
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous rhodium(I)-catalysis in de novo heterocycle syntheses
James D.
Neuhaus
and
Michael C.
Willis
 *
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
First published on 20th May 2016
Abstract
Recent research has led to the development of mild, efficient and selective catalytic systems based on Rh(I) complexes. This review summarises the applications of these catalysts to the synthesis of heterocycles from simple building blocks. Included herein are approaches based on cycloisomerisation, cycloaddition, hydroacylation and hydroarylation reactions, as well as various tandem and domino procedures and carbonylation processes.
 James D. Neuhaus | James Neuhaus received his MChem degree from Merton College, University of Oxford in 2012. During this he completed a Part II project under the supervision of Prof. Tim Donohoe, investigating asymmetric routes to saturated heterocycles. Since joining the Willis group in 2012, he has been involved in investigating various aspects of rhodium-catalysed hydroacylation, including application to the synthesis of N-containing heterocycles. |
 Michael C. Willis | Michael Willis received his undergraduate education at Imperial College London, and his PhD from the University of Cambridge working with Prof. Steven V. Ley, FRS. After a postdoctoral stay with Prof. David A. Evans at Harvard University, he was appointed to a lectureship at the University of Bath in November 1997. In January 2007 he moved to the University of Oxford, where he is a now a Professor of Chemistry and a Fellow of Lincoln College. His group's research interests are based on the development and application of new catalytic processes for organic synthesis. |
1. Introduction
Heterocyclic scaffolds represent one of the most important families of compounds in fields as far ranging as medicinal chemistry, materials science and natural product synthesis. The wide variability and tuneability of properties, such as polarity, co-ordination ability, UV absorbance and lipophilicity explains the prevalence of this architecture in this diverse range of fields.A variety of methods exist for the construction of heterocycles, with many methods developed in the early 20th century still employed today. Despite this rich history and range of published methods, there is still a desire to access new routes, improving on such factors as the level of molecular complexity and functional group tolerance in convergent and atom-economical approaches, utilising readily available starting materials under mild conditions; in short to fulfil many of the goals associated with sustainable synthesis.
Since their explosion onto the mainstream organic scene in the 1970s and 80s, transition metal catalysed reactions have represented a popular choice of method to achieve what were previously difficult, or even impossible transformations. With the great strides seen over the past few decades, a plethora of selective, mild, functional group tolerant transition metal catalysed reactions have been developed, and many of these methodologies have been tested in the field of heterocycle synthesis.1
Rhodium catalysis in particular, displays both a wide range of tuneable reactivity and a broad functional group tolerance that has allowed the development of highly efficient processes.2 Coupled with the ability to combine very different reaction pathways in cascade and tandem catalysis, a great deal of chemical space can be accessed using these highly effective reactions.
There exists a diverse array of rhodium complexes employed in synthetic organic chemistry, reflecting the multi-faceted nature of rhodium catalysis, with processes starting from four distinct oxidation states of rhodium: Rh(0), Rh(I), Rh(II) and Rh(III). By far the most widely applicable is rhodium(I) catalysis, and we shall see how the varied range of reaction types, often coupled together to achieve more complex transformations, is applied to a broad range of heterocycles. Rhodium(III) catalysis is dominated by the hugely successful field of directed ortho sp2 C–H activation, and this is reflected in the heterocyclic literature.3 Rh(0) carbonyl complexes are employed in the field of carbonylation chemistry although they have largely been superseded by Rh(I) and Rh(III) in recent years. Rh(II) catalysed processes typically involve the intermediacy of a rhodium carbenoid, predominantly generated from diazo compounds, although recent breakthroughs in the generation of iminocarbenes from sulfonyl triazoles, themselves easily accessible through Huisgen cycloaddition chemistry, have provided a versatile method for the synthesis of a range of heterocycles.4
For this review, we have chosen to focus on Rh(I)-catalysed processes that either directly form heterocycles, or are used in the preparation of heterocyclic precursors. The reactions covered fall under the broad headings of cycloisomerisations, cycloadditions, hydroacylation and hydroarylation based processes, in addition to various miscellaneous processes, and were published during the period from 2000 to the present, although in some cases earlier papers have been included where relevant. The field of asymmetric hydrogenation, whilst an invaluable route to the synthesis of partially or fully saturated heterocycles, is not a true de novo synthesis, and thus falls outside the purview of this review. For each transformation, we have provided a representative example, rather than using general structures or tables. Stereochemical notations for absolute and relative stereochemistry are as described by Maehr.5
2. Cycloisomerisation
Conceptually amongst the simplest rhodium-catalysed processes, cycloisomerisation represents a 100% atom-economical process. As an intramolecular reaction, the catalytic efficiency is often much higher than would be expected for similar intermolecular processes, enabling low catalyst loadings, and relatively mild conditions. An early application of rhodium to this type of process appeared in 2003, with a publication from the Trost group involving the synthesis of dihydrofurans and dihydropyrans from homopropargylic and bis-homopropargylic alcohols, respectively (Scheme 1a).6 In this report they invoke the presence of a rhodium vinylidene intermediate, which undergoes nucleophilic attack by the alkoxide, followed by protoderhodation to generate the enol ether. In 2006 Hartwig et al. published a related reaction, proceeding through an intramolecular endo-hydroamination mechanism.7 When using multisubstituted alkenes they observed high diastereoselectivity for syn-disubstituted piperidines (Scheme 1b). Later studies extended this methodology to the synthesis of pyrrolidines,8 and tetrahydroquinolines.9 In 2010, Buchwald applied chiral monophosphine ligands to the intramolecular hydroamination of alkenes, generating a range of chiral pyrrolidines in good to excellent enantiomeric ratios (Scheme 1c).10 The Messerle group have undertaken a body of research into the development and application of highly active intramolecular exo-hydroamination rhodium catalysts.11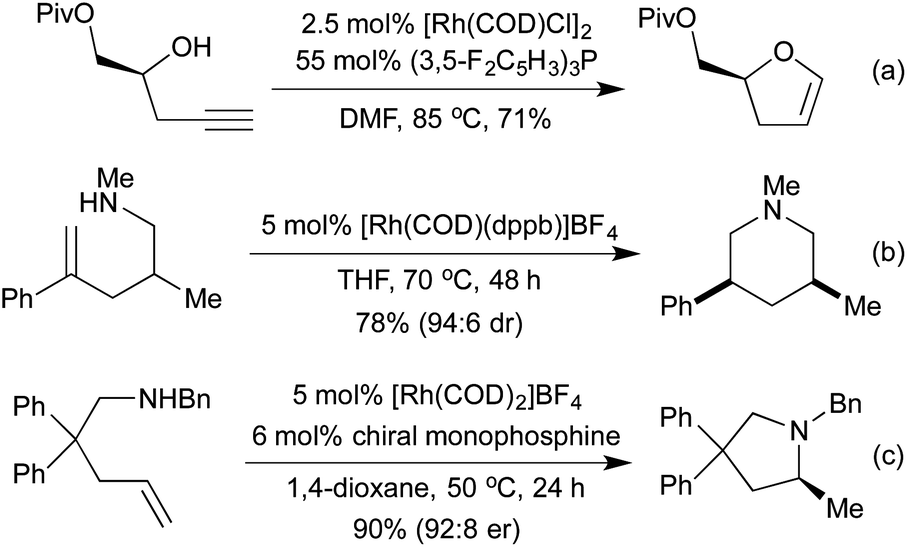 | ||
| Scheme 1 Cycloisomerisation to form saturated and partially saturated heterocycles. | ||
In 2007 Trost applied his cycloisomerisation conditions to 2-ethynyl anilines, generating indoles.12 A limitation of these initial conditions was the inability to use internal alkynes, affording only N-substitution, or substitution on the carbocyclic ring.
In 2009 Lautens extended this methodology to access not only benzofurans, but also substitution at the C(2) and C(3) positions (Scheme 2a).13 C(2) substitution arises from the use of internal alkynes, whereas the C(3) functionalisation results from the trapping of a C(3)-metallated intermediate with various Michael acceptors. This cascade methodology has since been extended to include various alternative trapping partners, such as 2-pyridyl alkynes,14 and isocyanates (Scheme 2b).15 This final example is particularly noteworthy due to the mild reaction conditions. A range of benzodipyrroles have also been synthesised from 1,4-phenylenediamines using a Rh(I) complex.11e
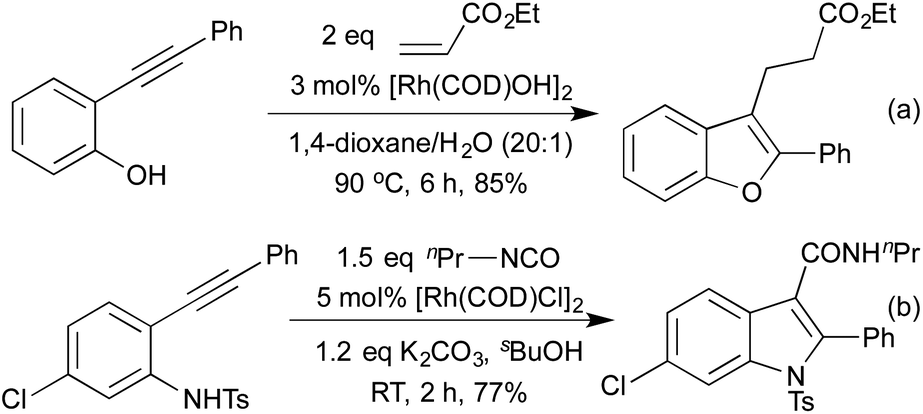 | ||
| Scheme 2 Cascade synthesis of C(3) functionalised indoles and benzofurans. | ||
A 2008 publication by Hanzawa et al. combined an initial [3,3]-sigmatropic rearrangement of N-propargylic anilines with intramolecular hydroamination of the generated o-allenyl aniline, to afford indoles via a formal C–H functionalisation procedure (Scheme 3).16 Electron-rich anilines were particularly reactive, although a broad scope was achievable by varying catalyst loadings. By generating the N-propargylic aniline in situ from propargyl bromide and the appropriate N-alkylaniline, a one-pot procedure direct from anilines to indoles was achieved.
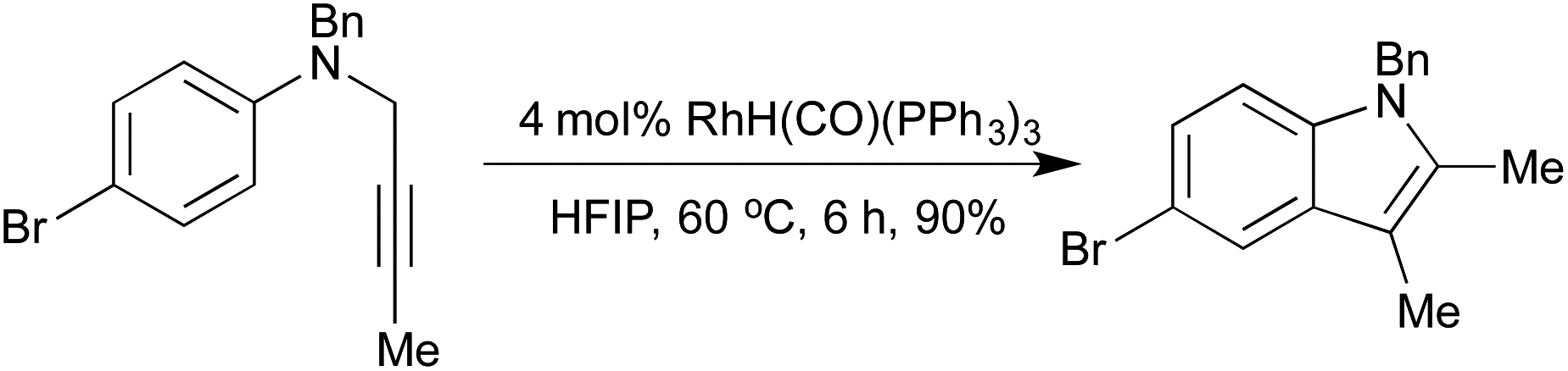 | ||
| Scheme 3 Cascade [3,3]-sigmatropic rearrangement/hydroamination indole synthesis. | ||
Several conceptually distinct cycloisomerisation approaches have also been developed in recent years. In 2007 Tanaka explored an approach where rhodium initially catalyses an isomerisation of a secondary propargyl alcohol to an enone (with opposite regioselectivity to the classical Meyer-Schuster rearrangement), followed by an intramolecular cyclisation to generate a range of simple 2,5-disubstituted furans (Scheme 4a).17 A similar intramolecular cyclisation under carbonylative conditions had previously been reported by Alper in 2000 (Scheme 4b).18 This method was later extended to the synthesis of pyrroles from the corresponding propargyl imines.19 The concept of combining rhodium carbonylations with other known modes of activation is an established concept, with reports from as early as 1993 of the synthesis of various lactams via a carbonylative hydroamination.20 The activation of a cyclopropane ring in Scheme 4c,21 followed by cycloisomerisation and carbonylation is one of a number of examples involving Rh-catalysed cascade processes in furan synthesis from the group of Zhang.22 Rh-catalysed enyne metathesis, followed by tautomerisation, has also been shown to generate furans, although the scope and efficiency of the reaction are limited (Scheme 4d).23
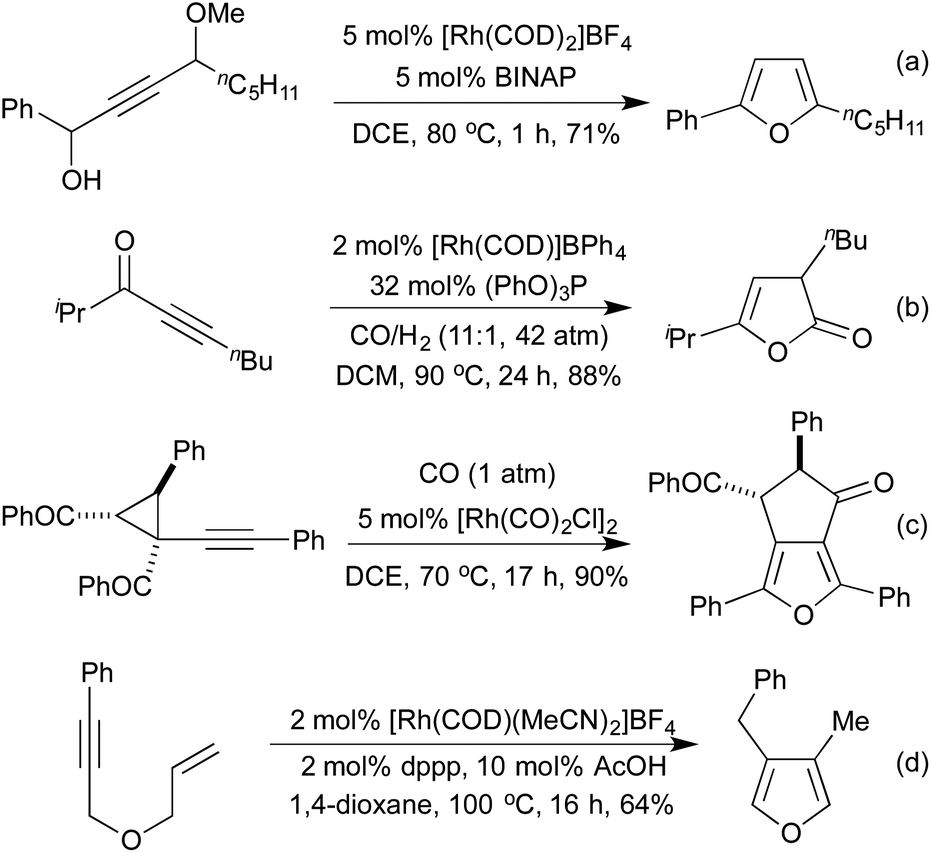 | ||
| Scheme 4 Cycloisomerisation routes to oxygen containing heterocycles. | ||
3. Cycloaddition
Rhodium(I) catalysts have long been the catalysts of choice for effecting a range of cycloaddition reactions. Whilst not restricted to these examples, the fields of Rh(I) catalysed [2 + 2 + 2] cycloadditions, [2 + 2 + 1] Pauson-Khand type carbonylations and [5 + 2] cycloadditions have contributed hundreds of publications over the last 20 years, which have been summarised in a number of comprehensive reviews.24Issues arising from regioselectivity have led to the use of formal tethers in many of these processes. The most common tethers employed often have either an ether linkage, or a CH2N(Ts)CH2 based system. Once the carbocyclic cycloadditions have occurred, the tether now forms a heterocycle, usually a dihydrofuran, or pyrroline moiety. In this review we have chosen instead to focus on approaches where the heteroatoms are directly involved in the cycloaddition, either as one of the partners, or directly bonded to one of the reaction partners.
As examples of formal carbocyclisations generating heterocycles where the heteroatom is directly conjugated into one of the reaction partners, the synthesis of dibenzofurans (Scheme 5a),25 carbazoles26 and phthalides27 all involve a Rh(I) catalysed [2 + 2 + 2] alkyne cross-cyclotrimerisation. Similarly, in a recent publication using ynamides as starting materials, Anderson and co-workers demonstrated an interesting [5 + 2] cyclisation to generate [5.3.0]-azabicycles where the enantioselectivity of the catalyst was able to overcome the inherent diastereoselectivity of the reaction (Scheme 5b).28
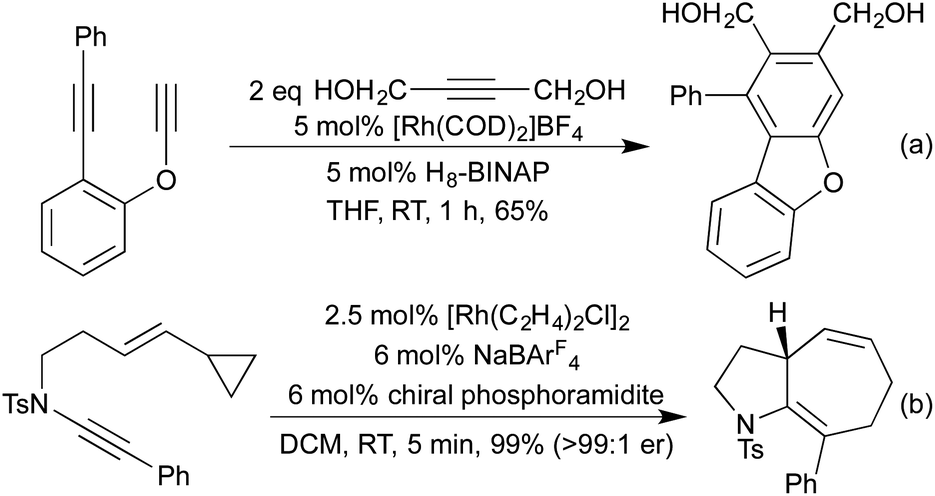 | ||
| Scheme 5 Carbocycle formation, resulting in heterocyclic products. | ||
Of more interest in the context of this review are cycloadditions that directly involve heteroatoms as part of one or more of the π-system components of the cycloaddition, for example [2 + 2 + 2] cross-cyclotrimerisations involving nitriles.29 First reported in 1987,30 this reaction proceeds via a 5-membered rhodacycle intermediate analogously to the alkyne cyclotrimerisation, which then annulates across a nitrile rather than another equivalent of alkyne, generating a pyridine (Scheme 6a). Since this initial report, the Tanaka group have used different methods to favour this reaction over unwanted cyclotrimerisation, as well as to control the regiochemistry of the process. A general trend has emerged that electron-rich alkynes and electron-poor nitriles are required for efficient catalysis. The most common method for controlling the regioselectivity is the use of a tether (Scheme 6a),31 however, even in the absence of this, some alkyne/nitrile combinations are unusually selective.32
 | ||
| Scheme 6 [2 + 2 + 2] Cycloadditions to generate pyridine derivatives. | ||
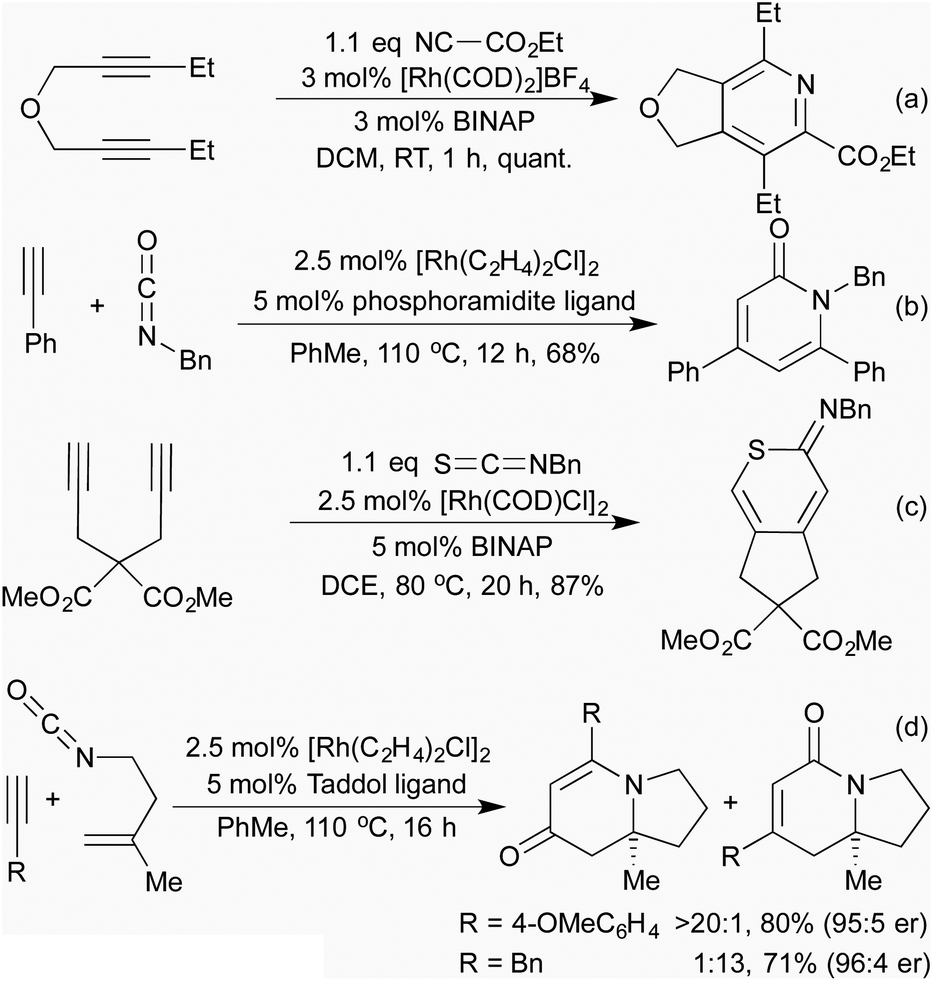
Building on their previous pyridine syntheses, in 2006 the Tanaka group showed that by using isocyanates as coupling partners it was possible to access pyridones, although a tether was still necessary.33 In 2009 Rovis et al. demonstrated that the use of a phosphoramidite ligand gave good levels of regiocontrol without the need for a tether (Scheme 6b).34 By using isothiocyanates or carbon disulfide, various sulfur containing heterocycles were obtained, although only with tethered alkynes (Scheme 6c).35 The use of in situ formed oximes as nitrile equivalents can lead to a 3-component system generating pyridines with environmentally favourable solvents.36 Whilst the generated heterocycles are necessarily planar, chiral catalytic systems have been shown to furnish axially chiral biaryls.33,37
When undertaking the [2 + 2 + 2]-cyclisations with tethered alkenyl isocyanates and alkynes, in 2006 Rovis and co-workers observed that, as well as the expected dihydropyridin-2-one products (which were observed in moderate yields for alkyl alkynes), when aryl alkynes were employed dihydropyridin-4-ones were the predominant product.38 This unexpected outcome was attributed to a decarbonylation-reinsertion mechanism, which sees the carbonyl unit migrate away from the nitrogen in the rhodacycle intermediate. This new isomeric rhodacycle then completes the [2 + 2 + 2] cyclisation to generate the observed product. Chiral phosphoramidite ligands were shown to be effective for achieving the asymmetric version of this process (Scheme 6d),39 and carbodiimides could also be employed in the place of isocyanates.40 Various mechanistic studies41 resulted in the development of a family of highly active catalysts that could switch the mode of reactivity for alkyl alkynes, previously unable to undergo the CO migration.42 The developed conditions were also applied to natural product synthesis, including the synthesis of the tricyclic core of the cylindricine alkaloids with excellent levels of chemoselectivity as well as regio- and enantiocontrol.39a,42
[2 + 2 + 2] cycloadditions have also been used to synthesise dihydropyrans, via the cross cyclotrimerisation of tethered 1,6-enynes and α-keto esters (Scheme 7a).43 This process proceeds in a highly regio-, diastereo- and enantioselective manner, and results in the formation of a quaternary stereogenic centre. The corresponding α-pyran formation resulting from [2 + 2 + 2] cycloaddition between two alkynes and an aldehyde does proceed, but the initially formed pyran undergoes rapid electrocyclic ring opening to form dienones.44 In 2014 Lee et al. published an interesting [2 + 2] cycloaddition between imines and ketenes, generated in situ by the oxidation of terminal alkynes, to form high-value β-lactams in good yields and excellent trans-diastereocontrol (Scheme 7b).45 Mechanistic studies suggest that the reaction proceeds through a metalloketene rather than a free ketene intermediate.
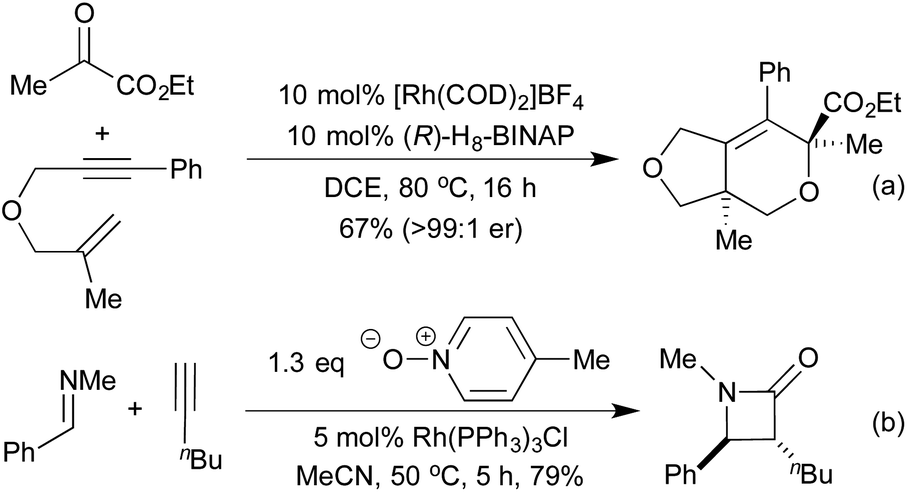 | ||
| Scheme 7 Other [2 + 2 + 2] and [2 + 2] cycloaddition routes to heterocycles. | ||
Vinyl cyclopropanes have long been used as 5-carbon synthons in [5 + 2] cycloaddition reactions, generating 7 membered carbocycles.28 Analogously to replacing alkynes with nitriles or isocyanates in the [2 + 2 + 2] cyclotrimerisation, there are a number of methods that employ N-containing synthons for aza-[5 + 2] cycloadditions. In 2002, Wender and co-workers demonstrated that cyclopropyl imines could readily undergo intermolecular [5 + 2] cycloadditions with alkynes to generate dihydroazepines (Scheme 8a).46 Later computational studies predicted that cyclopropyl aldehydes should undergo the same reaction, although no example of this reaction exists in the literature at this time.47 In 2015, the Zhang group pioneered the use of vinyl aziridines as another N-containing 5-carbon synthon. These intramolecular [5 + 2] cycloadditions with either alkenes48 or alkynes49 (Scheme 8b) proceeded at low temperatures and over short reaction times, with complete retention of chirality between the enantioenriched starting materials and the generated products. Interestingly, when the intermolecular reaction was attempted the [Rh(NBD)2]BF4 catalyst that had previously delivered the [5 + 2] product now selectively afforded the [3 + 2] product with complete chirality transfer (Scheme 8c).50 By switching to a [Rh(η6-C10H8)(COD)]SbF6 catalyst the [5 + 2] product could be obtained selectively.
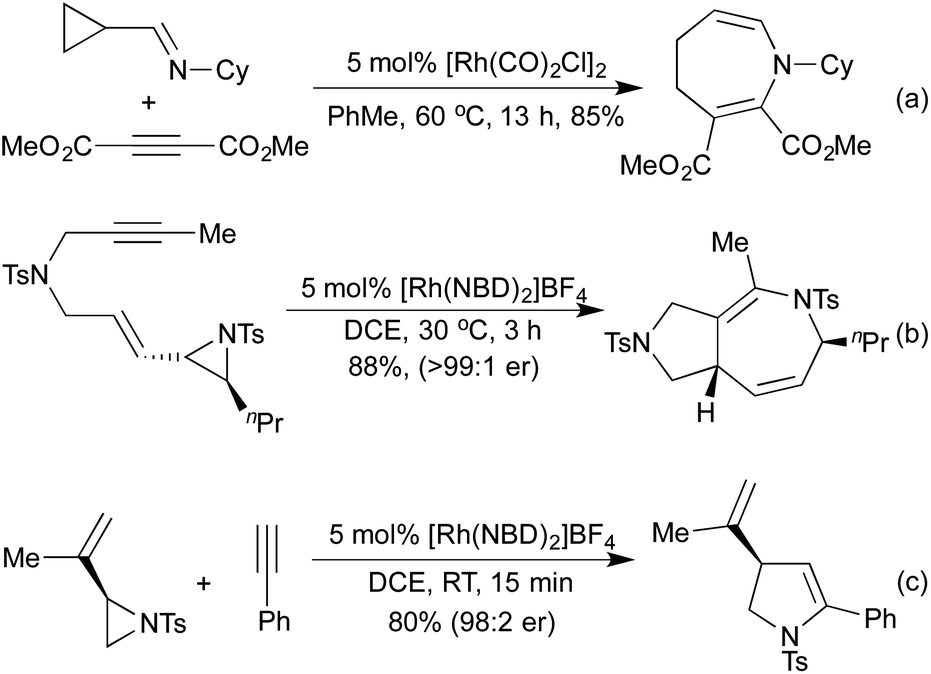 | ||
| Scheme 8 aza-[5 + 2] and [3 + 2] cycloadditions to generate azaheterocycles. | ||
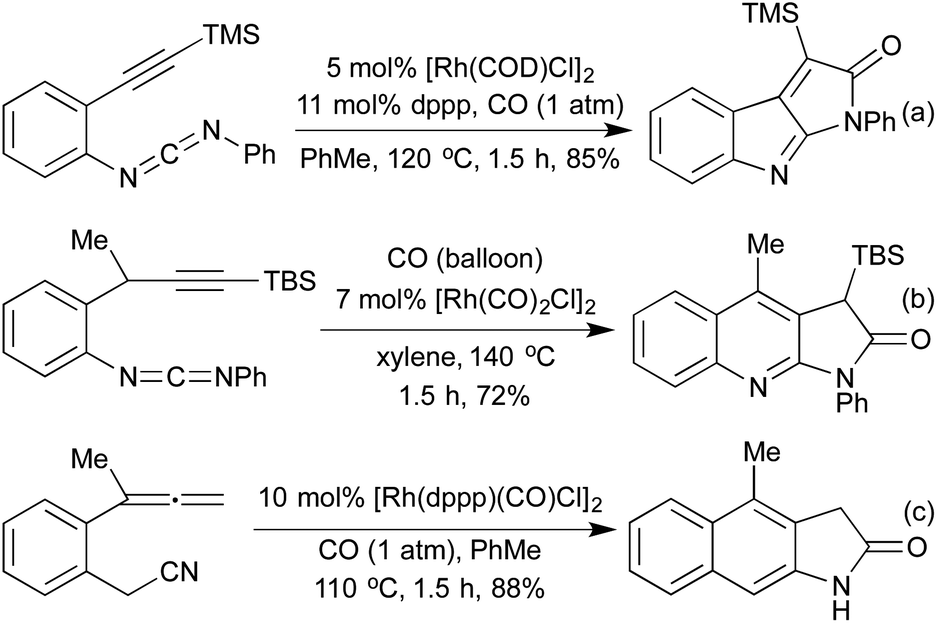
The Pauson-Khand-style (PKR) [2 + 2 + 1] carbonylative process, whilst classically achieved employing cobalt carbonyl clusters, has been extensively studied using rhodium catalysis. In 2007, Saito and co-workers showed that by using a carbodiimide starting material in place of the alkene component, interesting 5,5-fused heterocycles can be generated within short reaction times (Scheme 9a).51 The same group extended this methodology to the synthesis of cyclic thioesters by using an isothiocyanate derivative in the place of the carbodiimide.52 In 2016, Zhang and co-workers published an improved procedure whereby they generate the active carbodiimide in situ from an aryl azide and an isocyanide.53 By inserting a methylene spacer between the aryl unit and the alkyne, this methodology was also used to synthesise pyrrolo[2,3-b]quinolones (Scheme 9b).52 In 2013 the Mukai group set out to introduce N-functionality elsewhere on the substrate; by employing o-allenylbenzylnitriles, they were able to generate lactam fused naphthalene derivatives (Scheme 9c).54 They postulated that the benzylnitrile undergoes an initial tautomerisation to form a ketenimine, which then undergoes the [2 + 2 + 1] PKR to form the lactam directly. In the same publication, they demonstrate that the PKR of hexa-4,5-dienenitriles generated cyclopentadiene fused lactams in moderate to excellent yields.
 | ||
| Scheme 9 [2 + 2 + 1] Pauson-Khand cycloaddition approaches to heterocycles. | ||
4. Hydroacylation
Hydroacylation is formally the 100% atom-economical addition of a hydride and acyl moieties across alkenes and alkynes. Cationic rhodium(I) processes have been long shown to be effective promoters of both intra- and intermolecular hydroacylation processes.55 In recent years, the application of highly active small bite-angle bis-phosphine ligands has led to increased catalytic efficiency, on a wide range of terminal and internal alkenes and alkynes.56 Intermolecular alkyne hydroacylation was first applied to heterocycle synthesis by Willis et al. in 2011.57 By targeting γ-hydroxy-α,β-enones a 2-step synthesis affording a range of di- and tri-substituted furans was achieved. The high catalyst efficiency of this reaction was a result of using β-S chelating aldehydes, disfavouring a deleterious decarbonylation pathway. Whilst this does at first appear to be a limitation of the reaction, there have been a number of methods to mitigate this issue, or even to take advantage of this functionality. Research has been directed both at expanding the range of chelating tethers available,58 as well as ways to employ them as synthetic handles for further transformations. By combining S-directed hydroacylation with carbonyl directed C–S functionalisation reactions, hydroacylation products with no trace of the β-S tether are accessible.59 This tactic has been showcased in the Rh(I)-catalysed hydroacylation approaches to pyrroles and quinolines (Scheme 10).60 High yields were obtained for a range of substitution patterns, reflecting the wide functional group tolerance of Rh(I) catalysed reactions. By utilising these conditions on modified alkenes/alkynes various oxidation states such as pyrrolines, pyrrolidines and quinoline N-oxides were also accessed.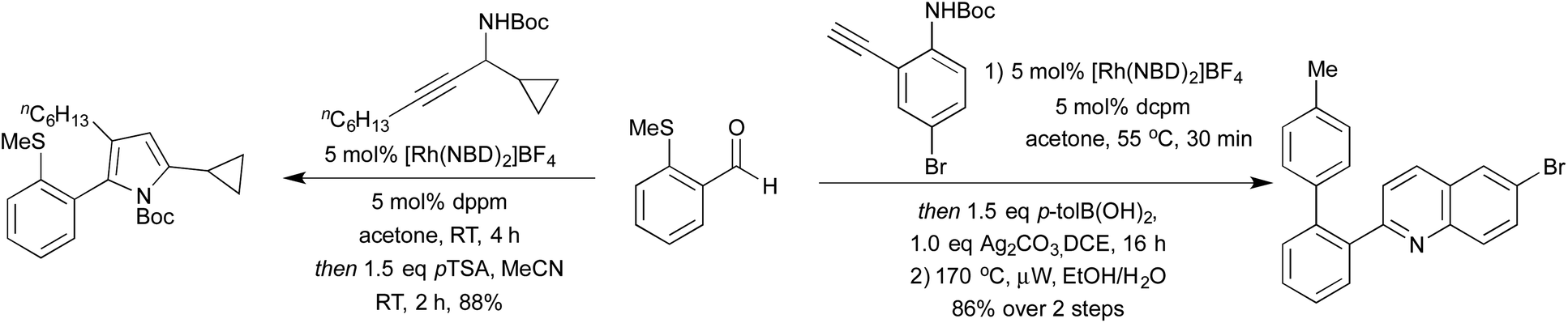 | ||
| Scheme 10 Intermolecular hydroacylation routes to highly functionalised aza-heterocycles. | ||
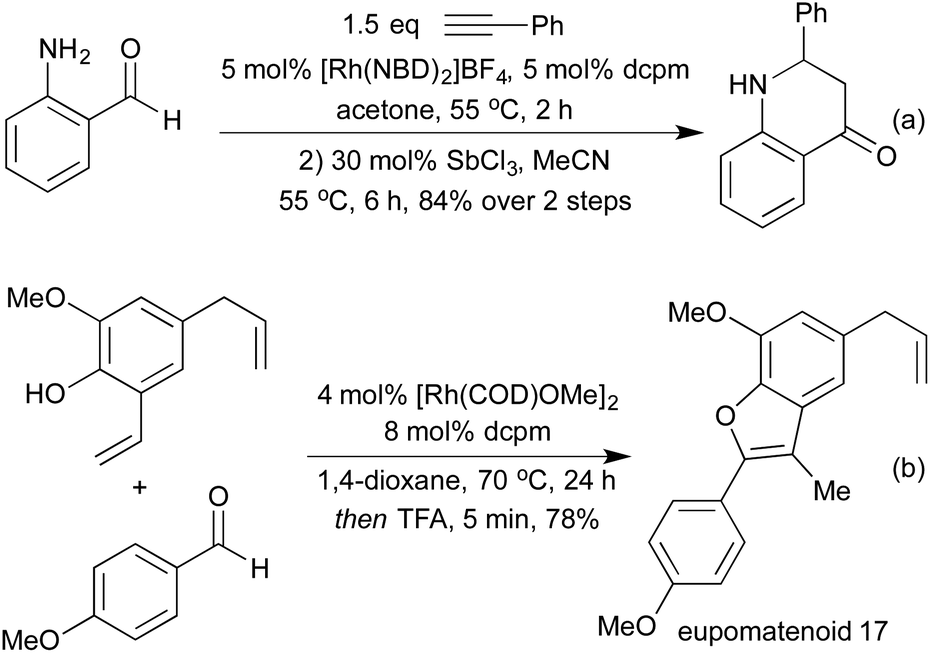
In the context of heterocycle synthesis, approaches that incorporate the heteroatom tether into the product heterocycle are also known. In 2013 the Willis group used N-tethered aldehydes leading to dihydroquinolone products (Scheme 11a).58b A related method to access chromanone derivatives was published in 2015 by the Stanley group, in which cyclisation occurred under the initial hydroacylation conditions without a separate promoter.61 In 2014 Dong and co-workers showed that instead of relying on an aldehyde tether, a co-ordinating group on the alkene partner could have a similar effect, although mechanistically distinct.62 This powerful reaction enabled the synthesis of benzofurans, and was utilised to synthesise a number of eupomatenoid natural products (Scheme 11b).
 | ||
| Scheme 11 Hydroacylation-based tether-incorporating heterocycle syntheses. | ||
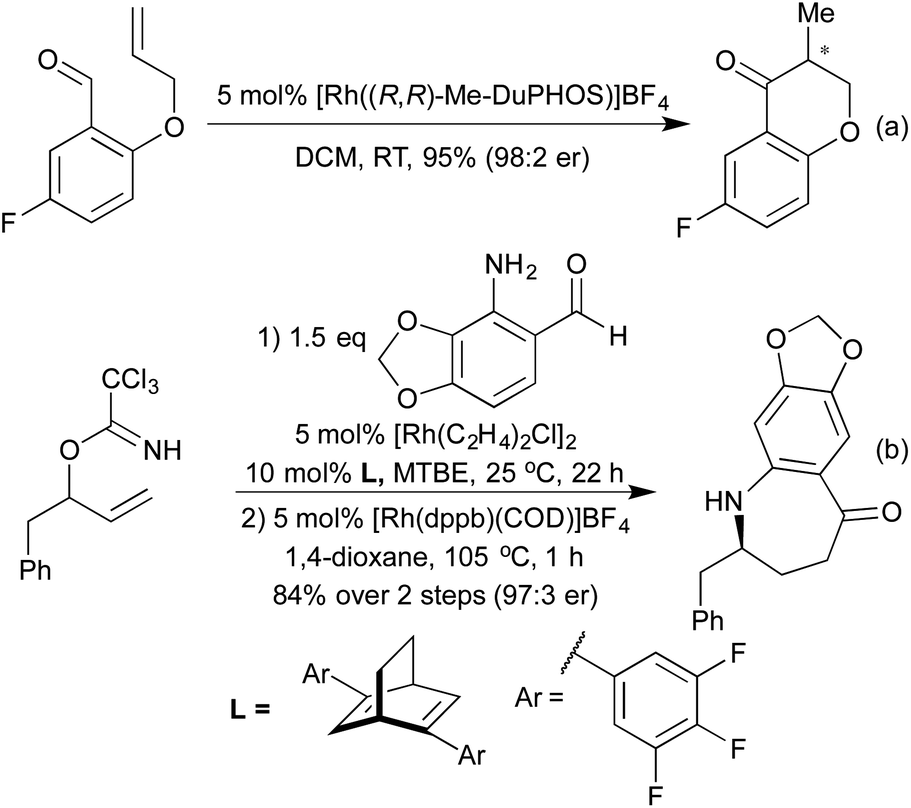
For many years intramolecular hydroacylation was restricted to the realm of carbocycle synthesis, but in 2002 Bendorf et al. demonstrated the first application to heterocycle synthesis.63 By including a S-tether to prevent decarbonylation, a range of medium-sized rings was made accessible. This tactic was expanded with N-64 and O-tethers,65 and by utilising chiral ligands it was possible to access enantioenriched medium rings (Scheme 12a). In 2014 Nguyen et al. combined an intramolecular hydroacylation with a preceding dynamic kinetic asymmetric amination step to generate 7-membered rings in high enantiomeric ratios over two separate Rh(I) catalysed steps (Scheme 12b).66
 | ||
| Scheme 12 Intramolecular alkene hydroacylation towards enantioenriched medium-ring heterocycles. | ||
In 2014 the Stanley group reported asymmetric tetherless intramolecular hydroacylation reactions that utilised 2-indole and 2-pyrrole aldehydes to access fused heterocyclic systems (Scheme 13).67 No tethers were necessary for these reactions due to the rapid intramolecular hydrometallation step outcompeting the potential decarbonylation processes.
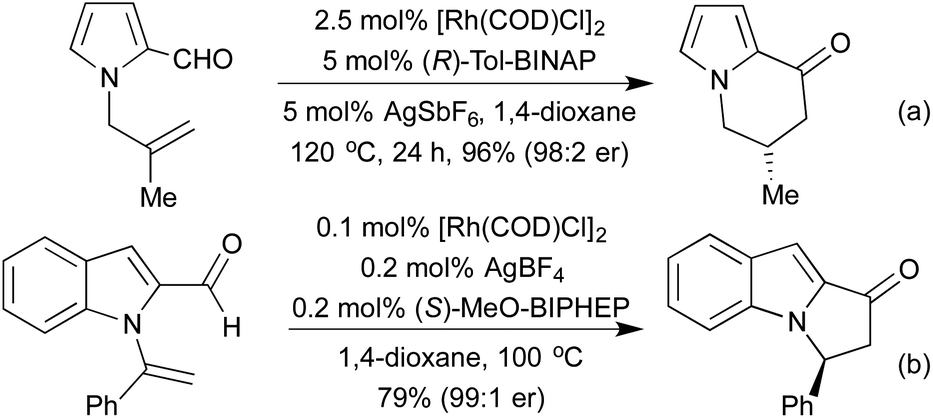 | ||
| Scheme 13 Intramolecular hydroacylation on indole/pyrrole backbones. | ||
Although alkene and alkyne hydroacylations dominate the field, some interesting examples of ketone hydroacylation provide access to a range of lactones. In a similar fashion to the previously mentioned medium ring syntheses, intramolecular ketone hydroacylation reactions forming enantioenriched 7- and 8-membered rings were enabled by utilising O- and N-tethers (Scheme 14a).68 A tether free process was also utilised in a concise asymmetric phthalide synthesis (Scheme 14b).69
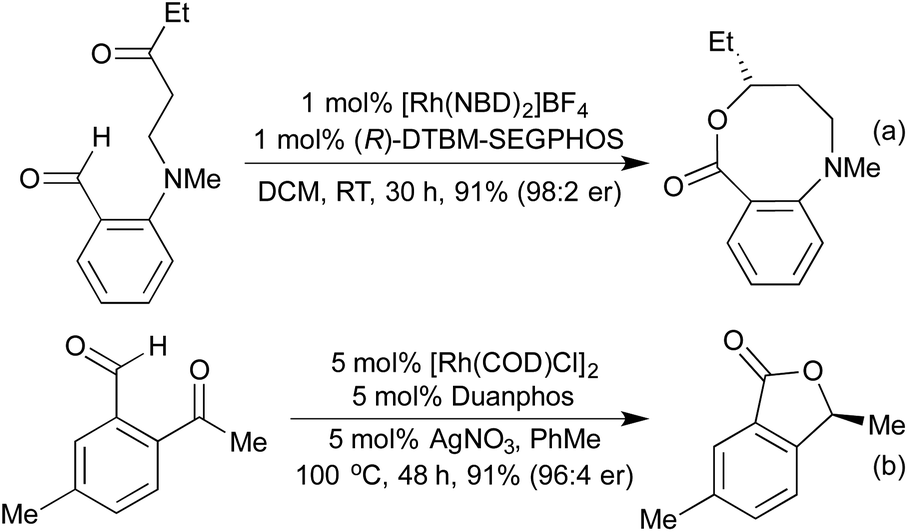 | ||
| Scheme 14 Intramolecular ketone hydroacylation based lactone syntheses. | ||
5. Hydroarylation
The addition of organometallic reagents across alkenes and alkynes has for a long time been one of the key reactions for achieving the formation of new C–C bonds. In particular, since the first reports by Hayashi and Miyaura in 1997,70 the addition of boronic acids across electron-deficient alkenes has been extensively investigated.71 The development of a range of ligand types, including phosphines and chiral dienes, has led to a toolkit of reliable catalyst architectures to effect hydroarylation reactions.72In the field of heterocycle synthesis, rhodium-catalysed hydroarylation, in the form of a conjugate addition, was first applied to the synthesis of quinolines and related derivatives. In 2006 Marinelli et al. published an approach to quinolines that involved addition of a boronic acid across an appropriately functionalised propargylic ketone to generate an enone in situ, which rapidly cyclised to form the quinoline ring system (Scheme 15a).73 Whilst an efficient process, this method was limited to the formation of 2,4-diaryl quinolines. In 2008 the Marsden group switched the disconnective pathway so as to use 2-aminophenylboronates as the source of the carbocyclic ring fragment.74 Although it necessitated an oxidative cyclisation, this simple change meant that the synthetic approach was no longer limited to aryl and vinyl substituted quinolines, and 2,4-dialkyl substrates were easily accessed. Further work in 2014 from the same group extended this protocol to include a reductive cyclisation step, generating diastereoenriched tetrahydroquinolines (Scheme 15b).75 By employing a chiral Rh(Duanphos) catalyst, excellent levels of enantiocontrol were achieved in the hydroarylation step, and a TFA-promoted silane reduction achieved a highly diastereoselective reductive amination. An earlier publication in 2009 had demonstrated that by utilising acrylate Michael acceptors instead of enones, dihydroquinolones were readily accessed.76
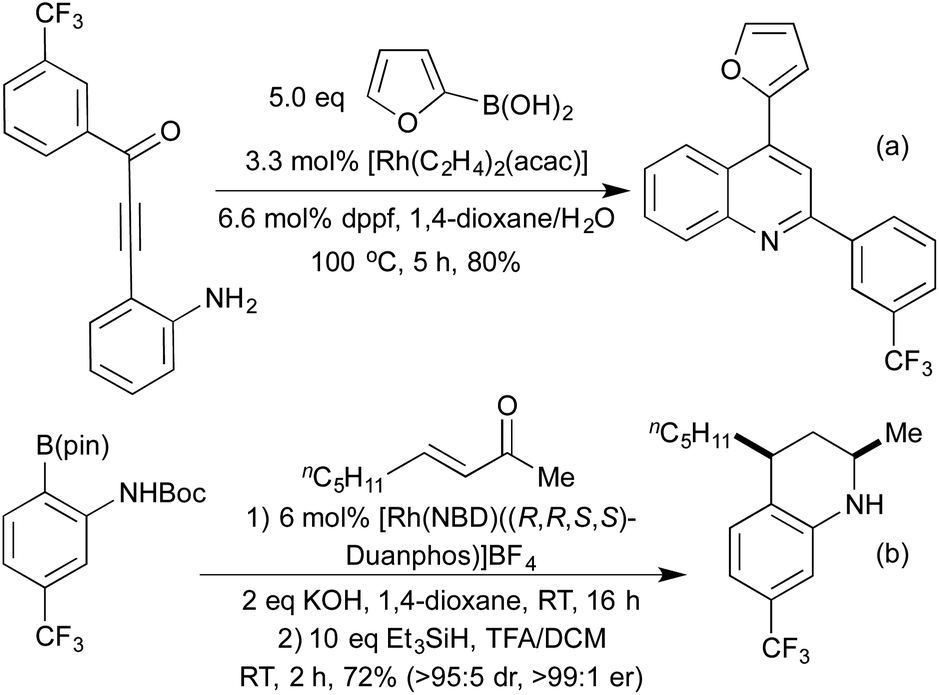 | ||
| Scheme 15 Access to quinoline derivatives via Rh(I) mediated hydroarylation. | ||
A conjugate addition strategy was used by Marinelli et al. in the formation of butenolides (Scheme 16a).77 The propargylic alcohols employed in this reaction were unreactive under standard palladium hydroarylation conditions, whereas the rhodium catalysts could be used in conjunction with a range of organoboron reagents. The same group used similar conditions as a method of constructing 7-membered benzolactones (Scheme 16b).78 In this instance the addition across the aryl alkyne could also be mediated by Pd, however the opposite regioselectivity was observed, with the two methods providing complementary routes to different substitution patterns. In 2010, Youn et al. expanded this strategy to the synthesis of dihydroquinolones (Scheme 16c), pyrrolidinones (Scheme 16d), and dihydrocoumarins in excellent yields with a wide scope on both the organoboron species and the Michael acceptor scaffold.79 Unlike the previous examples employing alkyne hydroarylation, this alkene hydroarylation generates a stereogenic centre, however, no chiral versions of these reactions have yet been reported.
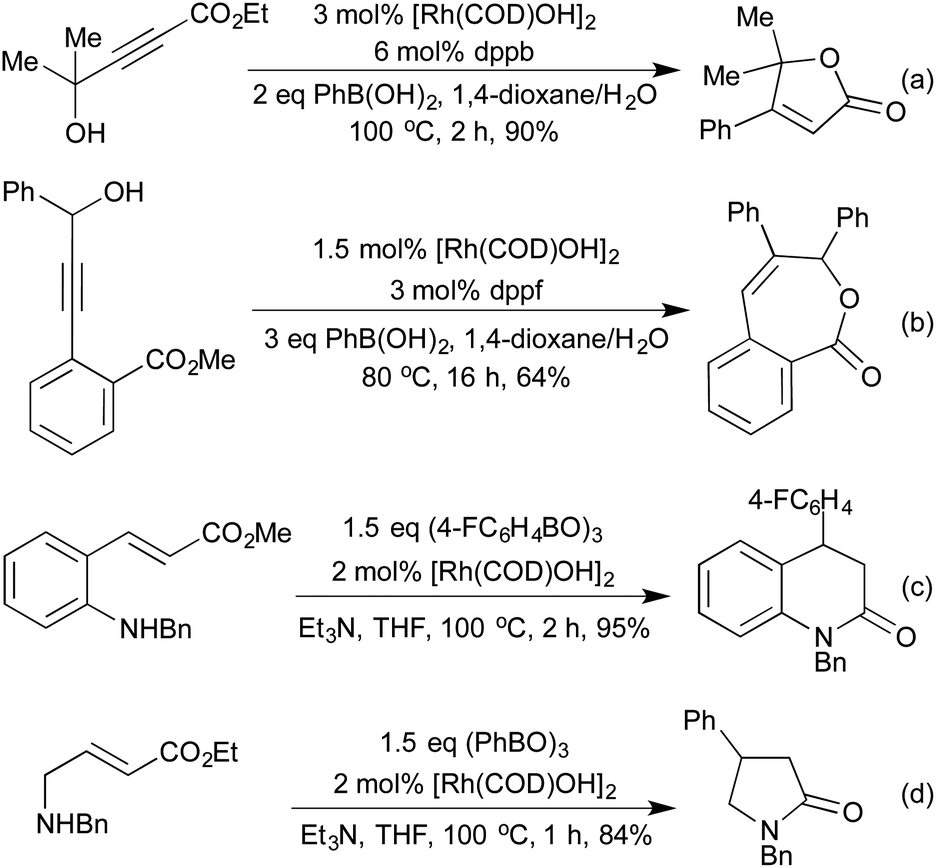 | ||
| Scheme 16 Rh hydroarylation routes to diverse heterocycles. | ||
By combining a Rh-hydroarylation step with a Pd-amination procedure, the Lautens group were able to synthesise dihydroquinolines in an orthogonal catalytic procedure (Scheme 17a).80 In the absence of rhodium, a Suzuki-Miyaura coupling between the boronic acid and the aryl chloride was the sole product, indicating that the Rh-hydroarylation is a significantly faster process, as has since been confirmed by further mechanistic studies.81 The Lautens group has extended the concept of combining Rh-hydroarylation with Pd coupling reactions to the synthesis of dihydrobenzofurans,82 dihydrodibenzoxepines,83 dibenzazepines,84 dihydroquinolones85 and dihydrophenanthridines.86 They were able to synthesise dihydroquinolones (Scheme 17b) in a multicomponent multicatalyst procedure, utilising Rh, Pd and Cu catalysis.87
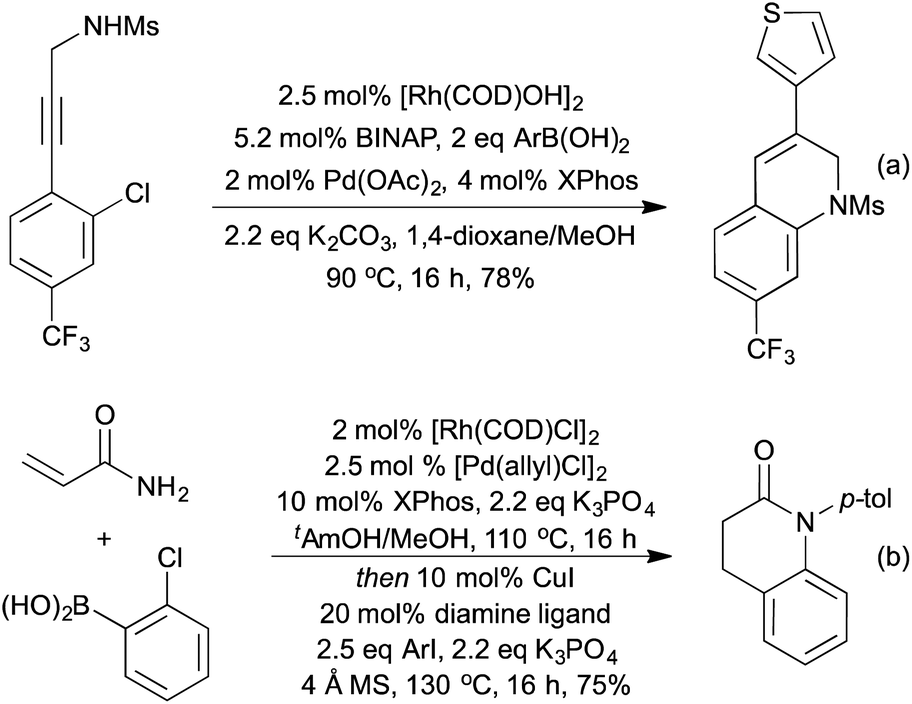 | ||
| Scheme 17 Tandem Rh/Pd catalysis in heterocycle synthesis. | ||
The previously discussed hydroarylation initiated cascades (Scheme 16) rely on the addition of a boronic acid across an alkyne or alkene to bring preinstalled nucleophiles and electrophiles into closer proximity, enabling a cyclisation reaction. A complimentary strategy has been developed that utilises the organorhodium compound generated from the initial carborhodation step as a reactive intermediate for further domino processes. An early publication to employ this strategy was from Murakami and co-workers in 2007 and related to the synthesis of 3-alkylideneoxindoles (Scheme 18a).88 Following the initial hydroarylation, the alkenyl rhodium species undergoes a facile second carborhodation step across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of an intramolecular isocyanate. Hydrolysis of the resulting product, followed by tautomerisation, generates the oxindole. In 2008 the Youn group switched the isocyanate trap for an endo cyclisation onto a preformed imine, thereby generating highly substituted tetrahydroquinolines (Scheme 18b).89 Diastereoselectivities between C(4) and C(3) were mixed, however the Zimmerman-Traxler nature of the intramolecular Mannich-type process led exclusively to generation of a syn-arrangement between the substituents at C(2) and C(3). A 3-step cascade reaction utilising o-bromo boronic acids generated various carbocycles in a pseudo [2 + 2 + 2] cyclisation, in which the boronic acid represents a benzyne synthon.90 Following hydroarylation of the initial alkyne, swift intramolecular carborhodation across the pendant alkene, followed by oxidative addition into the C–Br bond and reductive elimination generated a range of polycyclic compounds (Scheme 18c). Heteroatom containing tethers (from which the 5-membered heterocycle is generated) were used to increase the regioselectivity of the initial hydroarylation step. Building on an earlier publication by Hayashi et al.,91 the Lautens group published a route to oxindoles that proceeded via initial hydroarylation setting up an intramolecular enolate arylation (Scheme 18d).92 Whilst Hayashi's procedure required the use of zinc reagents, Lautens was able to employ trifluoroborate salts.
O bond of an intramolecular isocyanate. Hydrolysis of the resulting product, followed by tautomerisation, generates the oxindole. In 2008 the Youn group switched the isocyanate trap for an endo cyclisation onto a preformed imine, thereby generating highly substituted tetrahydroquinolines (Scheme 18b).89 Diastereoselectivities between C(4) and C(3) were mixed, however the Zimmerman-Traxler nature of the intramolecular Mannich-type process led exclusively to generation of a syn-arrangement between the substituents at C(2) and C(3). A 3-step cascade reaction utilising o-bromo boronic acids generated various carbocycles in a pseudo [2 + 2 + 2] cyclisation, in which the boronic acid represents a benzyne synthon.90 Following hydroarylation of the initial alkyne, swift intramolecular carborhodation across the pendant alkene, followed by oxidative addition into the C–Br bond and reductive elimination generated a range of polycyclic compounds (Scheme 18c). Heteroatom containing tethers (from which the 5-membered heterocycle is generated) were used to increase the regioselectivity of the initial hydroarylation step. Building on an earlier publication by Hayashi et al.,91 the Lautens group published a route to oxindoles that proceeded via initial hydroarylation setting up an intramolecular enolate arylation (Scheme 18d).92 Whilst Hayashi's procedure required the use of zinc reagents, Lautens was able to employ trifluoroborate salts.
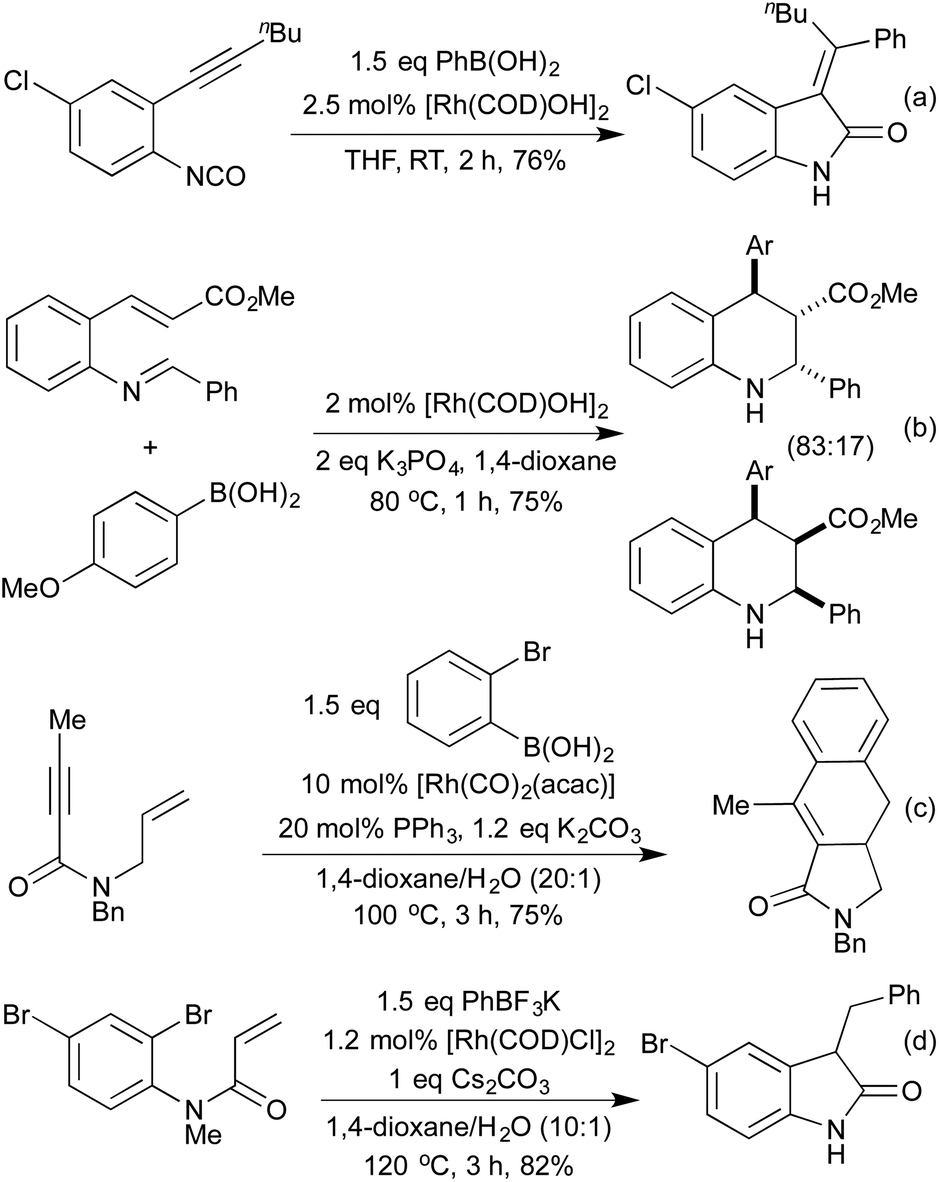 | ||
| Scheme 18 Rh hydroarylation initiated domino reactions. | ||
More recently, a number of groups have looked to synthesise highly substituted 5-membered heterocycles by combining alkyne hydroarylation with an intramolecular carborhodation in an enantioselective manner. Lautens and co-workers pioneered this approach in 2013, utilising ether linked alkynylcyclohexadienones (Scheme 19a).93 A more general procedure followed in 2014 from Xu and co-workers, where the ring-closing step involved cyclisation onto a ketone, generating a chiral tertiary alcohol stereocentre in addition to an all-carbon tetrasubstituted olefin (Scheme 19b).94 Using this method, they were able to generate a range of tetrahydrofuran and pyrrolidine derivatives. The Darses group has since extended this strategy to the synthesis of hydroxyl-substituted 6-membered heterocycles,95 and to include cyclisation onto Michael acceptors as an alternative ring-closing pathway (Scheme 19c).96 As well as addition across C–C multiple bonds, rhodium has also been used to add boronic acids to aldehydes and N-sulfonylimines, generating chiral alcohols and amines respectively. The use of chiral phosphine or diene ligands leads to high enantioselectivity in these reactions. Once generated, enantioenriched amines have been shown to undergo cyclisations in either a one- or two-pot process to synthesise pyrrolidines (Scheme 20a)97 and isoindolinones.98 The addition of boronic acids to aldehydes has also been used to generate enantioenriched phthalides.99 An improved synthesis of isoindolinones was published in 2012, where instead of relying on 2-(N-tosylimino)benzoates (which require a longer synthesis, limiting the ability to quickly generate a library of compounds), the authors used 2-halobenzaldimines.100 Following the Rh hydroarylation, a transfer carbonylation process, employing a sacrificial aldehyde as a carbonyl source generated the final isoindolines (Scheme 20b). This process is noteworthy for multiple reasons; not only is it an example of a CO-gas-free carbonylation reaction, but also it combines two distinct Rh(I) catalysed reactions in one pot, with a single addition of Rh pre-catalyst and only the addition of a bisphosphine ligand required to promote the transfer carbonylation step. In 2012 Lautens et al. synthesised 1,4-benzothiazine derivatives via sequential Rh(I) catalysed addition of a boronic acid to a nitrile to generate an electron deficient enamine, which can then undergo a base promoted SNAr reaction (Scheme 20c).101
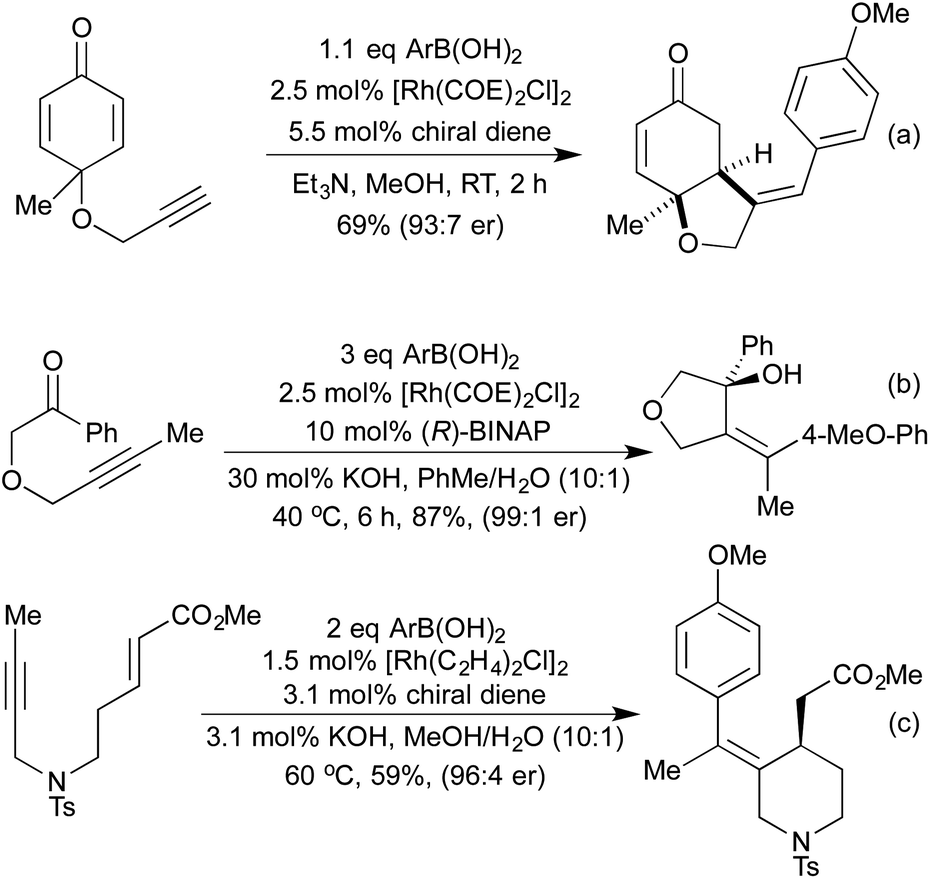 | ||
| Scheme 19 Enantioselective cascade routes to saturated heterocycles. | ||
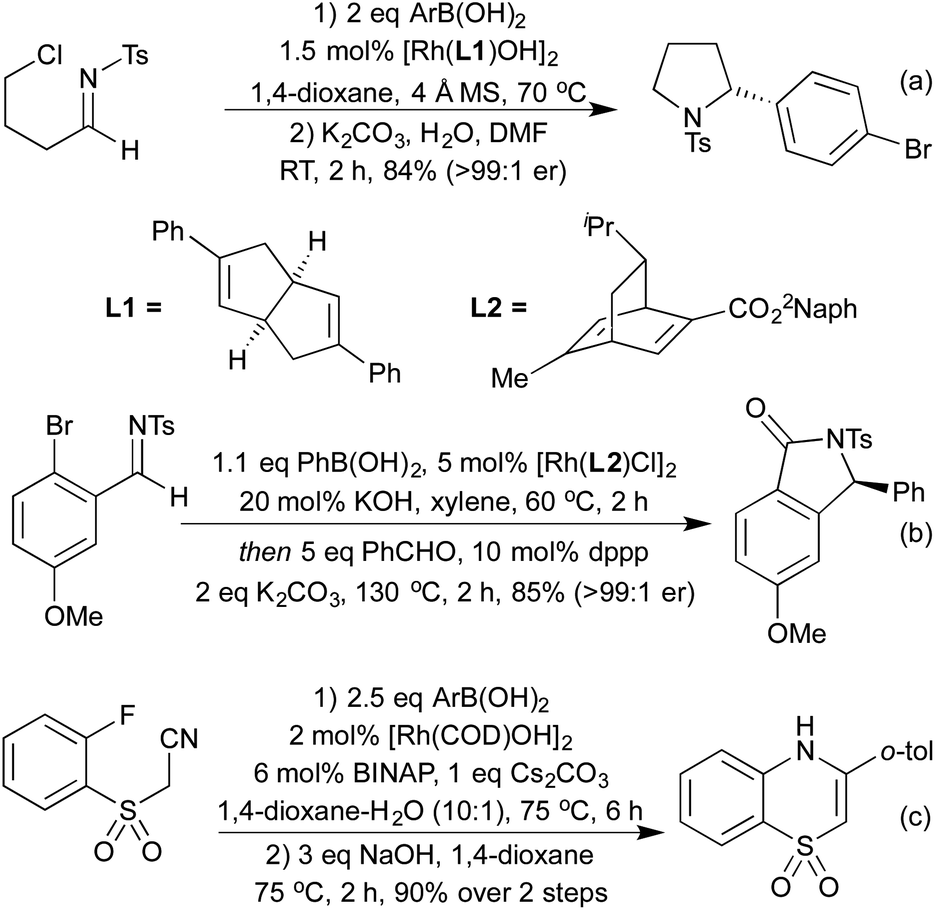 | ||
| Scheme 20 Synthesis of N-containing heterocycles via imine hydroarylation. | ||
6. Miscellaneous processes
6.1. Carbonylative processes
As has been demonstrated in many of the previous sections, a strength of rhodium catalysis is the ease with which existing methodology can be modified under carbonylative conditions to form high value CO containing products.102 As many classical heterocycle syntheses rely on the thermodynamically driven dehydration of carbonyl compounds to yield heteroaromatic systems, it is intuitive to see how carbonylation is a vital tool when targeting heterocycle precursors.One of the most common examples of homogenous transition-metal catalysis is hydroformylation, with over 10 million metric tons of aliphatic aldehydes synthesised each year using these reactions.103 Exposure of olefins to a range of Rh(I) catalysts under a CO/H2 atmosphere generates terminal olefins in extremely high efficiency, with asymmetric processes similarly well developed. As aldehydes have long been used as key building blocks in heterocycle synthesis, a range of tandem hydroformylation–heterocyclisation reactions have been published.104
By conducting hydroformylation in the presence of aryl hydrazines, a tandem hydroformylation/Fischer indole synthesis has been developed, and used by the Eilbracht group in the synthesis of a range of pharmaceutically relevant indoles (Scheme 21a).105 The same group have used similar conditions to mimic another classical heterocycle synthesis in the form of a hydroformylation/Pictet-Spengler cascade (Scheme 21b).106 The slow release of aldehyde from the hydroformylation reaction attenuated many of the issues of aldehyde dimerisation characteristic of the classical Pictet-Spengler process, which are usually tackled by employing high-dilution conditions. By combining hydroformylation with an in situ Wittig olefination it has been shown to be possible to generate Michael-acceptors which can be used in the synthesis of a range of pyrrolidines (Scheme 21c)107 and a number of tetrahydropyrans108 in high diastereoselectivity.
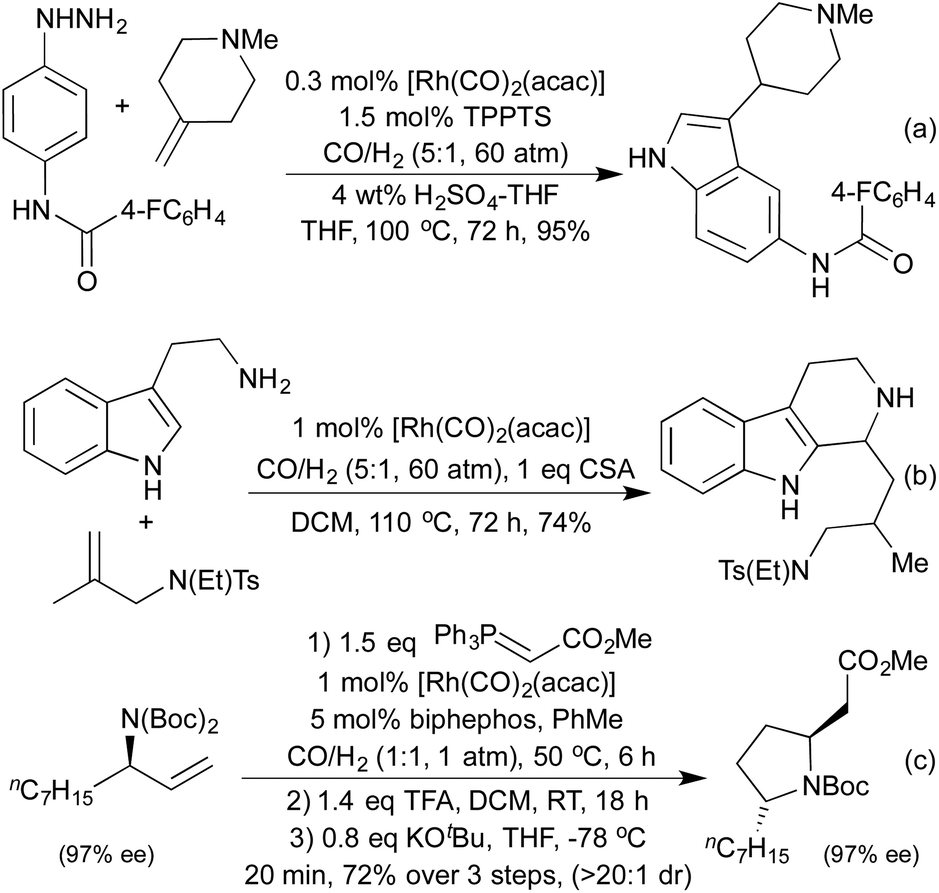 | ||
| Scheme 21 Tandem hydroformylation/heterocyclisation reactions. | ||
Various reductive amination-type cyclisations have been employed in the synthesis of alkaloids from enantioenriched olefins (Scheme 22a and b).109 In the absence of a reductant, it is also possible to isolate the cyclic enamines,110 or the resulting cyclic imine can be used in further cascades. A second pendant nucleophile can be used to form a bicyclic hemiaminal,111 or in the presence of alkyne functionality an aza-Prins cyclisation can be observed (Scheme 22c).112
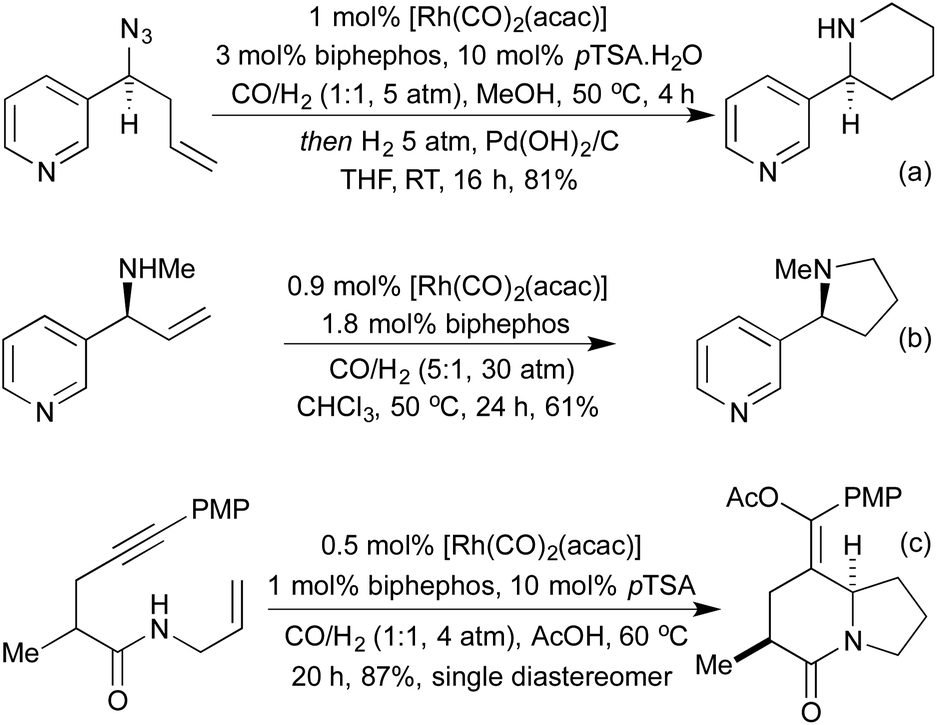 | ||
| Scheme 22 Tandem hydroformylation reactions in alkaloid synthesis. | ||
Combining the concept of boronic acid addition with carbonylation chemistry has enabled access to a broad family of heterocycle precursors. Carbonylation of the intermediate aryl–rhodium species prior to carbometallation allows for the generation of γ-hydroxy-α,β-enones from propargylic alcohols and boronic acids (Scheme 23a).113 These intermediates are readily cyclised to furans under the reaction conditions. In a similar sequence, using vinyl ketones as the coupling partner furnishes 1,4-dicarbonyls, which can be cyclised in a separate step to furans and pyrroles (Scheme 23b).114 By incorporating a second equivalent of CO, Artok et al. synthesised a range of tri-substituted butenolides from symmetrical alkynes (Scheme 23c).115 Their proposed mechanism relies on a second carbonylation event outcompeting the protoderhodation that would otherwise generate an enone product. This method compliments an earlier publication from Morimoto et al. on the generation of disubstituted butenolides, which uses formaldehyde as a carbonyl source.116 Carbonylation coupled with C–H activation on diaryldiazenes has produced a range of interesting N-fused heterocycles (Scheme 23d).117 The use of nitrobenzene as a stoichiometric oxidant is required, and the product formed is different to that obtained when they used orthogonal cobalt catalysis.
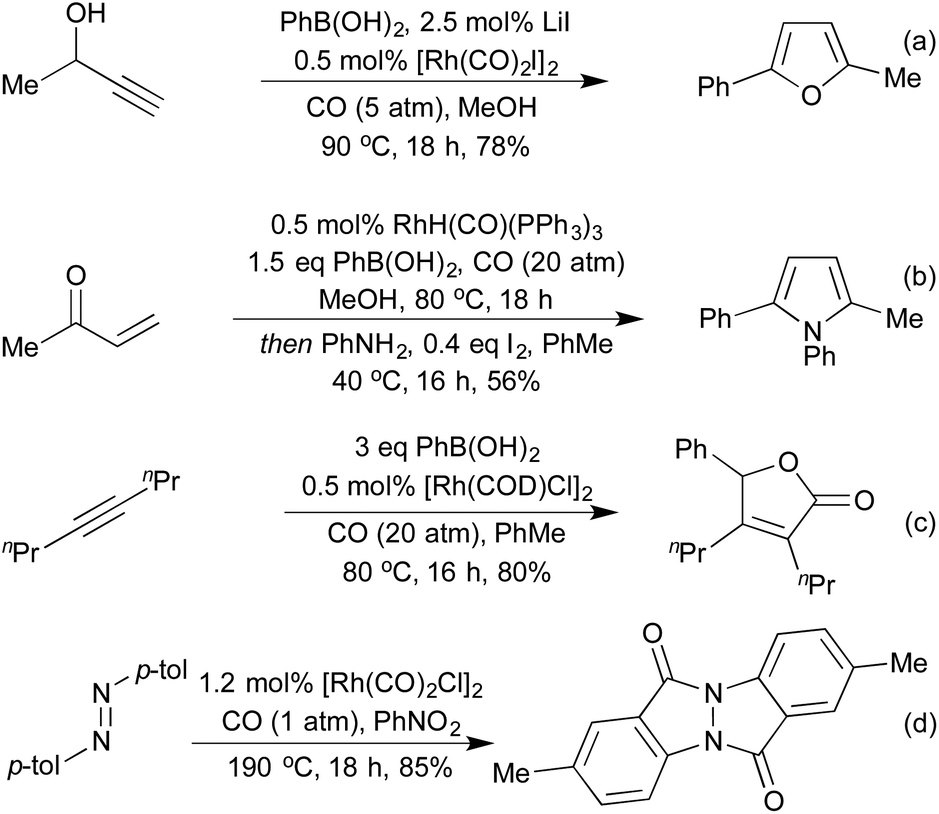 | ||
| Scheme 23 Carbonylative 3-component couplings to form 5-membered heterocycles. | ||
Carbonylation has also been used as a tool in ring expansion/ring rearrangement reactions. As early as 1994, Alper and co-workers demonstrated the carbonylative transformation of thiazolidines to thiazolidinones via a thiomorpholinone intermediate (Scheme 24a).118 In the absence of a KI additive the thiomorpholine could instead be isolated as the sole product. The same group has applied their carbonylation conditions to a variety of nitrogen and sulfur containing heterocycles (Schemes 24b and c).119 In the case of acetylenic thiazoles an unusual ring rearrangement was observed in addition to carbonylation and reduction. The carbonylative ring expansion of 2-aryl aziridines to β-lactams has been known since 1983, with [Rh(CO)2Cl]2 proving an efficient catalyst for the process.120 The stereochemistry of the aziridine is translated faithfully into the product, and by using menthol as a ligand it was possible to effect a kinetic resolution of racemic aziridines.121 In 2003 the use of a dendrimer bound Rh(I) catalyst enabled recycling of the active catalyst, with no loss of activity observed after three iterations, using a simple filtration to achieve separation (Scheme 24d).122 A 2006 computational study of the selectivity of the reaction for 2-aryl aziridines concluded that hyperconjugation was responsible for facilitating the initial C–N bond activation.123
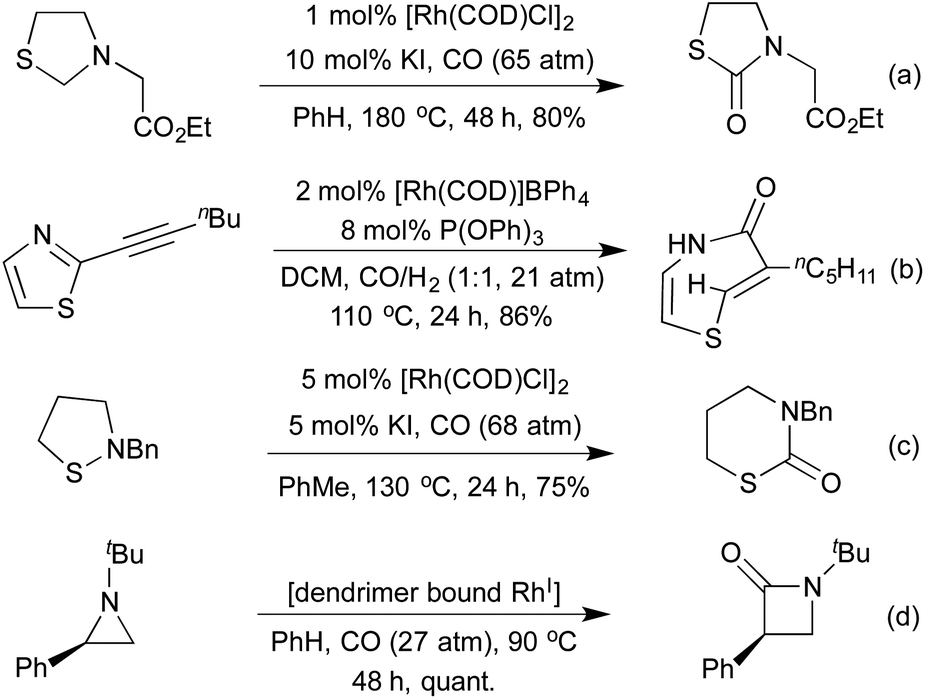 | ||
| Scheme 24 Carbonylative ring-expansion reactions. | ||
6.2. Non-carbonylative processes
Standard carbonylation conditions have also proved effective for the reduction of nitroarenes. In the presence of suitable ortho-functionality it is postulated that a rhodium-nitrene intermediate undergoes a rearrangement to form indoles124 or indazoles,125 from alkenes and imines, respectively (Scheme 25a and b). Krische demonstrated in 2006, that by using hydrogenation conditions on suitably functionalised alkynes, it was possible to isolate cyclic lactams produced via hydrogenation of a 5-membered rhodacycle intermediate (Scheme 25c).126 In the previous section we saw a number of hydroarylation initiated cascade reactions, which provided access to a diverse range of heterocycles. By employing Et2Zn and Wilkinson's complex to generate a Rh hydride active catalyst, Ando and co-workers achieved the reductive cyclisation of acrylates and imines to generate β-lactams (Scheme 25d).127 In this transformation, the rhodium-hydride effects a 1,4-reduction of the Michael-acceptor acrylates, which then adds into the imine prompting a cascade cyclisation. The rhodium alkoxide thereby generated is then reduced by Et2Zn to regenerate the rhodium hydride catalyst. | ||
| Scheme 25 Reductive cyclisation approaches to N-heterocycles. | ||
The Willis group has shown that Rh(I) hydroacylation catalysts are also efficient promoters of directed C–S activation processes.59a A slight modification of the catalyst from the hemilabile DPEphos to the pincer-type ligand Xantphos, produced a highly efficient catalyst system for carbothiolation reactions. By refluxing these alkenyl sulfide products with an NH3-equivalent in acetic acid, it was possible to obtain isoquinolines in excellent yields.128 A range of functional groups was tolerated, including an unusual ferrocenyl alkyne (Scheme 26a). Whilst normally within the realm of Rh(III) catalysis, directed C–H activation followed by alkenylation can be effected using Rh(I) catalysts, via a Rh(I)-Rh(III) redox cycle, as demonstrated by Bergman and Ellman.129 The resulting dienimine then undergoes a facile 6π-electrocyclisation to generate a dihydropyridine, which can be further functionalised for access to pyridines if required (Scheme 26b). By using oximes in place of imines, pyridines can be accessed directly, as demonstrated by Cheng and co-workers.130 An earlier report from Bergman and Ellman showed that the Rh(III) rhodacycle could hydroarylate a tethered olefin to generate dihydrobenzofurans or indolines depending on the identity of the tether.131 In a 2012 report, Terada et al. observed an unusual cycloisomerisation of O-propargylic cyclopropylcarbaldoximes to oxazepines (Scheme 26c).132 This has been shown to proceed via an initial rhodium-catalysed 2,3-migration, rhodacycle formation, followed by cyclopropyl ring opening and reductive elimination, not dissimilar to the mechanism of [5 + 2] cycloadditions involving cyclopropylimines previously discussed.
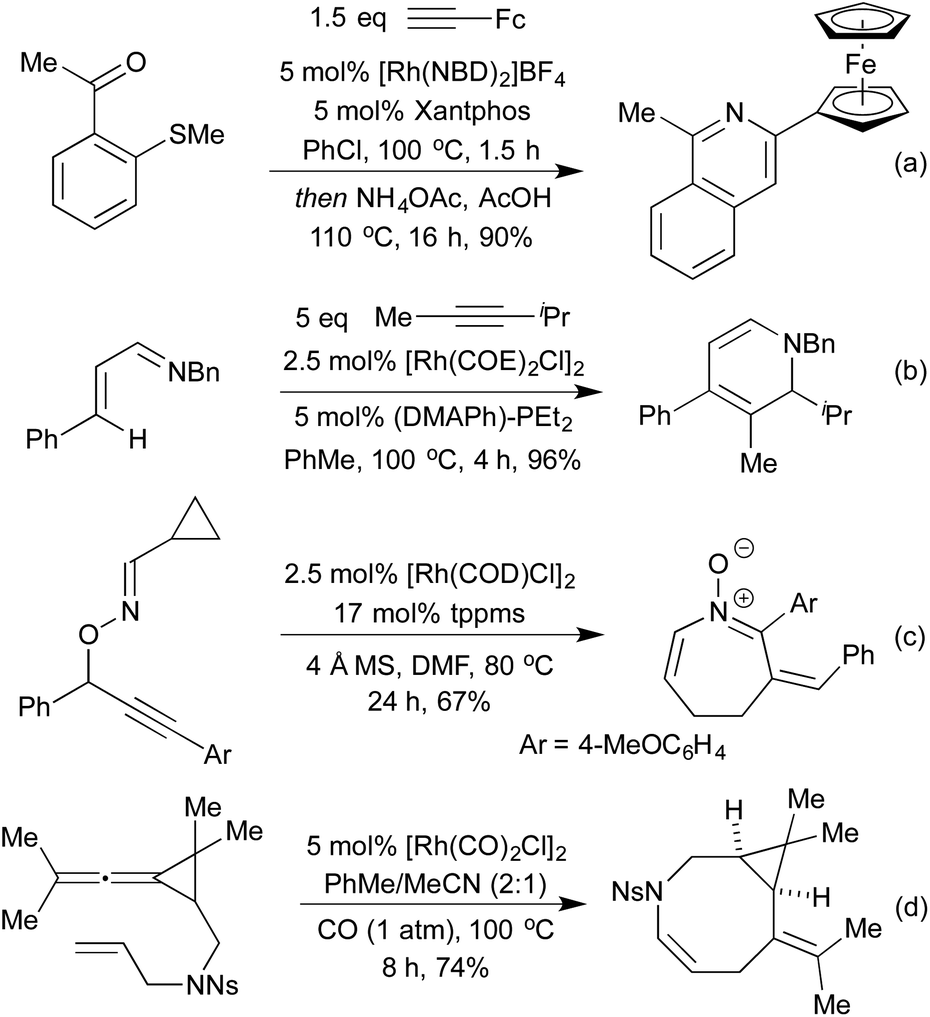 | ||
| Scheme 26 Miscellaneous Rh(I) catalysed heterocycle syntheses. | ||
Whilst investigating [2 + 2] cycloadditions with nitrogen containing tethers Shi et al. discovered an unusual 8-membered ring formation (Scheme 26d).133 They believe that this transformation occurs via an initial C–H activation at the allylic position adjacent to the N-functionality. The resulting Rh allyl hydride can add across one of the allene π-bonds in a carborhodation step, and reductive elimination affords the product.
7. Conclusion
In this review we have summarised the major fields of Rh(I) catalysis and how each has been applied to the synthesis of diverse heterocycles. The wide range of reaction pathways available, combined with the ability to combine one process with another (or carbonylation) in cascade or tandem reactions provides flexible approaches to a number of cyclic and acyclic scaffolds. The high functional group tolerance, ability to easily employ chiral ligand architectures and efficient catalysis are all attractive features of Rh(I) transformations in the context of any heterocycle synthesis. In addition, the orthogonality to other transition metal catalysts enables the synthesis of boronate and halide containing heterocycles, allowing for the rapid assembly of heterocyclic libraries via subsequent transition-metal-catalysed cross-coupling reactions, which is of great importance for medicinal chemistry in particular. We look forward to new developments in the field of Rh(I) catalysis and the application of these processes to the ever-expanding field of heterocycle synthesis.Notes and references
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- P. A. Evans, Modern Rhodium-Catalyzed Organic Reactions, Wiley-VCH, 2005 Search PubMed.
- X.-F. Wu, Transition Metal-Catalyzed Heterocycle Synthesis via C–H Activation, Wiley-VCH, 2016 Search PubMed.
- (a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS PubMed; (b) A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 1371–1373 CrossRef CAS PubMed; (c) S. C. Hockey and L. C. Henderson, Aust. J. Chem., 2015, 68, 1796–1800 CrossRef CAS; (d) K. Ouyang, W. Hao, W.-X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045–12090 CrossRef CAS PubMed.
- H. Maehr, J. Chem. Educ., 1985, 62, 114 CrossRef CAS.
- B. M. Trost and Y. H. Rhee, J. Am. Chem. Soc., 2003, 125, 7482–7483 CrossRef CAS PubMed.
- A. Takemiya and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 6042–6043 CrossRef CAS PubMed.
- (a) Z. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 1570–1571 CrossRef CAS PubMed; (b) L. D. Julian and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 13813–13822 CrossRef CAS PubMed.
- K. Okuro and H. Alper, Tetrahedron Lett., 2010, 51, 4959–4961 CrossRef CAS.
- X. Shen and S. L. Buchwald, Angew. Chem., Int. Ed., 2010, 49, 564–567 CrossRef CAS PubMed.
- (a) S. Burling, L. D. Field, H. L. Li, B. A. Messerle and P. Turner, Eur. J. Inorg. Chem., 2003, 3179–3184 CrossRef CAS; (b) L. D. Field, B. A. Messerle, K. Q. Vuong and P. Turner, Organometallics, 2005, 24, 4241–4250 CrossRef CAS; (c) S. Burling, L. D. Field, B. A. Messerle and S. L. Rumble, Organometallics, 2007, 26, 4335–4343 CrossRef CAS; (d) L. D. Field, B. A. Messerle, K. Q. Vuong, P. Turner and T. Failes, Organometallics, 2007, 26, 2058–2069 CrossRef CAS; (e) G. K. B. Clentsmith, L. D. Field, B. A. Messerle, A. Shasha and P. Turner, Tetrahedron Lett., 2009, 50, 1469–1471 CrossRef CAS; (f) L. D. Field, B. A. Messerle, K. Q. Vuong and P. Turner, Dalton Trans., 2009, 3599–3614 RSC; (g) S. R. Beeren, S. L. Dabb, G. Edwards, M. K. Smith, A. C. Willis and B. A. Messerle, New J. Chem., 2010, 34, 1200–1208 RSC; (h) S. L. Rumble, M. J. Page, L. D. Field and B. A. Messerle, Eur. J. Inorg. Chem., 2012, 2226–2231 CrossRef CAS; (i) K. Q. Vuong, M. G. Timerbulatova, M. B. Peterson, M. Bhadbhade and B. A. Messerle, Dalton Trans., 2013, 42, 14298–14308 RSC.
- B. M. Trost and A. McClory, Angew. Chem., Int. Ed., 2007, 46, 2074–2077 CrossRef CAS PubMed.
- N. Isono and M. Lautens, Org. Lett., 2009, 11, 1329–1331 CrossRef CAS PubMed.
- A. Boyer, N. Isono, S. Lackner and M. Lautens, Tetrahedron, 2010, 66, 6468–6482 CrossRef CAS.
- A. Mizukami, Y. Ise, T. Kimachi and K. Inamoto, Org. Lett., 2016, 18, 748–751 CrossRef CAS PubMed.
- A. Saito, S. Oda, H. Fukaya and Y. Hanzawa, J. Org. Chem., 2009, 74, 1517–1524 CrossRef CAS PubMed.
- K. Tanaka, T. Shoji and M. Hirano, Eur. J. Org. Chem., 2007, 2687–2699 CrossRef CAS.
- B. G. Van den Hoven, B. E. Ali and H. Alper, J. Org. Chem., 2000, 65, 4131–4137 CrossRef CAS PubMed.
- B. G. Van den Hoven and H. Alper, J. Am. Chem. Soc., 2001, 123, 10214–10220 CrossRef CAS PubMed.
- Z. Zhang and I. Ojima, J. Organomet. Chem., 1993, 454, 281–289 CrossRef CAS.
- Y. Zhang, Z. Chen, Y. Xiao and J. Zhang, Chem. – Eur. J., 2009, 15, 5208–5211 CrossRef CAS PubMed.
- (a) W. Zhao and J. Zhang, Chem. Commun., 2010, 46, 4384–4386 RSC; (b) W. Zhao and J. Zhang, Chem. Commun., 2010, 46, 7816–7818 RSC; (c) W. Zhao and J. Zhang, Org. Lett., 2011, 13, 688–691 CrossRef CAS PubMed.
- H. Kawai, S. Oi and Y. Inoue, Heterocycles, 2006, 67, 101–105 CrossRef CAS.
- (a) G. Dominguez and J. Perez-Castells, Chem. Soc. Rev., 2011, 40, 3430–3444 RSC; (b) D. L. J. Broere and E. Ruijter, Synthesis, 2012, 2639–2672 CAS; (c) K. Tanaka, in Transition-Metal-Mediated Aromatic Ring Construction, John Wiley & Sons, Inc., 2013, pp. 127–160 Search PubMed; (d) H.-W. Lee and F.-Y. Kwong, Eur. J. Org. Chem., 2010, 789–811 CrossRef CAS; (e) N. Jeong, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree, Elsevier, Oxford, 2007, pp. 335–365 Search PubMed; (f) R. R. Torres, in The Pauson-Khand Reaction, John Wiley & Sons, Ltd, 2012 Search PubMed; (g) H. Pellissier, Adv. Synth. Catal., 2011, 353, 189–218 CrossRef CAS; (h) H. Clavier and H. Pellissier, in Methods and Applications of Cycloaddition Reactions in Organic Syntheses, John Wiley & Sons, Inc., 2014, pp. 631–654 Search PubMed.
- Y. Komine, A. Kamisawa and K. Tanaka, Org. Lett., 2009, 11, 2361–2364 CrossRef CAS PubMed.
- B. Witulski and C. Alayrac, Angew. Chem., Int. Ed., 2002, 41, 3281–3284 CrossRef CAS.
- B. Witulski and A. Zimmermann, Synlett, 2002, 1855–1859 CrossRef CAS.
- R. N. Straker, Q. Peng, A. Mekareeya, R. S. Paton and E. A. Anderson, Nat. Commun., 2016, 7, 10109 CrossRef CAS PubMed.
- K. Tanaka, Heterocycles, 2012, 85, 1017–1043 CrossRef CAS.
- P. Cioni, P. Diversi, G. Ingrosso, A. Lucherini and P. Ronca, J. Mol. Catal., 1987, 40, 337–357 CrossRef CAS.
- K. Tanaka, N. Suzuki and G. Nishida, Eur. J. Org. Chem., 2006, 3917–3922 CrossRef CAS.
- (a) Y. Komine and K. Tanaka, Org. Lett., 2010, 12, 1312–1315 CrossRef CAS PubMed; (b) K. Kashima, M. Ishii and K. Tanaka, Eur. J. Org. Chem., 2015, 1092–1099 CrossRef CAS.
- K. Tanaka, Y. Takahashi, T. Suda and M. Hirano, Synlett, 2008, 1724–1728 CrossRef CAS.
- K. M. Oberg, E. E. Lee and T. Rovis, Tetrahedron, 2009, 65, 5056–5061 CrossRef CAS PubMed.
- K. Tanaka, A. Wada and K. Noguchi, Org. Lett., 2006, 8, 907–909 CrossRef CAS PubMed.
- F. Xu, C. Wang, H. Wang, X. Li and B. Wan, Green Chem., 2015, 17, 799–803 RSC.
- G. Nishida, N. Suzuki, K. Noguchi and K. Tanaka, Org. Lett., 2006, 8, 3489–3492 CrossRef CAS PubMed.
- R. T. Yu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 2782–2783 CrossRef CAS PubMed.
- (a) R. T. Yu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 12370–12371 CrossRef CAS PubMed; (b) E. E. Lee and T. Rovis, Org. Lett., 2008, 10, 1231–1234 CrossRef CAS PubMed; (c) R. K. Friedman and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10775–10782 CrossRef PubMed; (d) R. K. Friedman, K. M. Oberg, D. M. Dalton and T. Rovis, Pure Appl. Chem., 2010, 82, 1353 CrossRef CAS PubMed.
- R. T. Yu and T. Rovis, J. Am. Chem. Soc., 2008, 130, 3262–3263 CrossRef CAS PubMed.
- (a) D. M. Dalton, K. M. Oberg, R. T. Yu, E. E. Lee, S. Perreault, M. E. Oinen, M. L. Pease, G. Malik and T. Rovis, J. Am. Chem. Soc., 2009, 131, 15717–15728 CrossRef CAS PubMed; (b) M. E. Oinen, R. T. Yu and T. Rovis, Org. Lett., 2009, 11, 4934–4937 CrossRef CAS PubMed.
- (a) R. T. Yu, E. E. Lee, G. Malik and T. Rovis, Angew. Chem., Int. Ed., 2009, 48, 2379–2382 CrossRef CAS PubMed; (b) D. M. Dalton and T. Rovis, Org. Lett., 2013, 15, 2346–2349 CrossRef CAS PubMed.
- K. Tanaka, Y. Otake, H. Sagae, K. Noguchi and M. Hirano, Angew. Chem., Int. Ed., 2008, 47, 1312–1316 CrossRef CAS PubMed.
- K. Tanaka, Y. Otake, A. Wada, K. Noguchi and M. Hirano, Org. Lett., 2007, 9, 2203–2206 CrossRef CAS PubMed.
- I. Kim, S. W. Roh, D. G. Lee and C. Lee, Org. Lett., 2014, 16, 2482–2485 CrossRef CAS PubMed.
- P. A. Wender, T. M. Pedersen and M. J. C. Scanio, J. Am. Chem. Soc., 2002, 124, 15154–15155 CrossRef CAS PubMed.
- M. M. Montero-Campillo, E. M. Cabaleiro-Lago and J. Rodríguez-Otero, J. Phys. Chem. A, 2008, 112, 9068–9074 CrossRef CAS PubMed.
- J.-J. Feng, T.-Y. Lin, H.-H. Wu and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 15854–15858 CrossRef CAS PubMed.
- J.-J. Feng, T.-Y. Lin, H.-H. Wu and J. Zhang, J. Am. Chem. Soc., 2015, 137, 3787–3790 CrossRef CAS PubMed.
- J.-J. Feng, T.-Y. Lin, C.-Z. Zhu, H. Wang, H.-H. Wu and J. Zhang, J. Am. Chem. Soc., 2016, 138, 2178–2181 CrossRef CAS PubMed.
- T. Saito, K. Sugizaki, T. Otani and T. Suyama, Org. Lett., 2007, 9, 1239–1241 CrossRef CAS PubMed.
- T. Saito, N. Furukawa and T. Otani, Org. Biomol. Chem., 2010, 8, 1126–1132 CAS.
- Z. Zhang, F. Xiao, B. Huang, J. Hu, B. Fu and Z. Zhang, Org. Lett., 2016, 18, 908–911 CrossRef CAS PubMed.
- T. Iwata, F. Inagaki and C. Mukai, Angew. Chem., Int. Ed., 2013, 52, 11138–11142 CrossRef CAS PubMed.
- M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed.
- (a) A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS PubMed; (b) I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS; (c) A. Prades, M. Fernández, S. D. Pike, M. C. Willis and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 8520–8524 CrossRef CAS PubMed.
- P. Lenden, D. A. Entwistle and M. C. Willis, Angew. Chem., Int. Ed., 2011, 50, 10657–10660 CrossRef CAS PubMed.
- (a) S. R. Parsons, J. F. Hooper and M. C. Willis, Org. Lett., 2011, 13, 998–1000 CrossRef CAS PubMed; (b) M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 13280–13283 CrossRef CAS PubMed; (c) J. F. Hooper, S. Seo, F. R. Truscott, J. D. Neuhaus and M. C. Willis, J. Am. Chem. Soc., 2016, 138, 1630–1634 CrossRef CAS PubMed; (d) M. von Delius, C. M. Le and V. M. Dong, J. Am. Chem. Soc., 2012, 134, 15022–15032 CrossRef CAS PubMed.
- (a) J. F. Hooper, A. B. Chaplin, C. González-Rodríguez, A. L. Thompson, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 2906–2909 CrossRef CAS PubMed; (b) J. F. Hooper, R. D. Young, I. Pernik, A. S. Weller and M. C. Willis, Chem. Sci., 2013, 4, 1568–1572 RSC; (c) J. F. Hooper, R. D. Young, A. S. Weller and M. C. Willis, Chem. – Eur. J., 2013, 19, 3125–3130 CrossRef CAS PubMed; (d) J. Niu and M. C. Willis, Org. Chem. Front., 2016, 3, 625–629 RSC.
- (a) J. D. Neuhaus, S. M. Morrow, M. Brunavs and M. C. Willis, Org. Lett., 2016, 18, 1562–1565 CrossRef CAS PubMed; (b) M. K. Majhail, P. M. Ylioja and M. C. Willis, Chem. – Eur. J., 2016 DOI:10.1002/chem.201600311.
- X. W. Du and L. M. Stanley, Org. Lett., 2015, 17, 3276–3279 CrossRef CAS PubMed.
- S. K. Murphy, A. Bruch and V. M. Dong, Angew. Chem., Int. Ed., 2014, 53, 2455–2459 CrossRef CAS PubMed.
- H. D. Bendorf, C. M. Colella, E. C. Dixon, M. Marchetti, A. N. Matukonis, J. D. Musselman and T. A. Tiley, Tetrahedron Lett., 2002, 43, 7031–7034 CrossRef CAS.
- H. D. Bendorf, K. E. Ruhl, A. J. Shurer, J. B. Shaffer, T. O. Duffin, T. L. LaBarte, M. L. Maddock and O. W. Wheeler, Tetrahedron Lett., 2012, 53, 1275–1277 CrossRef CAS.
- M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932–6933 CrossRef CAS PubMed.
- J. S. Arnold, E. T. Mwenda and H. M. Nguyen, Angew. Chem., Int. Ed., 2014, 53, 3688–3692 CrossRef CAS PubMed.
- (a) A. Ghosh and L. M. Stanley, Chem. Commun., 2014, 50, 2765–2768 RSC; (b) X. W. Du, A. Ghosh and L. M. Stanley, Org. Lett., 2014, 16, 4036–4039 CrossRef CAS PubMed.
- (a) Z. Shen, H. A. Khan and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2916–2917 CrossRef CAS PubMed; (b) Z. Shen, P. K. Dornan, H. A. Khan, T. K. Woo and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 1077–1091 CrossRef CAS PubMed; (c) H. A. Khan, K. G. M. Kou and V. M. Dong, Chem. Sci., 2011, 2, 407–410 RSC.
- D. H. T. Phan, B. Kim and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 15608–15609 CrossRef CAS PubMed.
- M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229–4231 CrossRef CAS.
- (a) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95–119 CrossRef CAS; (b) M. Jean, B. Casanova, S. Gnoatto and P. van de Weghe, Org. Biomol. Chem., 2015, 13, 9168–9175 RSC; (c) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2093–2105 RSC; (d) D. Muller and A. Alexakis, Chem. Commun., 2012, 48, 12037–12049 RSC; (e) J. D. Hargrave, J. C. Allen and C. G. Frost, Chem. – Asian J., 2010, 5, 386–396 CrossRef CAS PubMed.
- (a) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed; (b) C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482–4502 CrossRef CAS PubMed; (c) H.-Q. Dong, M.-H. Xu, C.-G. Feng, X.-W. Sun and G.-Q. Lin, Org. Chem. Front., 2015, 2, 73–89 RSC.
- F. Marinelli, G. Abbiati, A. Arcadi, E. Rossi and M. Verdecchia, Synlett, 2006, 3218–3224 CrossRef.
- J. Horn, S. P. Marsden, A. Nelson, D. House and G. G. Weingarten, Org. Lett., 2008, 10, 4117–4120 CrossRef CAS PubMed.
- H. Y. Li, J. Horn, A. Campbell, D. House, A. Nelson and S. P. Marsden, Chem. Commun., 2014, 50, 10222–10224 RSC.
- J. Horn, H. Y. Li, S. P. Marsden, A. Nelson, R. J. Shearer, A. J. Campbell, D. House and G. G. Weingarten, Tetrahedron, 2009, 65, 9002–9007 CrossRef CAS.
- M. Alfonsi, A. Arcadi, M. Chiarini and F. Marinelli, J. Org. Chem., 2007, 72, 9510–9517 CrossRef CAS PubMed.
- A. Arcadi, G. Fabrizi, A. Goggiamani and F. Marinelli, J. Org. Chem., 2015, 80, 6986–6995 CrossRef CAS PubMed.
- J. O. Park and S. W. Youn, Org. Lett., 2010, 12, 2258–2261 CrossRef CAS PubMed.
- J. Panteleev, L. Zhang and M. Lautens, Angew. Chem., Int. Ed., 2011, 50, 9089–9092 CrossRef CAS PubMed.
- L. Zhang, J. Panteleev and M. Lautens, J. Org. Chem., 2014, 79, 12159–12176 CrossRef CAS PubMed.
- G. C. Tsui, J. Tsoung, P. Dougan and M. Lautens, Org. Lett., 2012, 14, 5542–5545 CrossRef CAS PubMed.
- A. A. Friedman, J. Panteleev, J. Tsoung, V. Huynh and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 9755–9758 CrossRef CAS PubMed.
- J. Tsoung, J. Panteleev, M. Tesch and M. Lautens, Org. Lett., 2014, 16, 110–113 CrossRef CAS PubMed.
- L. Zhang, Z. Qureshi, L. Sonaglia and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 13850–13853 CrossRef CAS PubMed.
- J. Ye, A. Limouni, S. Zaichuk and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 3116–3120 CrossRef CAS PubMed.
- L. Zhang, L. Sonaglia, J. Stacey and M. Lautens, Org. Lett., 2013, 15, 2128–2131 CrossRef CAS PubMed.
- T. Miura, Y. Takahashi and M. Murakami, Org. Lett., 2007, 9, 5075–5077 CrossRef CAS PubMed.
- S. W. Youn, J.-H. Song and D.-I. Jung, J. Org. Chem., 2008, 73, 5658–5661 CrossRef CAS PubMed.
- X. Fang, C. Li and X. Tong, Chem. Commun., 2009, 5311–5313 RSC.
- R. Shintani, T. Yamagami and T. Hayashi, Org. Lett., 2006, 8, 4799–4801 CrossRef CAS PubMed.
- Y. J. Jang, H. Yoon and M. Lautens, Org. Lett., 2015, 17, 3895–3897 CrossRef CAS PubMed.
- J. Keilitz, S. G. Newman and M. Lautens, Org. Lett., 2013, 15, 1148–1151 CrossRef CAS PubMed.
- Y. Li and M.-H. Xu, Org. Lett., 2014, 16, 2712–2715 CrossRef CAS PubMed.
- F. Serpier, J.-L. Brayer, B. Folléas and S. Darses, Org. Lett., 2015, 17, 5496–5499 CrossRef CAS PubMed.
- F. Serpier, B. Flamme, J.-L. Brayer, B. Folléas and S. Darses, Org. Lett., 2015, 17, 1720–1723 CrossRef CAS PubMed.
- Z. Cui, H. J. Yu, R. F. Yang, W. Y. Gao, C. G. Feng and G. Q. Lin, J. Am. Chem. Soc., 2011, 133, 12394–12397 CrossRef CAS PubMed.
- Z.-Q. Wang, C.-G. Feng, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2007, 129, 5336–5337 CrossRef CAS PubMed.
- C.-H. Xing, Y.-X. Liao, P. He and Q.-S. Hu, Chem. Commun., 2010, 46, 3010–3012 RSC.
- M. Fujioka, T. Morimoto, T. Tsumagari, H. Tanimoto, Y. Nishiyama and K. Kakiuchi, J. Org. Chem., 2012, 77, 2911–2923 CrossRef CAS PubMed.
- G. C. Tsui, Y. Singjunla and M. Lautens, Synthesis, 2012, 1359–1364 CAS.
- X.-F. Wu and H. Neumann, ChemCatChem, 2012, 4, 447–458 CrossRef CAS.
- A. Börner and R. Franke, Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2016 Search PubMed.
- B. P. Bondžić, J. Mol. Catal. A: Chem., 2015, 408, 310–334 CrossRef.
- (a) P. Köhling, A. M. Schmidt and P. Eilbracht, Org. Lett., 2003, 5, 3213–3216 CrossRef PubMed; (b) M. Ahmed, R. Jackstell, A. M. Seayad, H. Klein and M. Beller, Tetrahedron Lett., 2004, 45, 869–873 CrossRef CAS; (c) A. M. Schmidt and P. Eilbracht, Org. Biomol. Chem., 2005, 3, 2333–2343 RSC; (d) A. M. Schmidt and P. Eilbracht, J. Org. Chem., 2005, 70, 5528–5535 CrossRef CAS PubMed; (e) P. Linnepe, A. M. Schmidt and P. Eilbracht, Org. Biomol. Chem., 2006, 4, 302–313 RSC; (f) B. P. Bondzic, A. Farwick, J. Liebich and P. Eilbracht, Org. Biomol. Chem., 2008, 6, 3723–3731 RSC; (g) B. P. Bondzić and P. Eilbracht, Org. Lett., 2008, 10, 3433–3436 CrossRef PubMed; (h) M. Mentel, A. M. Schmidt, M. Gorray, P. Eilbracht and R. Breinbauer, Angew. Chem., Int. Ed., 2009, 48, 5841–5844 CrossRef CAS PubMed.
- B. P. Bondzic and P. Eilbracht, Org. Biomol. Chem., 2008, 6, 4059–4063 CAS.
- A. Farwick and G. Helmchen, Adv. Synth. Catal., 2010, 352, 1023–1032 CrossRef CAS.
- Q. Ruan, L. Zhou and B. Breit, Catal. Commun., 2014, 53, 87–90 CrossRef CAS.
- (a) T. Spangenberg, B. Breit and A. Mann, Org. Lett., 2009, 11, 261–264 CrossRef CAS PubMed; (b) P. Dübon, A. Farwick and G. Helmchen, Synlett, 2009, 1413–1416 Search PubMed.
- R. W. Bates, K. Sivarajan and B. F. Straub, J. Org. Chem., 2011, 76, 6844–6848 CrossRef CAS PubMed.
- W.-H. Chiou, N. Mizutani and I. Ojima, J. Org. Chem., 2007, 72, 1871–1882 CrossRef CAS PubMed.
- W.-H. Chiou, Y.-H. Lin, G.-T. Chen, Y.-K. Gao, Y.-C. Tseng, C.-L. Kao and J.-C. Tsai, Chem. Commun., 2011, 47, 3562–3564 RSC.
- J. Dheur, M. Sauthier, Y. Castanet and A. Mortreux, Adv. Synth. Catal., 2010, 352, 557–561 CrossRef CAS.
- H. Chochois, M. Sauthier, E. Maerten, Y. Castanet and A. Mortreux, Tetrahedron, 2006, 62, 11740–11746 CrossRef CAS.
- O. Aksin, N. Dege, L. Artok, H. Turkmen and B. Cetinkaya, Chem. Commun., 2006, 3187–3189 RSC.
- K. Fuji, T. Morimoto, K. Tsutsumi and K. Kakiuchi, Chem. Commun., 2005, 3295–3297 RSC.
- D.-Y. Zhou, T. Koike, S. Suetsugu, K. Onitsuka and S. Takahashi, Inorg. Chim. Acta, 2004, 357, 3057–3063 CrossRef CAS.
- K. Khumtaveeporn and H. Alper, J. Am. Chem. Soc., 1994, 116, 5662–5666 CrossRef CAS.
- (a) B. G. Van den Hoven and H. Alper, J. Am. Chem. Soc., 2001, 123, 1017–1022 CrossRef CAS PubMed; (b) C. Dong and H. Alper, Org. Lett., 2004, 6, 3489–3492 CrossRef CAS PubMed.
- H. Alper, F. Urso and D. J. H. Smith, J. Am. Chem. Soc., 1983, 105, 6737–6738 CrossRef CAS.
- S. Calet, F. Urso and H. Alper, J. Am. Chem. Soc., 1989, 111, 931–934 CrossRef CAS.
- S.-M. Lu and H. Alper, J. Org. Chem., 2004, 69, 3558–3561 CrossRef CAS PubMed.
- D. Ardura, R. López and T. L. Sordo, J. Org. Chem., 2006, 71, 7315–7321 CrossRef CAS PubMed.
- K. Okuro, J. Gurnham and H. Alper, J. Org. Chem., 2011, 76, 4715–4720 CrossRef CAS PubMed.
- K. Okuro, J. Gurnham and H. Alper, Tetrahedron Lett., 2012, 53, 620–622 CrossRef CAS.
- J. U. Rhee and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 10674–10675 CrossRef CAS PubMed.
- M. Isoda, K. Sato, M. Funakoshi, K. Omura, A. Tarui, M. Omote and A. Ando, J. Org. Chem., 2015, 80, 8398–8405 CrossRef CAS PubMed.
- M. Arambasic, J. F. Hooper and M. C. Willis, Org. Lett., 2013, 15, 5162–5165 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645–3651 CrossRef CAS PubMed.
- K. Parthasarathy, M. Jeganmohan and C.-H. Cheng, Org. Lett., 2008, 10, 325–328 CrossRef CAS PubMed.
- R. K. Thalji, K. A. Ahrendt, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2001, 123, 9692–9693 CrossRef CAS PubMed.
- I. Nakamura, M. Okamoto, Y. Sato and M. Terada, Angew. Chem., Int. Ed., 2012, 51, 10816–10819 CrossRef CAS PubMed.
- B.-L. Lu and M. Shi, Angew. Chem., Int. Ed., 2011, 50, 12027–12031 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |