
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An asymmetric approach to bicyclo[2.2.1]heptane-1-carboxylates via a formal [4 + 2] cycloaddition reaction enabled by organocatalysis†
Jian-Guo
Fu‡
,
Yi-Fan
Shan‡
,
Wang-Bin
Sun
,
Guo-Qiang
Lin
and
Bing-Feng
Sun
*
CAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai 200032, China. E-mail: bfsun@sioc.ac.cn
First published on 19th May 2016
Abstract
An organocatalytic formal [4 + 2] cycloaddition reaction has been realized that permits rapid access to a wide range of bicyclo[2.2.1]heptane-1-carboxylates in a highly enantioselective manner from simple starting materials under mild and operationally simple conditions.
A bicyclo[2.2.1]heptane scaffold is a privileged molecular structure embedded in numerous compounds with various functions (Fig. 1). Camphor,1 sordarins,2 α-santalol and β-santalol3,4 are bioactive natural products that contain this structural moiety. In addition, bicyclo[2.2.1]heptane is featured by drug candidates such as LMV-6015 and AMG 221.6 Moreover, the bicyclo[2.2.1]heptane scaffold provides the basis for asymmetric synthesis and catalysis. Bornanesultam is a well-known chiral auxiliary,7 while dibenzyldiene8 and diphonane9 are effective chiral ligands for transition-metal catalysis. The development of enantioselective approaches to functionalized bicyclo[2.2.1]heptanes is critical for target-oriented and diversity-oriented syntheses of related biologically significant molecules, and therefore it is highly desirable for relevant drug discovery.
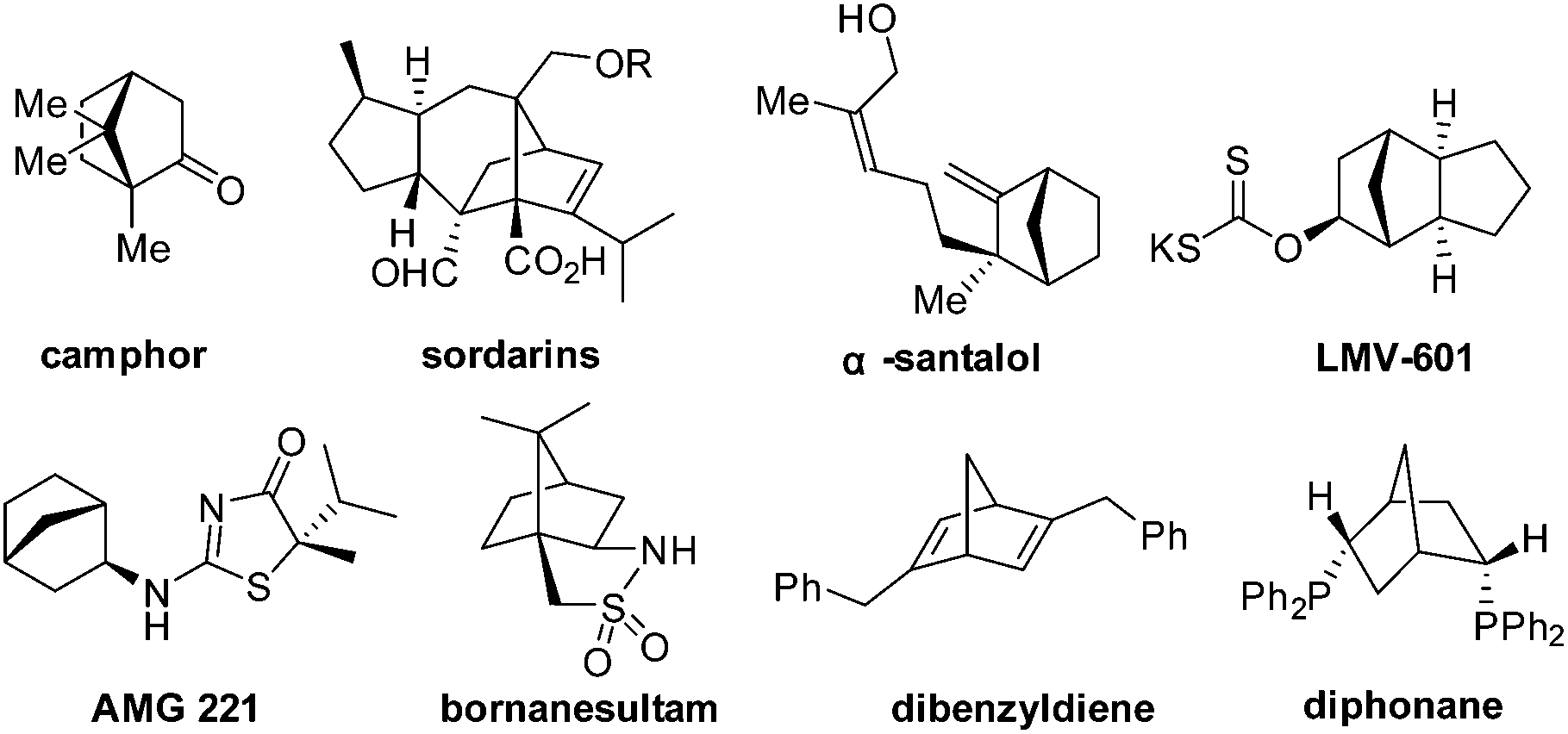 | ||
| Fig. 1 Selected compounds containing a [2.2.1] bicyclic core. | ||
From the perspective of molecular architecture, the molecules depicted in Fig. 1 may be classified into two main groups contingent on whether or not a bridgehead carbon is present. In this regard, camphor, sordarins, and bornanesultam belong to the same group. We are interested in the development of a synthetic approach that would allow direct access to bicyclo[2.2.1]heptanes with a functionalized bridgehead, particularly in view of the absence of such a method so far.10 We envisaged an enantioselective approach to bicyclo[2.2.1]heptane-1-carboxylates via a formal [4 + 2] cycloaddition reaction, with an understanding of the carboxylate group being a versatile function amenable to various transformations (Scheme 1). Mechanistically distinct from the preceding methods, which involve enamine catalysis10a–c or Brønsted acid catalysis,10d this reaction would be effected through a hydrogen bond catalysis.11,12 To this end, a chiral tertiary amine would be employed as the catalyst. Herein, we present the asymmetric approach to bicyclo[2.2.1]heptane-1-carboxylates by the reaction of α′-ethoxycarbonyl cyclopentenones with nitroolefins (Scheme 1).
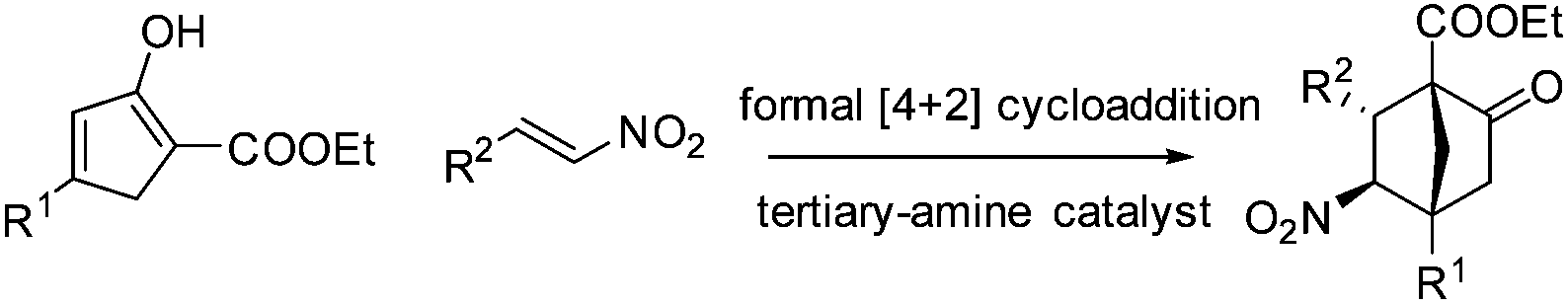 | ||
| Scheme 1 Formal [4 + 2] cycloaddition reaction. | ||
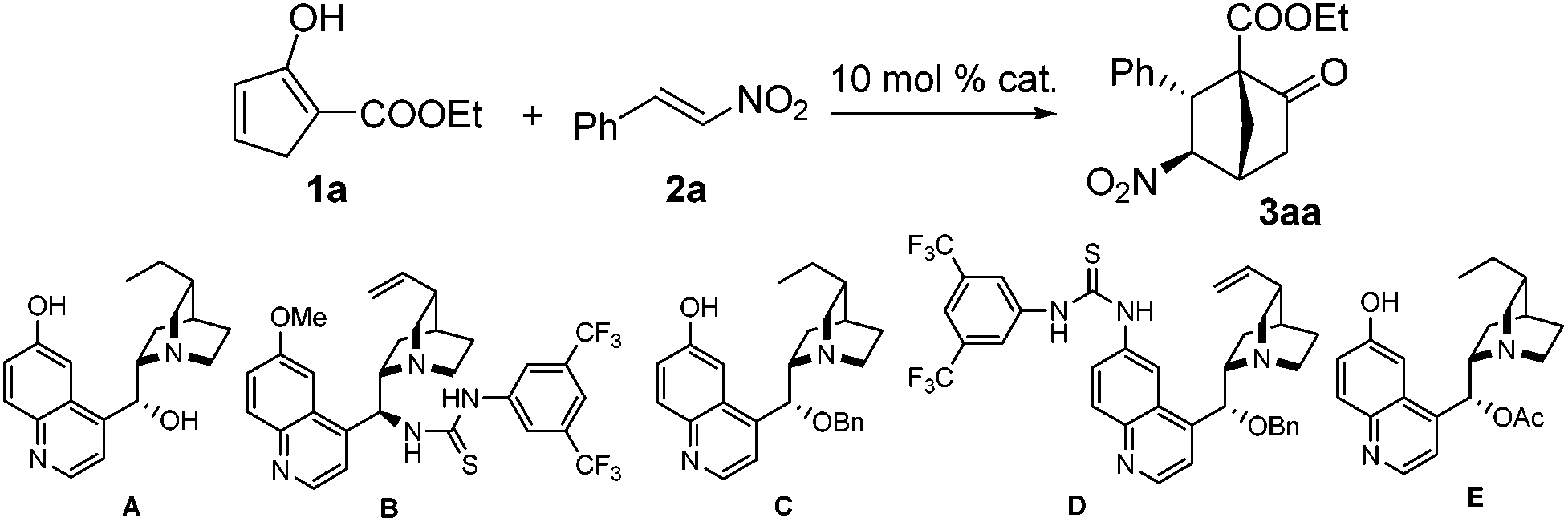
Our research commenced with investigating the reaction between 1a and 2a. When 10 mol% A was employed as the catalyst, a stepwise incomplete reaction was observed that produced a mixture containing both the Diels–Alder product and the Michael addition product.12 This problem was solved successfully by the addition of DBU after consumption of 1a to promote the second Michael addition reaction.13 Various reaction parameters were evaluated and selected results are listed in Table 1. The reaction of 1a and 2a catalyzed by 10 mol% A at room temperature produced a 2.3/1 diastereomeric mixture in 82% yield, with the major diastereomer 3a bearing an enantiopurity of 88% ee (entry 1). Lower temperatures of 0 °C and −20 °C appeared to give slightly higher diastereoselectivities and enantioselectivities (entries 2 and 3). The same reaction with catalyst B gave the best result at −40 °C, with 82% yield, 7.1/1 dr and 87% ee being achieved, however, further decreasing the temperature to −60 °C resulted in eroded selectivities (entries 4–7). The same was observed for catalyst C, which achieved the highest selectivities at −40 °C with the product being obtained in 84% yield with 3.2/1 dr and 94% ee (entries 8 and 9). Therefore, reactions with other catalysts were further tested at −40 °C. At this temperature, while the reaction with catalyst D was considerably less stereoselective, the enantioselectivity obtained with catalyst E was similar to that with C (entries 10 and 11). In light of these results, C was determined as the catalyst of choice and the solvent effect was then evaluated with this catalyst. The reactions in toluene, CH3CN, and THF, respectively, were similarly enantioselective as compared to that in dichloromethane. However, the diastereoselectivities and yields with these solvents were significantly lower (entries 12–14).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solv. | Temp. | Yld.b (%) | drc | eed (%) |
| a 1a (0.20 mmol), 2a (0.24 mmol), 10 mol% catalyst, and 1 mL of solvent were employed. After 1a was consumed as indicated by a TLC test, DBU (0.10 mmol) was added to the reaction mixture. b Isolated yield. c Determined by 1H NMR analysis of the crude product. d Determined by chiral HPLC analysis. | ||||||
| 1 | A | DCM | R.T. | 82 | 2.3/1 | 88 |
| 2 | A | DCM | 0 | 83 | 2.4/1 | 90 |
| 3 | A | DCM | −20 | 81 | 2.8/1 | 91 |
| 4 | B | DCM | R.T. | 86 | 3.2/1 | 81 |
| 5 | B | DCM | −20 | 80 | 5.6/1 | 84 |
| 6 | B | DCM | −40 | 82 | 7.1/1 | 87 |
| 7 | B | DCM | −60 | 69 | 3.7/1 | 69 |
| 8 | C | DCM | −40 | 84 | 3.2/1 | 94 |
| 9 | C | DCM | −60 | 73 | 2.5/1 | 75 |
| 10 | D | DCM | −40 | 77 | 2.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
−81 |
| 11 | E | DCM | −40 | 77 | 3.3/1 | 94 |
| 12 | C | PhMe | −40 | 34 | 3.5/1 | 93 |
| 13 | C | MeCN | −40 | 61 | 4.0/1 | 92 |
| 14 | C | THF | −40 | 41 | 2.5/1 | 95 |
With the optimal conditions in hand, we moved on to identify the substrate scope. β-Arylnitroalkenes 2b–2l were first examined as the electrophile (Table 2). Generally good yield, moderate diastereoselectivity and excellent enantioselectivity could be obtained for various nitrostyrenes bearing either electron donating or electron withdrawing groups (entries 2–10). The electron withdrawing groups appeared to be more favorable for the reaction to achieve higher yields and stereoselectivities (entries 2–4 vs. entries 5–10). The steric effect of the substituent did not play a significant role (entry 3 vs. entry 4, entry 5 vs. entry 6). For nitroolefins 2k and 2l bearing other aromatic substituents, the diastereoselectivity as well as the enantioselectivity dropped considerably (entries 11 and 12). Based on these results, it may be concluded for β-arylnitroalkene substrates that more electron-deficient nitroolefins are better substrates. Considering that more electron-deficient nitroolefins are more reactive electrophiles, it became a logical deduction for us that more reactive nitroolefins are better substrates for this reaction.
|
|
||||
|---|---|---|---|---|
| Entrya | R | Yld.b (%) | drc | eed (%) |
| a 1a (0.20 mmol), 2 (0.24 mmol), C (10 mol%), and 1 mL of DCM were employed. After 1a was consumed as indicated by a TLC test, DBU (0.10 mmol) was added to the reaction mixture. b Isolated yield. c Determined by 1H NMR analysis of the crude product. d Determined by chiral HPLC analysis. | ||||
| 1 | Ph (2a) | 84 | 3.2/1 | 94 |
| 2 | 4-MeC6H4 (2b) | 78 | 3.1/1 | 95 |
| 3 | 4-MeOC6H4 (2c) | 74 | 2.0/1 | 92 |
| 4 | 2-MeOC6H4 (2d) | 75 | 2.6/1 | 94 |
| 5 | 4-BrC6H4 (2e) | 84 | 3.5/1 | 97 |
| 6 | 2-BrC6H4 (2f) | 80 | 4.0/1 | 98 |
| 7 | 3-ClC6H4 (2g) | 84 | 4.7/1 | 98 |
| 8 | 4-FC6H4 (2h) | 86 | 2.8/1 | 96 |
| 9 | 3-NO2C6H4 (2i) | 82 | 4.2/1 | 99 |
| 10 | 4-CF3C6H4 (2j) | 80 | 4.2/1 | 97 |
| 11 | 2-Furyl (2k) | 73 | 1.3/1 | 92 |
| 12 | 2-Naphthyl (2l) | 77 | 1.9/1 | 81 |

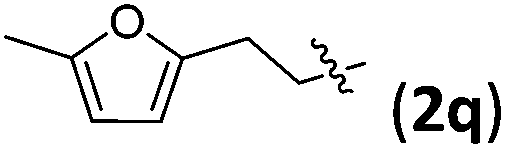
Accordingly, we then studied the reaction of 1a with β-alkylnitroalkenes. As summarized in Table 3, the various sterically undemanding β-alkylnitroalkenes are excellent substrates for this formal cycloaddition reaction. Nitroolefins with a saturated hydrocarbon chain provided the [2.2.1] bicyclic products in excellent yield with ca. 20/1 dr and extremely high ee (entries 1–3). An aromatic ring at the distal end of the chain was tolerable (entries 4–6). Importantly, nitroolefins with functional groups were excellent substrates for this reaction, providing the [2.2.1] products amenable for further elaborations (entries 7–10). Interestingly, 2w bearing a cyclopropyl group was also an effective substrate (entry 11). As the bulkiness of the substituent increased, the reactivity as well as the enantioselectivity dropped considerably (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | R | Yld.b (%) | drc | eed (%) |
| a 1a (0.20 mmol), 2 (0.24 mmol), C (10 mol%), and 1 mL of DCM were employed. After 1a was consumed as indicated by a TLC test, DBU (0.10 mmol) was added to the reaction mixture. b Isolated yield. c Determined by 1H NMR analysis of the crude product. d Determined by chiral HPLC analysis. | ||||
| 1 | n-Pr (2m) | 83 | 18/1 | 98 |
| 2 | n-Bu (2n) | 90 | 17/1 | 99 |
| 3 | i-Bu (2o) | 86 | >20/1 | 98 |
| 4 |
|
88 | >20/1 | 99 |
| 5 |
|
84 | >20/1 | 99 |
| 6 |
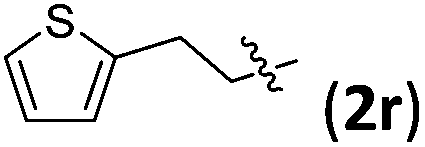
|
90 | 20/1 | 99 |
| 7 |
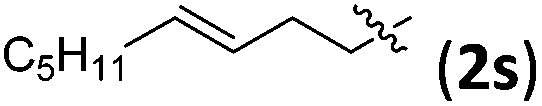
|
90 | >20/1 | 99 |
| 8 |

|
83 | >20/1 | 98 |
| 9 |
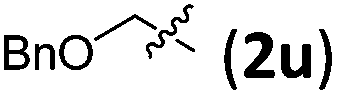
|
88 | 20/1 | 98 |
| 10 |

|
89 | >20/1 | 99 |
| 11 |
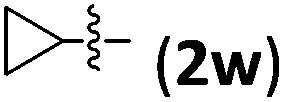
|
73 | 10/1 | 95 |
| 12 | i-Pr (2x) | 69 | 10/1 | 46 |
We further examined the reaction between 1b and various 2-alkyl-1-nitroethenes (Table 4). With 1b being employed as the nucleophile, products with the [2.2.1] bicyclic core decorated with two all-carbon quaternary stereocenters were expected to be garnered. To our delight, the reactions between 1b and 2 underwent smoothly and furnished the formal cycloaddition products in good yields with satisfactory selectivities (entries 1–7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Prod. | Yld.b (%) | drc | eed (%) |
| a 1b (0.20 mmol), 2 (0.24 mmol), C (10 mol%), and 1 mL of DCM were employed. After 1a was consumed as indicated by a TLC test, DBU (0.10 mmol) was added to the reaction mixture. b Isolated yield. c Determined by 1H NMR analysis of the crude product. d Determined by chiral HPLC analysis. | |||||
| 1 | i-Bu (2o) | 3ob | 78 | 6/1 | 84 |
| 2 |
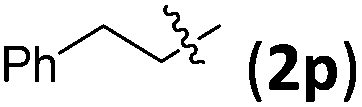
|
3pb | 73 | 6.5/1 | 84 |
| 3 |
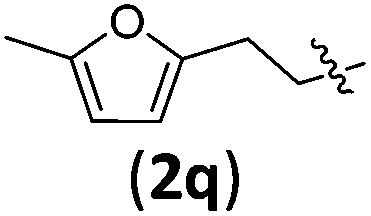
|
3qb | 80 | 13/1 | 95 |
| 4 |
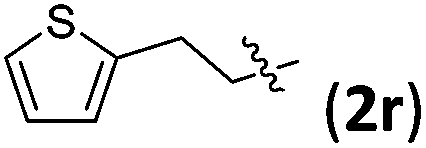
|
3rb | 74 | 8/1 | 88 |
| 5 |
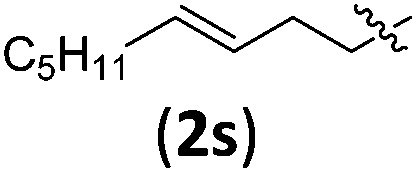
|
3sb | 74 | 6.3/1 | 83 |
| 6 |
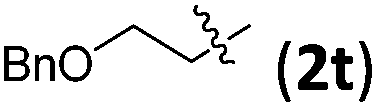
|
3tb | 76 | 8/1 | 75 |
| 7 |

|
3vb | 79 | >20/1 | 92 |
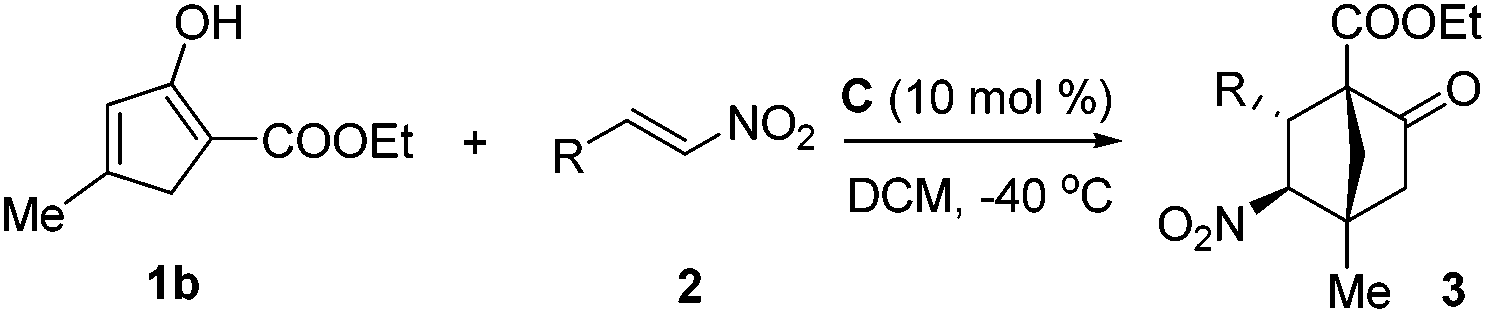
The structure as well as the absolute configuration of 3rb was established unambiguously through X-ray crystallographic analysis (Fig. 2).14
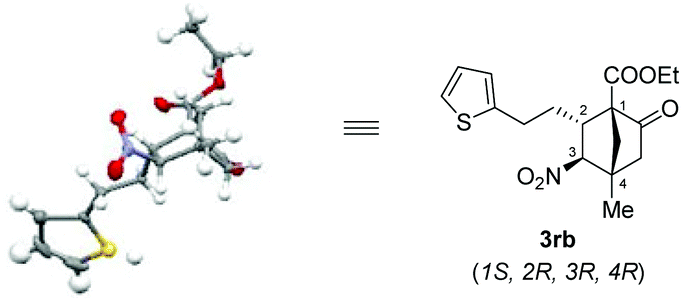 | ||
| Fig. 2 X-ray structure of 3rb (ORTEP drawing with thermal ellipsoids at 30% probability level). | ||
Based on the preceding results and in the light of our previous proposal,13 the stereochemical outcome of the tandem reaction could be rationalized, as exemplified by the reaction of 1b with 2r (Scheme 2). In the first step, 1b may form a complex with Cvia hydrogen-bonding interactions, thereby discriminating between the two faces of the enol. Directed by the hydrogen bonding interactions developing between the nitro and the quinolinol groups, the nitroolefin approaches the enol selectively from the α-face with the substituents of the two bond-forming carbons positioned in a staggered arrangement (4), leading to the formation of the intermediate compound 5. In the second step where DBU is used as the base, the open transition state 6 should be operative to minimize the steric repulsion between the nitro and the alkyl groups, delivering 3rb as the major product.
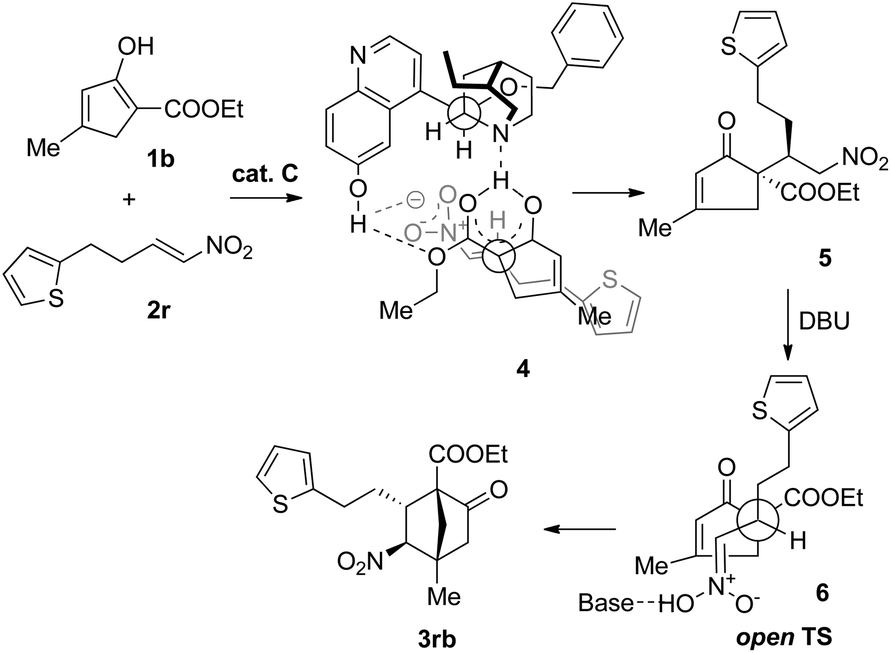 | ||
| Scheme 2 Proposed mechanism of the reaction. | ||
Eventually, to demonstrate the efficiency and synthetic utility of this newly developed formal [4 + 2] cycloaddition reaction, a gram-scale reaction was performed with 1a and 2p (Scheme 3). Thus, the reaction with 1.28 g of 1a (1.0 equiv.) and 1.79 g of 2p (1.2 equiv.) catalyzed by C (5 mol%) and DBU (0.5 equiv.) produced 2.55 gram of 3p in a yield of 91% with >20/1 dr and 99% ee.
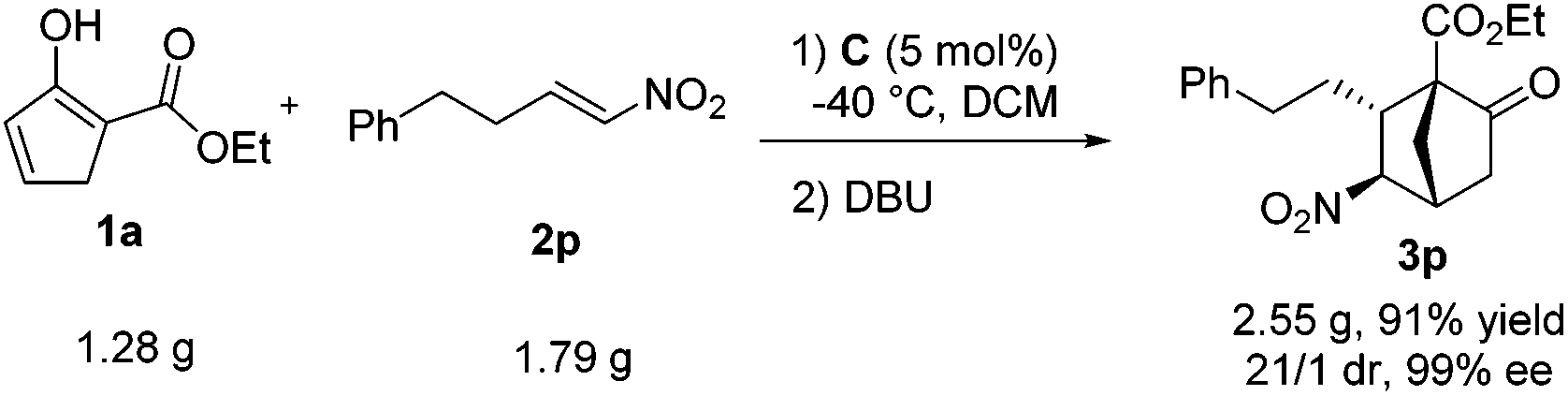 | ||
| Scheme 3 A gram-scale synthesis of 3pvia the formal [4 + 2] cycloaddition reaction of 1a and 2p. | ||
Conclusions
In summary, an organocatalytic formal [4 + 2] cycloaddition reaction has been realized that permits rapid access to a wide range of functionalized [2.2.1] bicyclic heptanes in a highly enantioselective manner from simple starting materials. The reaction features metal free, mild, and operationally simple conditions, providing synthetically useful bicyclo[2.2.1]heptane-1-carboxylates in good yields with excellent enantioselectivity. Importantly, this method is amenable to large scale preparation, thus facilitating relevant drug discovery and pharmaceutical activities. Efforts aiming at the application of this newly developed method are pursued in our laboratory and will be reported in due course.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21172246, 21290180, 21472210), and the Youth Innovation Promotion Association CAS.Notes and references
- K. C. Nicolaou and T. Montagnon, Molecules That Changed The World, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, p. 29 Search PubMed.
- H. Liang, Beilstein J. Org. Chem., 2008, 4, 31 CrossRef PubMed.
- (a) T. Satou, Y. Ogawa and K. Koike, Phytother. Res., 2015, 29, 1246 CrossRef CAS PubMed; (b) B. Lee, J. Bohmann, T. Reeves, C. Levenson and A. L. Risinger, J. Nat. Prod., 2015, 78, 1357 CrossRef CAS PubMed.
- Y. Matsuo, H. Sakagami and Y. Mimaki, Chem. Pharm. Bull., 2014, 62, 1192 CrossRef CAS PubMed.
- R. M. Adibhatla, J. F. Hatcher and A. Gusain, Neurochem. Res., 2012, 37, 671 CrossRef CAS PubMed.
- A. Li, C. C. Yuan, D. Chow, M. Chen, M. G. Emery, C. Hale, X. Zhang, R. Subramanian, D. J. St. Jean, R. Komorowski, M. Véniant, M. Wang and C. Fotsch, ACS Med. Chem. Lett., 2011, 2, 824 CrossRef CAS PubMed.
- (a) W. Oppolzer, Pure Appl. Chem., 1990, 62, 1241 CAS; (b) W. Oppolzer, Tetrahedron, 1987, 43, 1969 CrossRef CAS; (c) B. H. Kim and D. P. Curran, Tetrahedron, 1993, 49, 293 CrossRef CAS.
- T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508 CrossRef CAS PubMed.
- K. Vandyck, B. Matthys, M. Willen, K. Robeyns, L. V. Meervelt and J. V. D. Eycken, Org. Lett., 2006, 8, 363 CrossRef CAS PubMed.
- (a) H. Sundén, R. Rios, Y. M. Xu, L. Eriksson and A. Córdova, Adv. Synth. Catal., 2007, 349, 2549 CrossRef; (b) X. Feng, Z. Zhou, R. Zhou, Q. Q. Zhou, L. Dong and Y. C. Chen, J. Am. Chem. Soc., 2012, 134, 19942 CrossRef CAS PubMed; (c) R. Mose, M. E. Jensen, G. Preegel and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 13630 CrossRef CAS PubMed; (d) M. Hatano, Y. Goto, A. Izumiseki, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2015, 137, 13472 CrossRef CAS PubMed; (e) M. Rueping, A. Kuenkel and R. Fröhlich, Chem. – Eur. J., 2010, 16, 4173 CrossRef CAS PubMed; (f) B. Tan, Y. Lu, X. Zeng, P. J. Chua and G. Zhong, Org. Lett., 2010, 12, 2682 CrossRef CAS PubMed; (g) M. Tsakos, M. R. J. Elsegood and C. G. Kokotos, Chem. Commun., 2013, 49, 2219 RSC.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed.
- For selected examples of catalytic enantioselective conjugate addition reactions, see: (a) M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 5672 CrossRef CAS PubMed; (b) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed; (c) H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906 CrossRef CAS PubMed; (d) H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 105 CrossRef CAS PubMed; (e) Y. Chi, N. A. Kopf and S. H. Gellman, J. Am. Chem. Soc., 2008, 130, 5608 CrossRef CAS PubMed.
- Y.-Z. Li, J. Wang, W.-B. Sun, Y.-F. Shan, B.-F. Sun, G.-Q. Lin and J.-P. Zou, Org. Chem. Front., 2015, 2, 274 RSC and references therein.
- CCDC 1474474 (3rb) contains the supplementary crystallographic data for this paper.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1474474. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob00814c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |