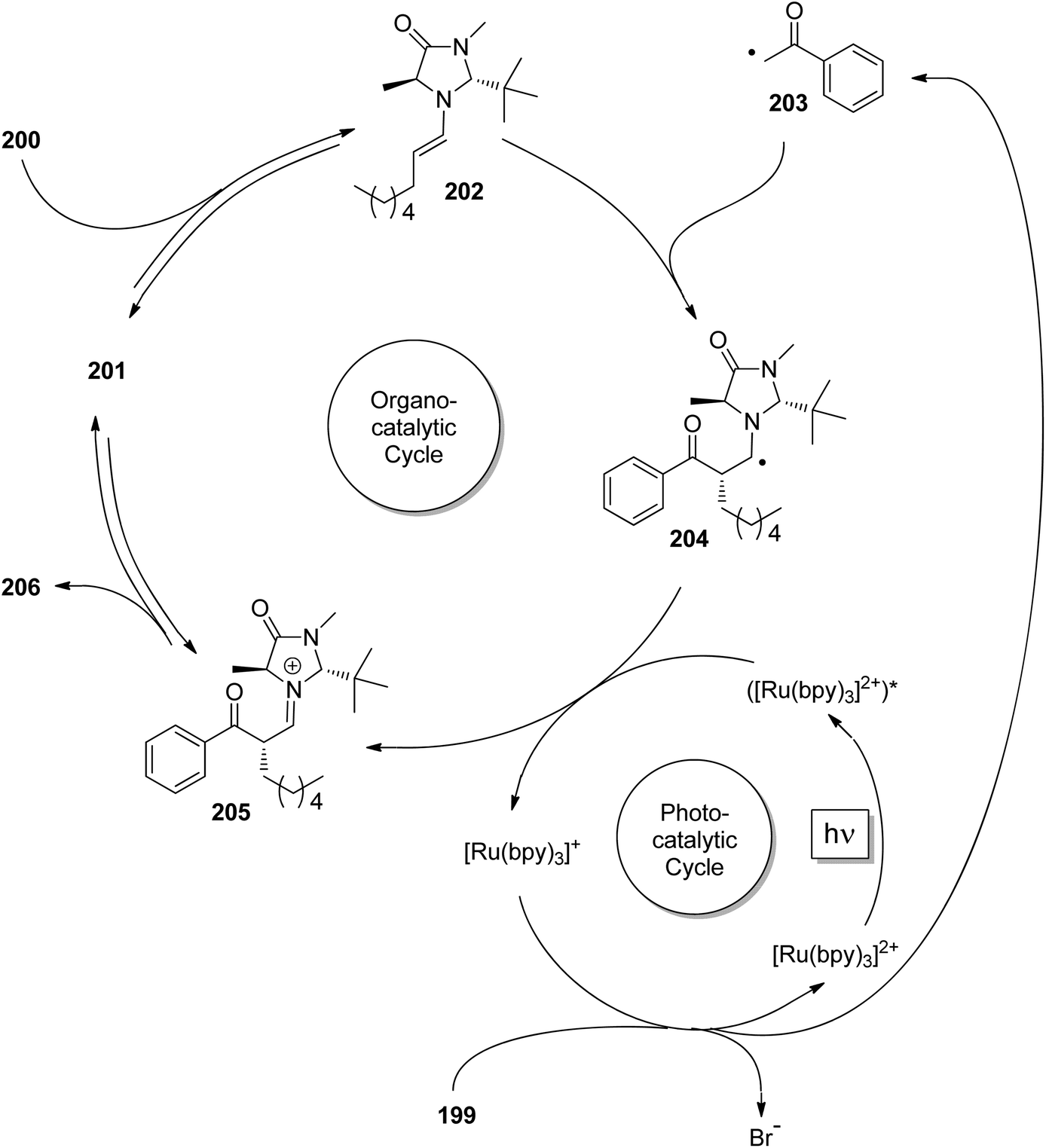 | ||
| Scheme 43 Mechanism of photoredox catalysis combined with asymmetric organocatalysis. | ||
The mechanism as is depicted in Scheme 43 needs an initiation step. Most probably, the reaction starts with an electron transfer from sacrificial enamine 202 to the excited ruthenium complex. Once, the first and more reactive α-aminoalkyl radicals 204 are formed, the oxidation of the enamine intermediate does not play an important role any more in the overall reaction. It must be pointed out that in the present example, both the oxidation and the reduction of the ruthenium complex are involved in the formation of the final product. A lot of other photoredox catalytic reactions have been reported in which only one of these redox processes is involved. In such cases, a stoichiometric amount of a sacrificial electron donor or acceptor is needed.
Similar reaction conditions have been applied to the asymmetric fluoro alkylations.169 In this case, somewhat better results were obtained when the iridium complex [Ir(ppy)2(dtb-bpy)]+ was used instead of [Ru(bpy)3]2+. This difference may be explained by the fact that the corresponding [Ir(ppy)2(dtb-bpy)] intermediate of the photoredox catalytic cycle is a stronger reductant (−1.51 V versus SCE in acetonitrile).170 Furthermore, such asymmetric alkylation combining photoredox catalysis and organocatalysis is also performed with organic dyes as photoredox catalysts with the same efficiency.171 Thus the whole catalytic system is organic and one may now distinguish in the catalytic cycles a part with closed shell steps and a part with open shell steps. Inorganic semiconductors such as photoredox catalysts have also been successfully applied.172
Interesting reactions have recently been performed with perylenetetracarbox-3,4:9,10-disimides. These dyes are particularly stable especially when electron transfer is involved. For this reason, many studies have been performed with such compounds.173 The photochemical and electrochemical properties have been intensively studied.174 Using the perylene derivative 207 as a photoredox catalyst, a cross-coupling reaction between ortho-chlorocyanobenzene 208 and pyrrolidine 209 was carried out (Scheme 44).175 The coupling product 210 was obtained in good yields. Under similar conditions, efficient hydrodehalogenation was also carried out. In such a reaction, the trifluoromethyl pyridine derivative 211 was transformed in high yields into 212. It must be pointed out that in these reactions aryl chlorides must be reduced and visible light irradiation in combination with the corresponding photo redox catalysts does not provide sufficient energy for such reaction steps. Thus such reaction conditions cannot be applied.
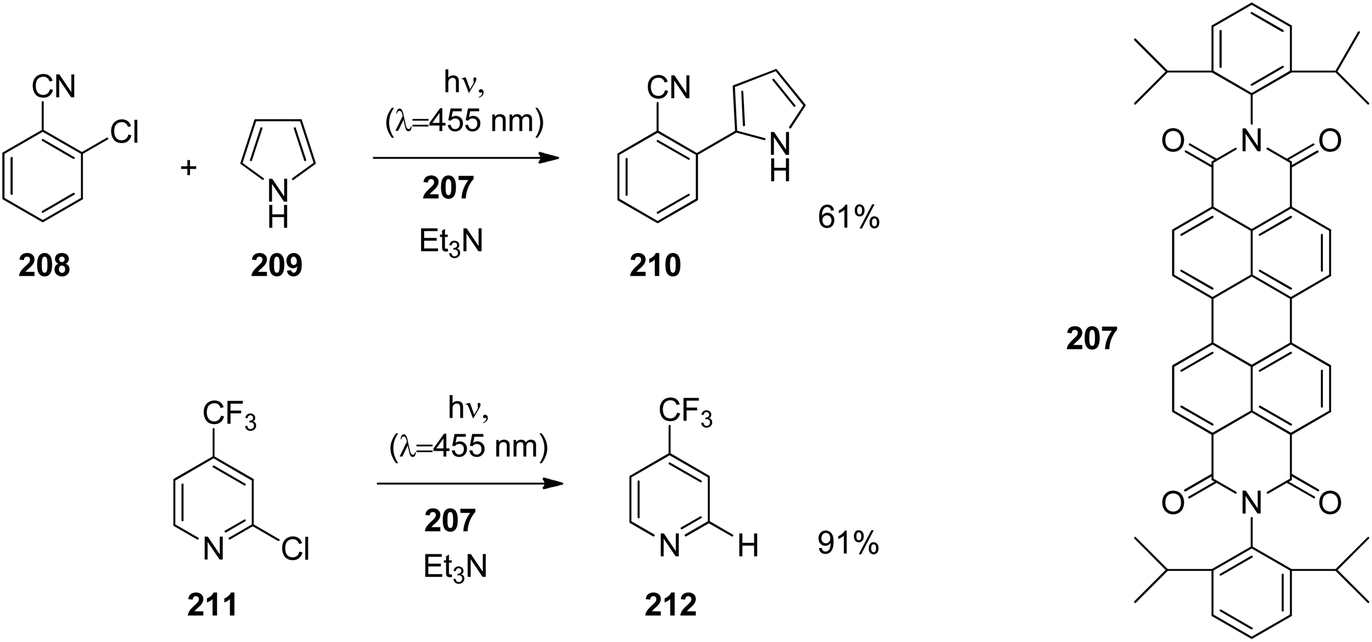 | ||
| Scheme 44 Photoredox catalytic reactions using perylenetetracarbox-3,4:9,10-disimides as catalysts. | ||
The mechanism depicted in Scheme 45 was suggested to explain the results. After photochemical excitation of the perylene derivative 207, electron transfer from the sacrificial electron donor trimethylamine (Et2N) takes place. The resulting radical anion is not capable of reducing substrates such as 208 or 211 in Scheme 44. The reduction redox potential for the formation of this radical anion is −0.88 V (vs. Fc+/Fc). In particular, in the absence of oxygen, such radical anions are remarkably stable and they have been well characterized.176 Possessing a blue color (characteristic absorption at λ ∼ 700 nm), these species are capable of absorbing a second photon in the visible spectrum range. The electronically excited radical ions reduce the arylchloride thus regenerating the photocatalyst. The resulting radical anion 213 of the arylchloride releases chloride and the aryl radical Ar˙ reacts either by hydrogen abstraction or by addition to pyrrole 209 leading to the coupling product 214. Under more standard photochemical reaction conditions, only UV light provides sufficient energy to generate intermediates for the cleavage of the C–Cl bond. In the present reaction, such a UV photon is replaced by two visible photons. This strategy might considerably increase the application of visible light in organic photochemical reactions.
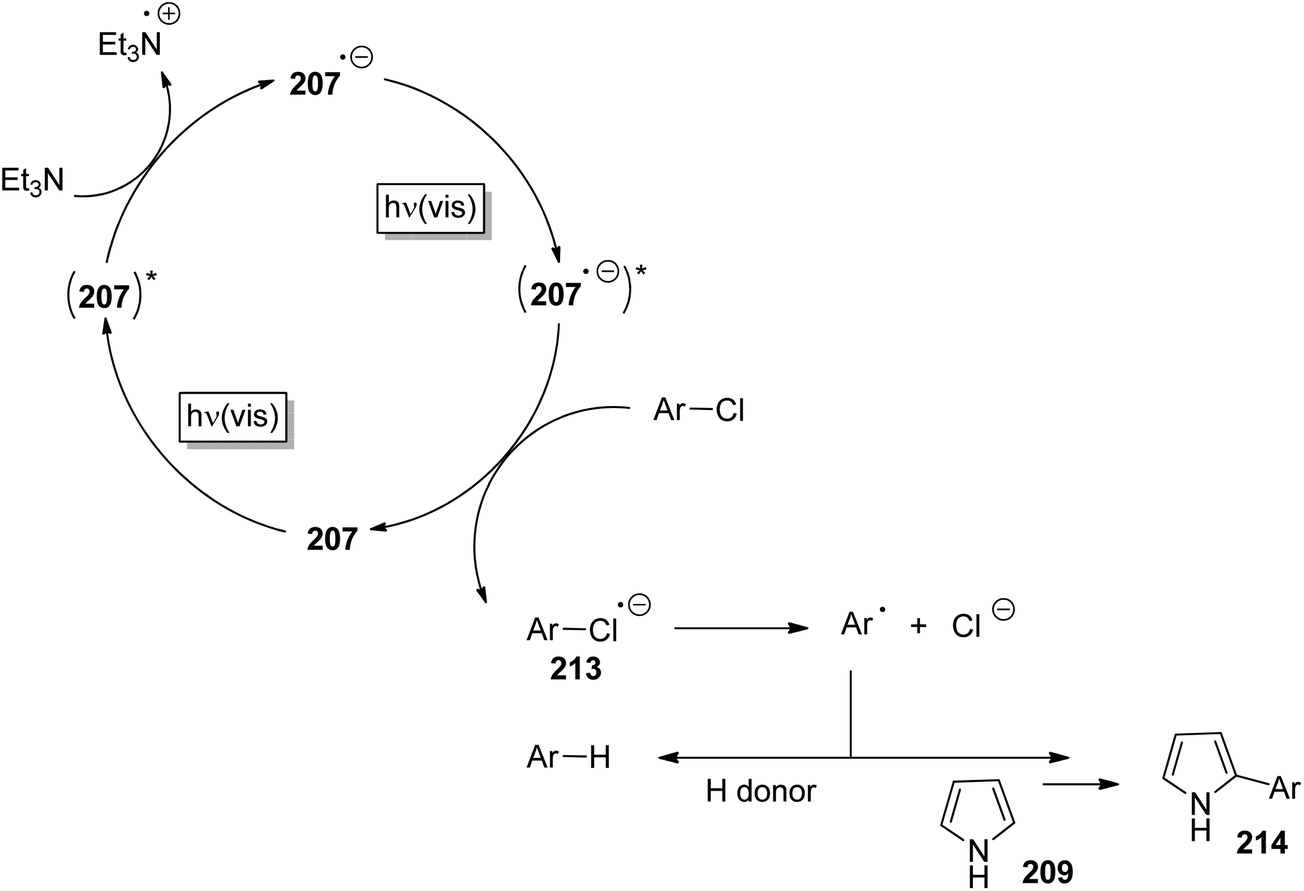 | ||
| Scheme 45 Mechanism of photoredox catalytic reactions using the perylenetetracarbox-3,4:9,10-disimide 207 involving two visible photon activation. | ||
A further process using two visible photons to generate a higher excited species is photon upconversion.177 Several types of processes have been reported. In one of them two triplet excited molecules interact with each other thus generating one molecular at its singlet ground state and a second one at its higher energetic first excited singlet state. Recently, this process has been applied to photoredox catalysis using metal free sensitizers.178
The previously discussed examples of photochemically supported or induced catalytic reactions involving organometallic or coordination compounds are based on electron transfer. In another type of metal catalysis, ligand exchange plays a key role. These steps are also affected by photochemical reaction conditions. A typical example is the Vollhardt reaction with inexpensive cobalt catalysts.179 Three acetylene moieties are combined in a [2 + 2 + 2] cycloaddition to form an aromatic compound. This reaction is an important method to synthesize a large variety of such compounds. Although the reaction occurs occasionally under thermal conditions, the best results are obtained when the reaction mixture is irradiated with visible light. The same reaction is also used to synthesize pyridines by combining two acetylene moieties with a nitrile.180 In this case, detailed investigations have been carried out in order to characterize the significant influence of light181 on the outcome. The enantiopure nitrile 215 was transformed with two equivalents of acetylene to yield the corresponding pyridine derivative 216 (Table 6).182 Under photochemical reaction conditions (irradiation with visible light) and when compared to thermal reaction conditions,183 the catalyst concentration was reduced. The reaction was carried out at room temperature and atmospheric pressure instead of high temperature and high pressure. The reaction was faster and the yields were increased. It must further be pointed out that in this particular example, heating of the acetylene containing reaction mixture under pressure causes an explosion risk. Under these soft photochemical reaction conditions, efficient asymmetric catalysis was also performed.184 As visible light is used to promote this catalysis, a similar reaction was performed with sunlight on an industrial scale.185
|
|
|||||
|---|---|---|---|---|---|
| Method | Conditions | Yield (%) | |||
| CpCo(cod) (mol%) | T (°C) | p (atm) | t (h) | ||
| hν | 0.5 | 25 | 1 | 4 | 90 |
| Thermal | 3.2 | 110 | 14 | 22 | 82 |
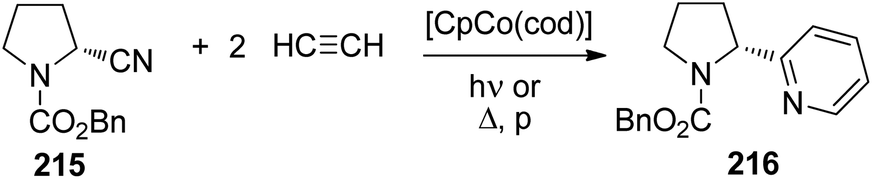
In order to characterize the influence of light on this catalytic reaction, detailed kinetic investigations have been carried out.186,187 The reaction starts with the generation of the catalytically active species CpCoI by release of cyclooctadiene from the cobalt complex 217 (Scheme 46). The addition of two molecules of acetylene generates the cobalacyclopentadiene intermediate 218.188 The addition of a nitrile molecule leads to the formation of the intermediate 219 and then to the final product 220. Under thermal reaction conditions the formation of the cobalacyclopentadiene intermediate 218 is the rate limiting step. The reaction is of zero order kinetics in nitrile consumption and of second order in acetylene consumption. Under photochemical conditions, first order kinetics is observed for the consumption of the nitrile. That means that the formation of the metallacyclic intermediate 218 is accelerated and the addition of the nitrile to 218 leading to the intermediate 219 has become the rate limiting step. Irradiation with visible light accelerates the generation of the catalytically active species CpCoI from the cobalt complex 217. Most probably, other mechanistic steps are also affected by the photochemical conditions. The formation of benzene is a competing reaction. In this case acetylene is added to the metallacyclic intermediate 218 leading to the intermediate 221. Due to the higher reactivity of the nitrile with intermediate 218, this side reaction generally plays only a minor role. However, due to the high acetylene pressure needed when thermal reaction conditions are applied, the formation of benzenes may become very disadvantageous. Using photochemical reaction conditions, the concentration of acetylene may be kept low which reduces the formation of benzene derivatives. Thus the transformations may also be carried out in solvents such as water in which the acetylene solubility is low.189
 | ||
| Scheme 46 Mechanism of the photochemically supported [2 + 2 + 2] cycloaddition leading to pyridines. | ||
Enzymatic catalysis may also be affected by photochemical reaction conditions. Recently, a number of investigations have been performed with peroxidases. Thus the chloroperoxidase (CPO) isolated from Caldariomuces fumago is capable of oxidizing thioethers such as thioanisole 222 (Scheme 47 and Table 7).190 The corresponding sulfoxide 223 is formed in high yield and high enantioselectivity when a photochemical redox reaction is involved as a co-catalytic process and when oxygen is used as the oxidant. Furthermore, no overoxidation is observed. Under normal conditions, this enzyme employs hydrogen peroxide as the oxidant. When the reaction is performed under these standard conditions with stoichiometric amounts of H2O2, conversion and turnover number (for CPO) remain low (Table 7). These observations are explained by the mechanism depicted in Scheme 48. In a photochemical redox reaction, flavin 224 is reduced. EDTA or HCO2Na are used as sacrificial electron donors. The reduced flavin derivative 225 is oxidized by molecular oxygen which leads to the formation of hydrogen peroxide. The advantage of this process using a photoredox process as co-catalysis is that the stationary concentration of H2O2 is kept low which prevents the enzyme from decomposition. Thus the high turnover numbers may be explained.
 | ||
| Scheme 47 Photochemical co-catalysis in an enzymatic CPO catalyzed oxidation. | ||
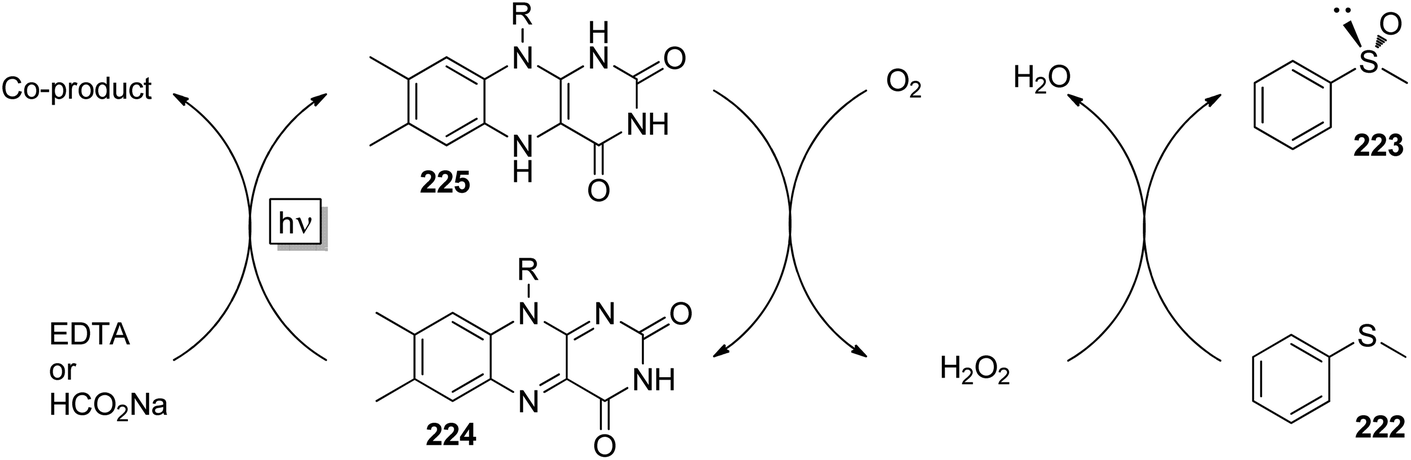 | ||
| Scheme 48 Mechanism of photochemical co-catalysis in an enzymatic CPO catalyzed oxidation. | ||
The particularly difficult activation of a C–H bond is involved in the highly enantioselective oxidation of tetrahydronaphthalene to tetrahydronaphthalen-1-ol which was accomplished with Agrocybe aegerita aromatic peroxygenase (AaeAPO).191 Using the same photoredox co-catalysis with flavin derivatives, a turnover number of 17520 and a turnover frequency of 366 min−1 (both values for the enzyme) were determined. Using the same catalytic system, alkenes are oxidized to epoxides. In biological systems, some of these oxygenases use a natural redox reaction based on enzyme fixed flavin derivatives to produce hydrogen peroxide. In this case, the reduced flavin derivative can be generated by NADPH which itself is generated from NADP+ by a dehydrogenase, for example glucose dehydrogenase. Thus, two enzymes are involved in the oxidation process. In the case of a phenyl acetone monooxygenase mutant, the NADPH cofactor system was replaced by an additional flavin based photoredox co-catalytic system.192 In such a system, the enzyme bound flavin is reduced by the added free flavin. Thus the expensive and inconvenient dehydrogenase co-catalysis is avoided.
6 Photooxygenation with singlet oxygen
In contrast to most organic molecules and many inorganic molecules, the ground state of oxygen possesses triplet spin multiplicity (Scheme 49).193,194 Two antibonding π orbitals are occupied by one electron. Two configurations are possible for energetically low singlet excited states. In the S1 state, one of the antibonding π orbitals is occupied by two electrons while the other is unoccupied. At this state, the oxygen is particular polarizable. Most of the reactions of singlet oxygen occur at this state. In the S2 state, the two antibonding orbitals are occupied, each by one electron with opposite spin. Despite its low electronegativity and due to spin forbidden interactions between triplet and singlet species, the chemical reactivity of oxygen at the ground state is reduced.194 Also for this reason, biomolecules along with many other molecules at a singlet ground state are stable in an oxygen containing atmosphere and most of the known living organisms could evolve under these conditions (for some recent studies on these properties see ref. 195). Reactions of triplet oxygen are often characterized by radical intermediates. In contrast to this, singlet oxygen should possess a much higher reactivity and polar intermediates should also play an important role in the reaction mechanism. Photochemically, singlet oxygen is most conveniently produced by sensitization (Scheme 50). Since the excitation energy is low (22.4 kcal mol−1 for the S1 state), dyes absorbing visible light are most frequently used.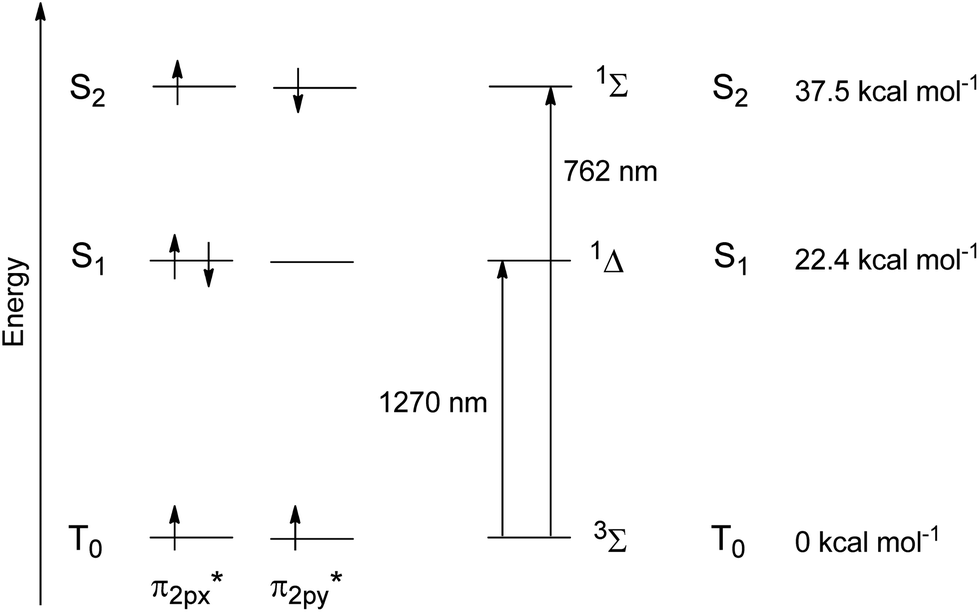 | ||
| Scheme 49 Electron configurations and energy levels of oxygen. | ||
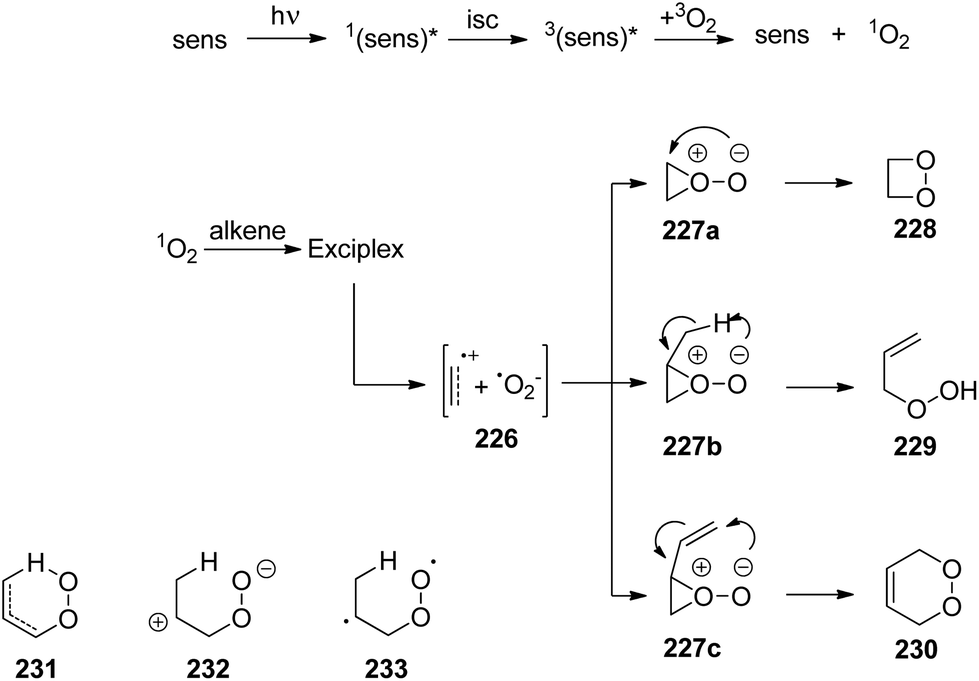 | ||
| Scheme 50 Intermediates of photooxygenations. | ||
Photooxygenation with singlet oxygen considerably enriches the oxidation methodology in organic synthesis.196–198 Furthermore, the simple and cheap oxidants, air or molecular oxygen, are used to perform sophisticated oxidations. In this context, the reactions with alkenes are of particular interest. A certain number of intermediates are involved in such transformations. First, exciplexes are formed (Scheme 50). Depending on the redox potentials of the reaction partners and the reaction medium more or less stable ion radicals 226 are formed. Formation of σ-bonds leads to intermediates such 227a,b,c depending on the structure of the alkenes (Scheme 50). In order to explain the outcome of organic photooxygenation, these intermediates are of particular interest.198,199 In particular, the regio- and stereoselectivity of many transformations may be explained when considering these structures. Three reactions of singlet oxygen and alkenes are most frequently observed. The tricyclic intermediate 227a may rearrange to form a dioxetane, the product of a [2 + 2] cycloaddition (228). The same type of intermediate (227b) also may undergo rearrangement leading to a hydroperoxide 229. In the case of dienes the corresponding intermediate (227c) leads to endoperoxides such as 230. Intermediates of transition states such as 231, 232, or 233 are also discussed.
Asymmetric synthesis with photochemical reactions is particularly interesting since highly energetic states and intermediates are involved.6,200,201 In the past, photochemical reactions were often considered as inherently unselective since it was supposed that the loss of the activation energy excess could only occur in an uncontrolled way via numerous reaction channels. It was concluded that photochemical reactions are only of little interest for application to organic synthesis. However in the large majority of photochemical reactions, the loss of the high electronic excitation energy occurs in a very controlled or defined way and the reactions become highly selective. This has already been shown by the previously discussed reactions. This topic is also well illustrated by the following example of an asymmetric photooxygenation of encarbamates.201 In this case, two processes, the ene reaction and the formal [2 + 2] photocycloaddition (dioxetane formation), are in competition (Scheme 51).202 In the case of Z-234, the dioxetane 235 is the major product. The ene product 236 is only formed in minor amounts. The ratio varies between 75:25 and 87:13 in favor of 235 depending on the substituent R. In the case of E-234, the formation of the ene product 236 is favored with respect to 235. The product ratios range between 64:36 and 92:8 in favor of 236. This product selectivity was mainly explained by frontier orbital interactions as depicted in structures 237 and 238 of the transition states for the approach of singlet oxygen. Other directing effects have been identified later. The nitrogen atom of 234 conducts the oxygen to the alkene. In the case of Z-234, hydrogen transfer from the distant methyl group is thus inhibited. In the case of E-234 the methyl group is close to the approaching singlet oxygen. Hydrogen transfer is facilitated and the ene reaction is favored. In the case of the ene reaction and the reaction of Z-234, the diastereoselectivity ranges from 53:47 (R = Me) to 95:5 (R = tBu) in favor of the ul attack. In the case of the reaction of E-234, it ranges from 56:44 (R = iPr) to 91:9 (R = tBu). In contrast to the diastereoselectivity of the ene reaction, that one of dioxetane formation is very high, around 99:1 and does not depend on the nature of R. Transition states of both reactions and their impact on the stereoselectivity resemble the corresponding epoxy like structures as depicted in Scheme 50 (227a,b).203
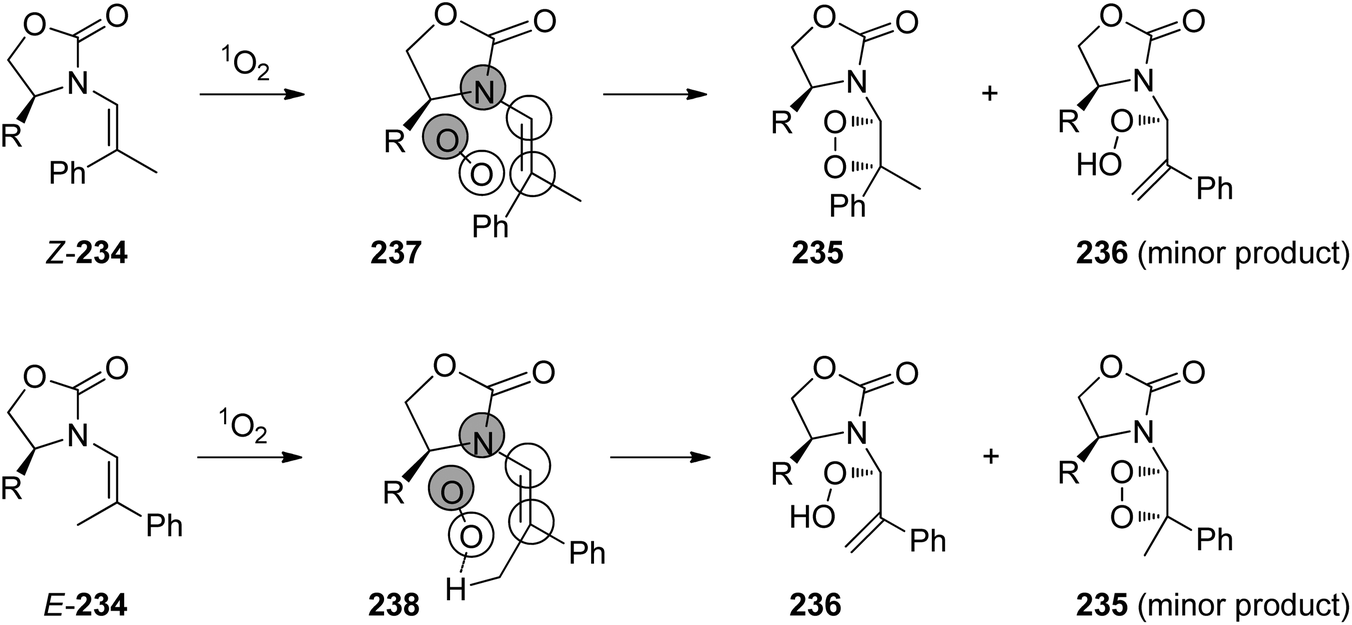 | ||
| Scheme 51 Photooxygenation of ene carbamates. | ||
Further remarkable effects were observed concerning the stereoselectivity. This topic was studied in detail for the formal [2 + 2] addition of singlet oxygen to ene carbamates leading to dioxetanes. Compounds Z-239 and E-239 carrying an additional chiral phenethyl substituent in position 2 have been transformed (Scheme 52). The absolute configurations of the dioxetanes 240 and 241 have been determined by chemical correlation transforming them into diols 242. In many cases in particular in the reactions of E-239, the resulting dioxetanes are not stable and decompose into diphenylpropanone 243 and the oxazolidinone derivative 244. As already pointed out and in contrast to the corresponding ene reaction, the diastereoselectivity in this transformation is very high and does not depend on the substituent R (Table 8). When R = Me (entry 1) the same diastereoselectivity is observed than in the cases of higher encumbering substituents such as R = iPr (entries 2 and 3), R = tBu (entry 5) or R = Ph (entry 6). This is surprising because the oxygen molecule is small and the stereodifferentiation of the attack in both diastereotopic half spaces should not be very different as is the case for the corresponding ene reaction. Only in the case of the deuterated substituent, the diastereoselectivity is lower. This effect will be discussed later. The configuration at the center 3 or the relative configuration of both stereo-centers has no influence on the diastereoselectivity (entries 2 and 3). At this stage, no effect of double induction204 is detected. Furthermore, it was found that the E-239b reacts with the same diastereoselectivity than the corresponding Z-239b which means that the chiral center in position 1 in 240 or 241 of the dioxetane ring is formed with the same relative configuration with respect to the chiral center of the carbamate ring (entries 7 and 2). Concerning the stereochemistry of the singlet oxygen approach to the enamine moiety, X-ray structure analytical data were discussed and it was assumed that similar privileged conformations exist in solution.201,202,205 It was found that in the case of the Z-isomer, the oxazolidinone ring is placed almost in the plane of the enamine moiety (Scheme 53). For steric reasons, the attack of the singlet oxygen occurs anti with respect to the R substituent of the oxazolidinone ring. In the case of the E-isomers, the oxazolidinone ring is almost orientated orthogonally with respect to the enamine plane. The singlet oxygen approaches preferentially from the same side leading to a dioxetane with the same relative configuration with respect to the chiral center in the oxazolidinone ring. In this case, the singlet oxygen is further conducted in this diastereomeric half-space by an interaction with the carbonyl group of oxazolidinone.206
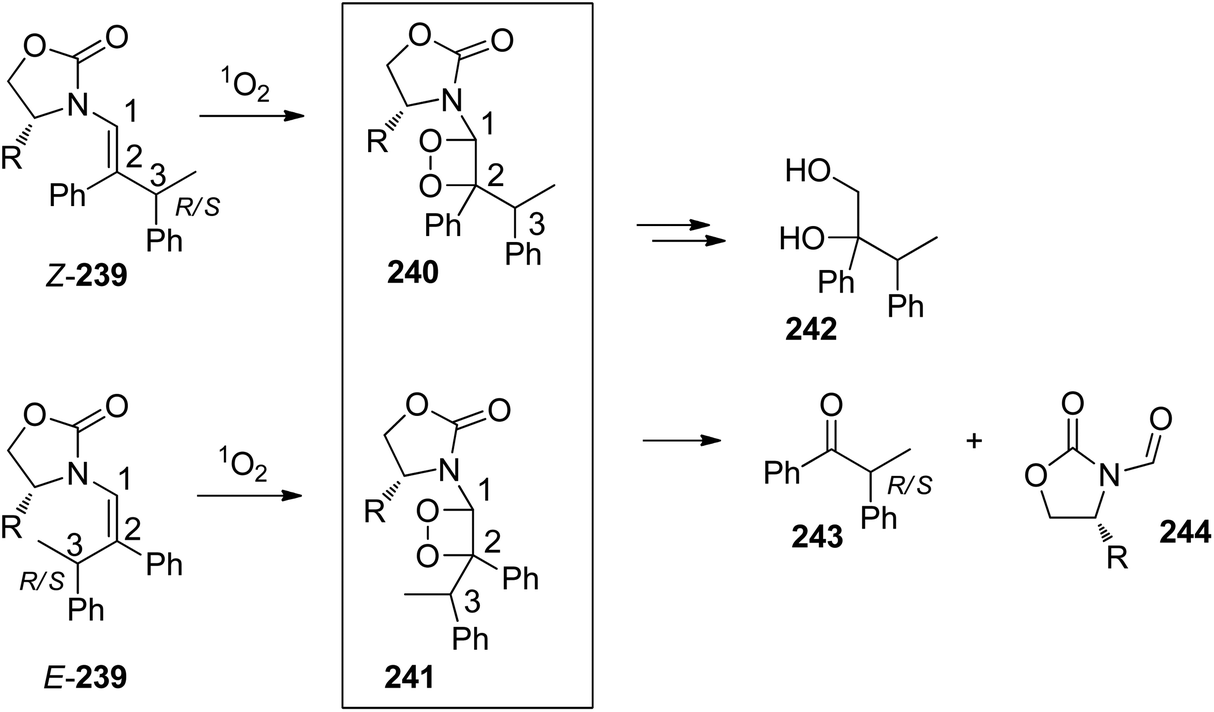 | ||
| Scheme 52 Chemical correlation for the determination of the chiral induction in the photooxygenation of ene carbamates, cleavage of dioxetanes leading to diphenylpropanone 243 and the oxazolidinone derivative 244. | ||
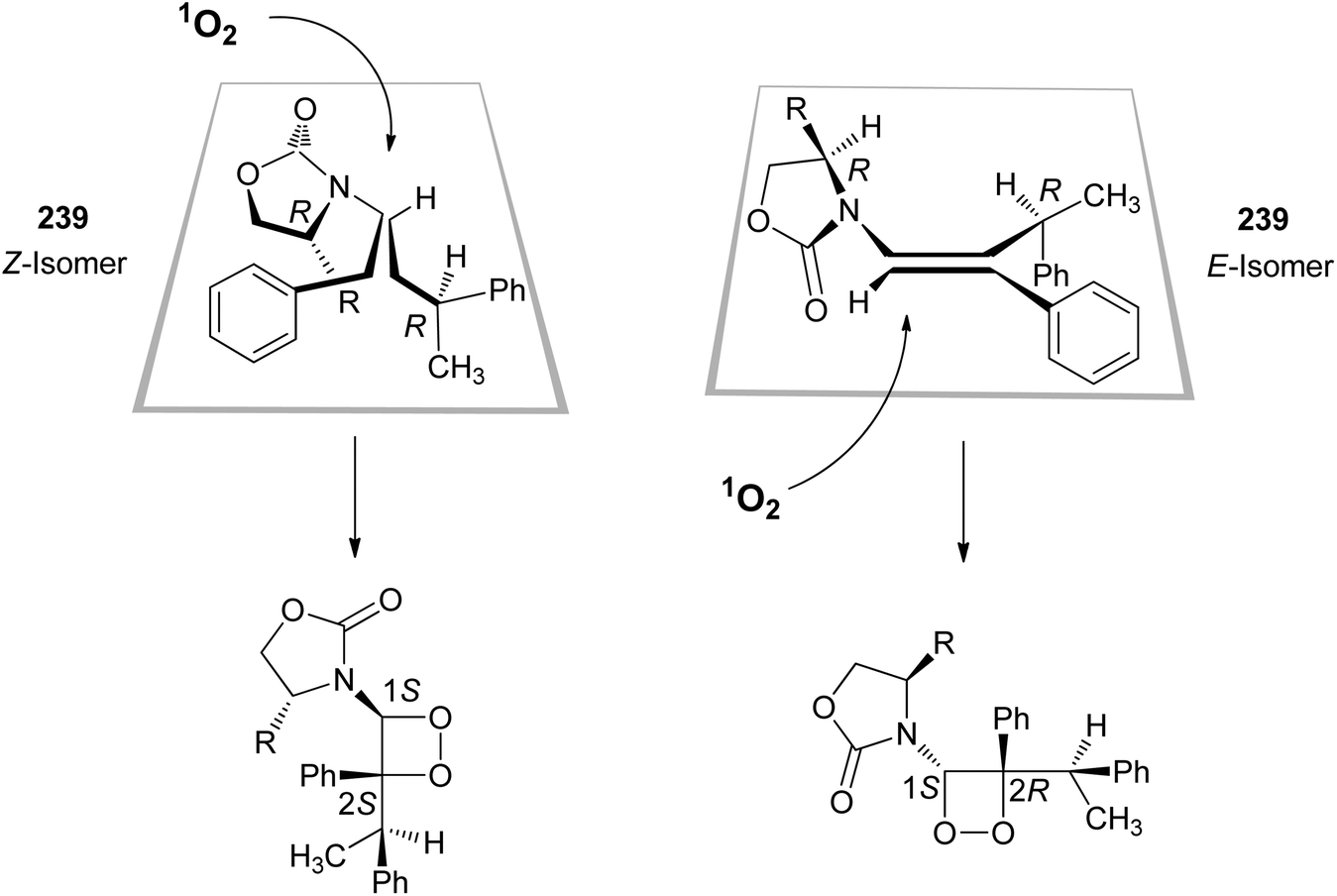 | ||
| Scheme 53 Diastereoselective photooxygenation of Z and E-ene carbamates 239. Structures of the substrates are based on X-ray structure analysis. | ||
| Entry | Substrate | Configuration | Diastereomeric ratio | |
|---|---|---|---|---|
| R | C-3 | (1S,2S)/(1R,2R) | ||
| a The transformations were carried out at −32 °C in CDCl3 until complete conversion. No competing ene reaction was detected. b The reactions were carried out in CD2Cl2 as the solvent. c The corresponding diastereomeric ratio is (1S,2R)/(1R,2S). | ||||
| 1 | Z-239a | (R)-Me | (S) | 99:1 |
| 2 | Z-239b | (R)-iPr | (S) | 99:1 |
| 3b | Z-239c | (S)-iPr | (S) | 1:99 |
| 4b | d8-Z-239c | d7-(S)-iPr | (S) | 10:90 |
| 5 | Z-239d | (S)-tBu | (R) | 1:99 |
| 6 | Z-239e | (S)-Ph | (S) | 1:99 |
| 7b | E-239b | (R)-iPr | (S) | 99:1c |
As has already been pointed out, the chiral center in position 3 of compounds such as 239 (Scheme 52) has no influence on the diastereoselectivity of dioxetane formation and consequently, the decomposition product 1,2-diphenylpropan-1-one 243 was obtained as a racemic mixture. A closer look at this transformation revealed however that the two diastereoisomers react with different rates.205,207,208 Consequently and depending on the difference of these two rates, the ketone 243 is obtained in an enantiomeric excess when the reaction is stopped before conversion is complete. For example, when a 1:1 mixture of the diastereoisomers E-239f and E-239b is photooxygenated, in CD3OD as the solvent, at 50 °C and when the reaction is stopped at a conversion of 30%, 243 is isolated with 70% ee in favor of the R enantiomer (Scheme 54).207 When the same mixture of diastereoisomers is photooxygenated in CD3CN as the solvent, at 50 °C and when the reaction is stopped at a conversion of 23%, 243 is isolated with 64% ee in favor of the S enantiomer. In view of the asymmetric synthesis of the chiral ketone 243, this transformation is a kinetic resolution.209 In such reactions the enantiomeric excess (ee) depends on the progress of the conversion C. The product ratio results from the rate ratio kR/kS which determines the stereoselectivity s of the reaction. These parameters are combined in eqn (2).
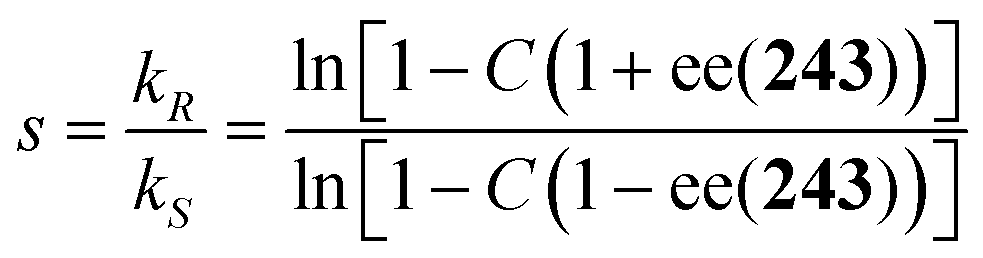 | (2) |
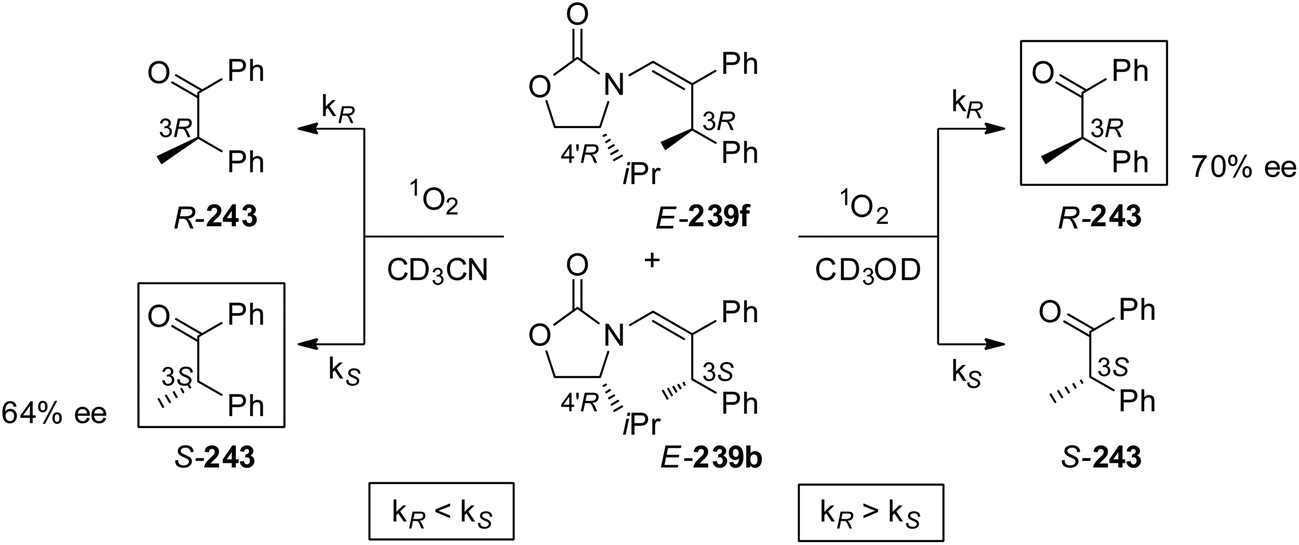 | ||
| Scheme 54 Kinetic resolution by photooxygenation of ene carbamates. | ||
Based on this equation, the selectivity s or the rate ratio kR/kS can be calculated when the conversion and enantioselectivity of a transformation are determined as is usually the case. In Fig. 9, this relation is depicted. In particular when the selectivity is high, best ee values are observed for conversions around 50%.
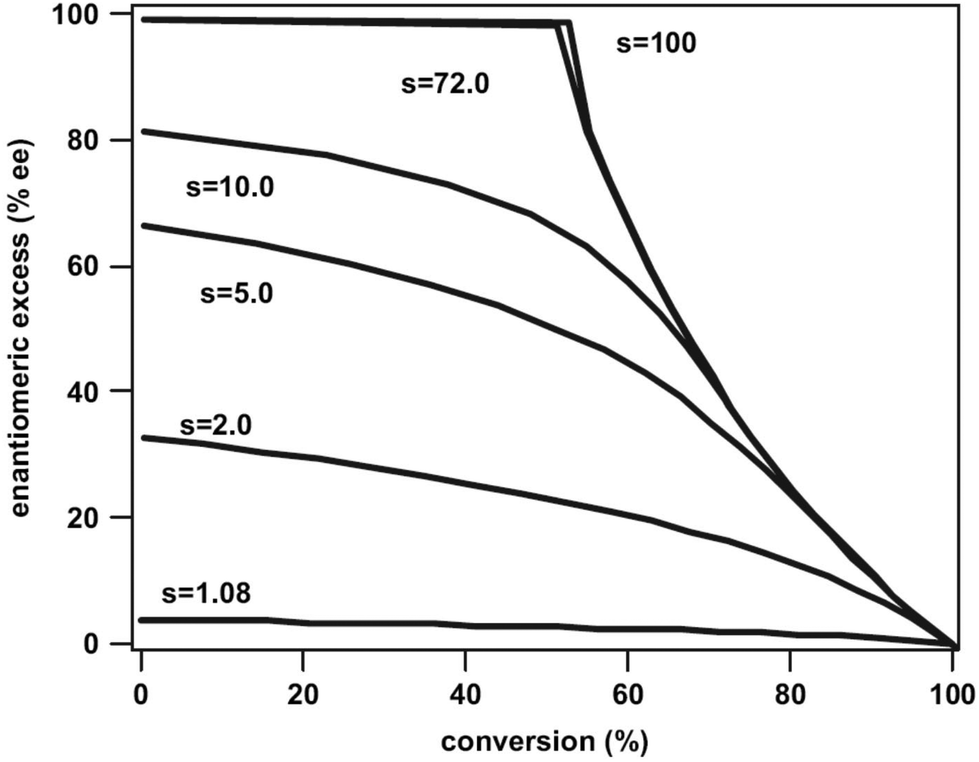 | ||
| Fig. 9 Dependence of enantiomeric excess of 243 on the conversion in the photooxygenation of ene carbamates (compare Scheme 54). (Reprinted with permission from: J. Sivaguru, M. R. Solomon, H. Saito, T. Poon, S. Jockusch, W. Adam, Y. Inoue and N. J. Turro, Tetrahedron, 2006, 62, 6707–6717. Copyright (2006) with permission of Elsevier.) | ||
To get a deeper insight into the mechanistic details of the stereoselectivity, the selectivity s or the rate ratio kR/kS was determined at different temperatures using different solvents.205 For the E isomeric substrates (Chart 1), a high dependence of the enantioselectivity of 243 from these parameters is observed. This is not the case for the corresponding Z isomers.207 Using the Eyring relation (eqn (3)) and a corresponding plot, the enthalpy and the entropy contribution to the free enthalpy ΔΔG≠ of the diastereoselectivity are determined.
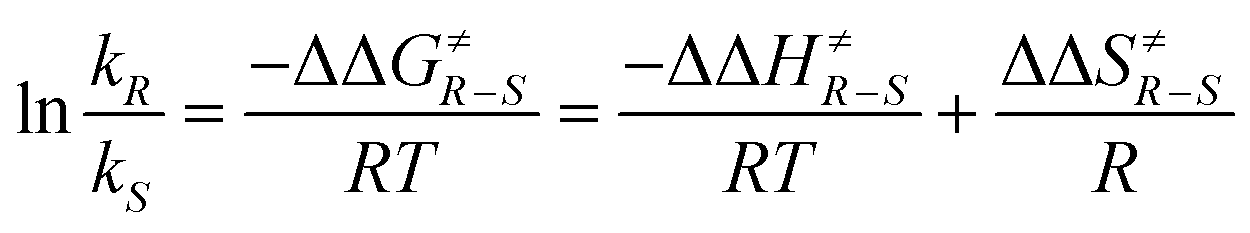 | (3) |
 | ||
| Chart 1 Compounds used to determine the influence of the reaction temperature and the solvent on the stereoselectivity. | ||
When s or the rate ratio ln(kR/kS) is plotted against T−1, linear relationships are observed and ΔΔH≠ and ΔΔS≠ values can be determined from the slope and the intersection with the y-axis respectively. Fig. 10 shows such plots for the photooxygenation of E-245a in different solvents and for E-245b in CD2Cl2. The highest enantioselectivity is observed in CD3OD at low temperatures. Particularly when the reaction is carried out in CD3CN or in CD2Cl2, it was observed that by increasing the temperature, the formation of R-245 is favored no more and instead that of S-243 is. According to the Eyring equation, at higher temperature, the entropy contribution increases and may become dominant. Thus the privileged formation of the S-243 is favored by the entropy while the privileged formation of R-243 is favored by the enthalpy. Such behavior is generally observed when conformational mobility plays a more important role than simple steric hindrance (in rigid structures). Indeed, these observations are in contrast to the more rigid structure of Z-245b or Z-245c for which the ΔΔH≠ and ΔΔS≠ are small and the temperature dependence of the enantioselectivity is low. Due to the fact that the chiral center at the oxazolidone moiety is changed in E-245b with respect to E-245a the corresponding lines for the photooxygenation in CD2Cl2 are mirroring. Their intersection is close to the x-axis and the corresponding ΔΔH≠ and ΔΔS≠ values have opposite signs.
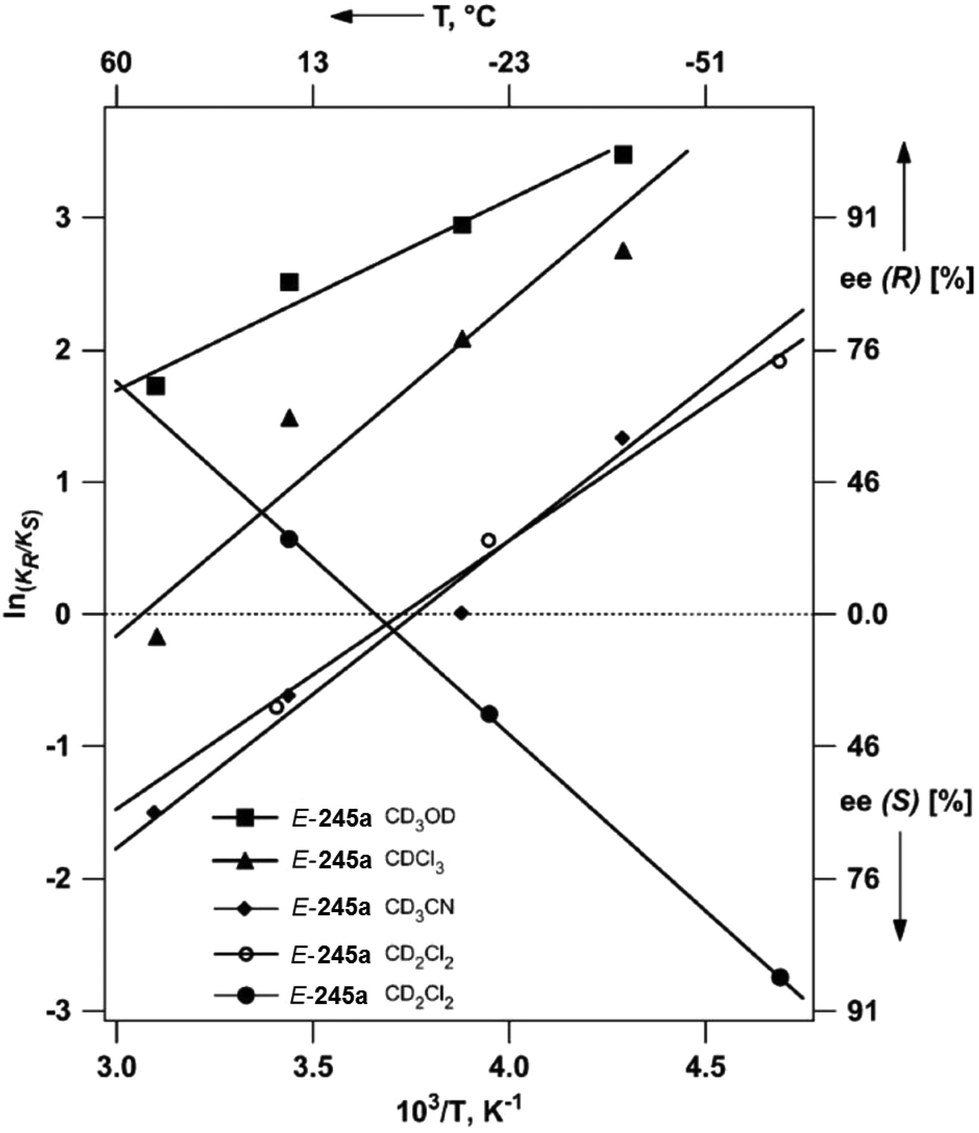 | ||
| Fig. 10 Photooxygenation of E-245a and E-245b (Chart 1) in different solvents. (Adapted with permission from: J. Sivaguru, M. R. Solomon, H. Saito, T. Poon, S. Jockusch, W. Adam, Y. Inoue, N. J. Turro, Tetrahedron, 2006, 62, 6707. Copyright (2006) with permission of Elsevier.) | ||
As pointed out above, the entropy (ΔΔS≠) and the enthalpy contribution (ΔΔH≠) to the free enthalpy (ΔΔG≠) of the enantioselectivity is different for each system (molecular structure, solvent). It is further observed that the difference of the enthalpy contribution between two systems is proportional to the corresponding difference of entropy contribution which means that there exists a linear relationship between ΔΔH≠ and ΔΔS≠. A change in entropy contribution is compensated by a change in enthalpy contribution. When activation enthalpies and activation entropies are involved such compensation phenomena are called isokinetic or isoselective relationships.51,81,88–90,210 The observation of such a relationship in the present case indicates that the photooxygenations of investigated enecarbamates205,207 possess the same reaction mechanism. In Fig. 11, the isoselective relationship is depicted. The photooxygenation of compounds E-245d and E-245a have been performed in 4 different solvents as indicated above while Z-245a and E-245b have been transformed only in CD2Cl2.
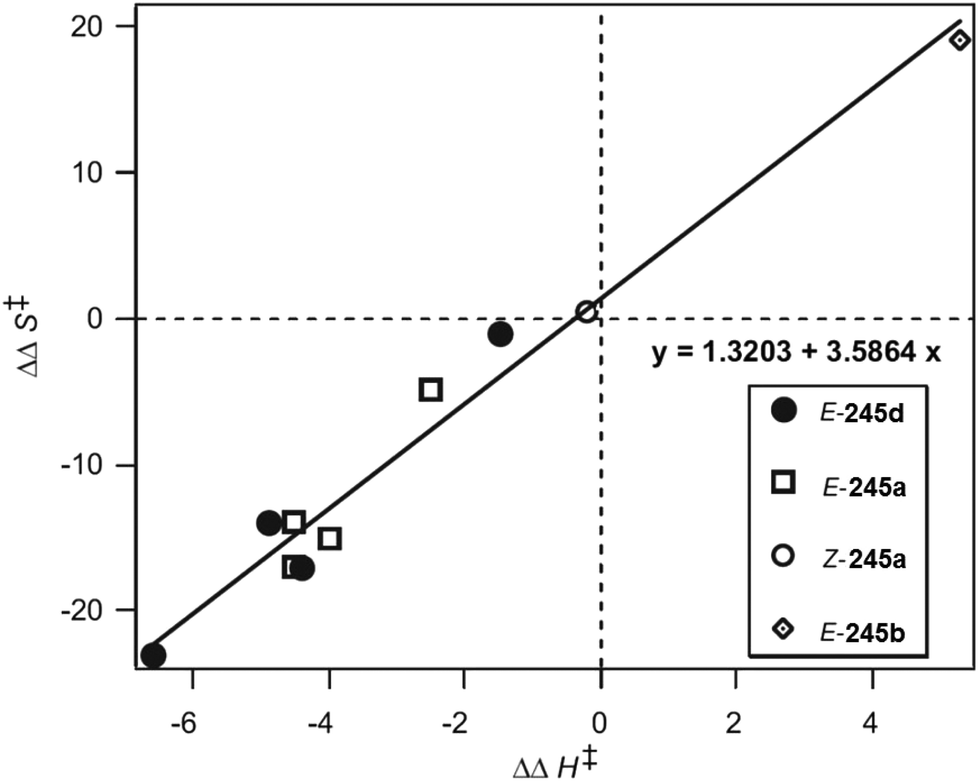 | ||
| Fig. 11 Isoselective relationship for the photooxygenation of various enecarbamates. (Adapted with permission from: J. Sivaguru, M. R. Solomon, H. Saito, T. Poon, S. Jockusch, W. Adam, Y. Inoue, N. J. Turro, Tetrahedron, 2006, 62, 6707. Copyright (2006) with permission of Elsevier.) | ||
It is surprising that the interaction of a small particle such as singlet oxygen can cause such high enantioselectivities, even in conformationally labile systems as just outlined above. In fact, 1O2 is the smallest oxidation reagent commonly used in organic synthesis. Oxidations of chiral ene carbamates with the somewhat more encumbering ozone were much less selective.208 Therefore, reasons other than only steric interactions have been suggested in order to explain the stereoselectivity increase of the photooxygenation of ene carbamates. In particular, deactivation of singlet oxygen has been discussed in this context. It is well known that the lifetime of the singlet oxygen is much higher in deuterated than in the corresponding non-deuterated solvents.197,211,212 Vibrations of a C–H bond more efficiently quench singlet oxygen than C–D bonds. This effect has an impact on the diastereoselectivity. In fact, the stereoselectivity of the photooxygenation of Z-239c is higher than that of d8-Z-239c (Table 8). The photooxygenation in a diastereotopic half-space containing a lot of C–H bonds is less efficient than the one in a diastereotopic half space with only few of such bonds since in the first case vibrationally induced deactivation of singlet oxygen is more efficient. Or, a diastereoisomer is slowly oxidized when it assembles a large number of C–H bonds in the volume element in which the singlet oxygen approaches the substrate. A competing diastereoisomer that assembles a lower number of C–H bonds in this volume element is rapidly oxidized. These effects have been discussed in the photooxidation of E-245c (Scheme 55 and Table 9).205 When carried out in acetonitrile as an aprotic solvent, the 3S-E-245c epimer was more rapidly transformed (kR < kS) at higher temperature (18 °C) while at low temperature (−40 °C) R-243 was preferentially formed (kR > kS). A closer inspection of the X-ray structures revealed that in the case of the 3R-E-245c epimer, the methyl group of the oxazolidone moiety and the phenyl substituent of the phenethyl moiety are oriented opposite to each other in two diastereotopic half-spaces. In the case of the 3S-E-245c epimer, the methyl groups of the oxazolidone and the phenethyl moieties are in the same diastereotopic half-space. When it is assumed that the crystalline structure resembles the privileged conformation in solution at low temperature, the reactive center of the epimer 3S-E-245c is better protected by C–H bonds and consequently the deactivation of singlet oxygen is more efficient. The photooxygenation of this stereoisomer is slower and the formation of R-243 from 3R-E-245c is faster. At higher temperatures, other conformations may be favored inducing the opposite stereoselectivity as was indeed observed at higher temperatures (18 °C). Furthermore, steric hindrance may become more important with respect to the deactivation rates of singlet oxygen.
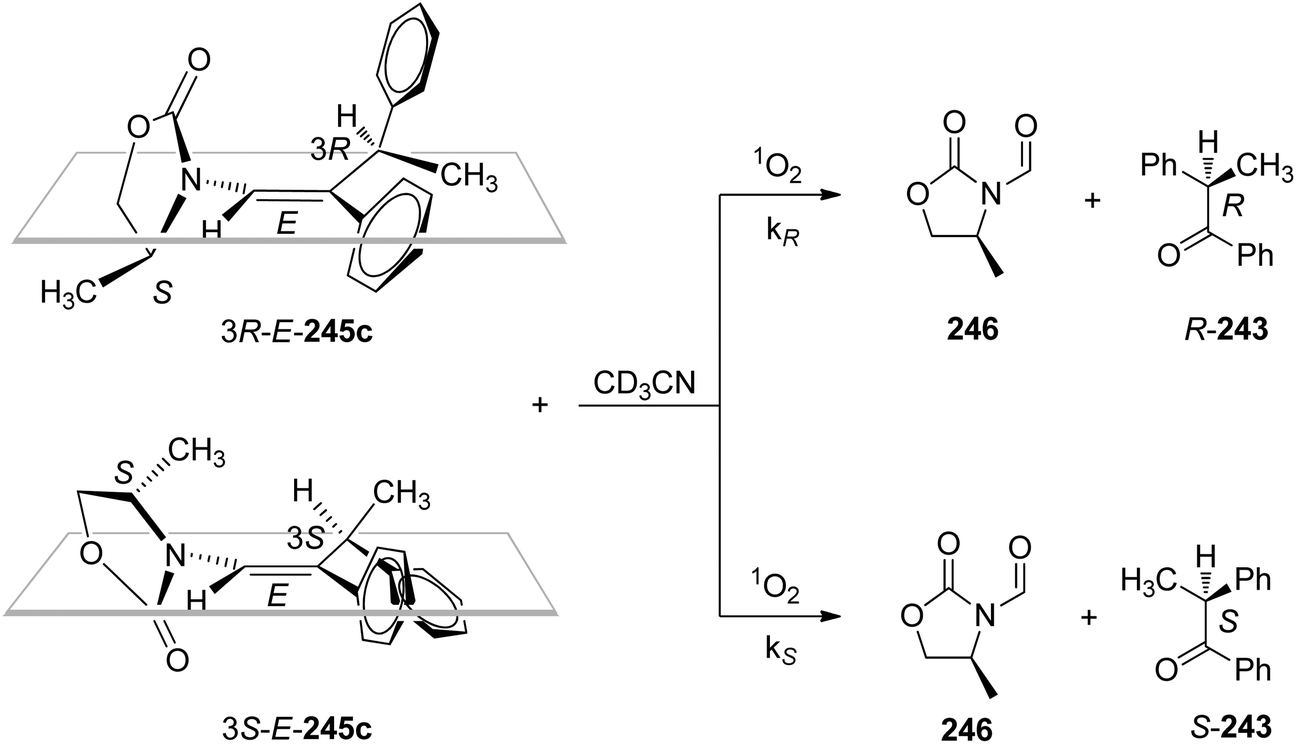 | ||
| Scheme 55 Stereoselective photooxygenation of two ene carbamate epimers. | ||
These investigations nicely show how excess energy of an electronically excited species can be released in a very controlled manner leading to highly selective transformations. As has already been pointed out, this property of photochemical reactions is essential for application to organic synthesis. Similar discussions may be performed for many other organic photochemical reactions.
7 Photochemical reactions in micro and continuous flow reactors
Photochemical reactions on the laboratory-scale are traditionally performed in batch mode using either immersion well or chamber reactors.213 Immersion well systems (Fig. 12a) enable individual irradiations of larger volumes (≤1 L) and comprise a central mercury pressure lamp, an immersion well (effectively a cooler for the lamp) and an outer reaction vessel. Chamber reactors utilize an outer array of fluorescent tubes and reaction vessels are inserted into the vacant inner chamber (Fig. 12b). These more flexible systems are typically used for small-scale synthesis (≤250 mL). | ||
| Fig. 12 (a) Immersion well batch reactor (UV-LRS-1; UV-Consulting Peschl, Germany). (b) Rayonet chamber reactor (RMR-600; Southern New England Ultraviolet Company, USA). | ||
Conventional batch photoreactors have a number of disadvantages that have restricted their applications on large, technical scales. As a consequence of the Beer–Lambert law,214 light is rapidly absorbed by the chromophoric reagent. Consequently, photochemical processes require high dilutions and narrow reaction vessels. The constant irradiation of the reaction mixture additionally favors degradations and polymerizations of light-sensitive products, thus reducing yields and necessitating resource-demanding purifications. Iron and polymer deposits on glass parts around the light source are also common and demand constant cleaning and maintenance. The intense heat generation of most mercury pressure lamps furthermore demands excessive cooling.
Recently, continuous microflow reactors have emerged as new, resource-efficient synthesis tools and have found widespread applications in synthetic organic chemistry.215 These devices are characterized by very narrow inner dimensions (by definition <1 mm in at least one direction, typically depth or diameter), which makes them suitable for photochemical applications.216,217 Fixed reactor models such as the dwell device by Mikroglas Chemtech have the reaction channel embedded in the reactor block (Fig. 13a). Complete commercially available microreactor systems have now been developed. The advanced UV-150 flow reactor by Vapourtec (Fig. 13b), for example, utilizes a microcapillary unit in combination with a 150 W mercury-lamp and allows for multi-gram scale synthesis.134c A flexible multi-microcapillary flow reactor (MμCFR) (Fig. 13c) for parallel microflow photochemistry operations has been recently described.218 This reactor has been adopted for process optimization, process validation, scale-up and parallel library synthesis.
 | ||
| Fig. 13 (a) Dwell device (Mikroglas Chemtech GmbH, Germany) under a UV-panel (Luzchem Research Inc., Canada). (b) UV-150 E-series flow chemistry system (Vapourtec Ltd, UK). (c) Multi-microcapillary flow reactor (MμCFR). | ||
Although earlier examples of flow-photochemical reactions exist, the photopinacolization of benzophenone 130 (0.5 M) in isopropanol is regarded as the first comprehensive microreactor comparison study (Scheme 56).219 The initial microchip reactor was fabricated by bonding a patterned silicon wafer to a Pyrex plate and incorporated a serpentine reaction channel. The device was coupled with a separate detection chip for on-line UV detection. An improved model consisted of a sandwich structure of a silicon wafer between two quartz plates. The chip incorporated a serpentine reaction channel with an integrated detection point for instant on-line UV analysis. The reaction mixtures were pumped through the chips with a syringe pump and irradiated with a small UVA lamp. With an optimum flow rate of approximately 4 μl min−1, high conversions of 130 of 60% (by HPLC) were achieved without blockage of the channel. The photodimer 247 readily crystallized in the product vessel, thus allowing easy isolation.
 | ||
| Scheme 56 Photopinacolization of 130 in isopropanol. | ||
The 4,4′-dimethoxybenzophenone (DMBP) sensitized addition of isopropanol to furanones 164 was chosen to investigate different flow reactor setups (Scheme 57).220 Reactor Type I consisted of a commercially available dwell device, which was placed under a UVA light panel (5 × 8 W). Reactor Type II combined a microchip with a UVA-LED array (6 × 75 mW), while reactor Type III utilized a dual polytetrafluoroethylene (PTFE) capillary tower equipped with a single UVA tube (8 W) at its center. The small UVA-LED microchip gave high conversions of 164 with a residence time of just 2.5 min, while all other microreactors and the conventional batch reactor required 5 min. Despite high space-time-yields (STY) and energy-efficiency estimations, however, the productivity of the microchip remained low. The flexible and open capillary system was thus further advanced into a 10-microcapillary flow reactor (MμCFR) for parallel microflow photochemistry.218
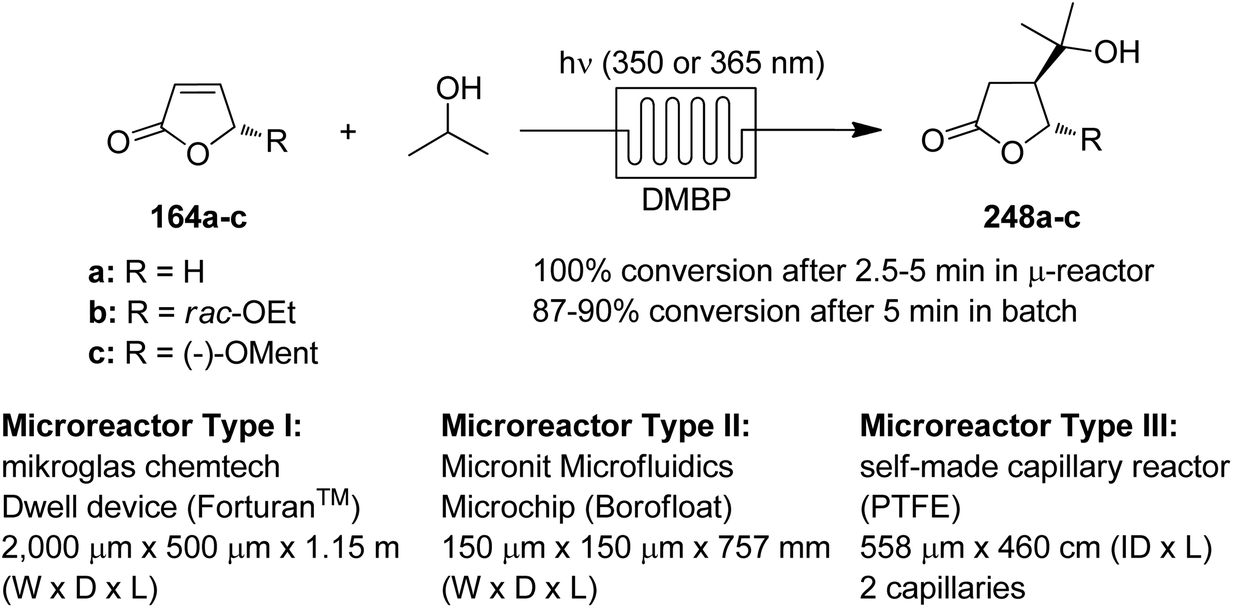 | ||
| Scheme 57 DMBP-sensitized addition of isopropanol to furanones. | ||
The steroid myriceric acid (251), a potent endothelin A receptor antagonist, is available from the intermediary nitrite 249via the Barton reaction. The conversion of 249 to the corresponding oxime 250 was therefore investigated in different microreactor setups (Scheme 58).221 A glass-covered, stainless steel microreactor (Type I) in combination with a 300 W high-pressure mercury lamp was chosen for process optimization. A soda lime cover gave the best yield of oxime 250 with 59% as its cut-off wavelength at >320 nm prevented further photodegradation. Yields of 71% and 70% (by HPLC) were increased when a 15 W black light (UVA) lamp or a UV-LED array (48 × 35 mW) in combination with a Pyrex glass top were used instead. Utilizing two stainless steel reactors in-series (Type II), a set of 8 × 20 W black light lamps, DMF and a residence time of 32 min, continuous operation for 20 h gave 3.1 g of pure 250 (60% yield). In an extension, a fully automated process for the synthesis of 250 was realized in a single multi-lane stainless steel reactor (Type III) equipped with six evenly positioned 15 W black light bulbs. After operation for 40 h and using a residence time of 20 min, the reactor plant produced 5.3 g of 250 (61% yield).
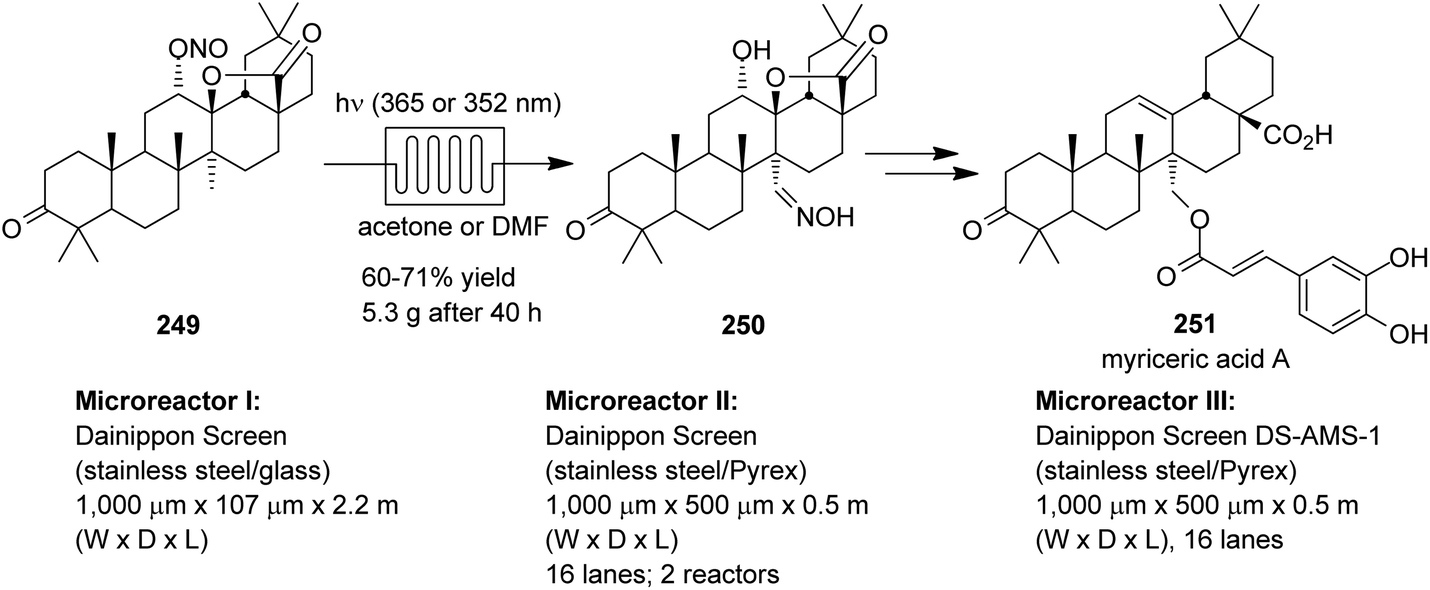 | ||
| Scheme 58 The Barton reaction as a key step in the synthesis of myriceric acid A (251). | ||
The synthesis of doxercalciferol (1α-hydroxyvitamin D2; 255) from ergocalciferol (252) has likewise been realized using a continuous flow photochemical key-step.222 The photoisomerization of the bis-tert-butyldimethylsilyl (bis-TBS) ether 253 was accomplished in a fluorinated ethylene propylene (FEP) microcapillary reactor using 9-acetylanthracene (9-AA) as a sensitizer (Scheme 59). The capillary was wrapped around an immersion well fitted with a Pyrex sleeve and a 450 W mercury pressure lamp. Removal of the sensitizer was achieved in-line via filtration over a short carbon/Celite (1:2 w/w) column. Consequent optimization of the reaction conditions produced 254 in up to 96% yield (by HPLC). Overall, 255 was obtained in about 10% from ergocalciferol (252) without purification by chromatography.
 | ||
| Scheme 59 Synthesis of doxercalciferol (255) from ergocalciferol (252). | ||
The synthesis of the antimalarial compound artemisinin (258) via a tandem photochemical–thermal continuous flow process has been recently achieved (Scheme 60).223 Dihydroartemisinic acid (256) is converted by photooxygenation with tetraphenylporphyrin (TPP) into the hydroperoxide intermediate 257. A slow stream of oxygen is injected into the liquid reagent stream creating a slug flow pattern inside a FEP capillary. Irradiation is conducted with a 450 W medium pressure mercury lamp. Subsequently, trifluoroacetic acid (TFA) is injected into the product stream from the photochemistry module. This mixture enters a second, thermal (60 °C) reactor module made of polytetrafluoroethylene (PTFE) tubing, where 257 is converted into artemisinin (256). Using this continuous flow plant, 258 is isolated in an overall yield of 39% with the capability of producing 200 g of 258 per day.
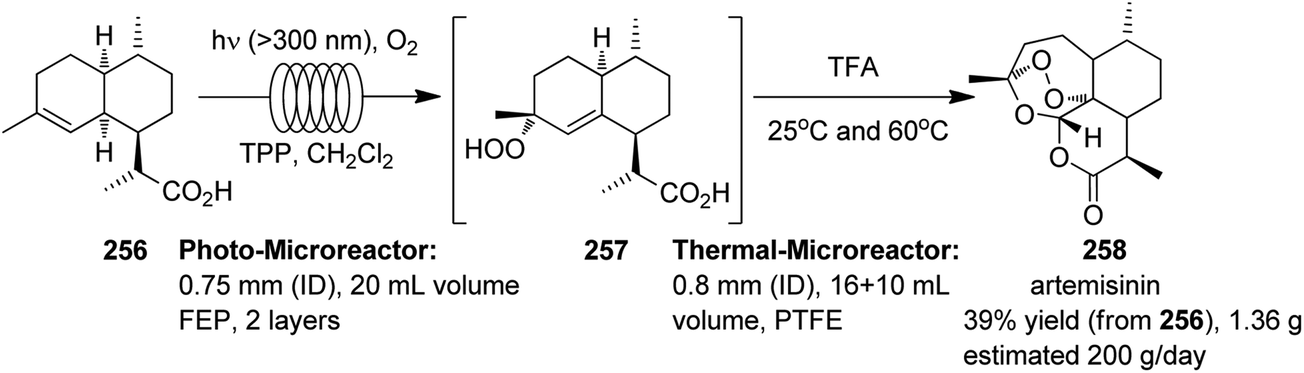 | ||
| Scheme 60 Continuous synthesis of artemisinin (258) via a tandem photochemical–thermal process. | ||
8 Conclusion
In conclusion, photochemistry represents an elegant enabling methodology for the synthesis of complex natural products, commodity chemicals or novel chemical entities. Photochemistry has thus expanded the synthesis portfolio of organic chemistry. The traditionally strong links between organic photochemistry and physical chemistry enable a high level of understanding of these reactions.1,2,4 Profound investigations of reaction mechanisms are frequently carried out in this domain which significantly facilitates the optimization and the application of these organic photochemical reactions. Light can be easily tuned and controlled with a ‘flick of a switch’, thus allowing safe operations. Despite these advantages, industrial photochemistry is limited to certain commodity chemicals.224 With the development of novel reactor systems and light sources,130,217,225,226 it is hoped that photochemical reactions will become more widespread in the future. The recently established industrial synthesis of artemisinin (A3) and the innovative Heraeus Noblelight process for the synthesis of anti-cancer precursors represent encouraging examples.227,228References
- (a) P. Klán and J. Wirz, Photochemistry of Organic Compounds, Wiley, Chichester, 2009 Search PubMed; (b) N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, 2010 Search PubMed.
- (a) N. J. Turro, Angew. Chem., Int. Ed. Engl., 1986, 25, 882–901 CrossRef; (b) M. Olivucci and F. Santoro, Angew. Chem., Int. Ed., 2008, 47, 6322–6325 CrossRef CAS PubMed; (c) I. Schapiro, F. Melaccio, E. N. Laricheva and M. Olivucci, Photochem. Photobiol. Sci., 2011, 10, 867–886 RSC.
- H. E. Zimmerman, Angew. Chem., Int. Ed. Engl., 1969, 8, 1–11 CrossRef CAS.
- (a) N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed; (b) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (c) C.-L. Ciana and C. G. Bochet, Chimia, 2007, 61, 650–654 CrossRef CAS.
- (a) CRC Handbook of Organic Photochemistry and Photobiology, ed. A. Griesbeck, M. Oelgemöller and F. Ghetti, CRC Press, Boca Raton, 3rd edn, 2012 Search PubMed; (b) Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH, Weinheim, 2010 Search PubMed; (c) Synthetic Organic Photochemistry, ed. A. G. Griesbeck and J. Mattay, Marcel Dekker, New York, 2005 Search PubMed.
- (a) Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS; (b) Chiral Photochemistry, ed. Y. Inoue and V. Ramamurthy, Marcel Dekker, New York, 2004 Search PubMed.
- H. Hopf, Classics in Hydrocarbon Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- N. Hoffmann, Photochem. Photobiol. Sci., 2012, 11, 1613–1641 CAS.
- (a) A. Albini and M. Fagnoni, Green Chem., 2004, 6, 1–6 RSC; (b) M. Oelgemöller, C. Jung and J. Mattay, Pure Appl. Chem., 2007, 79, 1939–1947 CrossRef; (c) S. Protti, D. Dondi, M. Fagnoni and A. Albini, Green Chem., 2009, 11, 239–249 RSC; (d) M. Oelgemöller, J. Chin. Chem. Soc., 2014, 61, 743–748 CrossRef; (e) M. Oelgemöller, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.5b00720.
- (a) P. Wessig and O. Mühling, in Synthetic Organic Photochemistry, ed. A. G. Griesbeck and J. Mattay, Marcel Dekker, New York, 2005, pp. 41–87 Search PubMed; (b) P. J. Wagner, in Synthetic Organic Photochemistry, ed. A. G. Griesbeck and J. Mattay, Marcel Dekker, New York, 2005, pp. 11–39 Search PubMed.
- (a) J. R. Scheffer, M. Garcia-Garibay and O. Nalamasu, Org. Photochem., 1987, 8, 249–347 CAS; (b) H. Ihmels and J. R. Scheffer, Tetrahedron, 1999, 55, 885–907 CrossRef CAS.
- M. Leibovich, G. Olovsson, J. R. Scheffer and J. Trotter, J. Am. Chem. Soc., 1998, 120, 12755–12769 CrossRef.
- (a) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed; (b) M. Abe, J. Ye and M. Mishima, Chem. Soc. Rev., 2012, 41, 3808–3820 RSC.
- (a) L. Salem and D. Rowland, Angew. Chem., Int. Ed. Engl., 1972, 11, 92–111 CrossRef CAS; (b) L. Carlacci, C. Doubleday Jr., T. R. Furlani, H. F. King and J. W. McIver Jr., J. Am. Chem. Soc., 1987, 109, 5323–5329 CrossRef CAS; (c) J. Michl, J. Am. Chem. Soc., 1996, 118, 3568–3579 CrossRef CAS. See also: (d) J. C. Scaiano, Tetrahedron, 1982, 38, 819–824 CrossRef CAS.
- A. G. Griesbeck, H. Mauder and S. Stadtmüller, Acc. Chem. Res., 1994, 27, 70–75 CrossRef CAS.
- A. G. Griesbeck, M. Abe and S. Bondock, Acc. Chem. Res., 2004, 37, 919–928 CrossRef CAS PubMed.
- (a) M. W. Nau, F. L. Cozens and J. C. Scaiano, J. Am. Chem. Soc., 1996, 118, 2275–2282 CrossRef. See also: (b) B. Giese, P. Wettstein, C. Stählin, F. Barbosa, M. Neuburger, M. Zehnder and P. Wessig, Angew. Chem., Int. Ed., 1999, 38, 2586–2587 CrossRef CAS.
- A. G. Griesbeck and H. Heckroth, J. Am. Chem. Soc., 2002, 124, 396–403 CrossRef CAS PubMed.
- (a) V. Ramamurthy and K. Venkateban, Chem. Rev., 1987, 87, 433–481 CrossRef CAS; (b) B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef; (c) M. Sakamoto, J. Photochem. Photobiol., C, 2006, 7, 183–196 CrossRef CAS; (d) C. Yang and W. Xia, Chem. – Asian J., 2009, 4, 1774–1784 CAS.
- M. Leibovich, G. Olovsson, J. R. Scheffer and J. Trotter, J. Am. Chem. Soc., 1997, 119, 1462–1463 CrossRef.
- I. Turowska-Tyrk, E. Trzop, J. R. Scheffer and S. Chen, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 128–134 Search PubMed.
- (a) P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed; (b) M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779–784 CrossRef CAS PubMed.
- N. R. Champness, Nat. Chem., 2014, 6, 757–759 CrossRef CAS PubMed.
- P. J. Wagner, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. Horspool and F. Lenci, CRC Press, Boca Raton, 2nd edn, 2004, pp. 58/1–58/70 Search PubMed.
- G. Descotes, Top. Curr. Chem., 1990, 154, 39–76 CrossRef CAS.
- A. Herrera, M. Rondón and E. Suárez, J. Org. Chem., 2008, 73, 3384–3391 CrossRef CAS PubMed.
- D. Álvarez-Dorta, E. I. León, A. R. Kennedy, C. Riesco-Fagundo and E. Suárez, Angew. Chem., Int. Ed., 2008, 47, 8917–8919 CrossRef PubMed.
- R. Breslow, Acc. Chem. Res., 1980, 13, 170–177 CrossRef CAS.
- (a) T. Bach, Synthesis, 1998, 683–703 CrossRef CAS; (b) M. T. Crimmins and T. L. Reinhold, Org. React., 1993, 44, 297–588 CAS; (c) J. Iriondo-Alberti and M. F. Greaney, Eur. J. Org. Chem., 2007, 4801–4815 CrossRef.
- (a) R. Alibés, J. L. Bourdelande, J. Font, A. Gregori and T. Parella, Tetrahedron, 1996, 52, 1267–1278 CrossRef; (b) G. Lejeune, J. Font, T. Parella, R. Alibés and M. Figueredo, J. Org. Chem., 2015, 80, 9437–9445 CrossRef CAS PubMed. See also: (c) P. de March, F. Figueredo, J. Font, J. Raya, A. Alvarez-Larena and J. F. Piniella, J. Org. Chem., 2003, 68, 2437–2447 CrossRef CAS PubMed.
- See for example: (a) H. Kreigh and F. S. Richardson, J. Chem. Soc., Perkin Trans. 2, 1976, 1674–1677 RSC; (b) J. K. Gawronski, A. van Oeveren, H. van der Deen, C. W. Leung and B. L. Feringa, J. Org. Chem., 1996, 61, 1513–1515 CrossRef CAS.
- R. Jahjah, A. Gassama, V. Bulach, C. Suzuki, M. Abe, N. Hoffmann, A. Martinez and J.-M. Nuzillard, Chem. – Eur. J., 2010, 16, 3341–3354 CrossRef CAS PubMed.
- (a) R. Bonneau, J. Am. Chem. Soc., 1980, 102, 3816–3822 CrossRef CAS; (b) A. C. Chan and D. I. Schuster, J. Am. Chem. Soc., 1986, 108, 4561–4567 CrossRef CAS; (c) S. Yamauchi, N. Hirota and J. Higuchi, J. Phys. Chem., 1988, 92, 2129–2133 CrossRef CAS; (d) D. I. Schuster, D. A. Dunn, G. E. Heibel, P. B. Brown, J. M. Rao, J. Woning and R. Bonneau, J. Am. Chem. Soc., 1991, 113, 6245–6255 CrossRef CAS; (e) M. Reguero, M. Olivucci, F. Bernardi and M. A. Robb, J. Am. Chem. Soc., 1994, 116, 2103–2114 CrossRef CAS; (f) S. Wilsey, L. González, M. A. Robb and K. N. Houk, J. Am. Chem. Soc., 2000, 122, 5866–5876 CrossRef CAS; (g) M. Fréneau, P. de Sainte-Claire, M. Abe and N. Hoffmann, J. Phys. Chem., 2016 DOI:10.1002/poc.3560.
- (a) K. Wiesner, Tetrahedron, 1975, 31, 1655–1658 CrossRef CAS; (b) G. Marini-Bettòlo, S. P. Sahoo, G. A. Poulton, T. Y. R. Tsai and K. Wiesner, Tetrahedron, 1980, 36, 719–721 CrossRef; (c) N. Braussaud, N. Hoffmann and H.-D. Scharf, Tetrahedron, 1997, 53, 14701–14712 CrossRef CAS; (d) D. Becker, M. Nagler, Y. Sahali and N. Haddad, J. Org. Chem., 1991, 56, 4537–4543 CrossRef CAS.
- N. Hoffmann, H. Buschmann, G. Raabe and H.-D. Scharf, Tetrahedron, 1994, 50, 11167–11186 CrossRef CAS.
- K. Hayday and R. D. McKelvey, J. Org. Chem., 1976, 41, 2222–2223 CrossRef CAS.
- (a) N. Hoffmann, J. Phys. Org. Chem., 2015, 28, 121–136 CrossRef CAS; (b) N. Hoffmann, Synthesis, 2016, 1782–1802 CrossRef CAS.
- (a) P. Wessig, in CRC Handbook of Organic Photochemistry and Photobiology, ed. A. Griesbeck, M. Oelgemöller and F. Ghetti, CRC Press, Boca Raton, 3rd edn, 2012, pp. 201–211 Search PubMed; (b) P. Wessig and O. Muehling, Eur. J. Org. Chem., 2007, 2219–2232 CrossRef CAS.
- (a) P. Wessig and O. Muehling, Angew. Chem., Int. Ed., 2005, 44, 6778–6781 CrossRef CAS PubMed; (b) P. Wessig and O. Muehling, Chem. – Eur. J., 2008, 14, 7951–7960 CrossRef PubMed.
- (a) G. Cainelli, D. Giacomini and P. Galletti, Chem. Commun., 1999, 567–572 RSC; (b) Y. Inoue, T. Wada, S. Asaoka, H. Sato and J.-P. Pete, Chem. Commun., 2000, 251–259 RSC.
- (a) F. Nerdel and W. Brodowski, Chem. Ber., 1968, 101, 1398–1406 CrossRef CAS; (b) M. Pfau, S. Combrisson, J. E. Rowe Jr. and N. D. Heindel, Tetrahedron, 1978, 34, 3459–3468 CrossRef CAS; (c) R. M. Wilson, K. Hannemann, W. R. Heineman and J. R. Kirchhoff, J. Am. Chem. Soc., 1987, 109, 4743–4745 CrossRef CAS.
- P. G. Sammes, Tetrahedron, 1976, 32, 405–422 CrossRef CAS.
- (a) P. Klán, J. Wirz and A. D. Gudmundsdottir, in CRC Handbook of Organic Photochemistry and Photobiology, ed. A. Griesbeck, M. Oelgemöller and F. Ghetti, CRC Press, Boca Raton, 3rd edn, 2012, pp. 627–652 Search PubMed; (b) P. Wagner and H.-S. Park, Org. Photochem., 1991, 11, 227–366 CAS.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- P. Wan and D. Shukla, Chem. Rev., 1993, 93, 571–584 CrossRef CAS.
- J. Wirz, Adv. Phys. Org. Chem., 2010, 44, 325–356 CrossRef CAS.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2015, 45, 169–202 RSC.
- N. Basarić, K. Mlinarić-Majerski and M. Kralj, Curr. Org. Chem., 2014, 18, 3–18 CrossRef.
- N. Hoffmann, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, ed. J. Reedijk, Elsevier, Waltham, 2014, DOI:10.1016/B978-0-12-409547-2.11017-0.
- (a) O. Piva, R. Mortezaei, F. Henin, J. Muzart and J.-P. Pete, J. Am. Chem. Soc., 1990, 112, 9263–9272 CrossRef CAS; (b) F. Hénin, S. Létinois and J. Muzart, Tetrahedron: Asymmetry, 2000, 11, 2037–2044 CrossRef.
- H. Buschmann, H.-D. Scharf, N. Hoffmann and P. Esser, Angew. Chem., Int. Ed. Engl., 1991, 30, 477–515 CrossRef.
- O. A. Mukhina, N. N. B. Kumar, T. M. Arisco, R. A. Valiulin, G. A. Metzel and A. G. Kutateladze, Angew. Chem., Int. Ed., 2011, 50, 9423–9428 CrossRef CAS PubMed.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- O. A. Mukhina, W. C. Cronk, N. N. B. Kumar, M. C. Sekhar, A. Samanta and A. G. Kutateladze, J. Phys. Chem. A, 2014, 118, 10487–10496 CrossRef CAS PubMed.
- (a) W. J. Umstead, O. A. Mukhina and A. G. Kutateladze, Eur. J. Chem., 2015, 2205–2213 CAS; (b) W. C. Cronk, O. A. Mukhina and A. G. Kutateladze, J. Org. Chem., 2014, 79, 1235–1246 CrossRef CAS PubMed; (c) N. S. Nandurkar, N. N. B. Kumar, O. A. Mukhina and A. G. Kutateladze, ACS Comb. Sci., 2013, 15, 73–76 CrossRef CAS PubMed; (d) N. N. B. Kumar, O. A. Mukhina and A. G. Kutateladze, J. Am. Chem. Soc., 2013, 135, 9608–9611 CrossRef CAS PubMed.
- (a) B. Li, C. Yang, W. Xia and W. Xia, Synlett, 2015, 1997–2000 CAS; (b) Y. Shao, C. Yang, W. Gui, Y. Liu and W. Xia, Chem. Commun., 2012, 48, 3560–3562 RSC.
- (a) I. V. Alabugin, W.-Y. Yang and R. Pal, in CRC Handbook of Organic Photochemistry and Photobiology, ed. A. Griesbeck, M. Oelgemöller and F. Ghetti, CRC Press, Boca Raton, 3rd edn, 2012, pp. 549–592 Search PubMed; (b) R. K. Mohamed, P. W. Peterson and I. V. Alabugin, Chem. Rev., 2013, 113, 7089–7129 CrossRef CAS PubMed. For a particular case of a thermal reaction see: (c) A. Baroudi, J. Mauldin and I. V. Alabugin, J. Am. Chem. Soc., 2010, 132, 967–979 CrossRef CAS PubMed.
- (a) M. Winkler and W. Sander, Angew. Chem., Int. Ed., 2000, 39, 2014–2016 CrossRef CAS; (b) M. Winkler and W. Sander, J. Org. Chem., 2006, 71, 6357–6367 CrossRef CAS PubMed.
- M. Fagnoni and A. Albini, Acc. Chem. Res., 2005, 38, 713–721 CrossRef CAS PubMed.
- V. Dichiarante and M. Fagnoni, Synlett, 2008, 787–800 CAS.
- H. Qrareya, C. Raviola, S. Protti, M. Fagnoni and A. Albini, J. Org. Chem., 2013, 78, 6016–6024 CrossRef CAS PubMed.
- S. Lazzaroni, D. Dondi, M. Fagnoni and A. Albini, J. Org. Chem., 2008, 73, 206–211 CrossRef CAS PubMed.
- (a) M. Freccero, M. Fagnoni and A. Albini, J. Am. Chem. Soc., 2003, 125, 13182–13190 CrossRef CAS PubMed; (b) K. K. Laali, G. Rasul, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 2002, 67, 2913–2918 CrossRef CAS PubMed; (c) M. Aschi and J. N. Harvey, J. Chem. Soc., Perkin Trans. 2, 1999, 1059–1062 RSC; (d) M. Slegt, H. S. Overkleeft and G. Lodder, Eur. J. Org. Chem., 2007, 5364–5375 CrossRef CAS.
- V. Dichiarante, M. Fagnoni and A. Albini, J. Org. Chem., 2008, 73, 1282–1289 CrossRef CAS PubMed.
- (a) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS PubMed; (b) H. Mayr and A. R. Ofial, J. Phys. Org. Chem., 2008, 21, 584–595 CrossRef CAS; (c) H. Mayr, Tetrahedron, 2015, 71, 5095–5111 CrossRef CAS.
- B. Guizzardi, M. Mella, M. Fagnoni and A. Albini, Tetrahedron, 2000, 56, 9383–9390 CrossRef CAS.
- S. Protti, M. Fagnoni and A. Albini, Org. Biomol. Chem., 2005, 3, 2868–2871 CAS.
- A. Fraboni, M. Fagnoni and A. Albini, J. Org. Chem., 2003, 68, 4886–4893 CrossRef CAS PubMed.
- S. Protti, M. Fagnoni and A. Albini, J. Am. Chem. Soc., 2006, 128, 10670–10671 CrossRef CAS PubMed.
- (a) E. Paternò and G. Chieffi, Gazz. Chim. Ital., 1909, 39, 341–361 Search PubMed; (b) G. Büchi, C. G. Inman and E. S. Lipinski, J. Am. Chem. Soc., 1954, 76, 4327–4331 CrossRef.
- (a) M. D'Auria, in CRC Handbook of Organic Photochemistry and Photobiology, ed. A. Griesbeck, M. Oelgemöller and F. Ghetti, CRC Press, Boca Raton, 3rd edn, 2012, pp. 653–681 Search PubMed; (b) T. Bach, Synthesis, 1998, 683–703 CrossRef CAS; (c) V. H. Rawal, A. Eschbach, C. Dufour and S. Iwasa, Pure Appl. Chem., 1996, 68, 675–678 CrossRef CAS; (d) J. A. Porco and S. L. Schreiber, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and L. A. Paquette, Plenum Press, New York, 1991, vol. 5, pp. 151–192 Search PubMed.
- M. Abe, in Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH, Weinheim, 2010, pp. 217–239 Search PubMed.
- D. R. Arnold, Adv. Photochem., 1968, 6, 301–423 CrossRef CAS.
- I. J. Palmer, I. N. Ragazos, R. Bernardi, M. Olivucci and M. A. Robb, J. Am. Chem. Soc., 1994, 116, 2121–2132 CrossRef CAS.
- J. Mattay, J. Gersdorf and K. Buchkremer, Chem. Ber., 1987, 120, 307–318 CrossRef CAS.
- J. Gersdorf, J. Mattay and H. Görner, J. Am. Chem. Soc., 1987, 109, 1203–1209 CrossRef CAS.
- M. Abe, Y. Shirodai and M. Nojima, J. Chem. Soc., Perkin Trans. 1, 1998, 3253–3260 RSC.
- A. G. Griesbeck and S. Stadtmüller, Chem. Ber., 1990, 123, 357–362 CrossRef CAS.
- A. G. Griesbeck, S. Buhr, M. Fiege, H. Schmickler and J. Lex, J. Org. Chem., 1998, 63, 3847–3854 CrossRef CAS.
- M. Abe, M. Ikeda and M. Nojima, J. Chem. Soc., Perkin Trans. 1, 1998, 3261–3266 RSC.
- H. Buschmann, H.-D. Scharf, N. Hoffmann, M. W. Plath and J. Runsink, J. Am. Chem. Soc., 1989, 111, 5367–5373 CrossRef CAS.
- N. J. Turro, J. C. Dalton, K. Dawes, G. Farrington, R. Hautala, D. Morton, M. Niemczyk and N. Schore, Acc. Chem. Res., 1972, 5, 92–101 CrossRef CAS.
- (a) A. G. Griesbeck and S. Stadtmüller, J. Am. Chem. Soc., 1991, 113, 6923–6928 CrossRef CAS; (b) A. G. Griesbeck, H. Mauder, K. Peters, E.-M. Peters and H. G. von Schnering, Chem. Ber., 1991, 124, 407–410 CrossRef CAS; (c) A. G. Griesbeck and S. Stadtmüller, J. Am. Chem. Soc., 1990, 112, 1281–1282 CrossRef CAS.
- S. Bondock and A. G. Griesbeck, Int. J. Photoenergy, 2005, 7, 23–25 CrossRef CAS.
- A. G. Kutateladze, J. Am. Chem. Soc., 2001, 123, 9279–9282 CrossRef CAS PubMed.
- A. G. Griesbeck, M. Fiege, S. Bondock and M. S. Gudipati, Org. Lett., 2000, 2, 3623–3625 CrossRef CAS PubMed.
- A. G. Griesbeck, S. Bondock and M. S. Gudipati, Angew. Chem., Int. Ed., 2001, 40, 4684–4687 CrossRef CAS.
- G. Cainelly, D. Giacomini and P. Galletti, Chem. Commun., 1999, 567–572 RSC.
- L. Liu and Q.-X. Guo, Chem. Rev., 2001, 101, 673–695 CrossRef CAS PubMed.
- (a) W. Linert and R. F. Jameson, Chem. Soc. Rev., 1989, 18, 477–505 RSC; (b) W. Linert, Chem. Soc. Rev., 1994, 23, 429–438 RSC.
- M. Abe, T. Kawakami, S. Ohata, K. Nozaki and M. Nojima, J. Am. Chem. Soc., 2004, 126, 2838–2846 CrossRef CAS PubMed.
- E. Juaristi and G. Cuevas, The Anomeric Effect, CRC Press, Boca Raton, 1995 Search PubMed.
- (a) W. Adam, K. Peters, E. M. Peters and V. R. Stegmann, J. Am. Chem. Soc., 2000, 122, 2958–2959 CrossRef CAS; (b) W. Adam and V. R. Stegmann, Synthesis, 2001, 1203–1214 CAS; (c) M. D'Auria, L. Emanuele, G. Poggi, R. Racioppi and G. Romaniello, Tetrahedron, 2002, 58, 5045–5051 CrossRef; (d) M. D'Auria, L. Emanuele and R. Racioppi, Photochem. Photobiol. Sci., 2004, 3, 927–932 RSC; (e) M. D'Auria, L. Emanuele, R. Racioppi and A. Valente, Photochem. Photobiol. Sci., 2008, 7, 98–103 RSC.
- T. Bach, H. Bergmann and K. Harms, J. Am. Chem. Soc., 1999, 121, 10650–10651 CrossRef CAS.
- (a) L. K. Sydnes, K. I. Hansen, D. L. Oldroyd, A. C. Weedon and E. Jørgensen, Acta Chem. Scand., 1993, 47, 916–924 CrossRef CAS; (b) S. McN. Sieburth and K. F. McGee Jr., Org. Lett., 1999, 1, 1775–1777 CrossRef CAS PubMed; (c) A. Yokoyama and K. Mizuno, Org. Lett., 2000, 2, 3457–2459 CrossRef CAS PubMed.
- S. Iwata and K. Morokuma, J. Am. Chem. Soc., 1973, 95, 7563–7575 CrossRef CAS.
- (a) U. Pischel and W. M. Nau, Photochem. Photobiol. Sci., 2002, 1, 141–147 RSC; (b) J. E. Del Bene and J. D. Watts, Int. J. Quantum Chem., 2000, 77, 187–191 CrossRef CAS; (c) C. N. R. Rao and A. S. N. Murthy, Theor. Chim. Acta, 1971, 22, 392–395 CrossRef CAS; (d) J. C. Scaiano, J. Photochem., 1973, 2, 81–118 CrossRef CAS.
- A. G. Griesbeck and S. Bondock, J. Am. Chem. Soc., 2001, 123, 6191–6192 CrossRef CAS PubMed.
- Y. Yabuno, Y. Hiraga, R. Takagi and M. Abe, J. Am. Chem. Soc., 2011, 133, 2592–2604 CrossRef CAS PubMed.
- (a) J. Mattay, R. Conrads and R. Hoffmann, in Houben-Weyl, Methods of Organic Chemistry, Vol. E21: Stereoselective Synthesis, ed. G. Helmchen, R. W. Hoffmann, J. Mulzer and E. Schaumann, Thieme Verlag, Stuttgart, 1993, pp. 3085–3132 Search PubMed; (b) J. P. Hehn, C. Müller and T. Bach, in Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH, Weinheim, 2010, pp. 171–215 Search PubMed; (c) D. I. Schuster, G. N. Lem and A. Kaprinidis, Chem. Rev., 1993, 93, 3–22 CrossRef CAS.
- (a) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS PubMed; (b) C. Müller and T. Bach, Aust. J. Chem., 2008, 61, 557–564 CrossRef.
- (a) R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS PubMed; (b) R. Brimioulle, A. Bauer and T. Bach, J. Am. Chem. Soc., 2015, 137, 5170–5176 CrossRef CAS PubMed.
- (a) F. D. Lewis and S. V. Barancyk, J. Am. Chem. Soc., 1989, 111, 8653–8661 CrossRef CAS; (b) H. Görner and T. Wolff, Photochem. Photobiol., 2008, 84, 1224–1230 CrossRef PubMed.
- H. Wang, X. Cao, X. Chen, W. Fang and M. Dolg, Angew. Chem., Int. Ed., 2015, 127, 14295–14298 CrossRef PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed.
- R. Telmesani, S. H. Park, T. Lynch-Colameta and A. B. Beeler, Angew. Chem., Int. Ed., 2015, 54, 11521–11525 CrossRef CAS PubMed.
- H. D. Roth, Top. Curr. Chem., 1990, 156, 1–19 CrossRef CAS.
- (a) S. L. Mattes and S. Farid, in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, New York, 1982, vol. 6, pp. 233–326 Search PubMed; (b) J. Mattay, Angew. Chem., Int. Ed. Engl., 1987, 26, 825–845 CrossRef; (c) J. Mattay, Synthesis, 1989, 233–252 CrossRef CAS; (d) A. G. Griesbeck and S. Schieffer, in Electron Transfer in Chemistry, ed. A. Balzani, 2000, vol. 2, pp. 457–493 Search PubMed.
- L. Eberson, Electron Transfer Reactions in Organic Chemistry, Springer, Berlin, 1987 Search PubMed.
- (a) G. J. Kovarnos and N. J. Turro, Chem. Rev., 1986, 86, 401–449 CrossRef; (b) J. Mattay and M. Vondenhof, Top. Curr. Chem., 1991, 159, 219–255 CrossRef CAS; (c) G. J. Kovarnos, Fundamentals of Photoinduced Electron Transfer, VCH Publishers, New York, 1993 Search PubMed; (d) Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 1 and 2 Search PubMed; (e) M. Oelgemöller, J.-O. Bunte and J. Mattay, in Synthetic Organic Photochemistry, ed. A. G. Griesbeck and J. Mattay, Marcel Dekker, New York, 2004, pp. 267–295 Search PubMed.
- (a) D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS; (b) D. Rehm and A. Weller, Ber. Bunsenges. Phys. Chem., 1969, 73, 834–839 CAS.
- P. J. Wagner, Acc. Chem. Res., 1983, 16, 461–467 CrossRef CAS.
- M. A. Fox, Adv. Photochem., 1986, 13, 237–272 CAS.
- T. Majima, C. Pac, A. Nakasone and H. Sakurai, J. Am. Chem. Soc., 1981, 103, 4499–4508 CrossRef CAS . Pac et al. were probably the first who introduced “redox photosensitization” which is synonymous to “co-sensitization”.
- J. Mattay, G. Trampe and J. Runsink, Chem. Ber., 1988, 121, 1991–2005 CrossRef CAS.
- (a) H. Görner, M. Oelgemöller and A. G. Griesbeck, J. Phys. Chem. A, 2002, 106, 1458–1464 CrossRef; (b) H. Görner, A. G. Griesbeck, T. Heinrich, W. Kramer and M. Oelgemöller, Chem. – Eur. J., 2001, 7, 1530–1538 CrossRef; (c) V. Wintgens, P. Valat, J. Kossanyi, L. Biczok, A. Demeter and T. Bérces, J. Chem. Soc., Faraday Trans., 1994, 90, 411–421 RSC; (d) A. G. Griesbeck and H. Görner, J. Photochem. Photobiol., A, 1999, 129, 111–119 CrossRef CAS; (e) H. Hayashi, S. Nagakura, Y. Kubo and K. Maruyama, Chem. Phys. Lett., 1980, 72, 291–294 CrossRef CAS; (f) J. D. Coyle, A. Harriman and G. L. Newport, J. Chem. Soc., Perkin Trans. 2, 1979, 799–802 RSC; (g) P. B. Filho, V. G. Toscano and M. J. Politi, J. Photochem. Photobiol., A, 1988, 43, 51–58 CrossRef.
- (a) M. Oelgemöller, A. G. Griesbeck, J. Lex, A. Haeuseler, M. Schmittel, M. Niki, D. Hesek and Y. Inoue, Org. Lett., 2001, 3, 1593–1596 CrossRef; (b) M. Oelgemöller, A. Haeuseler, M. Schmittel, A. G. Griesbeck, J. Lex and Y. Inoue, J. Chem. Soc., Perkin Trans. 2, 2002, 676–686 RSC; (c) D. W. Leedy and D. L. Muck, J. Am. Chem. Soc., 1971, 93, 4264–4270 CrossRef CAS; (d) G. Farnia, A. Romanin, G. Capobianco and F. Torzo, J. Electroanal. Chem., 1971, 33, 31–44 CrossRef CAS; (e) G. Capobianco, G. Farnia and F. Torzo, Ric. Sci., 1968, 38, 842–844 CAS.
- (a) M. Hovart, K. Mlinarić-Majerski and N. Basarić, Croat. Chem. Acta, 2010, 83, 179–188 Search PubMed; (b) G. McDermott, D. J. Yoo and M. Oelgemöller, Heterocycles, 2005, 65, 2221–2257 CrossRef CAS; (c) M. Oelgemöller and A. G. Griesbeck, J. Photochem. Photobiol., C, 2002, 3, 109–127 CrossRef; (d) M. Oelgemöller and A. G. Griesbeck, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and F. Lenci, CRC Press, Boca Raton, 2nd edn, 2004, pp. 84/1–84/19 Search PubMed; (e) M. Oelgemöller, A. G. Griesbeck, W. Kramer and F. Nerowski, J. Inf. Rec., 1998, 24, 87–94 Search PubMed; (f) J. D. Coyle, in Synthetic Organic Photochemistry, ed. W. M. Horspool, Plenum Press, New York, 1984, pp. 259–284 Search PubMed; (g) P. H. Mazzocchi, Org. Photochem., 1981, 5, 421–471 CAS; (h) Y. Kanaoka, Acc. Chem. Res., 1978, 11, 407–413 CrossRef CAS.
- N. J. Pienta, in Photoinduced Electron Transfer, ed. M. A. Fox and M. Chanon, Elsevier, Amsterdam, 1988, pp. 421–486 Search PubMed.
- (a) Y. Hatanaka, Y. Sato, H. Nakai, M. Wada, T. Mizoguchi and Y. Kanaoka, Liebigs Ann. Chem., 1992, 1113–1123 CrossRef CAS; (b) Y. Sato, H. Nakai, M. Wada, T. Mizoguchi, Y. Hatanaka, Y. Migata and Y. Kanaoka, Liebigs Ann. Chem., 1985, 1099–1118 CrossRef CAS.
- (a) Y. Sato, H. Nakai, M. Wada, T. Mizoguchi, Y. Hatanaka and Y. Kanaoka, Chem. Pharm. Bull., 1992, 40, 3174–3180 CrossRef CAS; (b) M. Wada, H. Nakai, K. Aoe, K. Kotera, Y. Sato, Y. Hatanaka and Y. Kanaoka, Tetrahedron, 1983, 39, 1273–1279 CrossRef CAS; (c) M. Wada, H. Nakai, Y. Sato, Y. Hatanaka and Y. Kanaoka, Chem. Pharm. Bull., 1983, 31, 429–435 CrossRef CAS.
- (a) M. Machida, H. Takechi and Y. Kanaoka, Chem. Pharm. Bull., 1982, 30, 1579–1587 CrossRef CAS; (b) J. D. Coyle, L. E. Smart, J. F. Challiner and E. J. Haws, J. Chem. Soc., Perkin Trans. 1, 1985, 121–129 RSC; (c) J. D. Coyle and G. L. Newport, Synthesis, 1979, 381–382 CrossRef CAS.
- (a) U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 2001, 34, 523–533 CrossRef CAS PubMed; (b) U. C. Yoon, Y. X. Jin, S. W. Oh, D. W. Cho, K. H. Park and P. S. Mariano, J. Photochem. Photobiol., A, 2002, 150, 77–84 CrossRef CAS; (c) A. G. Griesbeck, M. Oelgemöller, J. Lex, A. Haeuseler and M. Schmittel, Eur. J. Org. Chem., 2001, 1831–1843 CrossRef CAS.
- (a) K. Maruyama, T. Ogawa, Y. Kubo and T. Araki, J. Chem. Soc., Perkin Trans. 1, 1985, 2025–2031 RSC; (b) M. Machida, K. Oda and Y. Kanaoka, Tetrahedron, 1985, 41, 4995–5001 CrossRef CAS; (c) P. H. Mazzocchi and G. Fritz, J. Org. Chem., 1986, 51, 5362–5364 CrossRef; (d) K. Maruyama and Y. Kubo, J. Org. Chem., 1981, 46, 3612–3622 CrossRef CAS.
- (a) P. H. Mazzocchi, D. Shook and L. Liu, Heterocycles, 1987, 26, 1165–1167 CrossRef CAS; (b) K. Maruyama and Y. Kubo, Chem. Lett., 1978, 7, 851–854 CrossRef.
- J. Xue, L. Zhu, H.-K. Fun and J.-H. Xu, Tetrahedron Lett., 2000, 41, 8553–8557 CrossRef CAS.
- L. Eberson, in Electron Transfer Reactions in Organic Chemistry (Reactivity and Structure-Concepts in Organic Chemistry), ed. K. Hafner, Springer-Verlag, Berlin, 1987, vol. 25, pp. 39–66 Search PubMed.
- (a) A. G. Griesbeck, W. Kramer and M. Oelgemöller, Synlett, 1999, 1169–1178 CrossRef CAS; (b) A. G. Griesbeck, A. Henz, W. Kramer, J. Lex, F. Nerowski, M. Oelgemöller, K. Peters and E.-M. Peters, Helv. Chim. Acta, 1997, 80, 912–933 CrossRef CAS; (c) W. Kramer, A. G. Griesbeck, F. Nerowski and M. Oelgemöller, J. Inf. Rec., 1998, 24, 81–85 CAS.
- (a) A. G. Griesbeck, T. Heinrich, M. Oelgemöller, A. Molis and A. Heidtmann, Helv. Chim. Acta, 2002, 85, 4561–4578 CrossRef CAS; (b) A. G. Griesbeck, F. Nerowski and J. Lex, J. Org. Chem., 1999, 64, 5213–5217 CrossRef CAS; (c) D. J. Yoo, E. Y. Kim, M. Oelgemöller and S. C. Shim, Heterocycles, 2001, 54, 1049–1055 CrossRef CAS; (d) Y.-J. Lee, D.-H. Ahn, K.-S. Lee, A. R. Kim, D. J. Yoo and M. Oelgemöller, Tetrahedron Lett., 2011, 52, 5029–5031 CrossRef CAS; (e) A. R. Kim, K.-S. Lee, C.-W. Lee, D. J. Yoo, F. Hatoum and M. Oelgemöller, Tetrahedron Lett., 2005, 46, 3395–3398 CrossRef CAS.
- (a) A. G. Griesbeck, N. Maptue, S. Bondock and M. Oelgemöller, Photochem. Photobiol. Sci., 2003, 2, 450–451 RSC; (b) A. G. Griesbeck, W. Kramer and M. Oelgemöller, Green Chem., 1999, 1, 205–207 RSC.
- (a) O. Shvydkiv, K. Nolan and M. Oelgemöller, Beilstein J. Org. Chem., 2011, 7, 1055–1063 CrossRef CAS PubMed; (b) O. Shvydkiv, S. Gallagher, K. Nolan and M. Oelgemöller, Org. Lett., 2010, 12, 5170–5173 CrossRef CAS PubMed; (c) M. Oelgemöller, S. Gallagher and K. McCarthy, Processes, 2014, 2, 158–166 CrossRef; (d) H. M. Pordanjani, C. Faderl, J. Wang, C. A. Motti, P. Junk and M. Oelgemöller, Aust. J. Chem., 2015, 68, 1662–1667 CrossRef CAS.
- (a) A. G. Griesbeck, W. Kramer and J. Lex, Angew. Chem., Int. Ed., 2001, 40, 577–579 CrossRef CAS; (b) A. G. Griesbeck, W. Kramer and J. Lex, Synthesis, 2001, 1159–1166 CAS; (c) A. G. Griesbeck, W. Kramer, T. Heinrich and J. Lex, Photochem. Photobiol. Sci., 2002, 1, 237–239 RSC; (d) A. G. Griesbeck, W. Kramer, A. Bartoschek and H. Schmickler, Org. Lett., 2001, 3, 537–539 CrossRef CAS PubMed.
- T. Šumanovac Ramljak, M. Sohora, I. Antol, D. Kontrec, N. Basarić and K. Mlinarić-Majerski, Tetrahedron Lett., 2014, 55, 4078–4081 CrossRef.
- (a) V. Belluau, P. Noeureuil, E. Ratzke, A. Skvortsov, S. Gallagher, C. A. Motti and M. Oelgemöller, Tetrahedron Lett., 2010, 51, 4738–4741 CrossRef CAS; (b) F. Hatoum, J. Engler, C. Zelmer, J. Wißen, C. A. Motti, J. Lex and M. Oelgemöller, Tetrahedron Lett., 2012, 53, 5573–5577 CrossRef CAS; (c) S. Josland, S. Mumtaz and M. Oelgemöller, Chem. Eng. Technol., 2016, 39, 81–87 CrossRef; (d) F. Hatoum, S. Gallagher, L. Baragwanath, J. Lex and M. Oelgemöller, Tetrahedron Lett., 2009, 50, 6335–6338 CrossRef CAS; (e) M. Oelgemöller, P. Cygon, J. Lex and A. G. Griesbeck, Heterocycles, 2003, 59, 669–684 CrossRef.
- R. Jahjah, A. Gassama, F. Dumur, S. Marinković, S. Richert, S. Landgraf, A. Lebrun, C. Cadiou, P. Sellès and N. Hoffmann, J. Org. Chem., 2011, 76, 7104–7118 CrossRef CAS PubMed.
- L. Strekowski, M. Hojjat, S. E. Patterson and A. S. Kiselyov, J. Heterocycl. Chem., 1994, 31, 1413–1416 CrossRef CAS.
- (a) M. Periasamy, N. Kishorebabu and K. N. Jayakumar, Tetrahedron Lett., 2003, 44, 8939–8941 CrossRef CAS; see also: (b) W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996–8002 CrossRef CAS PubMed.
- S. Barata-Vallejo, M. M. Flesia, B. Lantaño, J. E. Argüello, A. Peñéñory and A. Postigo, Eur. J. Org. Chem., 2013, 981–1008 Search PubMed.
- (a) Homogeneous Photocatalysis, ed. M. Chanon, Wiley, Chichester, 1997 Search PubMed; (b) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed; (c) D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC; (d) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC; (e) R. E. Galian and J. Pérez-Prieto, Energy Environ. Sci., 2010, 3, 1488–1498 RSC.
- M. Julliard and M. Chanon, Chem. Rev., 1983, 83, 425–506 CrossRef CAS.
- (a) A. Heumann and M. Chanon, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2nd edn, 2002, pp. 1030–1978 Search PubMed; (b) H. Henning, R. Billing and H. Knoll, in Photosensitization and Photocatalysis Using Inorganic and Organometallic Compounds, ed. K. Kayanasundaram and M. Gräzel, Kluwer Academic Publishers, Dordrecht, 1993, pp. 51–69 Search PubMed; (c) H. Kisch, in Photocatalysis, Fundamentals and Applications, ed. N. Serpone and E. Pelizzetti, Wiley, New York, 1989, pp. 1–8 Search PubMed; (d) A. Albini, J. Chem. Educ., 1986, 63, 383–386 CrossRef CAS; (e) F. Chanon and M. Chanon, in Photocatalysis, Fundamentals and Applications, ed. N. Serpone and E. Pelizzetti, Wiley, New York, 1989, pp. 489–540 Search PubMed.
- N. Hoffmann, J. Photochem. Photobiol., C, 2008, 9, 43–60 CrossRef CAS.
- (a) N. Hoffmann, S. Bertrand, S. Marinković and J. Pesch, Pure Appl. Chem., 2006, 78, 2227–2246 CrossRef CAS; (b) A. G. Griesbeck, N. Hoffmann and K. D. Warzecha, Acc. Chem. Res., 2007, 40, 128–140 CrossRef CAS PubMed.
- (a) S. Bertrand, C. Glapski, N. Hoffmann and J.-P. Pete, Tetrahedron Lett., 1999, 40, 3169–3172 CrossRef CAS; (b) S. Bertrand, N. Hoffmann and J.-P. Pete, Eur. J. Org. Chem., 2000, 2227–2238 CrossRef CAS.
- See also: E. Santiago de Alvarenga and J. Mann, J. Chem. Soc., Perkin Trans. 1, 1993, 2141–2142 RSC.
- S. G. Cohen, A. H. Parola and G. H. Parsons Jr., Chem. Rev., 1973, 73, 141–161 CrossRef CAS.
- N. Hoffmann and H. Görner, Chem. Phys. Lett., 2004, 383, 451–455 CrossRef CAS.
- H. Fischer and L. Radom, Angew. Chem., Int. Ed., 2001, 40, 1340–1371 CrossRef CAS.
- (a) S. Marinković and N. Hoffmann, Chem. Commun., 2001, 1576–1578 RSC; S. Marinković and N. Hoffmann, Int. J. Photoenergy, 2003, 5, 175–182 Search PubMed.
- A. Bauer, F. Westkämper, S. Grimme and T. Bach, Nature, 2005, 436, 1139–1140 CrossRef CAS PubMed.
- S. Z. Zard, Angew. Chem., Int. Ed. Engl., 1997, 36, 672–685 CrossRef.
- (a) S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981–894 CrossRef CAS; (b) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS; (c) C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- D. Harakat, D. Pesch, S. Marinković and N. Hoffmann, Org. Biomol. Chem., 2006, 4, 1202–1205 CAS.
- A. Gassama, C. Ernenwein and N. Hoffmann, ChemSusChem, 2009, 2, 1130–1137 CrossRef CAS PubMed.
- (a) E. Riguet and N. Hoffmann, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley-VCH, Chichester, 2012, vol. 2, pp. 1217–1241 Search PubMed; (b) U. Wille, in CRC Handbook of Organic Photochemistry and Photobiology, ed. A. Griesbeck, M. Oelgemöller and F. Ghetti, CRC Press, Boca Raton, 3rd edn, 2012, pp. 329–345 Search PubMed; (c) Radicals in Organic Synthesis, ed. P. Renaud and M. Sibi, Wiley-VCH, Weinheim, 2001, vol. 1 and 2 Search PubMed; (d) L. F. Tietze, G. Brasche and K. M. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed.
- S. Bertrand, N. Hoffmann, S. Humbel and J.-P. Pete, J. Org. Chem., 2000, 65, 8690–8703 CrossRef CAS PubMed.
- S. Marinković, C. Brulé, N. Hoffmann, E. Prost, J.-M. Nuzillard and V. Bulach, J. Org. Chem., 2004, 69, 1646–1651 CrossRef PubMed.
- (a) S. Marinković and N. Hoffmann, Eur. J. Org. Chem., 2004, 3102–3107 CrossRef; (b) N. Hoffmann, Aust. J. Chem., 2015, 68, 1621–1639 CrossRef CAS.
- J. Tang, G. Grampp, Y. Liu, B.-X. Wang, F.-F. Tao, L.-J. Wang, X.-Z. Liang, H.-Q. Xiao and Y.-M. Shen, J. Org. Chem., 2015, 80, 2724–2732 CrossRef CAS PubMed.
- (a) G. Pandey and S. R. Gadre, ARKIVOC, 2003, 45–54 CAS; (b) G. Pandey, Top. Curr. Chem., 1993, 168, 175–221 CrossRef CAS; (c) G. Pandey, Synlett, 1992, 546–552 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) M. Reckenthähler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef; (c) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC; (d) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (e) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (f) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed; (g) Chemical Photocatalysis, ed. B. König, Walter de Gruyter, Berlin, 2013 Search PubMed.
- (a) N. Hoffmann, ChemSusChem, 2012, 5, 352–371 CrossRef CAS PubMed; (b) F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859–917 CrossRef.
- (a) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belzer and A. von Zelewski, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS; (b) S. Champagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef; (c) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2013, 77, 1617–1622 CrossRef PubMed.
- For some older examples see: (a) T. J. van Bergen, D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, J. Org. Chem., 1979, 44, 4953–4962 CrossRef CAS; (b) H. Cano-Yelo and A. Deronzier, J. Chem. Soc., Perkin Trans. 2, 1984, 1093–1098 RSC; (c) C. Pac, Y. Miyauchi, O. Ishitani, M. Ihama, M. Yasuda and H. Sakurai, J. Org. Chem., 1984, 49, 26–34 CrossRef CAS; (d) R. Maidan, Z. Goren, J. Y. Becker and I. Willner, J. Am. Chem. Soc., 1984, 106, 6217–6222 CrossRef CAS; (e) K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705–1709 RSC.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- D. D. M. Wayner, J. J. Dannenberg and D. Griller, Chem. Phys. Lett., 1986, 131, 189–191 CrossRef CAS.
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, J. Am. Chem. Soc., 1979, 101, 4815–4824 CrossRef CAS.
- S. Fukuzumi, S. Mochizuki and T. Tanaka, J. Phys. Chem., 1990, 94, 722–726 CrossRef CAS.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- (a) J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS PubMed; (b) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- (a) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed; (b) K. Fidaly, C. Ceballos, A. Falguières, M. S.-I. Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293–1297 RSC.
- M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062–4066 CrossRef CAS PubMed.
- (a) H. Zollinger, Color Chemistry, Verlag Helvetica Chimica Acta, Zürich, 2003 Search PubMed; (b) H. Langhals, Helv. Chim. Acta, 2005, 88, 1309 CrossRef CAS; (c) L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938 RSC; (d) C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed; (e) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS PubMed; (f) F. Würthner, Chem. Commun., 2004, 1564 RSC; (g) D. Wöll, E. Braeken, A. Deres, F. C. De Schryver, H. Uji-i and J. Hofkens, J. Chem. Soc. Rev., 2009, 38, 313–328 RSC.
- For recent systematic studies on a large compound family see: (a) M. Queste, C. Cadiou, B. Pagoaga, L. Giraudet and N. Hoffmann, New J. Chem., 2010, 34, 2537–2545 RSC; (b) B. Pagoaga, L. Giraudet and N. Hoffmann, Eur. J. Org. Chem., 2014, 5178–5195 CrossRef CAS; (c) B. Pagoaga, O. Mongin, M. Caselli, D. Vanossi, F. Momicchioli, M. Blanchard-Desce, G. Lemercier, N. Hoffmann and G. Ponterini, Phys. Chem. Chem. Phys., 2016, 18, 4924–4941 RSC.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- (a) D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545–6551 CrossRef CAS; (b) M. J. Tauber, R. F. Kelley, J. M. Giaimo, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2006, 128, 1782–1783 CrossRef CAS PubMed.
- (a) G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed; (b) F. Auzel, Chem. Rev., 2004, 104, 139–174 CrossRef CAS PubMed; (c) T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC; (d) T. W. Schmidt and F. N. Castellano, J. Phys. Chem. Lett., 2014, 5, 4062–4072 CrossRef CAS PubMed.
- M. Majek, U. Faltermeier, B. Dick, R. Pérez-Ruiz and A. Jacobi von Wangelin, Chem. – Eur. J., 2015, 21, 15496–15501 CrossRef CAS PubMed.
- (a) K. P. C. Vollhardt, Angew. Chem., Int. Ed. Engl., 1984, 23, 539–556 CrossRef; (b) V. Gandon, C. Aubert and M. Malacria, Chem. Commun., 2006, 2209–2217 RSC; (c) P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS; (d) G. Dominguez and J. Pérez-Castells, Chem. Soc. Rev., 2011, 40, 3430–3444 RSC; (e) N. Weding and M. Hapke, Chem. Soc. Rev., 2011, 40, 4525–4538 RSC.
- (a) H. Bönnemann, Angew. Chem., Int. Ed. Engl., 1985, 24, 248–262 CrossRef; (b) G. Chelucci, Tetrahedron: Asymmetry, 1995, 6, 811–826 CrossRef CAS; (c) B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085–1094 RSC; (d) J. A. Varela and C. Saa, Synlett, 2008, 2571–2578 CAS.
- W. Schulz, H. Pracejus and G. Oehme, Tetrahedron Lett., 1989, 30, 1229–1232 CrossRef CAS.
- B. Heller, Nachr. Chem., Tech. Lab., 1999, 47, 9–14 CrossRef CAS.
- G. Chelucci, M. Falorni and G. Giacomelli, Synthesis, 1990, 1121–1122 CrossRef CAS.
- A. Gutnov, B. Heller, C. Fischer, H.-J. Drexler, A. Spannenberg, B. Sundermann and C. Sundermann, Angew. Chem., Int. Ed., 2004, 43, 3795–3797 CrossRef CAS PubMed.
- (a) G. Oehme, B. Heller and P. Wagler, Energy, 1997, 22, 327–336 CrossRef CAS; (b) P. Wagler, B. Heller, J. Ortner, K.-H. Funken and G. Oehme, Chem. Ing. Tech., 1996, 68, 823–826 CrossRef CAS.
- (a) B. Heller, D. Heller and G. Oehme, J. Mol. Catal. A: Chem., 1996, 110, 211–219 CrossRef CAS; (b) B. Heller, D. Heller, P. Wagler and G. Oehme, J. Mol. Catal. A: Chem., 1998, 136, 219–233 CrossRef CAS.
- J. A. K. Du Plessis and J. S. Viljoen, J. Mol. Catal. A: Chem., 1995, 99, 71–76 CrossRef CAS.
- H. Bönnemann, Angew. Chem., Int. Ed. Engl., 1985, 24, 248–262 CrossRef.
- B. Heller and G. Oehme, J. Chem. Soc., Chem. Commun., 1995, 179–180 RSC.
- D. I. Perez, M. M. Grau, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2009, 6848–6850 RSC.
- E. Churakova, M. Kluge, R. Ullrich, I. Arends, M. Hofrichter and F. Hollmann, Angew. Chem., Int. Ed., 2011, 50, 10716–10719 CrossRef CAS PubMed.
- F. Hollmann, A. Taglieber, F. Schulz and M. T. Reetz, Angew. Chem., Int. Ed., 2007, 46, 2903–2906 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, 2010, pp. 1003–1008 Search PubMed.
- B. F. Minaev, Russ. Chem. Rev., 2007, 76, 988–1010 CrossRef.
- (a) B. F. Minaev and H. Ågren, THEOCHEM, 1998, 434, 193–206 CrossRef CAS; (b) A. Riahi, J. Muzart, M. Abe and N. Hoffmann, New J. Chem., 2013, 37, 2245–2249 RSC.
- (a) G. O. Schenck, Naturwissenschaften, 1948, 35, 28–29 CrossRef CAS; (b) H. H. Wasserman and J. L. Ives, Tetrahedron, 1981, 37, 1825–1852 CrossRef CAS; (c) E. L. Clennan, Tetrahedron, 2000, 56, 9151–9179 CrossRef CAS; (d) M. Stratakis and M. Orfanopoulos, Tetrahedron, 2000, 56, 1595–1615 CrossRef CAS; (e) T. Montagnon, D. Noutsias, I. Alexopoulou, M. Tofi and G. Vassilikogiannakis, Org. Biomol. Chem., 2011, 9, 2031–2039 RSC; (f) W. J. Kinart, C. M. Kinart and M. Sendecki, Curr. Org. Synth., 2013, 10, 337–346 CrossRef CAS; (g) M. Zamadar and A. Greer, in Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH, Weinheim, 2010, pp. 353–386 Search PubMed.
- C. S. Foot, Acc. Chem. Res., 1968, 1, 104–110 CrossRef.
- W. Fudickar and T. Linker, Aust. J. Chem., 2014, 67, 320–327 CrossRef CAS.
- K. Yamaguchi, Y. Ikeda and T. Fueno, Tetrahedron, 1985, 41, 2099–2107 CrossRef CAS.
- (a) H. Rau, Chem. Rev., 1983, 83, 535–347 CrossRef CAS; (b) A. G. Griesbeck and U. J. Meierhenrich, Angew. Chem., Int. Ed., 2002, 41, 3147–3154 CrossRef.
- J. Sivaguru, M. R. Solomon, T. Poon, S. Jockusch, S. G. Bosio, W. Adam and N. J. Turro, Acc. Chem. Res., 2008, 41, 387–400 CrossRef CAS PubMed.
- W. Adam, S. G. Bosio, N. J. Turro and B. T. Wolff, J. Org. Chem., 2004, 69, 1704–1715 CrossRef CAS PubMed.
- S. Inagaki, H. Fujimoto and K. Fukui, Chem. Lett., 1976, 749–752 CrossRef CAS.
- S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed. Engl., 1985, 24, 1–30 CrossRef.
- J. Sivaguru, M. R. Solomon, H. Saito, T. Poon, S. Jockusch, W. Adam, Y. Inoue and N. J. Turro, Tetrahedron, 2006, 62, 6707–6717 CrossRef CAS.
- M. Prein and W. Adam, Angew. Chem., Int. Ed. Engl., 1996, 35, 477–494 CrossRef CAS.
- T. Poon, J. Sivaguru, R. Franz, S. Jockusch, C. Martinez, I. Washington, W. Adam, Y. Inoue and N. J. Turro, J. Am. Chem. Soc., 2004, 126, 10498–10499 CrossRef CAS PubMed.
- J. Sivaguru, T. Poon, C. Hooper, H. Saito, M. R. Solomon, S. Jockusch, W. Adam, Y. Inoue and N. J. Turro, Tetrahedron, 2006, 46, 10647–10659 CrossRef.
- H. B. Kagan and J. C. Fiaud, Top. Stereochem., 1988, 18, 249–330 CAS.
- (a) J. E. Leffler, J. Org. Chem., 1955, 20, 1202 CrossRef CAS; (b) J. E. Leffler and E. Grunwald, Rates and Equilibria of Organic Reactions, J. Wiley & Sons, New York, 1963, ch. 9, pp. 318–402 Search PubMed; (c) O. Exner, Collect. Czech. Chem. Commun., 1975, 40, 2762–2780 CrossRef CAS; (d) O. Exner, Collect. Czech. Chem. Commun., 1975, 40, 2781–2791 CrossRef CAS; (e) W. Linert, Inorg. Chim. Acta, 1988, 141, 233–242 CrossRef CAS; (f) B. Giese, Acc. Chem. Res., 1984, 17, 438–442 CrossRef CAS; (g) N. Hoffmann, H. Buschmann, G. Raabe and H.-D. Scharf, Tetrahedron, 1994, 50, 11167–11186 CrossRef CAS; (h) Y. Inoue and T. Wada, Adv. Supramol. Chem., 1997, 4, 55–96 CAS.
- N. J. Turro, Tetrahedron, 1985, 41, 2089–2098 CrossRef CAS.
- (a) G. Peters and M. A. J. Rodgers, J. Am. Chem. Soc., 1981, 103, 6759–6761 CrossRef CAS; (b) M. A. J. Rodgers, J. Am. Chem. Soc., 1983, 105, 6201–6205 CrossRef CAS.
- (a) A. E. Cassano, C. A. Martin, R. J. Brandi and O. M. Alfano, Ind. Eng. Res., 1995, 34, 2155–2201 CrossRef CAS; (b) A. M. Braun, M. Maurette and E. Oliveros, Photochemical Technology, Wiley, Chichester, 1991 Search PubMed.
- J. M. Parnis and K. B. Oldham, J. Photochem. Photobiol., A, 2013, 267, 6–10 CrossRef CAS.
- (a) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS; (b) C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC; (c) J.-i. Yoshida, H. Kim and A. Nagaki, ChemSusChem, 2011, 4, 331–340 CrossRef CAS PubMed; (d) P. T. Baraldi and V. Hessel, Green Process. Synth., 2012, 1, 149–167 CAS; (e) L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef CAS PubMed; (f) N. G. Anderson, Org. Process Res. Dev., 2012, 16, 852–869 CrossRef CAS; (g) K. S. Elvira, X. Casadevall i Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed; (h) K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef PubMed.
- (a) T. Aillet, K. Loubière, O. Dechy-Cabaret and L. Prat, Int. J. Chem. React. Eng., 2014, 12, 257–269 Search PubMed; (b) T. Aillet, K. Loubière, O. Dechy-Cabaret and L. Prat, Chem. Eng. Process., 2013, 64, 38–47 CrossRef CAS; (c) K. Loubière, M. Oelgemöller, T. Aillet, O. Dechy-Cabaret and L. Prat, Chem. Eng. Process., 2016, 104, 120–132 CrossRef.
- (a) K. Gilmore and P. H. Seeberger, Chem. Rec., 2014, 14, 410–418 CrossRef CAS PubMed; (b) M. Oelgemöller, N. Hoffmann and O. Shvydkiv, Aust. J. Chem., 2014, 67, 337–342 CrossRef; (c) M. Oelgemöller, Chem. Eng. Technol., 2012, 35, 1144–1152 CrossRef; (d) M. Oelgemöller and O. Shvydkiv, Molecules, 2011, 16, 7522–7550 CrossRef PubMed; (e) E. E. Coyle and M. Oelgemöller, Photochem. Photobiol. Sci., 2008, 7, 1313–1322 RSC.
- A. Yavorskyy, O. Shvydkiv, N. Hoffmann, K. Nolan and M. Oelgemöller, Org. Lett., 2012, 14, 4342–4345 CrossRef CAS PubMed.
- H. Lu, M. A. Schmidt and K. F. Jensen, Lab Chip, 2001, 1, 22–28 RSC.
- (a) O. Shvydkiv, A. Yavorskyy, K. Nolan, A. Youssef, E. Riguet, N. Hoffmann and M. Oelgemöller, Photochem. Photobiol. Sci., 2010, 9, 1601–1603 RSC; (b) A. Yavorskyy, O. Shvydkiv, K. Nolan, N. Hoffmann and M. Oelgemöller, Tetrahedron Lett., 2011, 52, 278–280 CrossRef CAS; (c) O. Shvydkiv, A. Yavorskyy, S. B. Tan, K. Nolan, N. Hoffmann, A. Youssef and M. Oelgemöller, Photochem. Photobiol. Sci., 2011, 10, 1399–1404 RSC.
- (a) A. Sugimoto, Y. Sumino, M. Takagi, T. Fukuyama and H. Ryu, Tetrahedron Lett., 2006, 47, 6197–6200 CrossRef CAS; (b) A. Sugimoto, T. Fukuyama, Y. Sumino, M. Takagi and I. Ryu, Tetrahedron, 2009, 65, 1593–1598 CrossRef CAS.
- B. G. Anderson, W. E. Bauta and W. R. Cantrell, Org. Process Res. Dev., 2012, 16, 967–975 CrossRef CAS.
- (a) F. Lévesque and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 1706–1709 CrossRef PubMed; (b) D. Kopetzki, F. Lévesque and P. H. Seeberger, Chem. – Eur. J., 2013, 19, 5450–5456 CrossRef CAS PubMed.
- (a) M. Fischer, Angew. Chem., Int. Ed. Engl., 1978, 17, 16–26 CrossRef; (b) K. Gollnick, Chim. Ind., 1982, 63, 156–166 Search PubMed.
- (a) G. Shama, C. Peppiatt and M. Biguzzi, J. Chem. Technol. Biotechnol., 1996, 65, 56–64 CrossRef CAS; (b) S. D. Pask, O. Nuyken and Z. Cai, Polym. Chem., 2012, 3, 2698–2707 RSC; (c) V. Pareek, Chem. Eng., 2005, 764, 28 Search PubMed.
- (a) E. A. Sosnin, T. Oppenländer and V. F. Tarasenko, J. Photochem. Photobiol., C, 2006, 7, 145–163 CrossRef CAS; (b) N. T. Kalyania and S. J. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696–2723 CrossRef; (c) G. Kreisel, S. Meyer, D. Tietze, T. Fidler, R. Gorges, A. Kirsch, B. Schäfer and S. Rau, Chem. Ing. Tech., 2007, 79, 153–159 CrossRef CAS.
- J. Turconi, F. Griolet, R. Guevel, G. Oddon, R. Villa, A. Geatti, M. Hvala, K. Rossen, R. Göller and A. Burgard, Org. Process Res. Dev., 2014, 18, 417–422 CrossRef CAS.
- S. Werner, R. Seliger, H. Rauter and F. Wissmann, EP2065387A2, 2009 Search PubMed; Chem. Abstr. 2009 150 376721 Search PubMed.
- M. Oelgemöller and M. Bolte, Green Process. Synth., 2014, 3, 163–165 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |