
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C2-Alkenylation of N-heteroaromatic compounds via Brønsted acid catalysis†
Giacomo E. M.
Crisenza
,
Elizabeth M.
Dauncey
and
John F.
Bower
*
School of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: john.bower@bris.ac.uk
First published on 3rd May 2016
Abstract
Substituted heteroaromatic compounds, especially those based on pyridine, hold a privileged position within drug discovery and medicinal chemistry. However, functionalisation of the C2 position of 6-membered heteroarenes is challenging because of (a) the difficulties of installing a halogen at this site and (b) the instability of C2 heteroaryl-metal reagents. Here we show that C2-alkenylated heteroaromatics can be accessed by simple Brønsted acid catalysed union of diverse heteroarene N-oxides with alkenes. The approach is notable because (a) it is operationally simple, (b) the Brønsted acid catalyst is cheap, non-toxic and sustainable, (c) the N-oxide activator disappears during the reaction, and (d) water is the sole stoichiometric byproduct of the process. The new protocol offers orthogonal functional group tolerance to metal-catalysed methods and can be integrated easily into synthetic sequences to provide polyfunctionalised targets. In broader terms, this study demonstrates how classical organic reactivity can still be used to provide solutions to contemporary synthetic challenges that might otherwise be approached using transition metal catalysis.
Introduction
Nitrogen heterocycles are core motifs in the majority of small molecule pharmaceuticals and drug candidates. Within this context, substituted pyridine based systems, including quinolines and isoquinolines, are by far the most common.1 Consequently, modular C–C bond forming strategies for the functionalisation of 6-membered heteroarenes are highly prized. The low nucleophilicity of these ring systems means that electrophilic aromatic substitution approaches are not suitable, and so predominant methodologies rely on metal-catalysed cross coupling (Suzuki, Stille, Heck etc.). Within the context of C2-alkenylated derivatives, two complementary strategies therefore emerge (Scheme 1A). The first involves the coupling of a C2-halogenated derivative with either a vinyl metal reagent2 or an alkene.3 The former approach provides greater control, whereas the latter option is more direct and atom economical, however, both options often suffer from modest yields.2,3 The second strategy relies on an inverse arrangement wherein a C2-metalated heteroarene, itself often prepared from a C2-halogenated precursor, is cross-coupled with an appropriated vinyl halide partner.4 This approach is hampered by the well-established propensity of the heteroaryl metal reagent to undergo competitive proto-demetalation during cross-coupling.4 For both approaches, the C2-halogenated precursor usually cannot be prepared directly from the parent heteroarene, with the most general approaches involving a two-step sequence of oxidation to the N-oxide and halogenation.5,6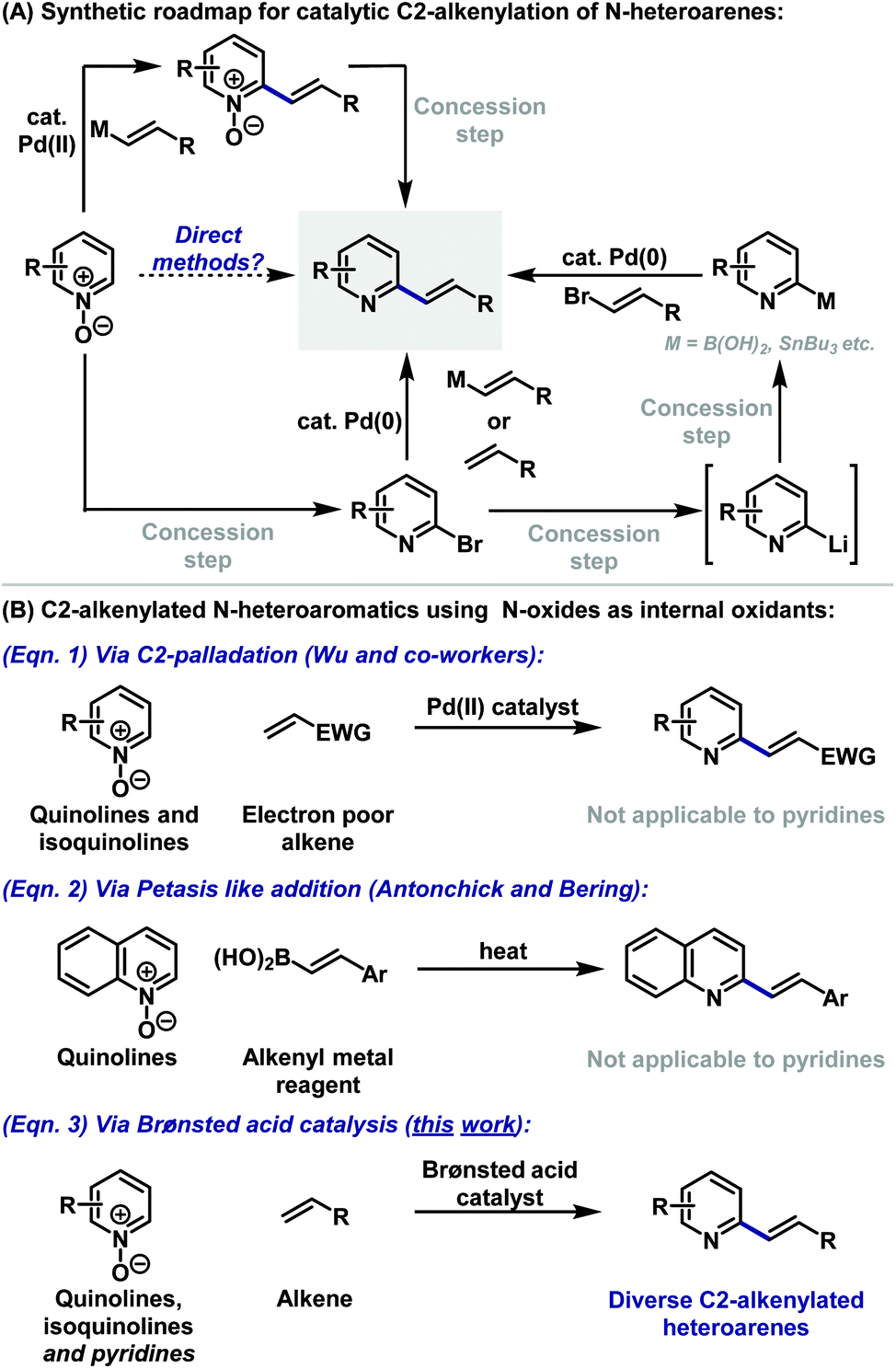 | ||
| Scheme 1 | ||
To circumvent the inefficiencies and atom/step economy issues outlined above, efforts have focussed on the use of N-oxides as heteroaryl metal surrogates.7,8 This functionality renders the heteroarene π-system electron-rich, such that selective C2 metalation (especially palladation) can be exploited.7 However, the requirement to remove the N-oxide activator in a subsequent reduction step detracts from the utility of this approach.7,9 In an ideal scenario, the N-oxide would serve as both an activator and internal oxidant in the C–C bond forming process, such that the target heteroarene is generated directly. There are limited examples where this has been achieved.10 In the context of this study, the most relevant work is that of Wu, who showed that C2-alkenylation of sparsely functionalised quinoline and isoquinoline N-oxides with electron deficient olefins can be achieved under Pd(II)-catalysed conditions (Scheme 1B, eqn (1)); the protocol was not applicable to pyridines.10a Additionally, Antonchick and Bering have developed a Petasis-like approach to the C2-alkenylation of quinolines (Scheme 1B, eqn (2)).11 The protocol was not applied to other classes of heteroarene and requires prefunctionalisation of the alkene partner. Here we demonstrate a complementary approach that uses a simple Brønsted acid catalyst (TsOH) for the coupling of heteroarene N-oxides with alkenes to provide C2-alkenylated products, including pyridines, directly.12 Water is the sole stoichiometric byproduct, the protocol is operationally simple, avoiding the use of toxic and scarce precious metal catalysts, and the method offers orthogonal functional group compatibility versus existing approaches.
Results and discussion
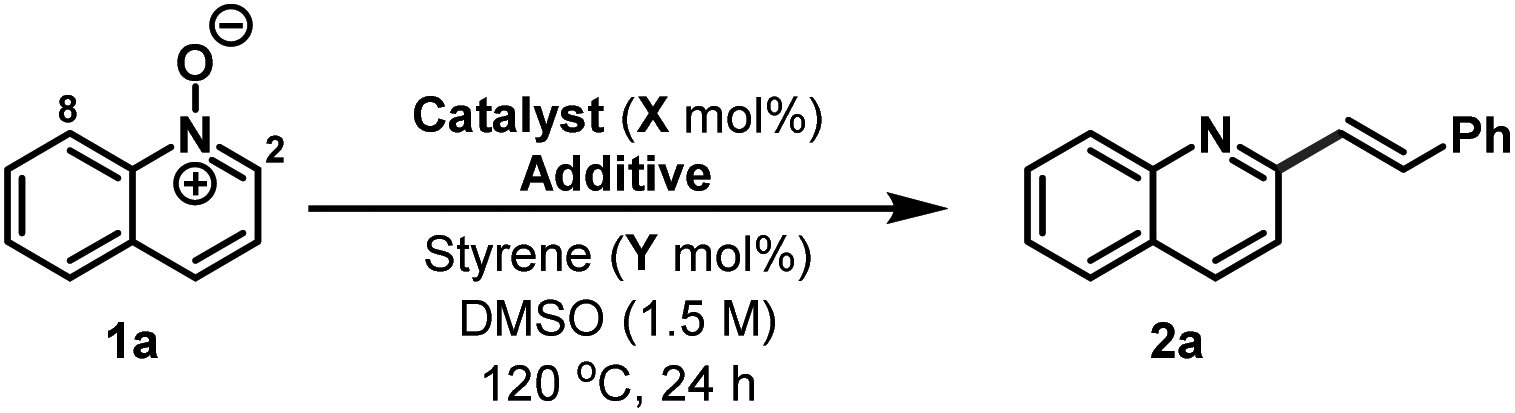
Our studies were initiated by observations made during attempts to promote iridium-catalysed ([IrCp*Cl2]2/AgPF6) C8-alkenylation of quinoline N-oxide 1a with styrene in DMSO.13 Instead of the target transformation, C2-alkenylated derivative 2a was formed in quantitative yield (Table 1, entry 1). Mindful of the studies of Wu and co-workers,10a we conducted a series of deuterium exchange experiments, which indicated that metalation occurred at both C8 and C2. However, further control experiments revealed that the iridium complex was not necessary for C–C bond formation and adduct 2a could be isolated in 71% yield using solely AgPF6 as catalyst (entry 2). Based on this, a range of Lewis and Brønsted acids were evaluated in place of AgPF6 (entries 3–9), leading to the observation that 5 mol% TsOH could promote the transformation in 81% yield (entry 9). Higher loadings of TsOH were detrimental, presumably due to competitive acid catalysed polymerisation of styrene (entry 10). However, further refinement was achieved by using a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of DMSO:water as solvent (entry 11). Under these conditions, the loading of styrene could be decreased to 300 mol% and 2a was generated in 82% yield (entry 12). In the absence of acid catalyst, 2a was formed in only 25% yield (entry 14), and, additionally, no product was observed when both water and the acid catalyst were omitted, with 1a being recovered intact (entry 15). The reaction can be run under air, using reagent grade DMSO, and efficiency is maintained (entry 13), however, lower reaction temperatures are not effective.
:1 ratio of DMSO:water as solvent (entry 11). Under these conditions, the loading of styrene could be decreased to 300 mol% and 2a was generated in 82% yield (entry 12). In the absence of acid catalyst, 2a was formed in only 25% yield (entry 14), and, additionally, no product was observed when both water and the acid catalyst were omitted, with 1a being recovered intact (entry 15). The reaction can be run under air, using reagent grade DMSO, and efficiency is maintained (entry 13), however, lower reaction temperatures are not effective.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | X | Additive | Y | Yield of 2a |
|
a 50:1 (v/v) mixture with DMSO.
b Run under air in reagent grade DMSO.
|
|||||
| 1 | [IrCp·Cl2]2/AgPF6 | 5 | None | 450 | Quantitative |
| 2 | AgPF6 | 5 | None | 450 | 71% |
| 3 | BzOH | 5 | None | 450 | 28% (+63% 1a) |
| 4 | CuI | 5 | None | 450 | 67% (+ 9% 1a) |
| 5 | Cu(OTf)2 | 5 | None | 450 | 70% |
| 6 | PdCI2 | 5 | None | 450 | 67% |
| 7 | Zn(TFA)2 | 5 | None | 450 | 27% (+18% 1a) |
| 8 | Sc(OTf)3 | 5 | None | 450 | 46% |
| 9 | TsOH·H20 | 5 | None | 450 | 81% |
| 10 | TsOH·H20 | 50 | None | 450 | 60% |
| 11 | TsOH·H20 | 5 | Watera | 450 | 96% |
| 12 | TsOH·H20 | 5 | Watera | 300 | 82% |
| 13b | TsOH·H20 | 5 | None | 300 | 84% |
| 14 | None | n/a | Watera | 300 | 25% |
| 15 | None | n/a | None | 450 | <5% (+>90% 1a) |
The scope of the new process with respect to the N-oxide component is outlined in Table 2A. Electronically and sterically diverse quinoline N-oxides all participated smoothly to provide the targets 2b–e in high yield. The protocol can be extended to isoquinoline N-oxides and this enables highly selective alkenylation of the C1 position, again providing targets 2f–i in good to excellent yield. Pyridine based N-oxides can also be accommodated as long as the arene possesses electron-withdrawing groups. Indeed, alkenylation of parent pyridine N-oxide to provide 2r was not successful, but 3-cyanopyridine N-oxide did undergo smooth conversion to afford target 2q with 3:1 C2 vs. C6 selectivity, favouring the indicated isomer. Other electron-withdrawing groups such as halides, trifluoromethyl groups and esters also render pyridine N-oxides sufficiently reactive for the process, thereby providing access to range of valuable derivatives, which in many cases have suitable handles for further functionalisation.
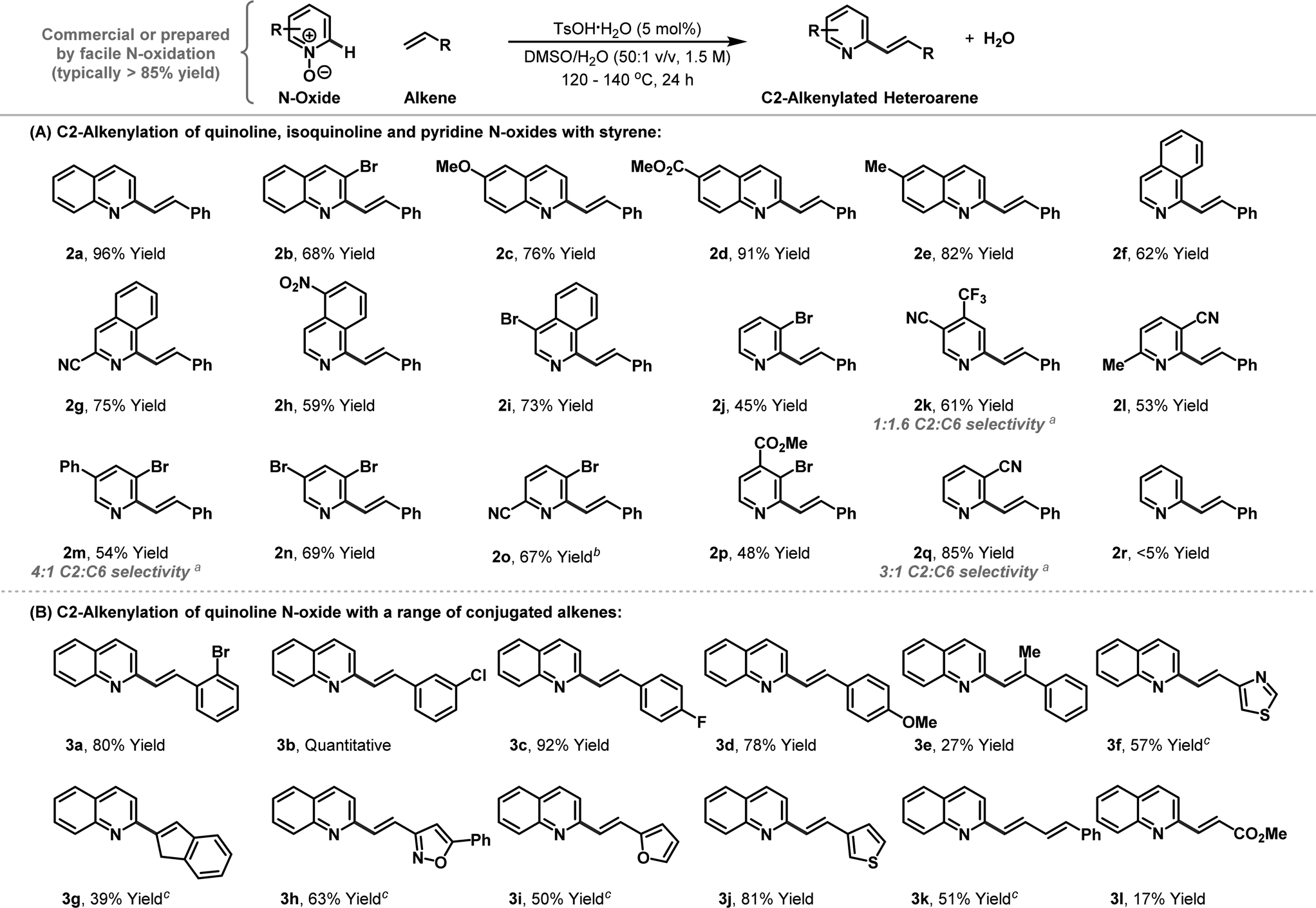
The scope of the alkene component has been assessed using quinoline N-oxide 1a as the heteroarene partner (Table 2B). Diverse mono-substituted aryl- and heteroaryl-alkenes are tolerated, with acceptable to high yields achieved in each case. More heavily substituted alkenes also participate, but generate the targets with lower efficiency. For example, alkenylation of quinoline N-oxide with indene provide adduct 3g in 39% yield. Likewise, α-methyl styrene did participate, but target 3e was generated in only 27% yield, albeit as a single geometric isomer. The directness of the approach may make it suitable for combinatorial synthesis were the operational simplicity of the protocol can offset any inherent inefficiencies. Preliminary results suggest that other classes of alkene may also be effective; for example, 1-phenyl butadiene provided adduct 3k in 51% yield. Using the current conditions, electron deficient alkenes are not suitable and methyl acrylate generated β-heteroarylated system 3l in only 17% yield.14
The protocol seems well suited to multistep synthetic sequences and we have demonstrated this in contexts where the method is used in tandem with transition metal catalysis (Scheme 2A). Pd-catalysed C8-selective arylation of quinoline N-oxide 1a, using iodobenzene, proceeded smoothly under conditions reported by Larionov and co-workers.13f Adduct 4, in which the N-oxide is still intact, then underwent Brønsted acid catalysed C2-alkenylation to provide disubstituted quinoline 5 in 65% yield. In this approach, serial metal-Brønsted acid catalysis is used to achieve sequential C–C bond forming C–H functionalisations, where the N-oxide first acts as a directing group and then as an internal oxidant. The method also has the potential to streamline syntheses of natural product targets. For example, Brønsted acid catalysed union of commercially available styrene 6 and N-oxide 1a afforded alkenylation product 7 in 90% yield (Scheme 2B). Hydrogenation of the alkene and pyridine units was then achieved under asymmetric Ir-catalysed conditions reported by Zhou and co-workers to provide tetrahydroquinoline 8.15 To maximise use of the valuable Ir-catalyst, this intermediate was not isolated, and instead the vessel was depressurised and a solution of aqueous formaldehyde in methanol was added. Re-pressurisation with hydrogen then facilitated iridium-catalysed reductive amination to install the N-methyl group of (−)-cuspareine, which was formed in 79% yield and 94% ee, thereby constituting the most efficient synthesis of this antimalarial alkaloid reported to date.16,17 Note that the only stoichiometric byproduct of the entire sequence is water.
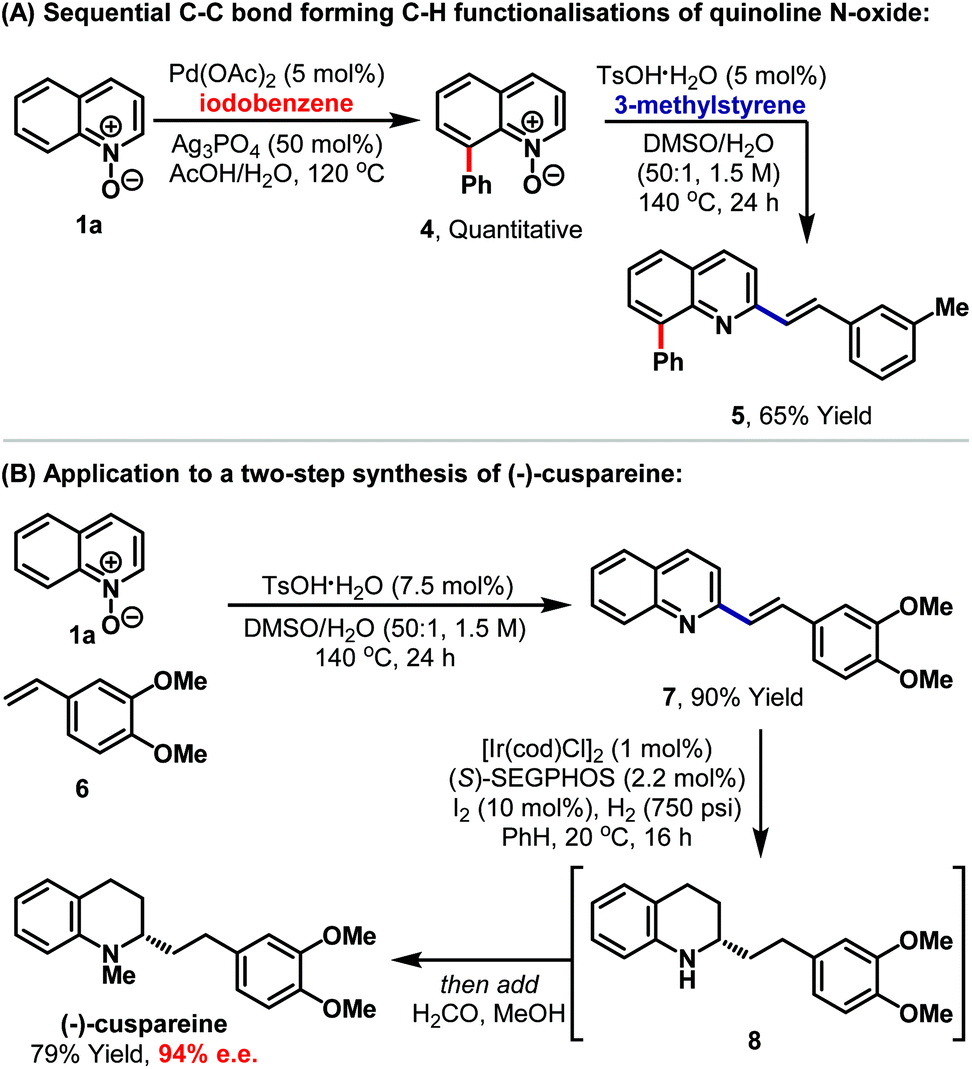 | ||
| Scheme 2 Integration of the Brønsted acid catalysed C2-alkenylation protocol into synthetic sequences. | ||
The C2-alkenylation process likely proceeds via initial dearomatising 1,3-dipolar cycloaddition between the N-oxide and the alkene (Scheme 3A).18,19 This provides intermediate I, which can undergo acid catalysed elimination via either the N–O (Path A) or C–O bond (Path B). The former seems more likely, given that N–O bonds are generally weaker and because elimination via this pathway directly re-establishes aromaticity. Acid catalysed elimination of the hydroxyl group may be facilitated by tautomerisation to enamine III rather than occurring directly from II.20 In most cases we have not observed the intermediate alcohol during the course of the reaction, which indicates that this process occurs reasonably fast. The proposed elimination mechanism is based on the observations that (a) only mild acidic conditions are required, (b) elimination still occurs for processes involving acrylates (see 3l) (which should disfavour E1 elimination from II) and (c) the efficiency of elimination is dependent on the heteroarene moiety. This latter assertion is based on the observation that for 2o significant quantities of intermediate alcohol (approximately 65%) remained and this necessitated addition of acetic anhydride to effect complete elimination. To demonstrate the feasibility of the proposed elimination, we prepared alcohol 9 (see the ESI†). Subjection of this to optimised alkenylation conditions (in the absence of styrene) resulted in smooth dehydration to quinoline 1a (Scheme 3B).
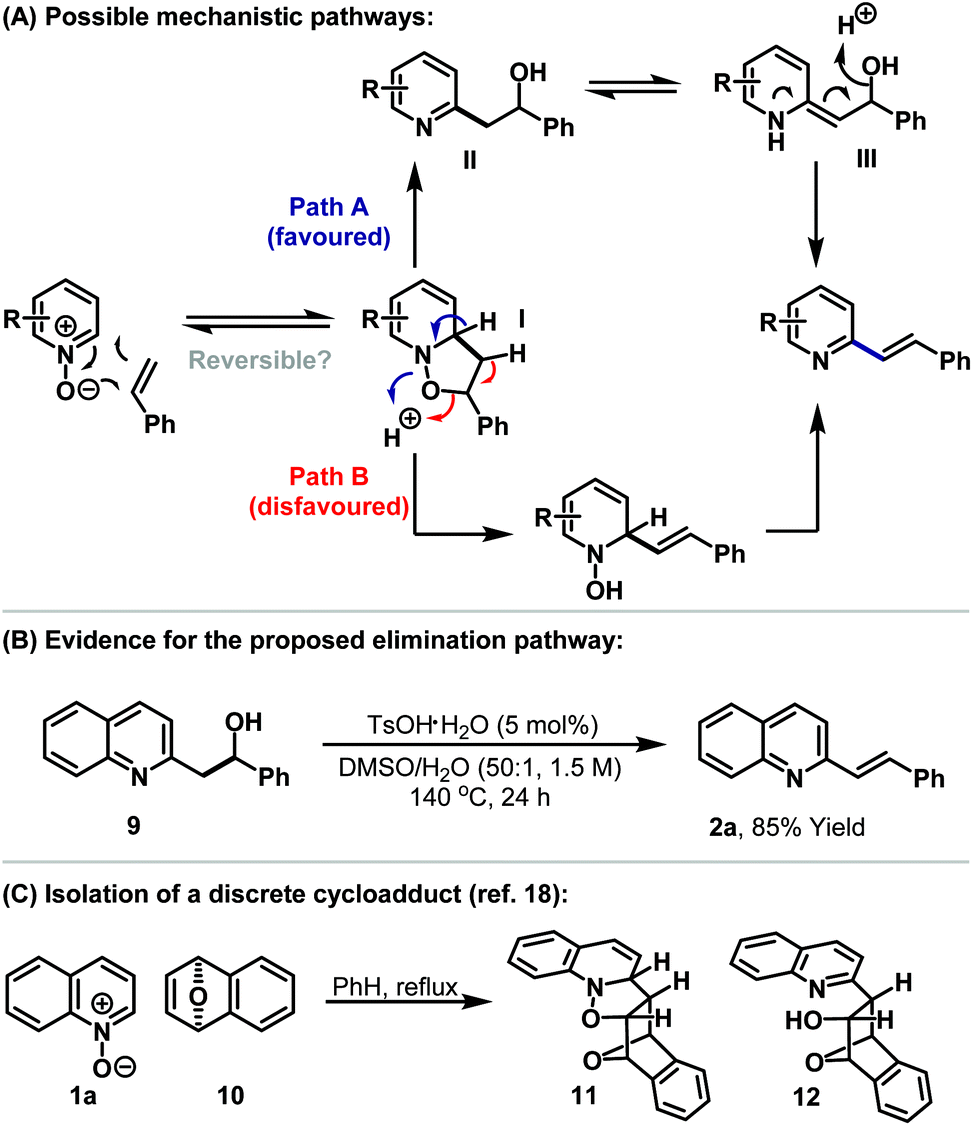 | ||
| Scheme 3 Mechanistic considerations for the C2-alkenylation process. | ||
We suggest that the success of the alkenylation process relies upon the ability to trap efficiently isoxazolidine I. 1,3-Dipolar cycloadditions between aromatic N-oxides and alkenes to form discrete initial cycloadducts are rare, with the only documented cases involving strained alkenes.18,19 Building on earlier work by Wittig and Steinhoff,18a Hisano and co-workers succeeded in isolating and characterising cycloaddition product 11, derived from quinoline 1a and alkene 10 (Scheme 3B).18b In the Wittig study ring cleavage product 12 was also observed (cf.I to II) although its stereochemistry was not confirmed.18a In other cases examined by Hisano, subsequent rearrangement processes were fast, which indicates that successful exploitation of I requires productive methods to divert it to target products.19 For the processes outlined here this is achieved by acid catalysed capture of I to afford II. Significantly, in the absence of acid/water, no cycloaddition is observed and starting material is recovered intact (see Table 1, entry 15). This suggests that the cycloaddition step may be reversible. If reversibility is a factor then a key issue is the energetic penalty involved in the dearomatising cycloaddition step. The heterocyclic ring systems of quinolines and isoquinolines possess less aromatic stabilisation than pyridines,21 and for this latter class, more electron deficient variants are more susceptible to dearomatisation.22 These trends in stabilisation provide an appealing rationalisation for the relative ease of the alkenylation reactions outlined in Table 2. A more sophisticated interpretation, which might also explain the regioselectivities observed for pyridine and isoquinoline N-oxides, must take into account the orbital energy match during the cycloaddition step (i.e. the kinetic feasibility of the process).23
Conclusions
In summary, we report a simple Brønsted acid catalysed approach to the synthesis of diverse C2-alkenylated N-heteroaromatic compounds. The protocol relies on thermally promoted (3 + 2) dearomatising cycloaddition between an N-oxide and an alkene to generate an isoxazolidine. The Brønsted acid catalyst then promotes N–O cleavage and dehydration to the target. We believe the method has high utility because (a) it is operationally simple, (b) the Brønsted acid catalyst is cheap, non-toxic and sustainable, (c) the N-oxide activator disappears during the reaction, (d) water is the sole stoichiometric byproduct of the process and (e) the functional group tolerance is orthogonal to metal-catalysed methods. Furthermore, we have demonstrated that the protocol can be integrated easily into synthetic sequences to provide polyfunctionalised targets, as exemplified by a highly concise synthesis of (−)-cuspareine and sequential N-oxide directed C–C bond forming C–H functionalisations of quinoline. Although inherently valuable, C2-alkenylated heteroarenes also function as precursors to C2-alkylated derivatives and as substrates for an emerging range of metal catalysed C–C/C–H bond forming processes.24 Given these considerations and the simplicity of our approach, which takes advantage of an intermediate routinely used in other contexts,1a it is likely that the protocol described here will find wide utility in synthesis and pharmaceutical chemistry. In broader terms, this study demonstrates how classical organic reactivity can still be used to provide solutions to contemporary synthetic challenges that might otherwise be approached using transition metal catalysis.Acknowledgements
G. E. M. C. thanks the Bristol Chemical Synthesis Doctoral Training Centre, funded by the EPSRC (EP/G036764/1) for a PhD studentship. J. F. B. is indebted to the Royal Society for the provision of a University Research Fellowship.Notes and references
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC; (b) V. Bonnet, F. Mongin, F. Trécourt, G. Breton, F. Marsais, P. Knochel and G. Quéguiner, Synlett, 2002, 1008 CrossRef CAS.
- For example, see: Y. Zou, G. Yue, J. Xu and J. Zhou, Eur. J. Org. Chem., 2014, 5901 CrossRef CAS.
- For example, see: M. L. Kantam, P. V. Reddy, P. Srinivas and S. Bhargava, Tetrahedron Lett., 2011, 52, 4490 CrossRef CAS.
- Contemporary approaches rely on modified 2-pyridyl boron-based reagents. Selected examples: (a) K. L. Billingsley and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 4695 CrossRef CAS PubMed; (b) G. R. Dick, D. M. Knapp, E. P. Gillis and M. D. Burke, Org. Lett., 2010, 12, 2314 CrossRef CAS PubMed; (c) G. R. Dick, E. M. Woerly and M. D. Burke, Angew. Chem., Int. Ed., 2012, 51, 2667 CrossRef CAS PubMed; (d) S. Sakashita, M. Takizawa, J. Sugai, H. Ito and Y. Yamamoto, Org. Lett., 2013, 15, 4308 CrossRef CAS PubMed.
- S. E. Wengryniuk, A. Weickgenannt, C. Reiher, N. A. Strotman, K. Chen, M. D. Eastgate and P. S. Baran, Org. Lett., 2013, 15, 792 CrossRef CAS PubMed and references cited therein.
- An alternative method involves metal catalysed condensation of 2-methyl N-heteroarenes with aldehydes or imines. This approach requires prior installation of a methyl substituent. Selected recent examples: (a) B. Qian, P. Xie, Y. Xie and H. Huang, Org. Lett., 2011, 13, 2580 CrossRef CAS PubMed; (b) Z. Jamal and Y.-C. Teo, Synlett, 2014, 2049 CAS.
- Seminal studies: (a) L.-C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020 CrossRef CAS PubMed; (b) L.-C. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266 CrossRef CAS PubMed; (c) L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291 CrossRef CAS PubMed.
- Examples of C2 selective C–H alkenylations of pyridine N-oxides: (a) K. S. Kanyiva, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8872 CrossRef CAS PubMed; (b) S. H. Cho, S. J. Hwang and S. Chang, J. Am. Chem. Soc., 2008, 130, 9254 CrossRef CAS PubMed; (c) L. Ackermann and S. Fenner, Chem. Commun., 2011, 47, 430 RSC.
- C2 selective C–H alkenylation of pyridines have also been achieved. Selected processes: (a) Y. Nakao, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 2448 CrossRef CAS PubMed; (b) P. Wen, Y. Li, K. Zhou, C. Ma, X. Lan, C. Ma and G. Huang, Adv. Synth. Catal., 2012, 354, 2135 CrossRef CAS.
- Examples where N-oxides direct metal catalysed C–H activation and serve as the internal oxidant: (a) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef CAS PubMed; (b) X. Huang, J. Huang, C. Du, X. Zhang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12970 CrossRef CAS PubMed; (c) B. Zhou, Z. Chen, Y. Yang, W. Ai, H. Tang, Y. Wu, W. Zhu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 12121 CrossRef CAS PubMed. See also ref. 13c and g. For a complementary strategy involving initial nucleophilic addition, see: (d) O. V. Larionov, D. Stephens, A. Mfuh and G. Chavez, Org. Lett., 2014, 16, 864 CrossRef CAS PubMed.
- L. Bering and A. P. Antonchick, Org. Lett., 2015, 17, 3134 CrossRef CAS PubMed.
- During the preparation of this manuscript Gou, Wang and co-workers reported a conceptually similar approach: H. Xia, Y. Liu, P. Zhao, S. Gou and J. Wang, Org. Lett., 2016, 18, 1796 CrossRef CAS PubMed . This protocol was strictly limited to quinoline-based systems, and required super-stoichiometric acetic acid (300 mol%) in combination with a large excess of the alkene (1000 mol%) (cf. this study: C2-alkenylation of quinoline, isoquinoline and pyridine N-oxides using just 5 mol% TsOH with 300–500 mol% of the alkene partner). See also: R. Kumar, I. Kumar, R. Sharma and U. Sharma, Org. Biomol. Chem., 2016, 14, 2613 Search PubMed.
- Selected examples of quinoline N-oxide directed C8-functionalisation with Rh or Ir catalysts: (a) H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770 CrossRef CAS PubMed; (b) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598 CrossRef CAS PubMed; (c) X. Zhang, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2014, 53, 10794 CrossRef CAS PubMed; (d) T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516 CrossRef CAS; (e) S. Konishi, S. Kawamorita, T. Iwai, P. G. Steel, T. D. Marder and M. Sawamura, Chem. – Asian J., 2014, 9, 434 CrossRef CAS PubMed; (f) D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Ateşin, G. Chavez, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167 CrossRef CAS PubMed; (g) R. Sharma, R. Kumar, I. Kumar and U. Sharma, Eur. J. Org. Chem., 2015, 7519 CrossRef CAS; (h) K. Shin, S.-W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584 CrossRef CAS PubMed; (i) D. E. Stephens, J. Lakey-Beitia, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, Chem. Commun., 2015, 51, 9507 RSC, For a review, see: (j) T. Iwai and M. Sawamura, ACS Catal., 2015, 5, 5031 CrossRef CAS.
- See ref. 10a and 12 for protocols that partially address this issue. Aliphatic alkenes are not suitable reaction partners.
- (a) W.-B. Wang, S.-M. Lu, P.-Y. Yang, X.-W. Han and Y.-G. Zhou, J. Am. Chem. Soc., 2003, 125, 10536 CrossRef CAS PubMed; (b) D.-W. Wang, X.-B. Wang, D.-S. Wang, S.-M. Lu, Y.-G. Zhou and Y.-X. Li, J. Org. Chem., 2009, 74, 2780 CrossRef CAS PubMed.
- J. H. Rakotoson, N. Fabre, I. Jacquemond-Collet, S. Hannedouche, I. Fourasté and C. Moulis, Planta Med., 1998, 64, 762 CrossRef CAS PubMed.
- For a review on syntheses of (−)-cuspareine and related alkaloids, see: G. D. Muñoz and G. B. Dudley, Org. Prep. Proced. Int., 2015, 47, 179 CrossRef.
- (a) G. Wittig and G. Steinhoff, Justus Liebigs Ann. Chem., 1964, 676, 21 CrossRef CAS; (b) T. Hisano, K. Harano, T. Matsuoka, T. Suzuki and Y. Murayama, Chem. Pharm. Bull., 1990, 38, 605 CrossRef CAS and references cited therein.
- For further selected processes where rearrangement (often [1,5]-sigmatropic) occurs after cycloaddition, see: (a) R. A. Abramovitch and I. Shinkai, J. Am. Chem. Soc., 1974, 96, 5265 CrossRef CAS; (b) R. E. Banks, R. N. Haszeldine and J. M. Robinson, J. Chem. Soc., Perkin Trans. 1, 1976, 1226 RSC; (c) T. Matsuoka, T. Hasegawa, K. Harao and T. Hisano, Heterocycles, 1992, 33, 179 CrossRef CAS; (d) H. J. Lindner, A. Krebs, J.-H. Förster and V. Sinnwell, Heterocycles, 1997, 45, 811 CrossRef CAS; (e) P. Jeschke, A. Harder, W. Etzel, W. Gau, A. Göhrt, J. Benet-Buchholz and G. Thielking, Bioorg. Med. Chem. Lett., 2005, 15, 2375 CrossRef CAS PubMed; (f) C. Raminelli, Z. Liu and R. C. Larock, J. Org. Chem., 2006, 71, 4689 CrossRef CAS PubMed; (g) R. Loska and M. Mąkosza, Chem. – Eur. J., 2008, 14, 2577 CrossRef CAS PubMed.
- Similar acid-catalysed tautomerisations (II to III) are known to be facile: M. Rueping and N. Tolstoluzhsky, Org. Lett., 2011, 13, 1095 CrossRef CAS PubMed . The proposed mechanism is akin to that determined for acid-catalysed aldol condensation of acetaldehyde to crotonaldehyde: L. M. Baigrie, R. A. Cox, H. Slebocka-Tilk, M. Tencer and T. T. Tidwell, J. Am. Chem. Soc., 1985, 107, 3640 CrossRef . The beneficial effects that water provides may be due to it facilitating tautomerisation or acting as a general catalyst for other steps. At the present stage, direct elimination of water from II cannot be discounted.
- M. Mandado, N. Otero and R. A. Mosquera, Tetrahedron, 2006, 62, 12204 CrossRef CAS.
- For a review, see: Q. Ding, X. Zhou and R. Fan, Org. Biomol. Chem., 2014, 12, 4807 CAS.
- Computational studies on the processes described here will be a focus of future work.
- For examples of metal catalysed C–C and C–H bond forming manipulations of 2-vinyl pyridines, see: (a) V. Komanduri, C. D. Grant and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 12592 CrossRef CAS PubMed; (b) L. Rupnicki, A. Saxena and H. W. Lam, J. Am. Chem. Soc., 2009, 131, 10386 CrossRef CAS PubMed; (c) G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all compounds. See DOI: 10.1039/c6ob00705h |
| This journal is © The Royal Society of Chemistry 2016 |