
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA systematic exploration of the effects of flexibility and basicity on sigma (σ) receptor binding in a series of substituted diamines†
Trent
Conroy‡
a,
Madhura
Manohar‡
a,
Yu
Gong
a,
Shane M.
Wilkinson
a,
Michael
Webster
a,
Brian P.
Lieberman
b,
Samuel D.
Banister
c,
Tristan A.
Reekie
a,
Robert H.
Mach
b,
Louis M.
Rendina
a and
Michael
Kassiou
*a
aSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: michael.kassiou@sydney.edu.au
bDepartment of Radiology, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA
cDepartment of Radiation Oncology, Stanford University School of Medicine, CA 94305, USA
First published on 13th September 2016
Abstract
The sigma-1 receptor (S1R) has attracted a great deal of attention as a prospective drug target due to its involvement in numerous neurological disorders and, more recently, for its therapeutic potential in neuropathic pain. As there was no crystal structure of this membrane-bound protein reported until 2016, ligand generation was driven by pharmacophore refinements to the general model suggested by Glennon and co-workers. The generalised S1R pharmacophore comprises a central region where a basic amino group is preferred, flanked by two hydrophobic groups. Guided by this pharmacophore, S1R ligands containing piperazines, piperazinones, and ethylenediamines have been developed. In the current work, we systematically deconstructed the piperazine core of a prototypic piperazine S1R ligand (vide infra) developed in our laboratories. Although we did not improve the affinity at the S1R compared to the lead, we identified several features important for affinity and selectivity. These included at least one basic nitrogen atom, conformational flexibility and, for S1R, a secondary or tertiary amine group proximal to the anisole. Furthermore, S2R selectivity can be tailored with functional group modifications of the N-atom proximal to the anisole.
Introduction
The existence of sigma receptors was proposed by Martin and co-workers 40 years ago.1 Two sigma receptor subtypes, the sigma-1 receptor (S1R) and sigma-2 receptor (S2R), have since been discovered.2 The S1R is abundantly distributed in the central and peripheral nervous systems.3 The S2R is typically found in the lungs, liver and kidney,4 but can also be highly expressed in malignant tumours.5The molecular structure of S1R was elucidated in the late 1990s.6 The S1R is a small protein (25.3 kDa) predominantly located at the mitochondria-associated endoplasmic reticulum membrane (MAM).7 Here the S1R is indirectly responsible for calcium homeostasis by acting as a molecular chaperone for inositol 1,4,5-triphosphate receptor type 3 (ITPR3).8 S1Rs also respond to ligand activation and undergo translocation to the cellular membrane, where they modulate the activity of ion channels and signalling molecules.7 Consequently, S1Rs are involved in numerous neurological disorders including depression, anxiety, substance abuse, schizophrenia, psychosis, memory deficit disorders (such as dementia and Alzheimer's disease), and motor disorders (including Parkinson's disease).9,10 The S1R has also experienced a recent surge of interest due to its involvement in analgesia,11,12 with a noteworthy ligand (E-52862) progressing to Phase II clinical trials for the treatment of pain.13
Due to the high expression of S2R in malignant tumours, S2R ligands have been utilised diagnostically for radiolabelling tumour sites, and have also been investigated therapeutically for their ability to induce apoptosis in cancer cells.14 No crystal structure exists for S2R. Compared to S1R, relatively fewer S2R ligands are known, and as such, there is limited information regarding S2R pharmacology.14,15 A study published in 2011 reported that the progesterone receptor membrane component 1 (PGRMC1) and S2R were each labelled irreversibly following treatment with a known S2R ligand (WC-21). Co-identity of S2R and PGRMC1 was proposed when S2R binding was found to correlate to PGRMC1 expression.15 However, more recently, an in vitro PGRMC1 knockout NSC34 cell line (designed using CRISPR/Cas9) showed no difference in the binding of a known radiolabelled S2R ligand ([3H]-DTG), in addition to DTG-masked [125I]IAF photolabelling, when compared with a wild type cell line.16
A recent milestone in S1R research was achieved in early 2016, when the crystal structure of the receptor bound to known S1R ligands (PD144418 and 4-IBP) was elucidated. The crystal structure of the S1R depicted a trimeric configuration with a single transmembrane domain in each protomer. The binding site itself was found to be buried suggesting that the nature of the binding site is highly hydrophobic. Furthermore, it was revealed that the basic amine of the ligands form hydrogen bonds with Glu172. Moreover, the distinct structures of PD144418 and 4-IBP reveal the plasticity of the binding site.17 Preceding this discovery, ligand design was propelled by pharmacophore refinements. A myriad of functional S1R ligands have now been identified,12 and common features for binding have been elucidated.18–20 Glennon and co-workers identified the ideal distance between a basic amine and two tethered hydrophobic groups (named primary and secondary hydrophobic regions) for prototypic aminergic S1R ligands.18,19 Since this discovery, several other groups have elaborated on this pharmacophore.21–23 Suggestions of a proton acceptor site, situated about 4 Å from the amine core, as an additional requirement for optimal binding have been proposed.21,23
In contrast to the models of S1R binding, the S2R pharmacophore has received little attention. One study assessed 19 compounds to elucidate the features necessary for affinity and selectivity of S2R binding ligands. Requisite features identified included: a hydrophobic aromatic and aliphatic component; a positive ionisable atom; a hydrogen bond acceptor; and a generic hydrophobic group. These features were initially determined in silico and the binding affinities of the compounds synthesised reflected the predicted binding profiles in most cases.24
Studies in our laboratory have focused on the development of structurally diverse S1R ligands with efficacy in various animal models of human disease.25–34 We have previously reported structure–activity relationships surrounding disubstituted piperazine S1R ligand 1.27 The effects of linker elongation26 and size of the diamine core,28 as well as modifications to the ether substituent have been explored.27 This work resulted in suitably high affinity ligands that when labelled with carbon-11 or fluorine-18, produced tracers for imaging S1R using positron emission tomography (PET).35,36 Owing to the high affinity and selectivity achieved with 1 for S1R in vitro (as well as in vivo), we proceeded to build our library of this class of ligands. The present work aims to introduce amide functionality at various positions on the piperazine and ethylenediamine analogues to evaluate the effects of varying the basicity and flexibility on S1R binding and selectivity. These modifications will provide further insights into the structure activity relationships of this library of diaminergic ligands. Furthermore, the nature of the alkyl amine core of the S1R pharmacophore can be understood through this body of work.
Results and discussion
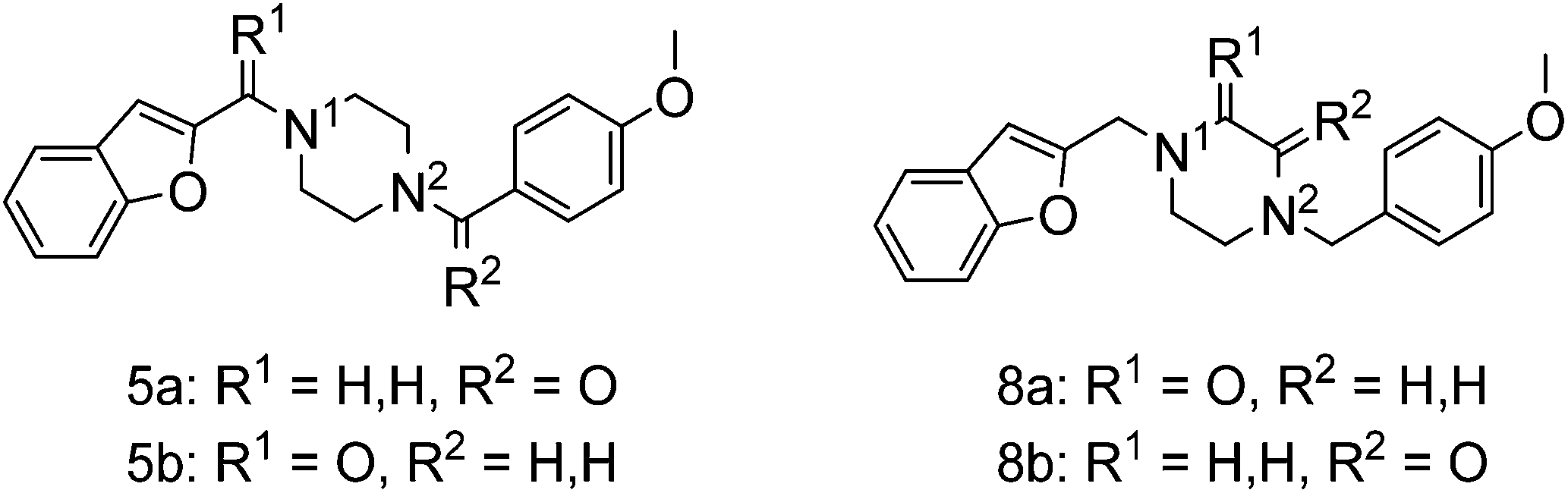
To study the effect of amine basicity on sigma receptor affinity and selectivity as well as to explore the effect of further rigidification of the piperazine core, we introduced a carbonyl moiety at various positions of lead structure 1, to give compounds 5a,b and 8a,b (Fig. 1, piperazine series). Similarly, we prepared compounds containing an ethylenediamine core in place of piperazine, representing analogues with increased conformational freedom. In addition, methyl groups and a carbonyl moiety were to be systematically moved around the core to better understand the effect of basicity on affinity and selectivity in this series (Fig. 1, ethylenediamine Series 1–3).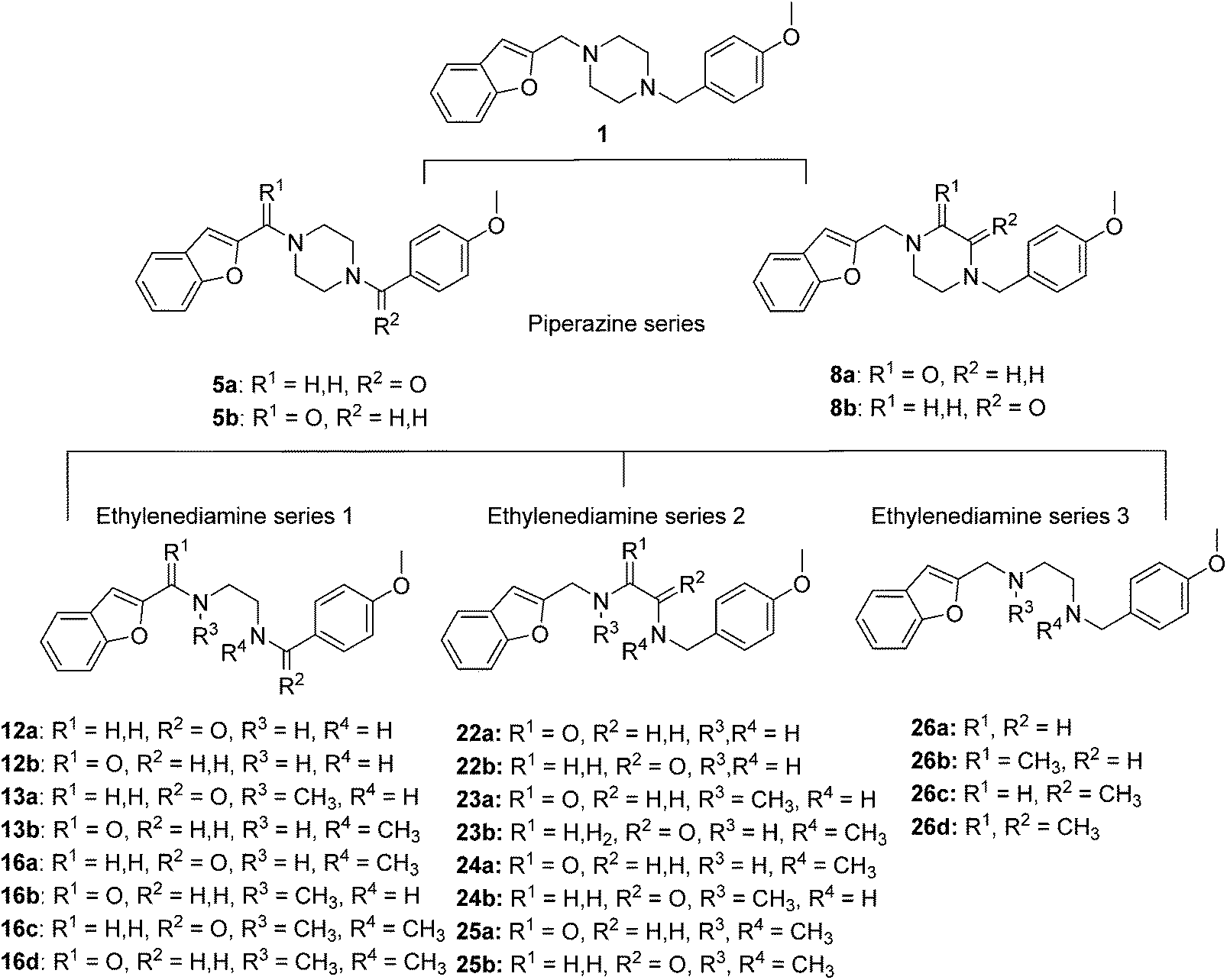 | ||
| Fig. 1 Structure of the lead compound 1 and the library of compounds derived from the lead. | ||
To this end, amide coupling reactions between Boc-protected piperazine and the appropriate acid chlorides furnished amides 3a and 3b (Scheme 1). Following Boc deprotection, the piperazines were subjected to reductive alkylation with suitable aryl aldehydes to afford the final compounds 5a,b.
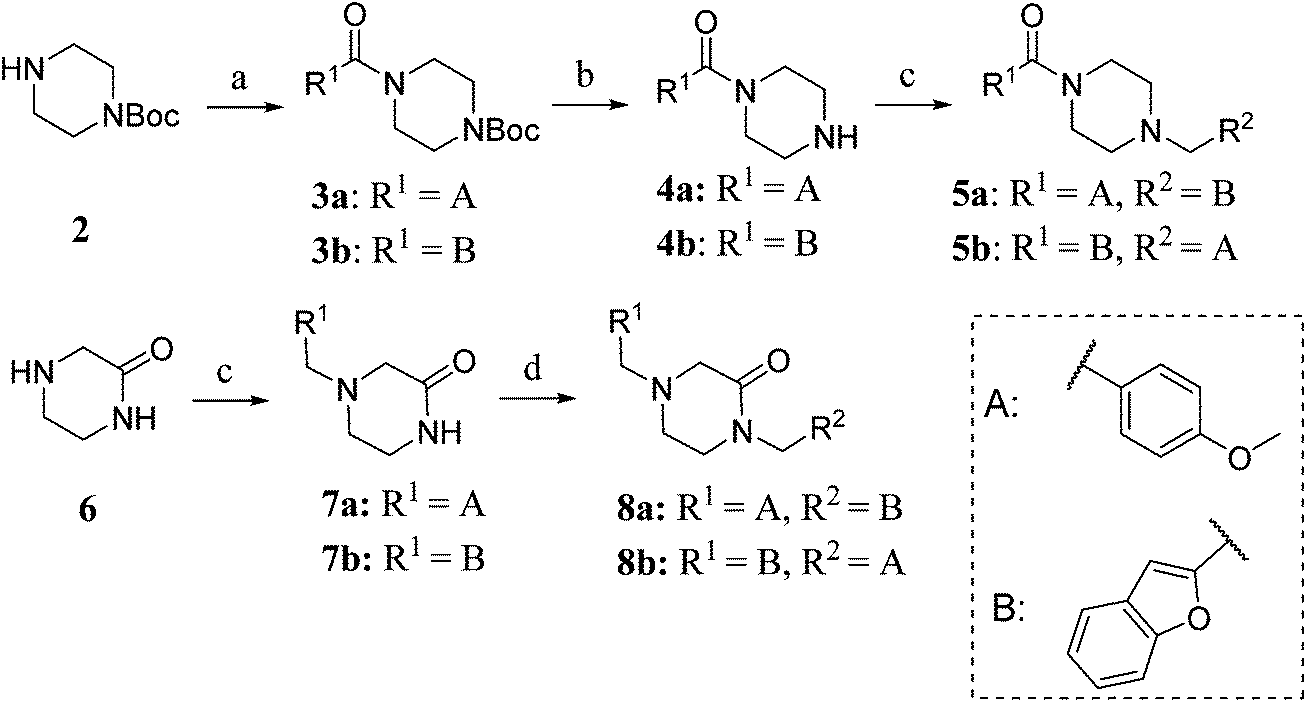 | ||
| Scheme 1 (a) acid chloride, Et3N, CH2Cl2, 0 °C – rt, 20 h (3a: 70%; 3b: 89%); (b) 4 M HCl in 1,4-dioxane, rt, 2 h (4a: 73%; 4b: 79%); (c) 2-benzofurancarboxaldehyde or anisaldehyde, NaBH(OAc)3, ClCH2CH2Cl, rt, 10 h (5a: 75%; 5b: 74%; 7a: 64%; 7b: 86%); (d) (2-benzofuranyl)methyl chloride or 4-methoxybenzyl chloride, NaH, THF, 0 °C – rt, 18 h (8a: 90%; 8b: 34%). | ||
Piperazinone 6 was reductively alkylated to provide 7a and 7b (Scheme 1). N-Alkylation of the amides of 7a and 7b was achieved by sodium hydride deprotonation and treatment with 2-benzofuranyl methyl chloride or p-methoxybenzyl chloride (synthesised according to the method reported by Ferorelli et al.37) to give 8a and 8b.
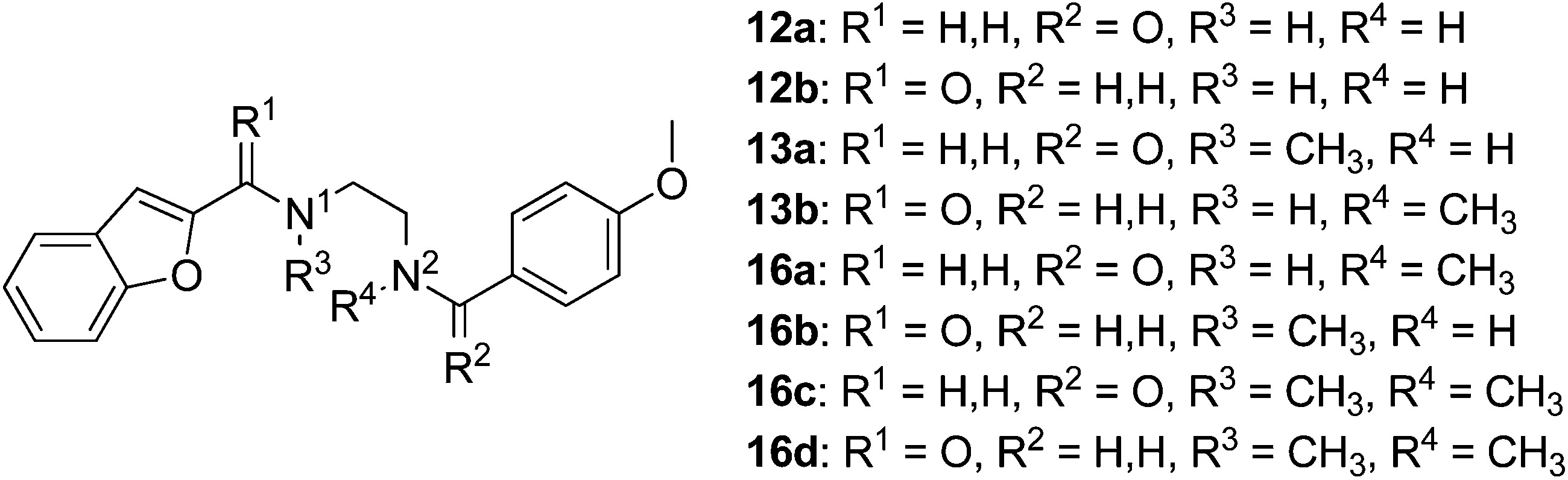
Synthesis of the ethylenediamine series began by monoprotection of ethylenediamine to give 9b (Scheme 2), and subsequent amide coupling between the free amine of 9b and the appropriate freshly prepared acyl chloride gave the desired disubstituted products (10a,b). The Boc group was cleaved under acidic conditions to give 11a and 11b, the corresponding free amines were subjected to reductive alkylation with the appropriate aldehydes to afford 12a and 12b in modest yields due to competing dialkylation. Methylation of the secondary amines (12a,b) using aqueous formaldehyde and sodium triacetoxyborohydride provided the desired tertiary amines 13a and 13b. Alternatively, monoprotection of the remaining nucleophilic amines (12a,b) furnished amides 14a and 14b, which were alkylated with iodomethane to give 15a and 15b. Cleavage of the Boc group of 15a and 15b provided 16a and 16b, respectively, and subsequent reductive alkylation with formaldehyde furnished ligands 16c and 16d.
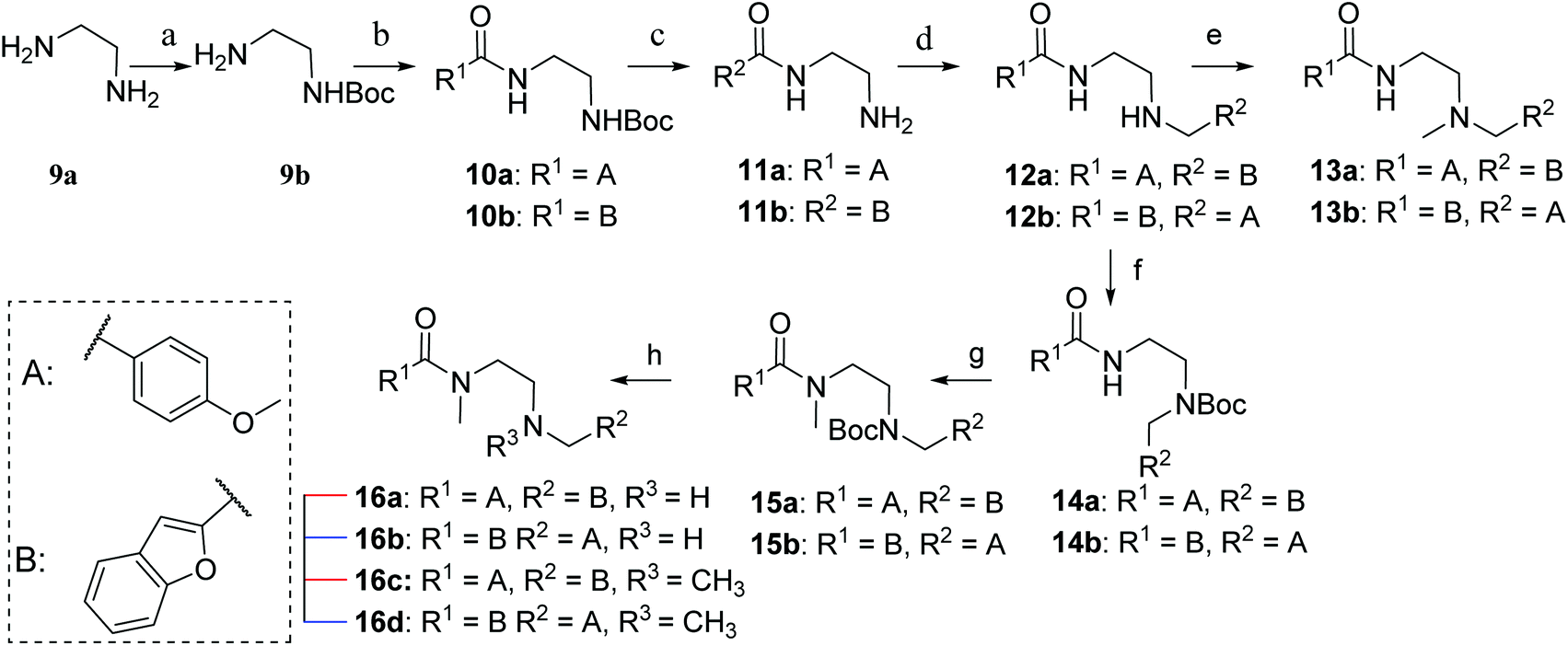 | ||
| Scheme 2 Reagents and conditions: (a) Boc2O, CH2Cl2, rt, 12 h (13: 91%); (b) acid chloride, Et3N, CH2Cl2, 0 °C – rt, 20 h (10a: 83%, 10b: 87%); (c) 4 M HCl in 1,4-dioxane, rt, 2 h, then. NaOH (11a: 70%; 11b: 91%); (d) aldehyde, NaBH(OAc)3, ClCH2CH2Cl, rt, 10 h (12a: 52%; 12b: 37%); (e) formalin, NaBH(OAc)3, ClCH2CH2Cl, rt, 10 h (13a: 48%; 13b: 25%); (f) Boc2O, CH2Cl2, rt, 12 h (14a: 97%; 14b: quant.); (g) NaH, DMF, 0 – rt °C, 10 min, cooled to 0 °C, CH3I, stirred at 0 °C, 30 min, rt, 30 min (15a: quant.; 15b: 96%); (h) 4 M HCl in 1,4-dioxane, rt, 2 h, then. NaOH, for 16c and 16d; formalin, NaBH(OAc)3, ClCH2CH2Cl, rt, 10 h (16a: 80%; 16b: 77%; 16c: 93%; 16d: quant.). | ||
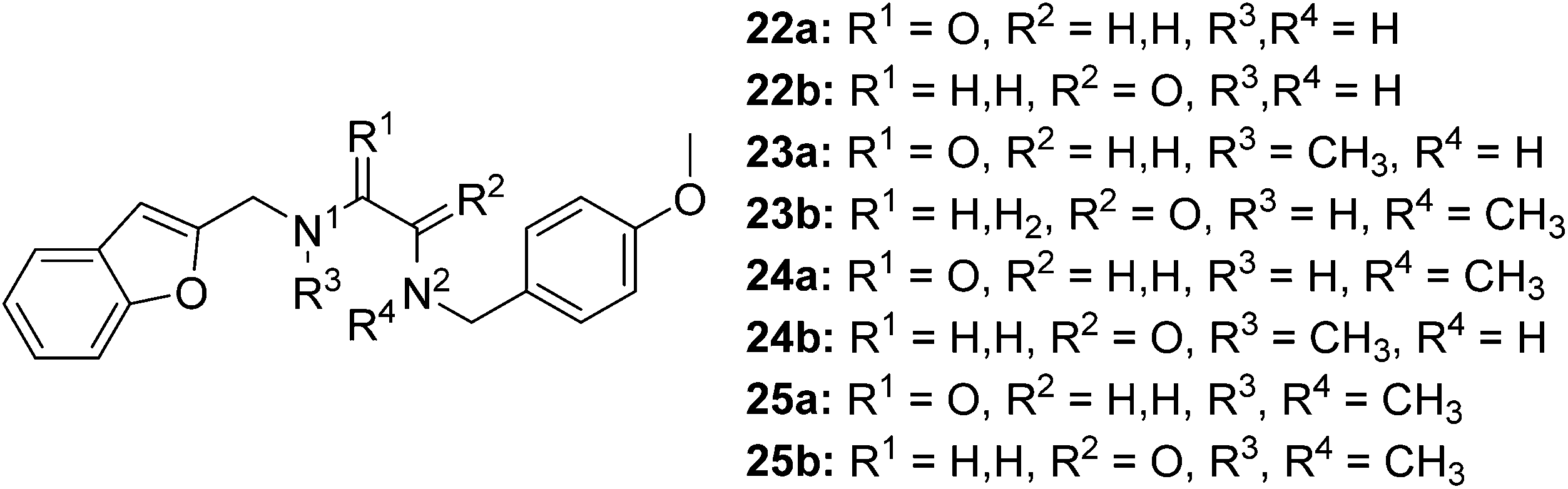
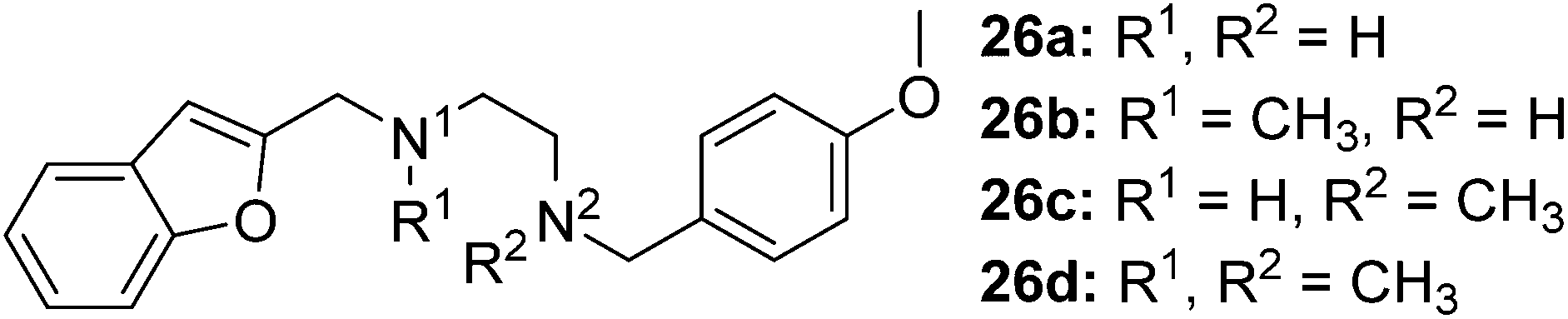
The synthesis of compounds containing a carbonyl moiety on the ethylenediamine linker required a different approach, as depicted in Scheme 3. Methyl ester-protected glycine (17) was condensed with anisaldehyde and the imine formed reduced with sodium borohydride to give 18a. Saponification of the ester was followed by Boc-protection of the secondary amine (19a), and the acids coupled with amines benzofuran-2-yl-methanamine or 1-(benzofuran-2-yl)-N-methylmethanamine using HBTU to give 20a and 21a. Boc deprotection yielded 22a and 23a and the resulting amines were N-alkylated under reductive amination conditions with formaldehyde to give 24a and 25a in good yield. The amides of compounds: 22a, 23a, 24a and 25a were then reduced with LiAlH4 to provide the requisite compounds 26a–26d.
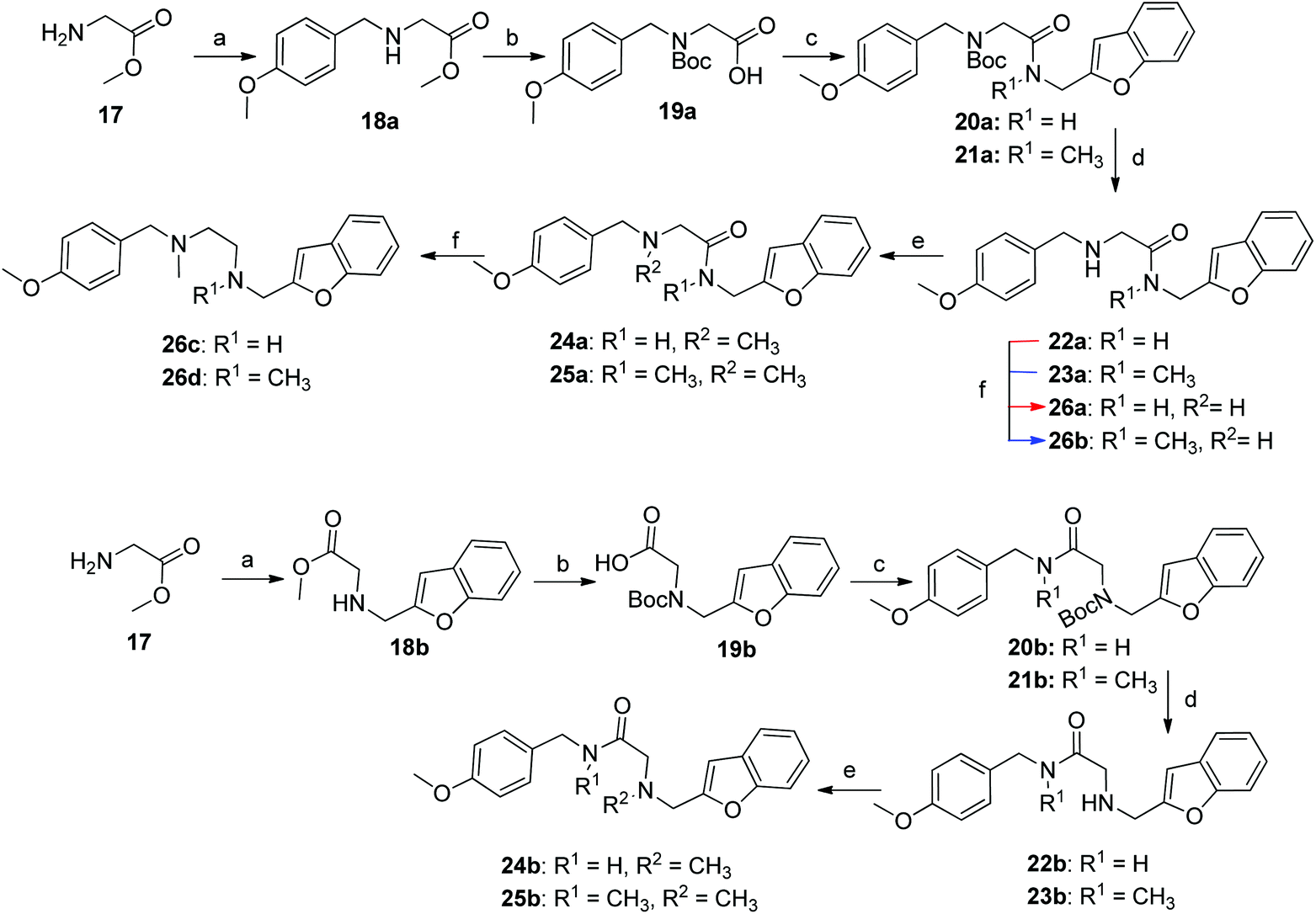 | ||
Scheme 3 Reaction condition and reagents: (a) (i) aldehyde, CH3OH, Et3N, 14 h, rt, (ii) NaBH4, CH3OH, 0 °C, 1 h, rt, 1 h (18a, 18b: 75%; 60% (b) 1 M aq. NaOH, THF, 1.5 h, 6 M aq. HCl, Na2CO3, Boc2O, 14 h (19a, 19b: 96%; 97%) (c) Et3N, HBTU, CH2Cl2/DMF, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, amine*, rt, 14 h (20a, 21a: 54%; 72%, 20b, 21b: 71%; 73%) (d) 4 M HCl in 1,4-dioxane, rt, 2 h, then. NaOH (22a, 23a: 84%; 98%, 22b, 23b: 93%; 88%) (e) formalin, CH3OH, Et3N, NaBH3CN, 14 h (24a, 25a: 86%; 74%, 24b, 25b: 91%; 83%) (f) LiAlH4, THF, 0 °C – rt, reflux, 16 h (26a–d: 68–84%). *Amines: PMBNH2, PMBNHMe, benzofuran-2-ylmethanamine and 1-(benzofuran-2-yl)-N-methylmethanamine, (ESI†). :1, v/v, amine*, rt, 14 h (20a, 21a: 54%; 72%, 20b, 21b: 71%; 73%) (d) 4 M HCl in 1,4-dioxane, rt, 2 h, then. NaOH (22a, 23a: 84%; 98%, 22b, 23b: 93%; 88%) (e) formalin, CH3OH, Et3N, NaBH3CN, 14 h (24a, 25a: 86%; 74%, 24b, 25b: 91%; 83%) (f) LiAlH4, THF, 0 °C – rt, reflux, 16 h (26a–d: 68–84%). *Amines: PMBNH2, PMBNHMe, benzofuran-2-ylmethanamine and 1-(benzofuran-2-yl)-N-methylmethanamine, (ESI†). | ||
The synthesis of the ligands with the amide moiety proximal to the anisole began in an analogous manner, with a two-step reductive amination between glycine methyl ester and 2-benzofurancarboxaldehyde (Scheme 3) to afford 18b. Saponification of the methyl ester, was followed by Boc protection of the secondary amine to afford 19b. Reaction between N-(4-methoxybenzyl)-N-methylamine or 4-methoxybenzylamine and the protected glycine derivative 19b using HBTU as a coupling reagent, yielded the requisite amides 20b and 21b. Acidic carbamate deprotection provided amines 22b and 23b, which were then alkylated under reductive conditions with formalin to generate 24b and 25b.
The binding affinities of these ligands were determined by using competitive radioligand binding assays for S1R and S2R, and are presented in Tables 1–4. Binding assays were performed in two rotations, compounds in Table 1 were independently assessed with respect to those compounds in Tables 2–4. Tissue homogenates were prepared by processing whole guinea pig brain samples for the S1R, while fresh rat liver was used for the S2R. [3H](+)-pentazocine (6 nM) was used for S1R assays, while [3H]DTG (5 nM) was used for S2R assays. For all S2R studies conducted, 100 nM of (+)-pentazocine was added to mask S1R binding sites (see Experimental section for further details).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Affinities Ki (nM ± SEM) | Subtype selectivity S1R/S2R | Predicted pKa | N 1 | N 2 | Predicted microspecies distribution (%) | ||
| S1R | S2R | N 1 | N 2 | |||||
| 1 | 2.7 | 103 | 38 | 7.39 | 1.56 | N | N | 50.4 |
| NH+ | N | 35.2 | ||||||
| N | NH+ | 14.4 | ||||||
| 5a | 850 ± 22 | >105 | >118 | 6.49 | — | N | N | 88.9 |
| NH+ | N | 11.1 | ||||||
| 5b | 472 ± 145 | >35000 ± 200 |
75 | — | 6.14 | N | N | 94.8 |
| N | NH+ | 5.2 | ||||||
| 8a | 2830 ± 66 | >61000 ± 8000 |
22 | — | 5.56 | N | N | 98.6 |
| N | NH+ | 1.4 | ||||||
| 8b | 2670 ± 214 | >105 | >37 | 5.96 | — | N | N | 96.5 |
| NH+ | N | 3.5 | ||||||
| Haloperidol | 3.19 | n/a | n/a | |||||
| DTG | n/a | 19.6 | n/a | |||||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Affinities Ki (nM ± SEM) | Subtype selectivity (S1R/S2R) | Predicted pKa | N 1 | N 2 | Predicted microspecies distribution (%) | ||
| S1R | S2R | N 1 | N 2 | |||||
| 1 | 0.72 ± 0.09 | 47.8 ± 1.9 | 66 | 7.39 | 1.56 | N | N | 50.4 |
| NH+ | N | 35.2 | ||||||
| N | NH+ | 14.4 | ||||||
| 12a | >1000 | >1000 | n/a | 8.14 | — | N | N | 15.5 |
| NH+ | N | 84.5 | ||||||
| 12b | 30.9 ± 4.2 | 145 ± 23 | 4.7 | — | 8.58 | N | N | 6.2 |
| N | NH+ | 93.8 | ||||||
| 13a | 207 ± 20 | 106 ± 2 | 0.5 | 7.80 | — | N | N | 28.7 |
| NH+ | N | 71.3 | ||||||
| 13b | 2.83 ± 0.21 | 78.7 ± 6.5 | 28 | — | 8.00 | N | N | 20.0 |
| N | NH+ | 80.0 | ||||||
| 16a | 242 ± 2 | 73.5 ± 11.2 | 0.3 | 8.17 | — | N | N | 14.5 |
| NH+ | N | 85.5 | ||||||
| 16b | 13.4 ± 1.6 | 9.51 ± 0.86 | 0.7 | — | 8.60 | N | N | 5.9 |
| N | NH+ | 94.1 | ||||||
| 16c | 510 ± 61 | 975 ± 44 | 1.9 | 7.84 | — | N | N | 26.8 |
| NH+ | N | 73.2 | ||||||
| 16d | 6.56 ± 0.75 | 35.8 ± 1.1 | 5.5 | — | 8.03 | N | N | 19.1 |
| N | NH+ | 80.9 | ||||||
| Haloperidol | 3.19 | n/a | n/a | — | — | — | — | — |
| DTG | n/a | 19.6 | n/a | — | — | — | — | — |
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Affinities Ki (nM ± SEM) | Subtype selectivity (S1R/S2R) | Predicted pKa | N 1 | N 2 | Predicted microspecies distribution (%) | ||
| S1R | S2R | N 1 | N 2 | |||||
| 1 | 0.72 ± 0.09 | 47.8 ± 1.9 | 66 | 7.74 | 2.01 | N | N | 31.2 |
| NH+ | N | 54.7 | ||||||
| N | NH+ | 14.0 | ||||||
| 22a | 2.28 ± 0.40 | >5000 | >2000 | — | 7.86 | N | N | 25.5 |
| N | NH+ | 74.5 | ||||||
| 22b | 516 ± 76 | >10000 |
>19 | 7.44 | — | N | N | 47.7 |
| NH+ | N | 52.3 | ||||||
| 23a | 8.32 ± 0.46 | >1000 | >120 | — | 7.90 | N | N | 24.1 |
| N | NH+ | 75.9 | ||||||
| 23b | 299 ± 18 | >1000 | >3.3 | 7.48 | — | N | N | 45.2 |
| NH+ | N | 54.8 | ||||||
| 24a | >1000 | >1000 | n/a | — | 7.09 | N | N | 67.1 |
| N | NH+ | 32.9 | ||||||
| 24b | >1000 | >1000 | n/a | 6.91 | — | N | N | 75.6 |
| NH+ | N | 24.4 | ||||||
| 25a | 146 ± 5 | 720 ± 37 | 4.9 | — | 7.13 | N | N | 64.9 |
| N | NH+ | 35.1 | ||||||
| 25b | 628 ± 75 | >1000 | >1.6 | 6.96 | — | N | N | 73.2 |
| NH+ | N | 26.8 | ||||||
| Haloperidol | 3.19 | n/a | n/a | — | — | — | ||
| DTG | n/a | 19.6 | n/a | — | — | — | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Affinities Ki (nM ± SEM) | Subtype selectivity (S1R/S2R) | Predicted pKa | N 1 | N 2 | Predicted microspecies distribution (%) | ||
| S1R | S2R | N 1 | N 2 | |||||
| 1 | 0.72 ± 0.09 | 47.8 ± 1.89 | 66 | 7.74 | 2.01 | N | N | 31.2 |
| NH+ | N | 54.7 | ||||||
| N | NH+ | 14.0 | ||||||
| 26a | 20.7 ± 1.4 | 626 ± 57 | 30 | 5.71 | 8.69 | N | N | 4.8 |
| NH+ | N | 24.8 | ||||||
| N | NH+ | 68.5 | ||||||
| NH+ | NH+ | 1.9 | ||||||
| 26b | 27.5 ± 4.7 | 294 ± 40 | 11 | 5.49 | 9.00 | N | N | 2.4 |
| NH+ | N | 3.4 | ||||||
| N | NH+ | 93.0 | ||||||
| NH+ | NH+ | 1.2 | ||||||
| 26c | 6.09 ± 0.76 | 53.5 ± 4.3 | 8.8 | 5.86 | 9.02 | N | N | 2.3 |
| NH+ | N | 8.1 | ||||||
| N | NH+ | 86.9 | ||||||
| NH+ | NH+ | 2.8 | ||||||
| 26d | 4.8 ± 0.5 | 77.4 ± 0.8 | 16 | 4.29 | 8.50 | N | N | 7.4 |
| NH+ | N | 36.3 | ||||||
| N | NH+ | 56.2 | ||||||
| NH+ | NH+ | 0.1 | ||||||
| Haloperidol | 3.19 | n/a | n/a | — | — | — | ||
| DTG | n/a | 19.6 | n/a | — | — | — | ||
The introduction of an amide functionality in the piperazine series reduced affinity for both S1R/S2R. An exocyclic carbonyl moiety proximal to the anisole in 5a imparted a two-fold decrease in binding affinity for S1R (Ki = 850 nM), when compared to an exocyclic carbonyl moiety near the benzofuranyl substituent (5b: S1R Ki = 472 nM). Selectivity for S1R was also reduced for 5a and 5b compared to 1, suggesting that a basic amino group close to the anisole confers affinity for S1Rs, as demonstrated by the introduction of carbonyl moieties at either exocyclic positions.
Interestingly, when the basicity of the N-atoms were reduced by introduction of an endocyclic carbonyl moiety, the S1R affinities dropped drastically by more than 103-fold (8a and 8b, Ki = 2830 and 2670 nM, respectively), and the compound with the amide in proximity to the benzofuran group presented greater selectivity. Unfavourable rigidity in the ring, steric factors, or a further reduction in the basicity of both the amine groups is postulated to cause this drop in affinity.
The protonation states of the current library of S1R ligands at physiological pH (7.4) were predicted using ChemAxon's Marvin software suite,38 and the predicted pKa and microspecies distributions for the ligands in the series are also shown in Tables 1–4. The microspecies distribution represents the % protonation of the N-atom(s) at a given pH (7.4 is reported herein). The pKa values for the amide functionality were calculated with protonation occurring at the oxygen atom. Secondary and tertiary amide values were typically around −2 or lower and were never calculated as being microspecies at pH 7.4, therefore they have been excluded from the tables. Potentiometric titrations were also conducted to support these predicted values. Experimental pKa values were acquired for benzylamine, 13b and 25b, with values calculated at 9.28 ± 0.038, 7.31 ± 0.01 and 8.01 ± 0.012, respectively (see ESI† for conditions). The experimentally determined pKa value for benzylamine is in agreement with the value predicted via ChemAxon (9.30) and a literature potentiometric value (9.33).39 The experimental pKa values for 13b and 25b exhibited a discrepancy in comparison to the predicted values (8.00 and 6.96, respectively). These differences can be explained by a number of factors. Firstly, the experimental values were obtained with the compounds dissolved in 40% ethanol/water, while the calculated values are for aqueous environments. The complexity of the molecule in comparison to the standard (benzylamine) would also be a contributing factor. Particularly with 25b where lone pairs of adjacent oxygen atoms in both the benzofuran and amide are likely to be playing a role in protonation. Interestingly, an older version of MarvinSketch (15.7.6.0) predicted a pKa of 8.02 for 25b, while for 13b a value of 7.80 was predicted. We have included in the paper the most recent values calculated, but since the older values seem to better correlate with the experimental values these are also tabulated in the ESI.† Nevertheless, for the purposes of this study, we believe the predicted values are sufficient aids to describe basicity trends.
For 1, the predicted pKa value of N1 (amine nearest to the benzofuran) was 7.39 and pKa of N2 was 1.56. 5a and 5b possess a tertiary amine at the benzofuran N1 (pKa 6.49) and benzyl N2 (pKa 6.14), respectively. Despite having the basic amino group proximal to the benzofuran, 5a displayed a two-fold decrease in S1R binding affinity compared to 5b. This could suggest that the N-atom proximal to the anisole might be most important for S1R affinity, indicating that the anisole may have a preference for the secondary hydrophobic pocket. Alternatively, it could be the result of other binding modes of these diaminergic ligands.
8a and 8b had slightly lower pKa values for their respective tertiary amines (5.56 and 5.96, respectively). This is likely a result of the proximity of the carbonyl moiety to both N-atoms. This lowered basicity corresponds with the reduced affinity obtained experimentally. The predicted microspecies distribution for 1 revealed that the fully deprotonated species is the major species at pH 7.4 (50.4%), with single protonation of the N-atom proximal to the benzofuran also prevalent (31.17%). The predicted microspecies distributions of the fully deprotonated forms of 5a, 5b, 8a, and 8b were 88.9%, 94.8%, 98.6% and 96.5% respectively. The program used to calculate pKa values herein was revealed to have an r2 value of 0.763 after analysing 197 pharmaceutical substances.40 While another source claims Marvin is better suited for acid pKa determinations rather than base pKa determinations due to a smaller error in experimental and calculated pKa values.41
These findings prompted us to investigate an open linker series (Table 2). Ethylenediamine was selected as an alternative flexible analogue of piperazine. This series contained carbonyl moieties on the periphery (ethylenediamine series one – Table 2), within the ethylenediamine linker (ethylenediamine series two – Table 3) and lacking carbonyl functionality (ethylenediamine series three – Table 4).
Similarly to the piperazine and piperazinone series, carbonyl moieties were tolerated proximal to the benzofuran and were less preferred near the anisole. This library suggested that the piperazine core was not crucial in the design of high affinity S1R ligands, however the flexible linker is thought to affect the selectivity of these ligands since higher affinities for the S2R was also observed. Hence, it is postulated that the ligands with a flexible linker can position themselves within the binding pocket of the S2R as well.
The binding affinities for 12a and 12b were examined. A carbonyl moiety proximal to the anisole produced a drop in S1R/S2R affinity analogous to 5a (12a, S1R and S2R Ki ≥ 1000). 12b displayed drastic improvements in affinities for both S1R and S2R with Ki = 30.9 nM and 145 nM respectively. The subtype selectivity appears to have improved for 16b, at 4.7 for the S1R. The introduction of tertiary amines in 13a and 13b produced higher S1R and S2R affinity. 13a in particular presented a greater subtype selectivity for S2R of S1R/S2R = 0.5 (S1R and S2R Ki = 207 and 106 nM). Again there was a distinct preference for a carbonyl moiety proximal to the benzofuran in 13b for S1R affinity and selectivity (Ki = 2.8 nM and S2R affinity of Ki = 78.7 nM). Surprisingly, comparisons of the pKa for the N-atom proximal to the benzofuran and anisole for compounds 12a and 13a showed similar pKa values, however 12a showed negligible activity, while 13a showed some preference for S2R. This could indicate that alternative factors besides basicity, such as the steric environment around the N-atom, are important for activity.
The next set of ligands investigated were the tertiary amides 16a–d. Analogous to 13a, 16a possessed an almost identical binding profile. This was reflected in the pKa of N1, which was also similar. 16c, while similar to 13a and 16a in the proximity of the carbonyl moiety to the anisole, it lacked greater selectivity for S2R than S1R as observed previously. 16b and 16d presented peculiar binding profiles also. Both ligands possess a tertiary amide proximal to the benzofuran, but 16b differs in a secondary amine at the anisole terminal, while 16d holds a tertiary amine. This key difference altered the selectivity of the ligand significantly. 16b exhibited greater affinity for S2R, while incorporation of a methyl group at the amine reversed the selectivity, by favouring the S1R in 16d. Observing the basicity trends, it appears that again reducing the basicity is well tolerated at the benzofuran and less preferred at the anisole for a potent and selective S1R ligand. The predicted microspecies distribution in each case depicts the secondary and tertiary amines protonated with the highest distribution.
The effect of carbonyl migration to within the ethylenediamine linker was explored in compounds 22a–25b (Table 3). Compounds 22a and 22b were both identified as S1R ligands with Ki = 2.28 and 516 nM respectively. Selectivity was vastly improved with 22a (in contrast to 12b) in particular possessing S1R subtype selectivity of more than 2000 times. When compared to the carbonyl isomer 12b, 22a presented significantly higher affinity and selectivity for S1R. When comparing pKa values at the N-atoms, it was clear that 22a in fact possessed less basic N-atoms in comparison to 12b. 22b followed the general trend of lowered affinities with a carbonyl moiety proximal to the anisole and selectivity was lower at S1R/S2R = 19. Conversion of the secondary amide to a tertiary amide in 23a produced only a slightly lowered affinity and selectivity for S1R when compared to 22a. A tertiary amide in 23b improved the binding affinity at both sites. Introduction of a tertiary amine at the anisole in 24a resulted in a complete loss of affinity for both sites, likewise when a secondary amide was introduced in 25b. Incorporation of a tertiary amine as in 24b when compared with 23b produced an almost two-fold decrease in affinity for S1R. Likewise, when the secondary amine in 23a was methylated (25a) a drop in affinity from Ki = 8.32 nM to 146 nM was observed for S1R and 25a exhibited an increase in S2R affinity which was absent in 23a.
The final four ligands in the series (26a–26d, Table 4) illustrate the effect of secondary amines and tertiary amines in this flexible linker. These ligands all possessed high S1R affinity and selectivity. The highest affinity and selectivity of the four ligands was 26d, with methylation at both N-atoms to yield tertiary amines, these ligands resulted in a reduction in the selectivity for S1R as well when compared with its non-methylated counterpart 26a. A tertiary amine at the benzofuran terminal (26b) exhibited the lowest affinity for S1R in this sub-series and a slight improvement in S2R affinity in comparison to 26a. Conversely, 26c, where the tertiary amine was at the anisole terminus exhibited comparable S1R and S2R affinities with 26d. However, 26d presented the greatest affinity for S1R in the series.
In this study we have demonstrated the effects of inserting a carbonyl moiety at various positions. It has been duly noted that affinity for the S1R was not improved by the modifications reported here. Nevertheless, the purpose of this body of work was to explore the nature of the alkylamine core of S1R ligands and to make associations to the pharmacophore. This has been achieved by systematic modifications to the lead in this series. The structural features for optimal S1R binding as proposed by Glennon and co-workers focussed on the ideal distance between a basic amine and two tethered hydrophobic groups. We have expanded upon this work to show that for high S1R affinity carbonyls/amides are not ideal. This is observed even when a basic amine remains in the structure, confirming that two amine sites are beneficial. Amide introduction at the benzofuran is tolerated to a greater extent than when incorporated closer to the anisole, suggesting the latter is more important as the basic site. Both exocyclic and endocyclic carbonyls are not favoured in the piperazine series, additionally endocyclic carbonyl moieties are less favoured of the two types of cyclic amides, since the internal carbonyl position is capable of lowering the basicity of both N-atoms. Ethylenediamines with increased flexibility have been proven to be suitable substitutes for piperazine, though tis does reduce selectivity. Within this library the compounds lacking a carbonyl moiety (ethylenediamine series three) produced the greatest affinity for S1R, however the purpose of this library was to make associations from the known S1R pharmacophore and the current library. The most significant finding of this study is that the basic N-atom conferring S1R affinity is most likely the one proximal to the anisole and not the one proximal to the benzofuran group. Alternatively, our findings could indicate modes of binding hitherto unknown to us given the simplicity of the diaminergic ligand library.
Future directions for this library will look at the design of S2R selective, high affinity ligands as we have identified that amides at the anisole and the manipulation of this amide can produce higher affinity S2R ligands. This could help in the future design of drugs for the diagnosis of cancer, since S2R are abundant in malignant cells.
General procedure for biological assays
In vitro assays were performed at University of Pennsylvania in Professor R. Mach's laboratory. Tissue homogenates were prepared by processing whole guinea pig brain samples for the S1R, while fresh rat liver was used for the S2R. Homogenate samples were prepared in 50 mM Tris-HCl, pH 7.45 and stored at −80 °C until use. Competitive binding assays were performed in 96 well polypropylene plates (Fisher Scientific, Pittsburgh, PA) with a final volume of 0.2–0.25 mL. [3H](+)-pentazocine (6 nM) was used for S1R assays while [3H]DTG (5 nM) was used for S2R assays. For all S2R studies conducted, 100 nM of (+) pentazocine was added to mask S1R binding sites. A wide range of 10 concentrations (10−10 to 10−5 M) of competing test compounds were evaluated. Competing compounds were serially diluted with buffer (as above) and non-specific binding was defined with haloperidol (10 μM) or DTG (5 μM). Incubation was carried out at room temperature for a period of 120 minutes. After the 120 minute incubation period, separation of the bound from free radio-ligands was conducted by filtration through a 96 well gf per b unifilter (Perkin Elmer, Boston, MA). The filters were washed 3× with ice-cold wash buffer (10 mM Tris-HCl, 150 mM NaCl, pH 7.4). 7 mL of scintillation fluid was added to all filter samples and counted on a Wallac 1450 Microbeta liquid scintillation counter (Perkin Elmer, Boston, MA) with a counting efficiency of ∼45%.Solvents, reagents and conditions
Starting materials, reagents, and solvents were purchased from commercial suppliers and used without further purification, except when acknowledged. Anhydrous solvents were obtained using PureSolv™ (purification system) for MeOH, and via distillation over sodium wire or CaH2 for THF and CH2Cl2, respectively. An atmosphere of argon or nitrogen was used for oxygen or water sensitive reactions. Reaction flasks were oven-dried and flushed with inert gas before use. The progress of reactions was monitored by Thin Layer Chromatography (TLC) on 0.2 mm thick, aluminium-backed, pre-coated silica gel plates (Merck Silica gel 60 F254), with visualisation by KMnO4, vanillin, or ninhydrin solutions followed by heating to stain. Silica for flash column chromatography was of 230–400 mesh particle size. Melting points were determined using a Stuart SMP10 Melting Point apparatus and are uncorrected. Infrared absorption (IR) spectra were obtained using a Bruker ALPHA FT-IR spectrometer, samples (neat) were pressed against a selenium–zinc cell (SeZn). Noteworthy absorbance peaks are provided in wavenumbers (cm−1). 1H NMR and 13C spectra were recorded at a spectrometer frequency of 300, 400 or 500 MHz, 13C NMR spectra at 75, 101 or 125 MHz. NMR experiments were acquired at 300 K using either Bruker DPX200 (200 MHz), DPX300 (300 MHz), DRX400 (400 MHz) or Avance III 500 Ascend (500 MHz) spectrometers. Data reported in chemical shifts δ (ppm) relative to TMS = 0, using residual protons in deuterated solvents as an internal reference, relative integral, multiplicity (s = singlet, br s = broad singlet, d = doublet, dd = doublet of doublets, dt = double of triplets, t = triplet, m = multiplet), and coupling constants (J, Hz). The deuterated solvents: CDCl3, CD3OD, (CD3)2CO and d6-DMSO were used. Low resolution mass spectra (LRMS) were obtained from a ThermoQuest Finnigan LCQ-Deca ion trap mass spectrometer with electro-spray ionisation in positive (+ESI) or negative (−ESI) mode or atmospheric-pressure chemical ionisation in positive mode (+APCI). Data are expressed as observed mass to charge ratio (m/z) and are assigned as molecular ion (M). High resolution mass spectra (HRMS) were obtained from a Bruker 7T Apex Qe Fourier Transform Ion Cyclotron resonance mass spectrometer equipped with an Apollo II ESI/APCI/MALDI Dual source by the Mass Spectrometry Facility of the School of Chemistry at The University of Sydney. The purity of final compounds submitted for in vitro testing was evaluated by HPLC analysis with a final purity of ≥95% for all new compounds. High performance liquid chromatography (HPLC) analysis of final compounds was carried out on a Waters Alliance 2695 instrument using a SunFire™ C18 column (5 μm, 2.1 × 150 mm) and detected via a UV detector set at 254 nm. Separation was achieved using water (solvent A) and acetonitrile (solvent B) at a flow rate of 0.2 mL min−1 and a gradient of 0% B to 100% B over 50 minutes. Compounds were assessed in binding assays as hydrochloride salts. The data is reported as percentage purity and retention time (RT) in minutes. Compounds were assessed in binding assays as hydrochloride salts. Room temperature refers to ambient temperature in the range of 20–30 °C. The synthesis of intermediates 1A–1F are reported in the ESI.† pKa values were calculated at 298 K using ChemAxon's MarvinSketch software version 16.8.29.0.38General procedures for the chemistry
:3 v/v DMF/CH2Cl2 (40 mL) was added Et3N (24.7 mmol, 2 eq.) and the reaction was stirred for 20 min before the amine (13.6 mmol, 1.2 eq.) was added and the reaction was stirred for a further 2 h. The reaction was then diluted with EtOAc (150 mL), washed with 1 M aq. HCl (100 mL), 1 M aq. NaOH (100 mL), brine (150 mL) and was dried (MgSO4) and concentrated in vacuo to obtain the crude product, the resulting residue was purified by flash column chromatography on silica gel, yielding the desired product.
:1 v/v THF/H2O (20 mL) and Na2CO3 (5.54 mmol) was added followed by Boc2O (3.05 mmol) and the reaction was left to stir for 14 h. The reaction was then acidified to pH 3 with 1 M aq. HCl and extracted into EtOAc (3 × 50 mL), and the combined organic fractions washed with brine (30 mL), dried (MgSO4) and concentrated in vacuo to an oil, the resulting residue was purified by flash column chromatography on silica gel (eluent: 1:1 v/v EtOAc/hexane), yielding the desired product.
:0–0:1 v/v hexane:EtOAc), yielding the desired product.
:0–0:1 v/v hexane:EtOAc), yielding the desired product.
:0–0:1 v/v hexane:EtOAc), yielding the desired product.
Synthesis of tert-butyl 4-(4-methoxybenzoyl)piperazine-1-carboxylate 3a. Using general procedure A. 4-Methoxybenzoic acid (215 mg, 1.4 mmol, 1.1 eq.) was reacted with (COCl)2 (0.25 mL, 3.0 mmol, 2 eq.) to form the acid chloride, which was subsequently coupled to tert-butyl piperazine-1-carboxylate (240 mg, 1.3 mmol, 1 eq.) using Et3N (0.62 mL). The crude product was purified by flash column chromatography on silica gel (CH2Cl2
:MeOH, 95:5) giving the titled compound as a colourless oil (317 mg, 70%), which solidified upon drying in vacuo overnight. m.p. 116–117 °C1; IR (ZnSe cell): νmax 2979, 2921, 2844, 1679, 1623, 1414, 1306, 1247 cm−1; H NMR (200 MHz, CDCl3): δ 7.40–7.36 (2H, d, J = 8.8 Hz), 6.93–6.89 (2H, d, J = 8.8 Hz), 3.83 (3H, s), 3.58–3.57 (4H, d, J = 8.0 Hz), 3.46–3.44 (4H, d, J = 8.0 Hz), 1.47 (9H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 170.60 (C), 160.91 (C), 154.60 (C), 129.18 (CH), 127.43 (C), 113.80 (CH), 80.33 (C), 55.38 (CH2), 30.99 (CH2), 28.37 (CH3) ppm; LRMS (+ESI): m/z 343 [M + Na]+.
Synthesis of tert-butyl 4-(2-benzofuranoyl)piperazine-1-carboxylate 3b. 2-Benzofurancarboxylic acid (1.2 g, 7.2 mmol, 1.1 eq.) was reacted with tert-butyl piperazine-1-carboxylate 2 (1.2 g, 6.6 mmol, 1 eq.) according to general procedure A, yielding the titled compound (3b) as a yellow oil (1.95 g, 89%). m.p. 102–103 °C (lit. m.p. 97–98 °C).42IR (ZnSe cell): νmax 2976, 2927, 2862, 1683, 1641, 1556, 1419, 1248 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.59–7.57 (1H, d, J = 7.5 Hz), 7.45–7.44 (1H, d, J = 8.0 Hz), 7.35–7.32 (1H, dd, J = 4.7 Hz), 7.24–7.21 (1H, dd, 3J = 7.5 Hz, 3J = 7.5 Hz), 7.19 (1H, s), 3.77–3.74 (4H, t, J = 5.0 Hz), 3.49–3.47 (4H, t, J = 5.0 Hz), 1.43 (9H, s) ppm; 13C NMR (125 MHz, CDCl3); 160.19 (C), 155.00 (C), 154.82 (C), 149.35 (C), 127.25 (C), 126.77 (CH), 122.88 (CH), 122.48 (CH), 112.52 (CH), 112.04 (CH), 80.53 (C), 44.91 (CH2), 44.11 (CH2), 28.66 (CH3) ppm; LRMS (+APCI): m/z 230.93 [M − Boc + H]+. Spectroscopic data matched that reported in literature.42
Synthesis of 1-(4-methoxybenzoyl)piperazine 4a. Procedure E was used in the carbamate deprotection of N-Boc piperazine 3a (0.6 g, 1.9 mmol), resulting in a colourless oil (302 mg, 73%). IR (ZnSe cell): νmax 3324, 2951, 2914, 2851, 1606, 1513, 1425, 1243, 1173, 1118, 1018, 872, 838 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.38–7.36 (2H, d, J = 8.8 Hz), 6.92–6.89 (2H, d, J = 8.8 Hz), 3.83 (3H, s), 3.58–3.56 (4H, t, J = 4.2 Hz), 2.88–2.86 (4H, q, J = 3.3 Hz), 1.52 (1H, s, NH) ppm; 13C NMR (101 MHz, CDCl3): δ 170.54 (C), 160.85 (C), 129.23 (CH), 128.14 (C), 113.85 (CH), 55.48 (CH3), 46.93 (CH2), 46.42 (CH2) ppm; LRMS (+ESI): m/z 221 [M + H]+.
Synthesis of 1-(2-benzofuranoyl)piperazine 4b. Procedure E was used in the carbamate deprotection of N-Boc piperazine 3b (607 mg, 1.8 mmol) yielding a yellow oil (332 mg, 79%), which solidified upon standing. IR (ZnSe cell): νmax 3292, 2965, 2929, 2867, 1615, 1557, 1431, 1283, 1255, 1221, 1177, 1122, 1014, 915, 862, 819 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.63–7.61 (1H, d, J = 8.4 Hz), 7.52–7.49 (1H, d, J = 8.4 Hz), 7.39–7.34 (1H, t, J = 7.5 Hz), 7.28–7.23 (2H, m), 3.78 (4H, br s), 2.93–2.90 (4H, t, J = 4.8 Hz), 2.00 (1H, s, NH) ppm; 13C NMR (75 MHz, CDCl3): δ 159.64 (C), 154.38 (C), 148.90 (C), 126.80 (C), 126.21 (CH), 123.40 (CH), 122.01 (CH), 111.66 (CH), 111.50 (CH), 66.87 (CH2), 46.14 (CH2) ppm; LRMS (+ESI): m/z 231 [M + H]+.
Synthesis of 1-(4-methoxybenzoyl)-4-(benzofuran-2-ylmethyl)piperazine 5a. Procedure G was used in the reductive amination of 4a (180 mg, 0.8 mmol, 1 eq.) and 2-benzofurancarboxaldehyde (0.13 mL, 1.1 mmol, 1.2 eq.) The residue was purified by flash column chromatography on silica gel (EtOAc
:Hexane, 30:70) to give a colourless oil (216 mg, 75%), which solidified upon standing. IR (ZnSe cell): νmax 3001, 2934, 2814, 1710, 1606, 1426, 1246, 1173, 1022 cm−1; 1H NMR (300 MHz, (CD3OD)): δ 7.58–7.55 (1H, d, J = 7.2 Hz), 7.48–7.46 (1H, d, J = 8.2 Hz), 7.38–7.35 (2H, d, J = 8.2 Hz), 7.29–7.18 (2H, m), 6.96–6.93 (2H, d, J = 8.2 Hz), 3.82 (3H, s), 6.73 (1H, s), 3.74 (2H, s), 3.58 (4H, s), 2.60–2.53 (4H, q, J = 2.8 Hz) ppm; 13C NMR (75 MHz, (CD3OD)): δ 170.27 (C), 161.77 (C), 156.10 (C), 156.02 (C), 130.18 (CH), 129.54 (C), 129.44 (C), 124.93 (CH), 123.69 (CH), 121.84 (CH), 114.48 (CH), 111.90 (CH), 106.41 (CH), 55.82 (CH2), 55.71 (CH2), 53.78 (CH3) ppm; LRMS (+ESI): m/z 351 [M + H]+HRMS(ESI): calc. C21H22N2O3 [M + Na+] = 373.1523, found 373.1523, HPLC: 97.47%, RT: 16.99 min.
Synthesis of 1-(2-benzofuranoyl)-4-(4-methoxybenzyl)piperazine 5b. 4-Methoxybenzaldehyde (108 μL, 0.89 mmol, 1.2 eq.) reacted with benzofuranoyl piperazine 4b (170 mg, 0.74 mmol, 1 eq.) according to general procedure G, the residue was purified by flash column chromatography on silica gel (EtOAc
:Hexane, 30:70), yielding a yellow solid (193 mg, 74%). m.p. 115–116 °C; IR (ZnSe cell): νmax 2924, 2835, 2809, 2760, 1628, 1507, 1423, 1231, 1180, 1028 cm−1;1H NMR (300 MHz, (CD3OD)): δ 7.73–7.71 (1H, d, J = 7.7 Hz), 7.59–7.56 (1H, d, J = 8.2 Hz), 7.45–7.40 (1H, t, J = 7.8 Hz), 7.34–7.28 (3H, m), 6.90–6.88 (2H, d, J = 7.6 Hz), 3.78 (7H, m), 3.48 (2H, s), 2.50–2.46 (4H, t, J = 3.5 Hz) ppm; 13C NMR (75 MHz, (CD3OD)): δ 159.65 (C), 155.11 (C), 150.12 (C), 130.76 (CH), 130.64 (C), 127.81 (C), 126.89 (CH), 124.18 (CH), 122.87 (CH), 114.18 (CH), 112.27 (CH), 111.29 (CH), 62.40 (CH2), 55.23 (CH3), 53.55 (CH2) ppm, 1 carbon obstructed or unresolved; LRMS (+ESI): m/z 351 [M + H]+. HRMS(ESI): calc. C21H22N2O3 [M + Na+] = 373.1523, found 373.1523, HPLC: 99.63%, RT: 17.31 min.
Synthesis of 4-(4-methoxybenzyl)-2-piperazinone 7a. 4-Methoxybenzaldehyde (1.45 mL, 11.9 mmol, 1.2 eq.) reacted with 2-piperazinone 6 (1.05 g, 10.1 mmol, 1.0 eq.) according to general procedure G, yielding the titled compound as a yellow solid (1.48 g, 64%). m.p. 132–133 °C; IR (ZnSe cell): νmax 3238, 2920, 2881, 2839, 1611, 1496, 1229, 1081, 1033 cm−1;1H NMR (300 MHz, CDCl3): δ 7.23–7.20 (2H, d, J = 8.8 Hz), 6.86–6.84 (2H, d, J = 8.7 Hz), 6.34 (1H, br s, NH), 3.79 (3H, s), 3.51 (2H, s), 3.34–3.30 (2H, q, J = 7.9 Hz), 3.13 (2H, s), 2.63–2.59 (2H, t, J = 5.4 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 169.76 (C), 159.19 (C), 130.37 (CH), 129.01 (C), 113.96 (CH), 61.31 (CH2), 57.02 (CH2), 55.41 (CH3), 48.50 (CH2), 41.57 (CH2) ppm; LRMS (+ESI): m/z 463 [2M + H]+.
Synthesis of 4-(benzofuran-2-ylmethyl)-2-piperazinone 7b. 2-Benzofurancarboxaldehyde (150 μL, 1.2 mmol, 1.2 eq.) was reacted with 2-piperazinone 6 (105 mg, 1.0 mmol, 1.0 eq.) according to general procedure G, yielding the titled compound as a white solid (208 mg, 86%). m.p. 128–129 °C; IR (ZnSe cell): νmax 3193, 3085, 281, 1666, 1455, 1344, 980, 744 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.56–7.54 (1H, d, J = 7.2 Hz), 7.50–7.47 (1H, d, J = 8.1 Hz), 7.29–7.21 (2H, m), 6.90 (1H, s), 6.65 (1H, s), 3.80 (2H, s), 3.41–3.37 (2H, q, J = 4.2 Hz), 3.28 (2H, s), 2.79–2.75 (2H, t, J = 5.4 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 169.45 (C), 155.21 (C), 153.17 (C), 128.10 (C), 124.37 (CH), 122.93 (CH), 120.95 (CH), 111.36 (CH), 106.29 (CH), 56.52 (CH2), 54.08 (CH2), 48.59 (CH2), 41.26 (CH2) ppm; LRMS (+ESI): m/z 253 [M + Na]+.
Synthesis of 1-(benzofuran-2-ylmethyl)-4-(4-methoxybenzyl)-2-piperazinone 8a. 4-(4-Methoxybenzyl)-2-piperazinone 7a (380 mg, 1.76 mmol, 1 eq.) was reacted with 2-(chloromethyl)benzofuran (381 mg, 2.29 mmol, 1.3 eq.) according to general procedure F, the residue was purified by flash column chromatography on silica gel (EtOAc
:Hexane, 30:70) yielding the titled compound as a yellow oil (568 mg, 90%).
IR (ZnSe cell): νmax 2931, 2803, 1734, 1650, 1511, 1454, 1239, 1171, 1032 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.39–7.37 (1H, d, J = 7.4 Hz), 7.29–7.26 (1H, d, J = 8.0 Hz), 7.13–7.02 (4H, m), 6.73–6.70 (2H, d, J = 8.8 Hz), 6.56 (1H, s), 4.55 (2H, s), 3.60 (3H, s), 3.31 (2H, s), 3.23–3.19 (2H, t, J = 5.4 Hz), 2.47 (2H, s), 2.49–2.45 (2H, t, J = 5.7 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 169.20 (C), 160.62 (C), 156.40 (C), 154.10 (C), 131.57 (CH), 129.66 (C), 129.49 (C), 125.40 (CH), 123.98 (CH), 122.02 (CH), 114.83 (CH), 111.99 (CH), 106.46 (CH), 61.78 (CH2), 57.71 (CH3), 55.66 (CH2), 48.15 (CH2), 47.63 (CH2), 43.79 (CH2) ppm; LRMS (+ESI): m/z 373 [M + Na]+. HRMS(ESI): calc. C21H22N2O3 [M + Na+] = 373.1523, found 373.1522, HPLC: 96.68%, RT: 17.89 min.
Synthesis of 1-(4-methoxybenzyl)-4-(benzofuran-2-ylmethyl)-2-piperazinone 8b. 4-(2-Methylbenzofuranyl)-2-piperazinone 7b (100 mg, 0.44 mmol, 1 eq.) was N-alkylated using 4-methoxybenzyl chloride (71 μL, 0.52 mmol, 1.2 eq.) according to general procedure F. The resulting residue was purified by flash column chromatography on silica gel (EtOAc
:Hexane, 80:20) yielding a colourless film (54 mg, 34%). IR (ZnSe cell): νmax 3509, 3001, 2962, 2913, 1710, 1522, 1423, 1358, 1220, 1092 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.54–7.52 (1H, d, J = 7.2 Hz), 7.43–7.41 (1H, d, J = 7.8 Hz), 7.17–7.14 (2H, d, J = 8.8 Hz), 6.86–6.83 (2H, d, J = 8.4 Hz), 6.70 (1H, s), 4.84 (2H, s), 4.48 (2H, s), 3.74 (3H, s), 3.74 (2H, s), 3.67 (2H, s), 3.23–3.20 (2H, t, J = 4.5 Hz), 2.74–2.71 (2H, t, J = 5.6 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 170.75 (C), 159.87 (C), 155.77 (C), 155.12 (C), 130.13 (CH), 129.15 (C), 124.77 (CH), 123.45 (CH), 121.63 (CH), 111.64 (CH), 114.57 (CH), 106.39 (CH), 60.39 (CH2), 55.36 (CH3), 54.12 (CH2), 50.04 (CH2), 48.80 (CH2), 46.37 (CH2) ppm, 1 carbon obstructed or unresolved; LRMS (+ESI): m/z 351 [M + H]+. HRMS(ESI): calc. C21H22N2O3 [M + Na+] = 373.1523, found 373.1521, HPLC: 98.32%, RT: 14.42 min.
Synthesis of tert-butyl (2-aminoethyl)carbamate 9b. Ethylenediamine 9a (3.66 mL, 55.3 mmol, 6 eq.) was Boc protected according to general procedure D. The residue was purified by flash column chromatography on silica gel (EtOAc
:MeOH:H2O:NH4OH, 70:20:9.5:0.5) yielding a colourless oil (1.33 g, 91%).
IR (ZnSe cell): νmax 3358, 2983, 2934, 1682, 1526, 1455, 1366, 1162, 971 cm−1; 1H NMR (300 MHz, CDCl3): δ 5.93 (1H, s, NH), 3.16 (2H, br s), 2.83–2.80 (2H, t, J = 8.9 Hz), 1.34 (2H, s, NH2), 1.30 (9H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 156.44 (C), 79.20 (C), 41.70 (CH2), 40.53 (CH2), 28.30 (CH3) ppm; LRMS (+ESI): m/z 161 [M + H]+. The spectroscopic data matched that reported in the literature.43
Synthesis of N-tert-butoxycarbonyl -N′-(4-methoxybenzoyl)ethylenediamine 10a. 4-Methoxybenzoic acid (1.14 g, 7.5 mmol, 1.2 eq.) was reacted with 9b (1.00 g, 6.2 mmol, 1 eq.) according to general procedure A. The residue was purified by flash column chromatography on silica gel (EtOAc
:Hexane, 30:70), yielding the titled compound as white solid (1.53 mg, 83%). m.p. 111–112 °C; IR (ZnSe cell): νmax 3366, 3318, 2979, 2929, 1683, 1633, 1531, 1458, 1331, 1254, 1167, 1023, 848 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.75–7.72 (2H, d, J = 8.8 Hz), 7.39 (1H, s), 6.83–6.80 (2H, d, J = 8.8 Hz), 5.48–5.44 (1H, t, J = 5.7 Hz), 3.76 (3H, s), 3.49–3.44 (2H, q, J = 5.5 Hz), 3.32–3.28 (2H, q, J = 3.5 Hz,), 1.36 (9H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 167.53 (C), 162.01 (C), 157.34 (C), 128.82 (CH), 126.43 (C), 113.49 (CH), 79.53 (C), 55.24 (CH3), 41.54 (CH2), 40.03 (CH2), 28.27 (CH3) ppm; LRMS (+ESI): m/z 317 [M + Na]+. The spectroscopic data matched that reported in the literature.44
Synthesis of N-(benzofuran-2-oyl)-N′-tert-butoxycarbonyl-ethylenediamine 10b. 2-Benzofurancarboxylic acid (3.65 g, 22.5 mmol, 1.2 eq.) was reacted with 9b (3.17 g, 18.7 mmol, 1 eq.) according to general procedure A. The residue was purified by flash column chromatography on silica gel (EtOAc
:Hexane, 30:70), yielding the titled compound as a white solid (4.94 g, 87%). IR (ZnSe cell): νmax 3306, 2972, 2930, 1693, 1652, 1516, 1433, 1364, 1281, 1168, 1110, 939, 828 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.67–7.65 (1H, d, J = 8.0 Hz), 7.80 (1H, s), 7.48–7.38 (3H, m), 7.29–7.27 (1H, d, J = 4.8 Hz),), 5.02 (1H, s), 3.62–3.57 (2H, q, J = 3.7 Hz), 3.44–3.40 (2H, q, J = 3.6 Hz), 1.43 (9H, s) ppm; 13C NMR (75 MHz, CDCl3): 159.56 (C), 156.93 (C), 153.34 (C), 148.64 (C), 127.42 (C), 126.72 (CH), 123.53 (CH), 122.50 (CH), 111.65 (CH), 110.17 (CH), 79.49 (C), 40.35 (CH2), 40.10 (CH2), 28.29 (CH3) ppm; LRMS (+ESI): m/z 327 [M + Na]+. The spectroscopic data matched that reported in the literature.28
Synthesis of N-(4-methoxybenzoyl)ethylenediamine 11a. Boc deprotection of N-Boc ethylenediamine 10a (620 mg, 2.1 mmol) was carried out according to general procedure E, yielding a colourless oil (283 mg, 70%). IR (ZnSe cell): νmax 3289, 2915, 2840, 1632, 1505, 1444, 1248, 1181, 1029, 846 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.79–7.76 (2H, d, J = 8.8 Hz), 7.26 (1H, s, NH), 6.87–6.84 (2H, d, J = 8.8 Hz), 3.79 (3H, s), 3.48–3.42 (2H, q, J = 5.7 Hz), 2.91–2.87 (2H, t, J = 7.0 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 167.44 (C), 161.96 (C), 128.78 (CH), 126.66 (C), 113.52 (CH), 66.96 (CH2), 55.25 (CH3), 41.24 (CH2) ppm; LRMS (+ESI): m/z 195 [M + H]+. The spectroscopic data matched that reported in the literature.45
Synthesis of N-(2-benzofuranoyl)ethylenediamine 11b. Boc deprotection of N-Boc ethylenediamine 10b (1.34 g, 4.4 mmol) was carried out according general procedure E, yielding a colourless solid (0.82 g, 91%). IR (ZnSe cell): νmax 3305, 3147, 3028, 2927, 1644, 1596, 1520, 1287, 1257, 1227, 1175, 1106, 884 cm−1; 1H NMR (300 MHz, CDCl3): δ 8.04 (1H, s), 7.53–7.52 (1H, d, 4J = 1.6 Hz), 7.43–7.41 (1H, d, J = 7.8 Hz,), 7.28–7.03 (3H, m), 3.39–3.36 (2H, q, J = 3.2 Hz), 2.85–2.81 (2H, t, J = 5.9 Hz), 2.52 (2H, br s, NH2) ppm; 13C NMR (75 MHz, CDCl3): δ 159.45 (C), 154.86 (C), 148.84 (C), 127.69 (C), 126.94 (CH), 123.76 (CH), 122.79 (CH), 111.86 (CH), 110.47 (CH), 41.50 (CH2), 41.31 (CH2) ppm; LRMS (+ESI): m/z 205 [M + H]+. The spectroscopic data matched that reported in the literature.28
Synthesis of N-(benzofuran-2-ylmethyl)-N′-(4-methoxybenzoyl)-ethylenediamine 12a. 2-Benzofurancarboxaldehyde (1.20 mL, 10.2 mmol, 1 eq.) was reacted with 11a (1.97 g, 10.2 mmol, 1 eq.) according to the general procedure G. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97), yielding a white solid (1.72 g, 52%). m.p. 60–61 °C; IR (ZnSe cell): νmax 3343, 3290, 2889, 2831, 1622, 1504, 1454, 12534, 1174, 1102, 1020, 850 cm−1;1H NMR (300 MHz, CDCl3): δ 7.90 (1H, s), 7.80–7.77 (2H, d, J = 8.8 Hz), 7.53–7.50 (1H, d, J = 7.6 Hz), 7.40–7.38 (1H, d, J = 8.2 Hz), 7.22–7.17 (2H, m), 6.98–6.95 (2H, d, J = 8.8 Hz), 6.68 (1H, s), 3.95 (2H, s), 3.84 (3H, s), 3.54–3.50 (2H, t, J = 6.3 Hz), 2.89–2.85 (2H, t, J = 1.8 Hz) ppm, one NH not observed; 13C NMR (75 MHz, CDCl3) δ 167.29 (C), 162.11 (C), 156.41 (C), 154.94 (C), 128.83 (C), 128.38 (C), 126.89 (C), 123.93 (CH), 122.75 (CH), 120.80 (CH), 113.68 (CH), 111.05 (CH), 104.02 (CH), 55.37 (CH3), 47.88 (CH2), 46.14 (CH2), 39.44 (CH2) ppm; LRMS (+ESI): m/z 325 [M + H]+. HRMS(ESI): calc. C19H20N2O3 [M + Na+] = 347.1366, found 347.1369, HPLC: 99.27%, RT: 17.24 min.
Synthesis of N-(benzofuran-2-oyl)-N′-(4-methoxybenzyl)-ethylenediamine 12b. 4-Methoxybenzaldehyde (0.75 mL, 6.1 mmol, 1 eq.) was reacted with 11b (1.25 g, 6.1 mmol, 1 eq.) according the general procedure G. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97), yielding the titled compound as a white solid (0.73 g, 37%). m.p. 80–81 °C; IR (ZnSe cell): νmax 3342, 2952, 2835, 1643, 1595, 1506, 1448, 1240, 1174, 1104, 1032, 841, 819 cm−1;1H NMR (300 MHz, CD3OD): δ 7.69–7.67 (1H, d, J = 8.0 Hz), 7.56–7.53 (1H, d, J = 8.2 Hz), 7.48–7.31 (3H, m), 7.28–7.25 (2H, d, J = 7.2 Hz), 6.85–6.82 (2H, d, J = 8.4 Hz), 3.72 (3H, s), 3.71 (2H, s), 3.53–3.57 (2H, t, J = 6.5 Hz), 3.31 (1H, s), 2.84–2.80 (2H, t, J = 6.5 Hz) ppm; 13C NMR (75 MHz, CD3OD): δ 161.39 (C), 160.37 (C), 151.23 (C), 149.95 (C), 132.33 (C), 130.77 (CH), 128.76 (C), 128.13 (CH), 124.83 (CH), 123.71 (CH), 114.82 (CH), 112.73 (CH), 111.21 (CH), 53.49 (CH2), 48.16 (CH2), 39.74 (CH2) ppm, one carbon obscured or unresolved; LRMS (+ESI): m/z 325 [M + H]+. HRMS(ESI): calc. C19H20N2O3 [M + H+] = 325.1547, found 325.1546, HPLC: 98.66%, RT: 17.81 min.
Synthesis of N-(benzofuran-2-ylmethyl)-N′-(4-methoxybenzoyl)-N-methyl-ethylenediamine 13a. A suspension of diamine 12a (500 mg, 1.5 mmol, 1 eq.) was reacted with formalin (37% aq., 185 μL, 2.3 mmol, 1.5 eq.) according to general procedure G. The residue was purified by flash column chromatography on silica gel (MeOH–CHCl3, 1
:99), yielding the titled compound as colourless oil (225 mg, 48%). IR (ZnSe cell): νmax 3355, 2958, 2798, 1634, 1604, 1536, 1503, 1451, 1253, 1175, 1027, 939, 809 cm−1; 1H NMR (300 MHz, CD3OD): δ 7.78–7.75 (2H, d, J = 8.8 Hz), 7.53–7.50 (1H, d, J = 7.6 Hz), 7.26–7.25 (1H, d, J = 7.6 Hz), 7.40–7.38 (2H, m), 6.98–6.95 (2H, d, J = 8.8 Hz), 6.71 (1H, s), 3.85 (3H, s), 3.82 (2H, s), 3.57–3.53 (2H, t, J = 6.7 Hz), 2.74–2.70 (2H, t, J = 6.7 Hz), 2.41 (3H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 159.06 (C), 158.94 (C), 154.89 (C), 149.07 (C), 130.40 (CH), 129.91 (C), 127.77 (C), 126.85 (CH), 123.71 (CH), 122.79 (CH), 113.90 (CH), 111.83 (CH), 110.16 (CH), 61.68 (CH2), 55.28 (CH2), 54.97 (CH3), 41.83 (CH2), 36.47 (CH3) ppm; LRMS (+ESI): m/z 339 [M + H]+. HRMS(ESI): calc. C20H22N2O3 [M + Na+] = 361.1523, found 361.1525, HPLC: 99.36%, RT: 17.34 min.
Synthesis of N-(benzofuran-2-oyl)-N′-(4-methoxybenzyl)-N′-methyl-ethylenediamine 13b. 12b (400 mg, 1.2 mmol, 1 eq.) was reacted with formalin (37% aq., 145 μL, 1.9 mmol, 1.5 eq.) according to the general procedure G. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97), yielding the titled compound as colourless oil (106 mg, 25%). IR (ZnSe cell): νmax 3341, 2941, 2835, 2795, 1651, 1550, 1508, 1447, 1295, 1240, 1173, 1030, 847 cm−1; 1H NMR (300 MHz, CD3OD): δ 7.67–7.65 (1H, d, J = 7.6 Hz), 7.55–7.53 (1H, d, J = 7.6 Hz), 7.48–7.36 (2H, m), 7.29–7.24 (1H, t, J = 7.5 Hz), 7.21–7.18 (2H, d, J = 8.4 Hz), 6.78–6.75 (2H, d, J = 8.4 Hz), 3.51–3.49 (2H, t, J = 3.3 Hz), 3.66 (3H, s), 3.46 (2H, s), 2.59–2.55 (2H, t, J = 6.7 Hz), 2.25 (3H, s) ppm, NH proton not observed; 13C NMR (75 MHz, CD3OD): δ 160.93 (C), 160.31 (C), 156.27 (C), 149.96 (C), 131.57 (CH), 131.08 (C), 128.75 (C), 128.11 (CH), 124.82 (CH), 123.70 (CH), 114.59 (CH), 112.70 (CH), 110.40 (CH), 62.36 (CH2), 56.38 (CH2), 55.54 (CH2), 42.35 (CH3), 37.85 (CH3) ppm; LRMS (+ESI): m/z 339 [M + H]+. HRMS(ESI): calc. C20H22N2O3 [M + Na+] = 361.1523, found 361.1523, HPLC: 99.62%, RT: 17.28 min.
Synthesis of tert-butyl (benzofuran-2-ylmethyl)(2-(4-methoxybenzamido)ethyl)carbamate 14a. Amine 12a (1.00 g, 2.77 mmol) was carbamate protected using Boc2O (665 mg, 3.05 mmol) according to general procedure D. The crude viscous product was purified by flash column chromatography on silica gel (eluent: 1
:1 EtOAc/hexane v/v) to obtain 14a as an amorphous solid (1.10 g, 97%). 1H NMR (500 MHz, CDCl3): δ 7.77–7.76 (2H, d, J = 8.4 Hz), 7.51–7.49 (1H, d, J = 7.8 Hz), 7.41–7.39 (1H, d, J = 8.7 Hz), 7.26–7.18 (2H, m), 6.88–6.86 (2H, d, J = 7.9 Hz), 6.55 (1H, s), 4.53 (2H, s), 3.82 (3H, s), 3.62 (4H, br s), 1.45 (9H, s) ppm, NH proton not observed; 13C NMR (101 MHz, CDCl3) δ 167.23 (C), 162.17 (C), 157.14 (C), 155.00 (C), 154.34 (C), 128.86 (C), 128.22 (C), 126.63 (C), 124.25 (CH), 122.93 (CH), 120.97 (CH), 113.70 (CH), 111.24 (CH), 104.33 (CH), 81.11 (C), 55.44 (CH3), 46.12 (CH2), 45.09 (CH2), 40.18 (CH2), 28.41 (CH3) ppm; LRMS (+ESI): m/z 425 [M + H]+.
Synthesis of tert-butyl (2-(benzofuran-2-carboxamido)ethyl)(4-methoxybenzyl)carbamate 14b. Amine 12b (1.00 g, 2.77 mmol) was carbamate protected using Boc2O (665 mg, 3.05 mmol) according to general procedure D. The crude viscous product was purified by flash column chromatography (eluent: 3
:2 → 1:1 v/v hexane/EtOAc) to afford 14b as an amorphous solid (1.14 g, quant.). 1H NMR (400 MHz, CDCl3) δ 7.67–7.65 (1H, d, J = 7.8 Hz), 7.54–7.38 (3H, m), 7.29–7.26 (1H, m), 7.18 (2H, s), 6.85–6.83 (2H, d, J = 8.4 Hz) 4.41 (2H, s), 3.75 (3H, s), 3.57–3.52 (4H, m), 1.48 (9H, s) ppm, NH proton not observed; 13C NMR (101 MHz, CDCl3) δ 159.11 (C), 157.25 (C), 156.13 (C), 154.88 (C), 149.05 (C), 130.03 (C), 128.84 (C), 127.72 (CH), 126.82 (CH), 123.68 (CH), 122.72 (CH), 114.16 (CH), 111.88 (CH), 109.98 (CH), 80.68 (C), 55.31 (CH3), 50.47 (CH2), 45.00 (CH2), 39.12 (CH2), 28.48 (CH3) ppm; LRMS (+ESI): m/z 448 [M + Na]+.
tert-Butyl(benzofuran-2-ylmethyl)(2-(4-methoxy-N-methylbenzamido)ethyl)carbamate 15a. Amine 14a (0.921 g, 2.25 mmol) was alkylated according to general procedure F to obtain 15a as a colourless oil, which was purified by flash column chromatography on silica gel (eluent: 3
:2 → 1:1 v/v hexane/EtOAc) to give 15a as a colourless oil (0.950 g, quant.). 1H NMR (500 MHz, CDCl3): δ 7.51–7.49 (1H, d, J = 7.4 Hz), 7.42–7.435 (3H, m), 7.26–7.18 (2H, dt, J = 24.64, 7.38 Hz), 6.88–6.87 (2H, d, J = 8.9 Hz), 6.57 (1H, s), 4.61 (2H, s), 3.81 (3H, s), 3.74–3.48 (4H, m), 3.08 (3H, s), 1.45 (9H, s) ppm; 13C NMR (101 MHz, CDCl3) δ 171.63 (C), 160.69 (C), 155.72 (C), 155.00 (C), 129.21 (C), 128.31 (C), 124.13 (CH), 122.82 (CH), 120.91 (CH), 113.61 (CH), 111.19 (CH), 104.22 (CH), 80.67 (C), 55.39 (CH3), 49.56 (CH2), 45.32 (CH2), 44.23 (CH3), 38.35 (CH2), 28.47 (CH3) ppm, one obscured or unresolved carbon; LRMS (+ESI): m/z 439 [M + H]+.
tert-Butyl (4-methoxybenzyl)(2-(N-methylbenzofuran-2-carboxamido)ethyl)carbamate 15b. Amine 14b (0.956 g, 2.34 mmol) was alkylated according to general procedure F to obtain 15b as a colourless oil, which was purified by flash column chromatography on silica gel (eluent: 3
:2 → 1:1 v/v hexane/EtOAc) to give 15b as a colourless oil (0.948 g, 96%.). 1H NMR (500 MHz, CDCl3): δ 7.64–7.63 (1H, d, J = 8.2 Hz), 7.49 (1H, m), 7.39–7.37 (1H, t, J = 7.5 Hz), 7.29–7.28 (2H, m), 7.20 (1H, s), 7.08 (1H, s), 6.82 (2H, m), 4.43 (2H, s), 3.74 (3H, s), 3.49–3.11 (7H, m), 1.42 (9H, s) ppm; 13C NMR (125 MHz, CDCl3) δ 161.03 (C), 159.03 (C), 155.81 (C), 154.74 (C), 149.13 (C), 130.39 (C), 129.51 (C), 128.98 (CH), 127.09 (CH), 126.52 (CH), 123.62 (CH), 122.37 (CH), 114.03 (CH), 111.94 (CH), 80.09 (C), 55.29 (CH3), 50.29 (CH2), 46.54 (CH2), 43.20 (CH2), 37.39 (CH3), 28.48 (CH3) ppm; LRMS (+ESI): m/z 439 [M + H]+.
Synthesis of N-(2-((benzofuran-2-ylmethyl)amino)ethyl)-4-methoxy-N-methylbenzamide 16a. Amine 15a (0.850 g, 2.34 mmol) was carbamate deprotected according to general procedure E to obtain 16a as the white HCl salt (0.581 g, 80%). 1H NMR (400 MHz, CD3OD) δ 7.66–7.64 (1H, d, J = 7.71 Hz), 7.55–7.48 (3H, m), 7.39–7.26 (2H, dt, J = 34.87, 7.38 Hz), 7.11 (1H, s), 6.99–6.97 (2H, d, J = 8.72 Hz), 4.55 (2H, s), 3.87–3.83 (5H, m), 3.46 (2H, m), 3.10 (3H, s) ppm, NH protons not observed; 13C NMR (101 MHz, CD3OD) δ 175.19 (C), 162.91 (C), 156.87 (C), 148.79 (C), 130.67 (C), 128.98 (CH), 127.96 (C), 126.71 (CH), 124.57 (CH), 122.79 (CH), 114.76 (CH), 112.32 (CH), 110.40 (CH), 68.09 (CH2), 55.91 (CH3), 46.51 (CH2), 44.98 (CH3), 38.91 (CH2) ppm; LRMS (+ESI): m/z 361 [M + Na]+. HRMS(ESI): calc. C20H22N2O3 [M + Na+] = 361.1523, found 361.1524, HPLC: 97.38%, RT: 17.37 min.
Synthesis of N-(2-((4-methoxybenzyl)amino)ethyl)-N-methylbenzofuran-2-carboxamide 16b. Amine 15b (0.785 g, 1.79 mmol) was carbamate deprotected according to general procedure E to obtain 16b as the white HCl salt (0.516 g, 77%). 1H NMR (400 MHz, CD3OD) δ 7.74–7.72 (1H, d, J = 7.84 Hz), 7.58–7.43 (5H, m), 7.35–7.31 (1H, t, J = 7.84 Hz), 6.99–6.97 (2H, m), 4.24 (2H, s), 3.93 (2H, br s), 3.79 (3H, s), 3.42–3.39 (5H, m) ppm, NH protons not observed; 13C NMR (101 MHz, CD3OD) δ 162.17 (C), 156.21 (C), 132.62 (CH), 128.30 (C), 128.19 (C), 124.98 (CH), 124.17 (CH), 123.70 (CH), 115.55 (CH), 114.30 (CH), 112.76 (CH), 55.82 (CH3), 52.17 (CH3), 46.93 (CH2), 46.30 (CH2), 37.77 (CH2) ppm, two carbon signals obscured or unresolved; LRMS (+ESI): m/z 339 [M + H]+. HRMS(ESI): calc. C20H22N2O3 [M + Na+] = 361.1523, found 361.1523, HPLC: 95.07%, RT: 17.17 min.
N-(2-((Benzofuran-2-ylmethyl)(methyl)amino)ethyl)-4-methoxy-N-methylbenzamide 16c. Amine 16a (0.250 g, 0.67 mmol) alkylated according to general procedure G, and was purified by flash column chromatography on silica gel (eluent: EtOAc
:Et3N 1%), followed by general procedure E to obtain 16c as the white HCl salt (0.289 g, 93%). 1H NMR (400 MHz, CD3OD) δ 7.69–7.67 (1H, d, J = 7.60 Hz), 7.54–7.48 (3H, m), 7.41–7.37 (1H, m), 7.32–7.28 (1H, m), 7.24 (1H, s), 7.00–6.98 (2H, d, J = 8.80 Hz), 4.76 (2H, s), 3.95 (2H, s), 3.84 (3H, s), 3.56 (2H, s), 3.08–3.06 (6H, m) ppm; 13C NMR (101 MHz, CD3OD) δ 175.27 (C), 163.07 (C), 157.09 (C), 147.25 (C), 130.74 (CH), 128.78 (C), 127.94 (C), 127.17 (CH), 124.78 (CH), 123.03 (CH), 114.80 (CH), 112.83 (CH), 112.47 (CH), 55.94 (CH3), 54.94 (CH2), 53.29 (CH3), 45.16 (CH2), 41.70 (CH3), 38.89 (CH2) ppm; LRMS (+ESI): m/z 353 [M + H]+. HRMS(ESI): calc. C21H24N2O3 [M + Na+] = 375.1679, found 375.1679, HPLC: 96.64%, RT: 17.66 min.
Synthesis of N-(2-((4-methoxybenzyl)(methyl)amino)ethyl)-N-methylbenzofuran-2-carboxamide 16d. Amine 16b (0.250 g, 0.67 mmol) was alkylated according to general procedure G, and was purified by flash column chromatography on silica gel (eluent: EtOAc
:Et3N 1%), followed by general procedure E to obtain 16d as the white HCl salt (0.256 g, quant). 1H NMR (400 MHz, CD3OD) δ 7.78–7.76 (1H, d, J = 7.82 Hz), 7.62–7.47 (5H, m), 7.38–7.34 (1H, t, J = 7.45 Hz), 7.03–7.01 (2H, d, J = 8.57 Hz), 4.03 (2H, s), 3.81 (3H, s), 3.54 (2H, br s), 3.43 (2H, br s), 2.93 (3H, s), 1.31 (3H, s) ppm;13C NMR (101 MHz, CD3OD) δ 164.20 (C), 162.59 (C), 161.80 (C), 156.31 (C), 133.81 (CH), 128.43 (C), 128.17 (C), 125.05 (CH), 123.76 (CH), 122.34 (CH), 115.71 (CH), 114.61 (CH), 112.79 (CH), 60.73 (CH3), 55.85 (CH3), 54.62 (CH2), 45.82 (CH2), 40.89 (CH3), 37.78 (CH2) ppm, one carbon obscured or unresolved; LRMS (+ESI): m/z 353 [M + H]+. HRMS(ESI): calc. C21H24N2O3 [M + Na+] = 375.1679, found 375.1678, HPLC: 97.56%, RT: 17.51 min.
Synthesis of methyl (4-methoxybenzyl)glycinate 18a. To a solution of glycine methyl ester (2.76 g, 22.0 mmol) in MeOH (100 mL) was added Et3N (3.05 mL, 22.0 mmol) followed by anisaldehyde (1.78 mL, 14.7 mmol) and the reaction was stirred for 14 h at ambient temperature, before it was cooled to 0 °C and NaBH4 was added portionwise over 20 min. The reaction was subsequently stirred for 1 h at 0 °C, then 1 h at ambient temperature before being diluted with 1 M aq. HCl (100 mL). The mixture was then washed with Et2O (100 mL), and the aqueous layer was basified to pH >11 with 1 M aq. NaOH and extracted into CH2Cl2 (3 × 100 mL). The combined organic fractions were dried (MgSO4) and concentrated in vacuo to an oil which was purified by flash column chromatography on silica gel (eluent 1
:1 → 2:1 EtOAc/hexane + 1% Et3N) to afford amine 18a as a colourless oil (2.30 g, 75%). 1H NMR (500 MHz, CDCl3) δ 7.25–7.18 (2H, m), 6.86–6.85 (2H, d, J = 8.89 Hz), 3.89 (2H, s), 3.79 (3H, s), 3.72 (2H, s), 3.47 (3H, s) ppm, NH proton not observed; 13C NMR (125 MHz, CDCl3) δ 172.75 (C), 163.39 (C), 130.17 (CH), 127.16 (C), 114.46 (CH), 55.46 (CH3), 52.65 (CH2), 51.98 (CH3), 49.20 (CH2) ppm; LRMS (+ESI): m/z 232 [M + Na]+.
Synthesis of methyl (benzofuran-2-ylmethyl)glycinate 18b. Glycine methyl ester 17 (2.58 g, 20.5 mmol) was reacted with 2-benzofurancarboxaldehyde (1.66 mL, 13.7 mmol) according to general procedure H to obtain the crude oil, which was purified by flash column chromatography on silica gel (eluent 2
:1 → 1:1 hexane/EtOAc + 1% Et2NH) to afford amine 18b as a colourless oil (1.80 g, 60%). 1H NMR (500 MHz, CDCl3) δ 7.52–7.51 (1H, 2, J = 7.4 Hz), 7.45–7.44 (1H, d, J = 7.92 Hz), 7.26–7.18 (2H, m), 6.59 (1H, s), 3.99 (2H, s), 3.71 (3H, s), 3.48 (2H, s); 13C NMR (125 MHz, CDCl3) δ 172.41 (C), 155.45 (C), 155.18 (C), 128.37 (C), 124.18 (CH), 122.85 (CH), 120.94 (CH), 111.28 (CH), 104.69 (CH), 52.05 (CH3), 49.49 (CH2), 45.96 (CH2); LRMS (+ESI): m/z 220 [M + H]+.
Synthesis of N-(tert-butoxycarbonyl)-N-(4-methoxybenzyl)glycine 19a. To a solution of amine 18a (2.13 g, 10.2 mmol) in THF (15 mL) was added 1 M aq. NaOH (20.4 mL, 20.4 mmol), and the reaction stirred for 1.5 h. A solution of 6 M aq. HCl (3.4 mL, 20.4 mmol) was subsequently added, followed by Na2CO3 (2.16 g, 20.4 mmol), and then Boc2O (2.44 g, 11.2 mmol) and the reaction was left to stir for 14 h. The reaction was then acidified to pH 3 with 1 M aq. HCl (25 mL) and extracted into EtOAc (3 × 90 mL), and the combined organic fractions washed with brine (50 mL), dried (MgSO4) and concentrated in vacuo to an oil which was purified by flash column chromatography on silica gel (eluent: EtOAc + 1% AcOH) to afford acid 19a as a colourless oil which crystallised on standing (2.89 g, 96%). 1H NMR (400 MHz, CDCl3) δ 9.76 (1H, br s), 7.19–7.14 (2H, m); δ 6.87–6.85 (2H, d, J = 6.8 Hz); δ 4.48–4.44 (2H, d, J = 13.56 Hz), 3.93 (2H, s), 3.79 (3H, s), 1.49 (9H) ppm 13C NMR (101 MHz, CDCl3) δ 175.82 (C), 159.22 (C), 156.16 (C), 129.70 (C), 129.08 (CH), 114.16 (CH), 81.20 (C), 55.39 (CH3), 50.38 (CH2), 47.41 (CH2), 28.48 (CH3) ppm; LRMS (+ESI): m/z 296 [M + H]+.
Synthesis of N-(benzofuran-2-ylmethyl)-N-(tert-butoxycarbonyl)glycine 19b. To a solution of amine 18b (1.42 g, 6.47 mmol) in THF (10 mL) was added 1 M aq. NaOH (13.0 mL, 13.0 mmol), and the reaction stirred for 1.5 h. A solution of 6 M aq. HCl (2.17 mL, 13.0 mmol) was subsequently added, followed by Na2CO3 (1.37 g, 13.0 mmol), and then Boc2O (1.55 g, 7.12 mmol) and the reaction was left to stir for 14 h. The reaction was then acidified to pH 3 with 1 M aq. HCl (15 mL) and extracted into EtOAc (3 × 70 mL), and the combined organic fractions washed with brine (40 mL), dried (MgSO4) and concentrated in vacuo to an oil, which was purified by flash column chromatography on silica gel (eluent: EtOAc + 1% AcOH) to afford acid 19b as a viscous yellow oil (1.92 g, 97%). 1H NMR (400 MHz, CDCl3) δ 8.57 (1H, br s), 7.54–7.52 (1H, d, J = 7.27 Hz), 7.46–7.44 (1H, d, J = 7.56 Hz), 7.27–7.16 (2H, m), 6.66 (1H, s), 4.67 (2H, s), 4.03 (2H, s), 1.47 (9H, s) ppm; 13C NMR (101 MHz, CDCl3) δ 175.176 (C), 155.67 (C), 155.17 (C), 153.66 (C), 129.17 (CH), 125.44 (C), 124.43 (CH), 121.09 (CH), 111.32 (CH), 105.56 (CH), 81.47 (C), 47.99 (CH2), 44.55 (CH2), 28.35 (CH3); LRMS (+ESI): m/z 306 [M + H]+.
tert-Butyl (2-((benzofuran-2-ylmethyl)amino)-2-oxoethyl)(4-methoxybenzyl)carbamate 20a. Protected glycine 19a (325 mg, 1.10 mmol) was coupled with amine 1E (195 mg, 1.32 mmol) according to general procedure B. The product was purified by flash column chromatography on silica gel (eluent: 3
:2 hexane/EtOAc) to afford the title compound 20a as a pale yellow oil (251 mg, 54%). This compound was carried through to the next step.
tert-Butyl (benzofuran-2-ylmethyl)(2-((4-methoxybenzyl)amino)-2-oxoethyl)carbamate 20b. Protected glycine 19b (1.00 g, 3.28 mmol) was coupled with 4-methoxybenzylamine (511 μL, 3.94 mmol) according to general procedure B. The product was purified by flash column chromatography on silica gel (eluent: 3
:2 → 1:1 hexane/EtOAc) to afford the title compound 20b as a pale yellow oil (1.04 g, 71%). This compound was carried through to the next step.
tert-Butyl (2-((benzofuran-2-ylmethyl)(methyl)amino)-2-oxoethyl)(4-methoxybenzyl)carbamate 21a. Protected glycine 19a (540 mg, 1.83 mmol) was coupled with amine 1F (354 mg, 2.20 mmol) according to general procedure B. The product was purified by flash column chromatography on silica gel (eluent: 3
:2 → 1:1 hexane/EtOAc) to afford the title compound 21a as a pale yellow oil (576 mg, 72%). This compound was carried through to the next step.
tert-Butyl (benzofuran-2-ylmethyl)(2-((4-methoxybenzyl)(methyl)amino)-2-oxoethyl)carbamate 21b. Protected glycine 19b (336 mg, 1.10 mmol) was coupled with amine 4-methoxy-N-benzyl-methanamine (200 mg, 1.32 mmol) according to general procedure B. The product was purified by flash column chromatography on silica gel (eluent: 3
:2 → 1:1 hexane/EtOAc) to afford the title compound 21b as a pale yellow oil (365 mg, 73%). This compound was carried through to the next step.
Synthesis of N-(benzofuran-2-ylmethyl)-2-((4-methoxybenzyl)amino)acetamide 22a. Amine 20a (245 mg, 577 μmol) was Boc deprotected according to general procedure E to give 22a as the HCl salt (174 mg, 84%). The spectral data for the free amine is provided. 1H NMR (400 MHz, CDCl3) δ 7.51–7.49 (1H, m), 7.39–7.38 (1H, d, J = 8.24 Hz), 7.27–7.16 (4H, m), 6.86–6.83 (2H, m), 6.53 (1H, s), 4.36–4.35 (2H, d, J = 5.86 Hz), 3.88 (2H, s), 3.78 (3H, s), 3.37 (2H, s), 1.88 (1H, br s) ppm, one NH not observed; 13C NMR (101 MHz, CDCl3) δ 171.10 (C), 159.12 (C), 155.59 (C), 155.11 (C), 130.52 (C), 129.19 (CH), 128.30 (C), 124.26 (CH), 122.93 (CH), 121.01 (CH), 114.19 (CH), 111.17 (CH), 104.53 (CH), 55.42 (CH3), 51.76 (CH2), 46.71 (CH2), 42.66 (CH2) ppm; LRMS (+ESI): m/z 325 [M + H]+.HRMS(ESI): calc. C19H20N2O3 [M + Na+] = 347.1366, found 347.1366, HPLC: 99.89%, RT: 17.19 min.
Synthesis of 2-((benzofuran-2-ylmethyl)amino)-N-(4-methoxybenzyl)acetamide 22b. Amine 20b (505 mg, 1.2 mmol) was Boc deprotected according to general procedure E to give 22b as the HCl salt (399 mg, 93%). 1H NMR (400 MHz, CD3OD) δ 7.67–7.65 (1H, d, J = 7.42 Hz), 7.56–7.54 (1H, d, J = 7.42 Hz), 7.41–7.37 (1H, t, J = 7.42 Hz), 7.32–7.28 (1H, t, J = 7.62 Hz), 7.24–7.22 (2H, d, J = 7.62 Hz), 7.07 (1H, s), 6.90–6.87 (2H, d, J = 7.62 Hz), 4.53 (2H, s), 4.35 (2H, s), 3.91 (2H, s), 3.78 (3H, s) ppm, NH protons not observed; 13C NMR (101 MHz, CD3OD) δ 164.36 (C), 159.21 (C), 155.58 (C), 147.15 (C), 129.79 (C), 128.74 (CH), 127.56 (C), 125.39 (CH), 123.21 (CH), 121.42 (CH), 113.60 (CH), 110.98 (CH), 109.27 (CH), 54.33 (CH3), 43.14 (CH2), 42.37 (CH2) ppm. One obscured or unresolved carbon; LRMS (+ESI): m/z 347 [M + Na]+. HRMS(ESI): calc. C19H20N2O3 [M + Na+] = 347.1366, found 347.1366, HPLC: 99.49%, RT: 17.42 min.
Synthesis of N-(benzofuran-2-ylmethyl)-2-((4-methoxybenzyl)amino)-N-methylacetamide 23a. Amine 21a (575 mg, 1.3 mmol) was Boc deprotected according to general procedure E to give 23a as the HCl salt (482 mg, 98%). 1H NMR (500 MHz, d6-DMSO) δ 7.63–7.59 (1H, m), 7.53–7.47 (3H, m), 7.31–7.22 (2H, m), 6.97–6.93 (2H, m), 6.86 (1H, s), 4.74–4.71 (2H, d, J = 14.57 Hz), 4.11–4.07 (4H, m), 3.76–3.75 (3H, m), 2.99–2.95 (3H, d, J = 18.45 Hz) ppm, NH proton not observed; 13C NMR (125 MHz, d6-DMSO) δ 165.43 (C), 159.71 (C), 154.30 (C), 153.28 (C), 131.86 (C), 131.78 (CH), 127.82 (C), 124.23 (CH), 123.45 (CH), 120.95 (CH), 113.92 (CH), 110.98 (CH), 105.06 (CH), 55.17 (CH3), 49.49 (CH2), 46.08 (CH2), 43.81 (CH2), 33.97 (CH3) ppm; LRMS (+ESI): m/z 339 [M + H]+. HRMS(ESI): calc. C20H22N2O3 [M + Na+] = 361.1523, found 373.1523, HPLC: 95.27%, RT: 19.03 min.
Synthesis of 2-((benzofuran-2-ylmethyl)amino)-N-(4-methoxybenzyl)-N-methylacetamide 23b. Amine 21b (320 mg, 730 μmol) was Boc deprotected according to general procedure E to give 23b as the HCl salt (241 mg, 88%). 1H NMR (400 MHz, CD3OD) δ 7.69–7.65 (1H, m), 7.58–7.52 (1H, m), 7.42–7.38 (1H, t, J = 7.73 Hz), 7.38–7.29 (1H, t, J = 7.33 Hz), 7.23–7.21 (1H, d, J = 8.14 Hz), 7.16–7.05 (2H, m), 6.91–6.87 (2H, m), 4.56–4.48 (4H, m), 4.22–4.19 (2H, d, J = 14.06 Hz), 3.79 (3H, s), 2.92 (3H, s) ppm, NH protons not observed; 13C NMR (101 MHz, CD3OD) δ 164.70 (C), 159.41 (C), 155.59 (C), 147.32 (C), 129.13 (CH), 128.19 (C), 127.85 (C), 125.38 (CH), 123.20 (CH), 121.42 (CH), 113.71 (CH), 110.97 (CH), 109.24 (CH), 54.33 (CH3), 51.43 (CH2), 50.01 (CH2), 43.33 (CH2), 32.52 (CH3) ppm.; LRMS (+ESI): m/z 339 [M + H]+. HRMS(ESI): calc. C20H22N2O3 [M + Na+] = 361.1523, found 361.1523, HPLC: 98.09%, RT: 18.09 min.
Synthesis of N-(benzofuran-2-ylmethyl)-2-((4-methoxybenzyl)(methyl)amino)acetamide 24a. Amine 22a (100 mg, 277 μmol) was methylated according to general procedure G. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97) to afford 24a as a clear oil (81 mg, 86%). 1H NMR (300 MHz, d6-DMSO) δ 7.61–7.51 (2H, m), 7.48–7.45 (2H, d, J = 8.46 Hz), 7.30–7.20 (2H, m), 6.98–6.96 (2H, d J = 8.46 Hz), 6.79 (1H, s), 4.51 (2H, m), 4.31 (2H, br s), 3.93–3.87 (2H, d, J = 18.67 Hz), 3.76 (3H, s), 2.76 (3H, s) ppm, NH protons not observed; 13C NMR (75 MHz, d6-DMSO) δ 167.27 (C), 164.88 (C), 160.62 (C), 154.967 (C), 133.41 (CH), 128.438 (C), 124.605 (C), 123.3946 (CH), 121.85 (CH), 121.429 (CH), 114.60 (CH), 111.39 (CH), 104.45 (CH), 58.66 (CH2), 55.68 (CH3), 54.70 (CH3), 49.72 (CH2), 36.48 (CH2); LRMS (+ESI): m/z 361 [M + Na]+. HRMS(ESI): calc. C20H22N2O3 [M + Na+] = 361.1523, found 361.1522, HPLC: 96.20%, RT: 18.29 min.
2-((Benzofuran-2-ylmethyl)(methyl)amino)-N-(4-methoxybenzyl)acetamide 24b. Amine 22b (150 mg, 416 μmol) was methylated according to general procedure G. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97) to afford 24b as a clear oil (128 mg, 91%). 1H NMR (500 MHz, d6-DMSO) δ 9.14 (1H, br s), 7.73–7.71 (1H, d, J = 7.09 Hz), 7.60–7.58 (1H, d, J = 8.26 Hz), 7.41–7.38 (1H, t, J = 7.74 Hz), 7.32–7.29 (1H, t, J = 7.53 Hz), 7.21–7.20 (3H, m), 6.89–6.87 (2H, d, J = 8.61 Hz), 4.66 (2H, s), 4.26–4.25 (2H, d, J = 5.73 Hz), 4.02 (2H, s), 3.72 (3H, s), 2.87 (3H, s) ppm; 13C NMR (126 MHz, d6-DMSO) δ 163.88 (C), 158.41 (C), 154.87 (C), 146.80 (C), 130.27 (C), 128.82 (CH), 127.30 (C), 125.61 (CH), 123.37 (CH), 121.88 (CH), 113.76 (CH), 111.43 (CH), 111.41 (CH), 66.35 (CH2), 55.11 (CH3), 51.25 (CH2), 41.72 (CH2), 40.91 (CH3) ppm; LRMS (+ESI): m/z 339 [M + H]+. HRMS(ESI): calc. C20H22N2O3 [M + Na+] = 361.1523, found 361.1523, HPLC: 97.08%, RT: 17.83 min.
Synthesis of N-(benzofuran-2-ylmethyl)-2-((4-methoxybenzyl)(methyl)amino)-N-methylacetamide 25a. Amine 23a (100 mg, 267 μmol) was methylated according to general procedure G. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97) to afford 25a as a clear oil (70 mg, 74%). 1H NMR (500 MHz, d6-DMSO) δ 7.63–7.59 (1H, m), 7.53–7.47 (3H, m), 7.31–7.22 (2H, m), 6.90–6.95 (2H, m), 6.87 (1H, s), 4.76–4.70 (2H, m), 4.39–4.24 (4H, m), 3.76 (3H, s), 2.98 (3H, s), 2.76 (3H, s) ppm; 13C NMR (126 MHz, d6-DMSO) δ 164.85 (C), 160.18 (C), 154.32 (C), 153.18 (C), 132.88 (CH), 127.82 (C), 124.23 (C), 122.96 (CH), 121.54 (CH), 120.99 (CH), 114.15 (CH), 110.99 (CH), 105.13 (CH), 58.55 (CH2), 55.21 (CH3), 54.21 (CH2), 43.89 (CH2), 40.39 (CH3), 34.09 (CH3) ppm; LRMS (+ESI): m/z 353 [M + H]+. HRMS(ESI): calc. C21H22N2O3 [M + H+] = 353.1859, found 353.1859, HPLC: 98.22%, RT: 19.39 min.
2-((Benzofuran-2-ylmethyl)(methyl)amino)-N-(4-methoxybenzyl)-N-methylacetamide 25b. Amine 23b (100 mg, 255 μmol) was methylated according to general procedure G. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97) to afford 25b as a clear oil (78 mg, 83%). 1H NMR (500 MHz, CD3OD) δ 7.69–7.66 (1H, t, J = 6.94 Hz), 7.54–7.50 (1H, t, J = 7.71 Hz), 7.42–7.39 (1H, t, J = 6.94 Hz), 7.32–7.29 (1H, t, J = 6.94 Hz), 7.20–7.19 (2H, m), 7.14–7.13 (1H, m), 6.87–6.85 (2H, m), 4.69 (2H, s), 4.54–4.35 (4H, m), 3.77 (3H, s), 3.05 (3H, s), 2.89 (3H, s) ppm; 13C NMR (101 MHz, CD3OD) δ 165.45 (C), 160.80 (C), 157.16 (C), 147.21 (C), 130.58 (CH), 129.3741 (C), 128.59 (C), 127.17 (CH), 124.71 (CH), 123.02 (CH), 115.13 (CH), 112.97 (CH), 112.47 (CH), 57.11 (CH2), 55.74 (CH3), 53.82 (CH2), 51.46 (CH2), 42.67 (CH3), 33.97 (CH3); LRMS (+ESI): m/z 353 [M + H]+. HRMS(ESI): calc. C21H24N2O3 [M + Na+] = 375.1679, found 373.1679, HPLC: 95.36%, RT: 18.49 min.
Synthesis of N1-(benzofuran-2-ylmethyl)-N2-(4-methoxybenzyl)ethane-1,2-diamine 26a. Amine 22a (205 mg, 632 μmol) was reduced according to general procedure C. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97) to afford 26a as a faintly yellow oil (155 mg, 79%). 1H NMR (400 MHz, CDCl3) δ 7.49–7.47 (2H, d, J = 8.70 Hz), 7.11–6.99 (5H, m), 6.78–6.77 (2H, m), 4.22 (2H, br s), 3.82–3.81 (3H, s), 3.39 (4H, m), 3.09–3.06 (2H, t, J = 7.67 Hz), 2.75–2.72 (2H, t, J = 7.14 Hz) ppm; 13C NMR (101 MHz, CDCl3) δ 160.29 (C), 155.767 (C), 155.58 (C), 132.29 (C), 132.19 (CH), 130.39 (C), 127.77 (CH), 127.28 (CH), 124.41 (CH), 119.49 (CH), 115.64 (CH), 114.58 (CH), 55.82 (CH3), 50.25 (CH2), 47.39 (CH2), 43.56 (CH2), 43.16 (CH2) ppm; LRMS (+ESI) m/z 311 [M + H]+. HRMS(ESI): calc. C19H22N2O2 [M + Na+] = 333.1574, found 333.1573, HPLC: 99.50%, RT: 16.16 min.
Synthesis of N1-(benzofuran-2-ylmethyl)-N2-(4-methoxybenzyl)-N1-methylethane-1,2-diamine 26b. Amine 23a (250 mg, 667 μmol) was reduced according to general procedure C. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97) to afford 26b as a faintly yellow oil (181 mg, 84%) 1H NMR (500 MHz, CD3OD) δ 7.69–7.68 (1H, d, J = 6.96 Hz), 7.57–7.55 (1H, d, J = 7.65 Hz), 7.47–7.45 (2H, d, J = 8.35 Hz), 7.42–7.39 (1H, t, J = 6.96 Hz), 7.33–7.30 (1H, t, J = 6.94 Hz), 7.22 (1H, s), 7.00–6.99 (2H, s), 4.68 (2H, s), 4.23 (2H, s), 3.82 (3H, s), 3.56 (4H, s), 2.98 (3H, s) ppm, NH proton not observed; 13C NMR (76 MHz, CD3OD) δ 162.38 (C), 157.33 (C), 146.85 (C), 132.73 (CH), 128.78 (C), 127.28 (C), 124.83 (CH), 123.74 (CH), 123.08 (CH), 115.65 (CH), 113.22 (CH), 112.56 (CH), 55.87 (CH3), 53.73 (CH2), 52.47 (CH2), 52.25 (CH2), 42.65 (CH2), 41.17 (CH3); LRMS (+ESI): m/z 325 [M + H]+. HPLC: 99.83%, RT: 18.96 min.
Synthesis N1-(benzofuran-2-ylmethyl)-N2-(4-methoxybenzyl)-N2-methylethane-1,2-diamine 26c. Amine 24a (300 mg, 800 μmol) was reduced according to general procedure C. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97) to afford 26c as a faintly yellow oil (177 mg, 68%). 1H NMR (500 MHz, CD3OD) δ 7.56–7.54 (2H, d, J = 8.36 Hz), 7.10–7.00 (5H, m), 6.80–6.76 (2H, m), 3.81 (3H, s), 3.56 (4H, br s), 3.09–3.07 (2H, m), 2.83 (3H, s), 2.73–2.71 (2H, m) ppm, NH proton not observed; 13C NMR (125 MHz, CD3OD) δ 162.59 (C), 156.31 (C), 134.027 (CH), 131.23 (C), 128.66 (C), 127.64 (CH), 121.92 (CH), 120.75 (CH), 116.03 (CH), 115.68 (CH), 115.53 (CH), 62.78 (CH2), 61.31 (CH2), 55.91 (CH3), 52.08 (CH2), 43.11 (CH3), 39.98 (CH2) ppm, one obscured or unresolved carbon; LRMS (+ESI): m/z 325 [M + H]+. HRMS(ESI): calc. C20H24N2O2 [M + Na+] = 347.1730, found 347.1730, HPLC: 99.07%, RT: 18.06 min.
Synthesis of N1-(benzofuran-2-ylmethyl)-N2-(4-methoxybenzyl)-N1,N2-dimethylethane-1,2-diamine 26d. Amine 25a (150 mg, 426 μmol) was reduced according to general procedure C. The crude product was purified by flash column chromatography on silica gel (MeOH–CH2Cl2, 1
:99 → 3:97) to afford 26d as a faintly yellow oil (102 mg, 71%). 1H NMR (400 MHz, CD3OD) δ 7.69–7.68 (1H, d, J = 7.02 Hz), 7.58–7.51 (3H, m), 7.43–7.39 (1H, t, J = 7.02 Hz), 7.33–7.29 (1H, t, J = 7.02 Hz), 7.24 (1H, s), 7.01–6.99 (2H, m), 4.69 (2H, s), 4.41 (2H, s), 3.82 (3H, s), 3.74 (4H, br s), 2.97 (3H, s), 2.85 (3H, s) ppm 13C NMR (101 MHz, CD3OD) δ 162.76 (C), 157.14 (C), 134.08 (CH), 128.75 (C), 127.28 (C), 124.84 (CH), 123.11 (CH), 121.78 (CH), 115.81 (CH), 113.33 (CH), 112.58 (CH), 62.81 (CH2), 61.67 (CH2), 55.98 (CH3), 53.88 (CH2), 51.10 (CH2), 41.46 (CH3), 40.44 (CH3) ppm, one obscured or unresolved tertiary carbon; LRMS (+ESI): m/z 339 [M + H]+. HRMS(ESI): calc. C21H26N2O2 [M + H+] = 339.2069, found 339.2067, HPLC: 98.93%, RT: 15.78 min.
References
- W. R. Martin, C. G. Eades, J. A. Thompson, R. E. Huppler and P. E. Gilbert, J. Pharmacol. Exp. Ther., 1976, 197, 517 CAS.
- S. B. Hellewell and W. D. Bowen, Brain Res., 1990, 527, 244 CrossRef CAS PubMed.
- A. D. Weissman, T. P. Su, J. C. Hedreen and E. D. London, J. Pharmacol. Exp. Ther., 1988, 247, 29 CAS.
- I. S. Ahmed, C. Chamberlain and R. J. Craven, Expert Opin. Drug Metab. Toxicol., 2012, 8, 361 CrossRef CAS PubMed.
- B. J. Vilner, C. S. John and W. D. Bowen, Cancer Res., 1995, 55, 408 CAS.
- T. Hayashi, J. Pharmacol. Sci., 2015, 127, 2 CrossRef CAS PubMed.
- T.-P. Su, T. Hayashi, T. Maurice, S. Buch and A. E. Ruoho, Trends Pharmacol. Sci., 2010, 31, 557 CrossRef CAS PubMed.
- S.-Y. Tsai, T. Hayashi, T. Mori and T.-P. Su, Cent. Nerv. Syst. Agents Med. Chem., 2009, 9, 184 CrossRef CAS PubMed.
- S.-Y. A. Tsai, M. J. Pokrass, N. R. Klauer, N. E. De Credico and T.-P. Su, Expert Opin. Ther. Targets, 2014, 18, 1461 CAS.
- D. S. Banister and M. Kassiou, Curr. Pharm. Des., 2012, 18, 884 CrossRef PubMed.
- J. L. Diaz, D. Zamanillo, J. Corbera, J. M. Baeyens, R. Maldonado, M. A. Pericàs, J. M. Vela and A. Torrens, Cent. Nerv. Syst. Agents Med. Chem., 2009, 9, 172 CrossRef CAS PubMed.
- M. Manohar, S. D. Banister, C. Beinat, J. O'Brien-Brown and M. Kassiou, Aust. J. Chem., 2015, 68, 600 CrossRef CAS.
- J. L. Díaz, R. Cuberes, J. Berrocal, M. Contijoch, U. Christmann, A. Fernández, A. Port, J. Holenz, H. Buschmann, C. Laggner, M. T. Serafini, J. Burgueño, D. Zamanillo, M. Merlos, J. M. Vela and C. Almansa, J. Med. Chem., 2012, 55, 8211 CrossRef PubMed.
- Y.-S. Huang, H.-L. Lu, L.-J. Zhang and Z. Wu, Med. Res. Rev., 2014, 34, 532 CrossRef CAS PubMed.
- J. Xu, C. Zeng, W. Chu, F. Pan, J. M. Rothfuss, F. Zhang, Z. Tu, D. Zhou, D. Zeng, S. Vangveravong, F. Johnston, D. Spitzer, K. C. Chang, R. S. Hotchkiss, W. G. Hawkins, K. T. Wheeler and R. H. Mach, Nat. Commun., 2011, 2, 1386/1 CAS.
- U. B. Chu, T. A. Mavlyutov, M.-L. Chu, H. Yang, A. Schulman, C. Mesangeau, C. R. McCurdy, L.-W. Guo and A. E. Ruoho, EBioMedicine, 2015, 2, 1806 CrossRef PubMed.
- H. R. Schmidt, S. Zheng, E. Gurpinar, A. Koehl, A. Manglik and A. C. Kruse, Nature, 2016, 532, 527 CrossRef CAS PubMed.
- R. A. Glennon, A. M. Ismaiel, J. D. Smith, M. Yousif, M. El-Ashmawy, J. L. Herndon, J. B. Fischer, K. J. Burke Howie and A. C. Server, J. Med. Chem., 1991, 34, 1855 CrossRef CAS PubMed.
- R. A. Glennon, S. Y. Ablordeppy, A. M. Ismaiel, M. B. El-Ashmawy, J. B. Fischer and K. Burke Howie, J. Med. Chem., 1994, 37, 1214 CrossRef CAS PubMed.
- R. A. Glennon, Mini-Rev. Med. Chem., 2005, 5, 927 CrossRef CAS PubMed.
- T. M. Gund, J. Floyd and D. Jung, J. Mol. Graphics Modell., 2004, 22, 221 CrossRef CAS PubMed.
- C. Laggner, C. Schieferer, B. Fiechtner, G. Poles, R. D. Hoffmann, H. Glossmann, T. Langer and F. F. Moebius, J. Med. Chem., 2005, 48, 4754 CrossRef CAS PubMed.
- D. Zampieri, M. G. Mamolo, E. Laurini, C. Florio, C. Zanette, M. Fermeglia, P. Posocco, M. S. Paneni, S. Pricl and L. Vio, J. Med. Chem., 2009, 52, 5380 CrossRef CAS PubMed.
- E. Laurini, D. Zampieri, M. G. Mamolo, L. Vio, C. Zanette, C. Florio, P. Posocco, M. Fermeglia and S. Pricl, Bioorg. Med. Chem. Lett., 2010, 20, 2954 CrossRef CAS PubMed.
- S. D. Banister, I. A. Moussa, M. J. T. Jordan, M. J. Coster and M. Kassiou, Bioorg. Med. Chem. Lett., 2010, 20, 145 CrossRef CAS PubMed.
- I. A. Moussa, S. D. Banister, F. N. Akladios, S. W. Chua and M. Kassiou, Bioorg. Med. Chem. Lett., 2011, 21, 5707 CrossRef CAS PubMed.
- I. A. Moussa, S. D. Banister, C. Beinat, N. Giboureau, A. J. Reynolds and M. Kassiou, J. Med. Chem., 2010, 53, 6228 CrossRef CAS PubMed.
- I. A. Moussa, S. D. Banister, M. Manoli, M. R. Doddareddy, J. Cui, R. H. Mach and M. Kassiou, Bioorg. Med. Chem. Lett., 2012, 22, 5493 CrossRef CAS PubMed.
- X. Liu, S. D. Banister, M. J. Christie, R. Banati, S. Meikle, M. J. Coster and M. Kassiou, Eur. J. Pharmacol., 2007, 555, 37 CrossRef CAS PubMed.
- S. D. Banister, D. T. Yoo, S. W. Chua, J. Cui, R. H. Mach and M. Kassiou, Bioorg. Med. Chem. Lett., 2011, 21, 5289 CrossRef CAS PubMed.
- S. D. Banister, I. A. Moussa, C. Beinat, A. J. Reynolds, P. Schiavini, W. T. Jorgensen and M. Kassiou, Bioorg. Med. Chem. Lett., 2011, 21, 38 CrossRef CAS PubMed.
- S. D. Banister, I. A. Moussa, W. T. Jorgensen, S. W. Chua and M. Kassiou, Bioorg. Med. Chem. Lett., 2011, 21, 3622 CrossRef CAS PubMed.
- S. D. Banister, M. Manoli, M. R. Doddareddy, D. E. Hibbs and M. Kassiou, Bioorg. Med. Chem. Lett., 2012, 22, 6053 CrossRef CAS PubMed.
- S. D. Banister, M. Manoli, M. L. Barron, E. L. Werry and M. Kassiou, Bioorg. Med. Chem., 2013, 21, 6038 CrossRef CAS PubMed.
- S. Banister, D. Roeda, F. Dolle and M. Kassiou, Curr. Radiopharm., 2010, 3, 68 CrossRef CAS.
- S. D. Banister, M. Manoli and M. Kassiou, J. Labelled Compd. Radiopharm., 2013, 56, 215 CrossRef CAS PubMed.
- S. Ferorelli, C. Abate, M. P. Pedone, N. A. Colabufo, M. Contino, R. Perrone and F. Berardi, Bioorg. Med. Chem., 2011, 19, 7612 CrossRef CAS PubMed.
- MarvinSketch (16.8.29.0) ChemAxon, http://www.chemaxon.com, 2016.
- R. C. Weast and M. J. Astle, Handbook of data on organic compounds, CRC Press, 1985 Search PubMed.
- C. Liao and M. C. Nicklaus, J. Chem. Inf. Model., 2009, 49, 2801 CrossRef CAS PubMed.
- L. Settimo, K. Bellman and R. A. Knegtel, Pharm. Res., 2014, 31, 1082 CrossRef CAS PubMed.
- I. A. Moussa, S. D. Banister, C. Beinat, N. Giboureau, A. J. Reynolds and M. Kassiou, J. Med. Chem., 2010, 53, 6228 CrossRef CAS PubMed.
- J. P. Holland, V. Fisher, J. A. Hickin and J. M. Peach, Eur. J. Inorg. Chem., 2010, 2010, 48 CrossRef.
- P. R. Muktapuram, R. K. Gara, K. Sharma, C. Rohit, K. Srinivas, D. P. Mishra and S. R. Bathula, Eur. J. Med. Chem., 2012, 56, 400 CrossRef CAS PubMed.
- K. Bailey and E. W. Tan, Bioorg. Med. Chem., 2005, 13, 5740 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00615a |
| ‡ These authors made equal contributions to the paper. |
| This journal is © The Royal Society of Chemistry 2016 |