
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of new C-5-triazolyl-functionalized thymidine analogs and their ability to engage in aromatic stacking in DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :DNA and DNA:RNA duplexes†
:DNA and DNA:RNA duplexes†
Mick
Hornum
,
Alevtina
Djukina
,
Ann-Katrin
Sassnau
and
Poul
Nielsen
*
Nucleic Acid Center, Department of Physics, Chemistry & Pharmacy, University of Southern Denmark, Campusvej 55, DK-5230 Odense, Denmark. E-mail: pouln@sdu.dk
First published on 11th April 2016
Abstract
1-Phenyl-1,2,3-triazole scaffolds on the 5-position of pyrimidine nucleosides have previously shown to enhance nuclease stability and increase the duplex thermal stability (Tm) by engaging in duplex stacking interactions. In this study, we have introduced two new derivatives of this scaffold in DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DNA and DNA:RNA duplexes in order to explore the thermal effects of (1) using a 1,5-triazole instead of the usual 1,4-triazole, and (2) replacing the apolar phenyl substituent with a polar uracil-5-yl substituent.
:DNA and DNA:RNA duplexes in order to explore the thermal effects of (1) using a 1,5-triazole instead of the usual 1,4-triazole, and (2) replacing the apolar phenyl substituent with a polar uracil-5-yl substituent.
Introduction
The 5-position of pyrimidines has long been a popular site for fine-tuning the physical and biological properties of oligonucleotides and nucleic acid duplexes.1 In addition to their well-defined location in the major groove of duplexes, 5-substituents are known to influence the stacking of nucleobases in duplexes. For instance, a 5-methyl group increases the molecular polarizability of the pyrimidine, thereby increasing the base stacking,2 which is widely used in e.g. antisense therapeutics for increasing the affinity of oligonucleotides toward complementary RNA.3 The stacking interactions can be further augmented by employing small 5-alkynyl or 5-heteroaryl substituents, such as 5-propyn-1-yl4 and 5-thiazol-2-yl.5Elaboration of these structures has enabled us to design 5-triazole-substituted pyrimidine nucleosides, which were shown to provide dramatic improvements in duplex stability.6 In addition, oligodeoxynucleotides bearing these monomers maintain high base-pairing specificity,6,7 show improved stability toward 3′-exonucleoases7b and compatibility with RNase H enzymes,8 making these 5-triazole-substituted pyrimidine building blocks strong candidates in antisense therapeutics. Starting from 5-ethynyl pyrimidine nucleosides,9 the copper(I)-catalyzed alkyne–azide cycloaddition10 (CuAAC) has allowed us to employ an arsenal of different aliphatic or aromatic azides to yield an analogous set of 5-(1-substituted-1,2,3-triazole)-functionalized nucleoside products with relative ease.6,7,11 The studies showed that monomer X (Fig. 1) with a 1-phenyltriazol-4-yl moiety6,7a stacks more strongly than the triazole alone, and different small substituents on the phenyl group can enhance stacking interactions slightly.7a,11 While single incorporations of these monomers generally destabilize DNA:DNA and DNA:RNA duplexes, due to their displacement of the otherwise well-ordered water molecules within the major groove of the duplex, multiple incorporations evoke thermostabilizing effects that more than compensates for the solvation penalty, suggesting that the phenyltriazoles stack efficiently in the major groove.
 | ||
| Fig. 1 Structures of monomers X, Y and Z. | ||
Although aromatic stacking provides the dominating contribution to the overall stability of the nucleic acid duplexes in general,12 the nature of this interaction is not well understood. It is now well-established that π–π stacking is not precisely described by the mere interaction of π clouds above and below the planes of neighbouring aromatic rings.13 Both enthalpy-driven electrostatic interactions and van der Waals dispersion forces, as well as entropy-driven solvophobic effects have been suggested as the major components behind the stacking in aqueous media,14 but no clear unified picture regarding the relative importance of these basic physical contributions exists.12 While it is accepted that stacking correlates with the surface area, there is no satisfactory answer to the correlation between other physical properties such as hydrophobicity or geometry.
In the present study, we have introduced more radical changes to our 1-phenyl-1,2,3-triazole scaffold in order to investigate the scope of these stacking interactions in the aqueous media of the major groove. To this end, we hereby report our findings with monomers Y and Z (Fig. 1), where we have explored the thermostabilizing and structural consequences of, respectively, increasing the hydrophilicity of the 5-triazole substituent significantly, and altering the substitution geometry of the triazole ring (1,4- vs. 1,5-disubstituted triazole).
Monomer Y features a uracil–triazole moiety on the 5-position and is hereby designed to be a simple hydrophilic version of the traditional phenyl–triazole moiety of monomer X. Being roughly the same size as the phenyl substituent, the uracil substituent also possesses very large bond moments and is less polarizable, and so these two monomers are expected to stack energetically differently in the major groove. The other new monomer, Z, is designed to be a geometric analog of monomer X, where the phenyl substituent is projected non-linearly into the major groove as opposed to monomer X. This bent structure is interesting in terms of understanding what geometry of the 5-substituent is most suitable for engaging in stacking interactions in the major groove. While these two structures differ only in their substitution pattern of the 1,2,3-triazole ring, we speculated that their slightly different vectors of expanding into the major groove could exert diametrically opposed effects on the duplex structure.
Results
Chemical synthesis
The new monomers Y and Z were successfully incorporated into oligodeoxynucleotides on an automated solid-phase DNA synthesizer using the nucleoside phosphoramidites 3 and 8 (Scheme 1) when activated by 1H-tetrazole. The oligonucleotide sequences chosen for the present study is a T-rich 9-mer sequence (5′-dGTGTTTTGC) with one to four central incorporations of Y and Z in accordance with some of our previous studies with X.6,7a,11 | ||
| Scheme 1 Reagents and conditions: (a) 5-azidouracil, Na ascorbate, CuSO4, H2O, t-BuOH, rt, 75%; (b) P(N(i-Pr)2)O(CH2)2CN, diisopropylammonium tetrazolide, CH2Cl2, rt, 71% 3, 50% 8; (c) automated DNA synthesis; (d) phenylazide, Cp*RuCl(PPh3)2, MTBE, THF, MW, 100 °C; 80%; (e) NH3, MeOH, rt, 67%; (f) DMTCl, pyridine, 57%. | ||
The core structure of monomer Y was successfully obtained by treating the well-known 5-ethynyl-5′-O-DMT-2′-deoxy-uridine15 (1) with freshly synthesized 5-azidouracil16 under copper(I) catalysis, similar to our protocol for synthesis of X.17 This afforded nucleoside 2 in 75% yield. The structure of 2 was carefully identified by means of 2D HSQC, COSY and HMBC in order to confirm the precise structure of the 5-substituent. Subsequent phosphitylation of nucleoside 2 under standard conditions gave phosphoramidite 3. For the synthesis of monomer Z, the 1,5-disubstituted triazole scaffold (nucleoside 5) was obtained by treating acetyl-protected 5-ethynyl-2′-deoxyuridine (4) with phenylazide under a cooperative effect of microwave activation and ruthenium catalysis18 analogous to a procedure used by Agrofoglio and co-workers19 for the regioselective generation of similar 1,5-disubstituted 1,2,3-triazolo-nucleosides. Small amounts of the formed 1,4-regioisomer was removed by chromatography, and nucleoside 5 was isolated in 80% yield. Although this reaction type generally accepts unprotected nucleosides,18b we observed that protecting groups were necessary in order to minimize unspecific side reactions. Phosphoramidite 8 was obtained in good yield after deacetylation (to form 6), selective 5′-O-tritylation (to form 7) and finally 3′-O-phosphitylation; all under standard conditions.
UV spectroscopy
The oligonucleotides were mixed in a phosphate buffered saline solution with the complementary DNA and RNA sequences using 1.5 μM of each strand. The melting temperatures (Tm) of the resulting DNA:DNA and DNA:RNA duplexes (Table 1) were determined by means of a UV spectrometer using a Peltier Temperature Programmer. The Tm values were derived from the first derivatives of the melting curves recorded at 260 nm (as duplicate readings that agreed within ±0.5%).
| # | Sequence |
T
ma (ΔTm/mod.) [°C] |
|
|---|---|---|---|
| Compl. DNA | Compl. RNA | ||
| a Melting temperatures (Tm) are derived from the maxima of the first derivatives of the absorbance (260 nm) vs. temperature curves. The samples contained 1.5 μM of each oligonucleotide and 1.5 μM of complementary DNA or RNA (5′-GCAAAACAC) in PBS buffer (2.5 mM Na2HPO4, 5.0 mM NaH2PO4, 100 mM NaCl, 0.1 mM EDTA, pH 7). Values in brackets show changes in the Tm values per modification compared to the unmodified duplex. b Value taken from ref. 6. c Value taken from ref. 7a. d No clear transition was observed. | |||
| 1 | 5′-dGTG TTT TGC | 33.0 | 31.0 |
| 2 | 5′-dGTG TXT TGC | 28.0b (−5.0) | 29.0b (−2.0) |
| 3 | 5′-dGTG TXX TGC | 30.5c (−1.5) | 37.5c (+3.3) |
| 4 | 5′-dGTG XXX TGC | 30.0c (−1.0) | 43.0c (+4.0) |
| 5 | 5′-dGTG XXX XGC | 32.0b (−0.3) | 51.5b (+5.1) |
| 6 | 5′-dGTG TYT TGC | 31.3 (−1.7) | 29.9 (−1.1) |
| 7 | 5′-dGTG TYY TGC | 32.2 (−0.4) | 36.2 (+2.6) |
| 8 | 5′-dGTG YYY TGC | 35.0 (+0.7) | 43.1 (+4.0) |
| 9 | 5′-dGTG YYY YGC | 38.3 (+1.3) | 52.0 (+5.2) |
| 10 | 5′-dGTG TZT TGC | 19.7 (−13.3) | n.t.d |
| 11 | 5′-dGTG TZZ TGC | 19.0 (−7.0) | n.t.d |
| 12 | 5′-dGTG ZZZ TGC | n.t.d | n.t.d |
As shown in Table 1, a single incorporation of Y in the centre of DNA:DNA destabilizes the duplex by −1.7 °C (entry 6), which is, however, significantly less than that for X (ΔTm = −5.0 °C, entry 2), suggesting that the polar profile of Y is much better accommodated in the major groove than the non-polar residue of X. As expected, the degree of destabilization is compensated once several residues are installed: the DNA:DNA duplexes featuring three and four consecutive Y residues are thermally stabilized by +2.0 °C (entry 8) and +5.3 °C (entry 9), respectively, which is significantly better than what is achieved with 3× X (−3.0 °C, entry 3) and 4× X (−1.0 °C, entry 4). While the stacking between two X residues (entry 2) appears slightly better than between two Y residues (entry 7), the favorable stacking of multiple Y residues seems more consistent and, in general, better than multiple X residues (Fig. 2).
 | ||
| Fig. 2 Graphical illustration of correlation between Tm and the number of incorporations of X (red), Y (blue) and Z (green) in the DNA:DNA duplexes. | ||
For a single incorporation of monomer Z in the DNA:DNA duplex (entry 10), a relatively weak melting transition was observed at 19.7 °C, which represents a remarkable destabilization of −13.3 °C compared to the unmodified duplexes (entry 1) and −8.3 °C relative to that of its regioisomer X (entry 2). Similarly, the duplex bearing two Z residues was destabilized by −14.0 °C (or −7.0 °C per mod., entry 11), however, the melting transition for this entry was extraordinarily weak. No clear melting was observed for the entry with 3× Z (entry 12), which is an indication of either the dim hyperchromicity exerted by the presence of Z or the lack of any hybridization such that no denaturation occurs. In the study involving targeting complementary RNA, all entries with Z showed no clear melting transitions; even when measured down to 5 °C and at an increased salt concentration. Monomer Z was found to cause a slight redshift of the absorption maximum, but also no clear melting was detected when recorded at λmax = 252 nm.
In the case of Y in the DNA:RNA study, an initial destabilization of −1.1 °C was observed for the initial incorporation (entry 6) compared to the native duplex, but with each additional Y residue, the melting temperature increases almost linearly with a gradient of roughly +7 °C per incorporation (entries 7–9). Therefore, it follows the exact same tendency as X, and their thermostabilizing effects on DNA:RNA duplexes are indeed almost equivalent as evident from Fig. 3.
 | ||
| Fig. 3 Graphical illustration of the correlation between Tm and the number of incorporations of X (red), Y (blue) and Z (green; no entries) in the DNA:RNA duplexes. | ||
The high-affinity targeting of complementary RNA for oligonucleotides containing four incorporations of Y allowed us to study its base-pairing specificity toward mismatched RNA strands (Table 2). By introducing a Y:U mismatched base pair in the centre of the duplex (entry 2), we found that the Tm plummeted by −21.9 °C relative to the fully complementary DNA:RNA duplex, indicating that the A → U mutation is very well-discriminated similar to what was observed for the corresponding X:U mismatch (entry 1, ΔTm = −20.5 °C). A central Y:C mispair was also found to be very well-discriminated (ΔTm = −18.0 °C), albeit not as exceptional as the corresponding X:C mismatch (ΔTm = −27.5 °C). Traditionally, the A → G transition is the least discriminated mutation (ΔTm = −8.8 °C) as the Y:G mispair presumably adopts a wobble configuration.
| # | Sequences [*]= |
T
ma (ΔTm) [°C] |
|||
|---|---|---|---|---|---|
| A | U | G | C | ||
| a Melting temperatures (Tm) are derived from the maxima of the first derivatives of the absorbance (260 nm) vs. temperature curves. The samples contained 1.5 μM of each oligonucleotide strands in PBS buffer (2.5 mM Na2HPO4, 5.0 mM NaH2PO4, 100 mM NaCl, 0.1 mM EDTA, pH 7). Values in brackets show changes in the Tm values compared to the duplexes with [*] = A. b This value is taken from ref. 6. | |||||
| 1 | 5′-dGTG XXX XGC | 51.5b | 31.0b (−20.5) | 42.0b (−9.5) | 24.0b (−27.5) |
| 3′-rCAC A[*]A ACG | |||||
| 2 | 5′-dGTG YYY YGC | 52.0 | 30.1 (−21.9) | 43.2 (−8.8) | 34.0 (−18.0) |
| 3′-rCAC A[*]A ACG | |||||
CD spectroscopy
In order to investigate the overall structural changes imposed by Y and Z, the unmodified and modified duplexes were evaluated with circular dichroism (CD) spectroscopy. The CD spectra were recorded in the 200–350 nm range using 1.5 μM concentrations of each strand dissolved in PBS buffer. A static temperature of 10 °C was used for duplexes containing Y, and 5 °C was used for duplexes containing Z, in order to ensure that most of the oligonucleotides were in the hybridized form. The CD spectra of duplexes containing Y and Z are shown in Fig. 4, and the CD spectra of duplexes containing X can be found in ref. 6 and 7a. The CD spectrum of the single-stranded RNA (3′-rCACAAAACG) is also included in the DNA:RNA figures, since this oligonucleotide was found to show large Cotton effects on its own. The CD bands arising from the single-stranded DNA strands were found to be completely negligible, and are not included.
 | ||
| Fig. 4 CD spectra (200–350 nm) of (A) DNA:DNA duplexes featuring Y at 10 °C (entries 6–9), (B) DNA:DNA duplexes featuring Z at 5 °C (entries 10–12), (C) DNA:RNA duplexes featuring Y at 10 °C (entries 6–9), and (D) DNA:RNA duplexes featuring Z at 5 °C (entries 10–12). Recorded in PBS buffer (2.5 mM Na2HPO4, 5.0 mM NaH2PO4, 100 mM NaCl, 0.1 mM EDTA, pH 7) using 5 mm quartz cuvettes and a split width of 2 nm. | ||
As expected, the unmodified DNA:DNA duplex adopts bands that are typical of B-type helix such as CD maxima at ∼285 and ∼220 nm, CD minimum at ∼250 nm, and a clear cross-over point at the absorption maxima ∼260 nm.20 Upon a single incorporation of Y (Fig. 4A, entry 6), no significant change in the CD curve was observed. However, upon introducing multiple Y residues (entries 7–9), a progressive transition toward the A-type helix was observed, epitomized by a distinct blue shift of the CD maximum at 285 nm and an intensifying negative band at 210 nm, which are the spectral features of A-type helices. In addition, new bands evolve at ∼310 and ∼235 nm, reflecting local geometry changes in the structures that are neither typical of A- nor B-type helices, but possibly arise from the stacking uracil–triazole moieties. While the bands at ∼235 nm were also present upon multiple incorporations of X, it is much more intense for Y than Z, and the band at ∼310 nm was completely absent in the case of X. However, in general, the structural impacts by introducing X and Y in DNA:DNA duplexes are rather similar, and both induce a gradual shift towards an A-type duplex.6,7a
The CD curve of the DNA:DNA duplex containing a single incorporation of Z (Fig. 4B, entry 10) was largely similar to the unmodified duplex—like in the case of Y—suggesting that a single Z residue does not profoundly affect the secondary structure. With 2× Z (entry 11), a small shift toward the more compact A-type duplex was observed, however, with 3× Z (entry 12), the duplex structure appears essentially absent.
As shown in Fig. 4C and D, the unmodified DNA:RNA duplex adopts a conformation somewhere between the standard A- and B-type geometries with a sharp CD maximum at ∼265 nm (A-type), a shoulder at ∼285 nm (B-type), a positive band at ∼220 nm (B-type), and negative bands at ∼245 nm (B-type) and ∼210 (A-type). Upon a single incorporation of Y (Fig. 4C, entry 6) in the DNA:RNA duplex, no distinct change in the CD profile was observed, analogous to DNA:DNA. But with each additional incorporation of Y residues (entries 7–9), a gradual shift toward an A-type duplex was observed, portrayed by the drop in the intensities of the archetypal B-type bands at ∼285 and ∼220 nm, and an intensity gain at ∼265 nm. This shift towards an A-type duplex and the new band at 240 nm was also observed in the CD spectra of duplexes featuring X, although to a lesser degree.6,7 Like in the case of DNA:DNA, a weak band appears in the low-energy region (∼320 nm) of the duplexes containing 3× Y and 4× Y, which was completely absent for the duplexes containing X.
For the DNA:RNA duplexes containing Z (Fig. 4D), all CD spectra show that, in fact, no duplexes are formed upon mixing the complementary strands, which is particularly evident by the lack of any negative-to-positive Cotton effects in the 200–210 nm-region. The CD curves appear as duplicates of the recorded spectrum of the single-stranded RNA, except that they are slightly perturbed by the weak CD bands of the single-stranded DNA.
Molecular modelling
To better understand the thermal and structural changes induced by X, Y and Z in duplexes, the monomers were subjected to molecular mechanical conformational analyses. Within their base moiety, the monomers contain two rotational axes, i.e. the bond between the triazole and the nucleobase (φ), and the bond between the triazole and the phenyl/uracil substituents (ψ). To facilitate speedy calculation, the sugar portions were replaced by a methyl group. These model structures were energy-minimized by DFT calculations under vacuum using the B3LYP hybrid functional and the 6-31G** basis set, and then subjected to coordinate scans using OPLS-2005 force field parameters21 (MacroModel V10.4, release 2014-2) in GB/SA solvation.22 Scanning from 0–360° in 1° increments for both φ and ψ dihedral angles generated 3612 = 130321 structures per search, which were individually minimized, allowing all degrees of freedom to vary except φ and ψ. The energy plots of the model compounds in their (φ,ψ) spaces, and the global minimum structures in ball-and-stick models, are shown in Fig. 5.
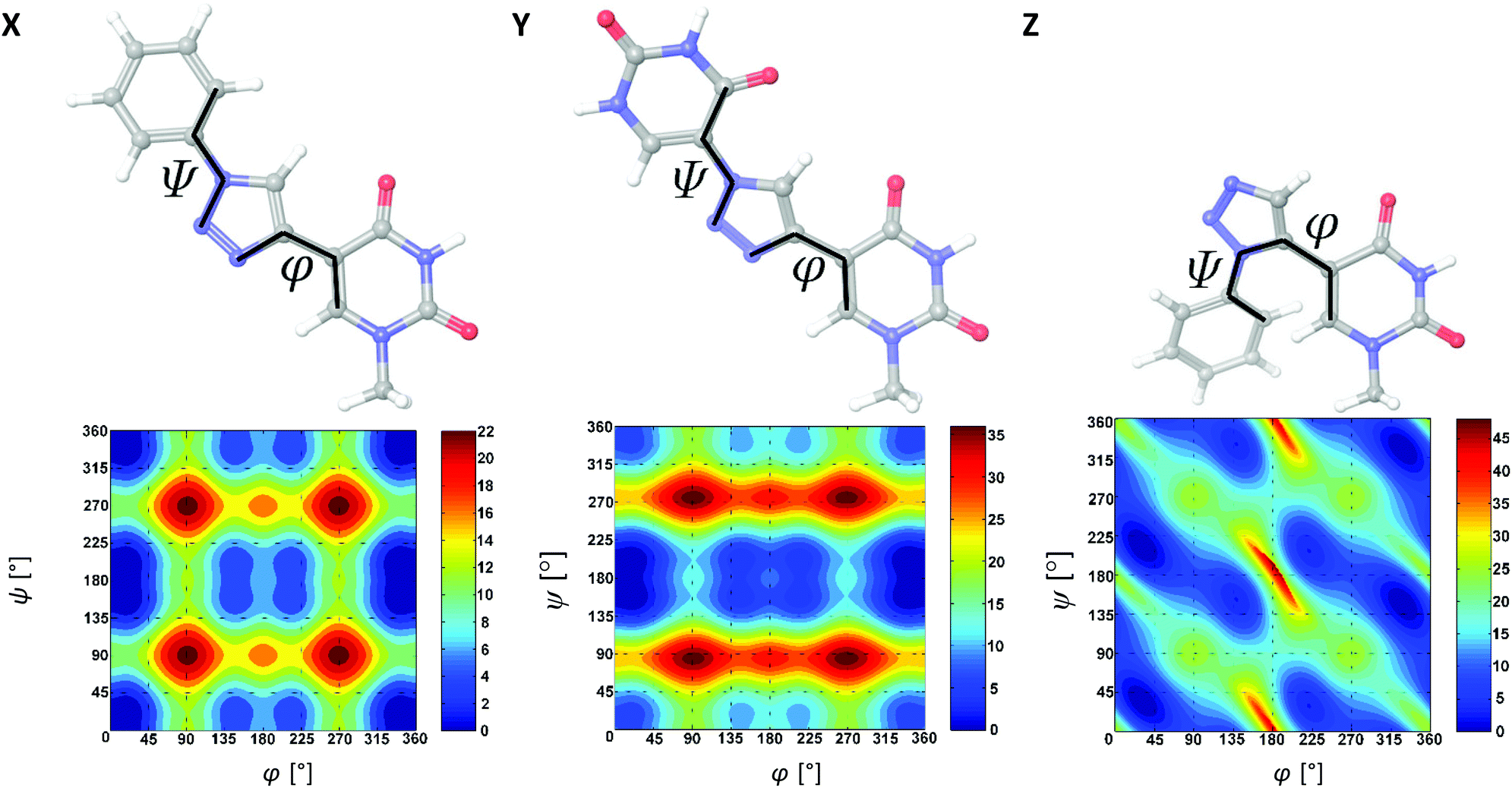 | ||
| Fig. 5 Lowest-energy ball-and-stick models, and relative potential energy contour plots (in units of kJ mol−1) of the model compounds of X, Y and Z in their (φ,ψ) spaces. The contour interval is set to 2 kJ mol−1 (is best viewed on screen). Calculations were carried out using OPLS-2005 parameters and using 1° increments from 0–360° for both φ and ψ. | ||
As shown in the (φ,ψ) contour maps, the conformational spaces for the model compounds of X and Y are somewhat similar and both contain a deep global minimum at (φ,ψ) = (0°,180°). The φ = 0° minimum signifies a strong conformational preference for a syn coplanar nucleobase–triazole torsion stabilized by a weak 2.5 Å CH…O interaction between the nucleobase O4 and the triazole H atom. The ψ = 180° minimum in Y corresponds to an anti coplanar orientation, seemingly stabilized by another weak 2.4 Å interaction between the distal uracil O4 atom and the triazole H atom. Therefore, both X and Y favor having the three ring systems in the same plane. Notably, the calculated barrier to conformational transition from the global minimum to various local minima is higher for Y than X, which is, at least in parts, due to this interaction.
In contrast to X and Y, the model compound of monomer Z favors a syn but non-coplanar orientation of the nucleoside–triazole bond. It has a pair of symmetry related global minima at (φ,ψ) = (32°,34° ± 180°) and (φ,ψ) = (328°,146° ± 180°). Both the two possible conformations with the three ring systems in the same plane are disfavored by steric hindrance, indicating that conjugation of π electrons is effectively broken between the aromatic moieties. Interestingly, monomer Z has a pair of symmetry related secondary minima (+2 kJ mol−1) at (φ,ψ) = (140°,154° ± 180°) and (φ,ψ) = (220°,206° ± 180°), which are entirely enclosed within the +8 kJ mol−1 contour of the global minimum.
In order to visualize how the 5-substituents of X, Y and Z influence the helical topology of the duplexes, and to establish the degree of stacking of the aromatic moieties in the duplex, the global minimum structures of the duplexes containing X, Y and Z were obtained from 5 ns molecular dynamics (MD) simulations using the all-atom AMBER* force field (MacroModel V10.4, release 2014-2) in GB/SA solvation. The unmodified duplexes and the duplexes featuring X and Y were modeled at a constant 25 °C, whereas duplexes featuring Z were modeled at 5 °C in order for the geometry minimizations to successfully converge. The global minimum structures from the MD simulations are shown in Fig. 6 and 7 for modeled DNA:DNA and DNA:RNA structures, respectively.
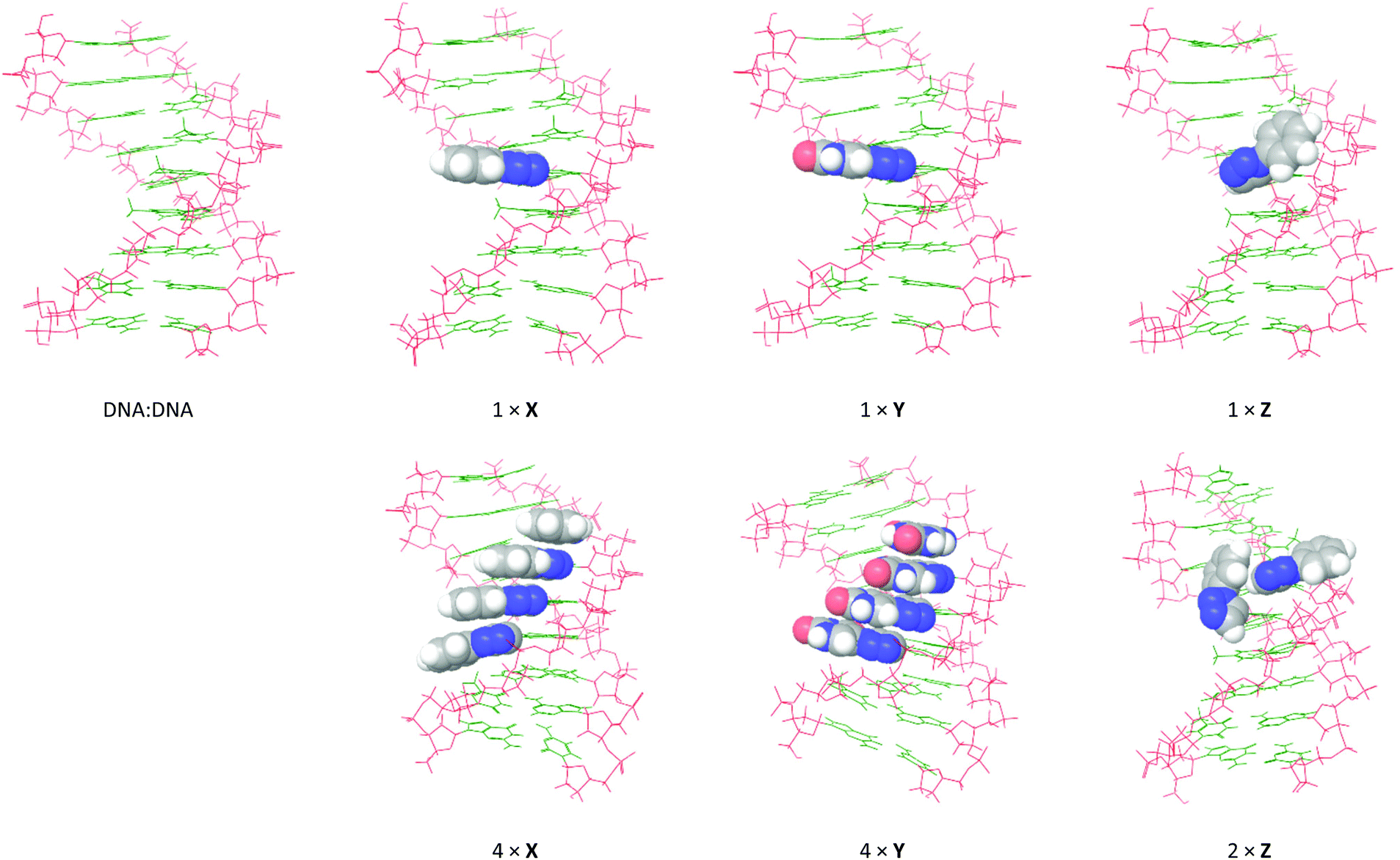 | ||
| Fig. 6 Global minimum structures obtained by 5 ns MD simulations of selected DNA:DNA duplexes from Table 1: monomer X (entries 2 + 5), monomer Y (entries 6 + 9), and monomer Z (entries 10 + 11). The calculations were performed using the all-atom AMBER* force field in GB/SA solvation (van der Waals 8 Å and electrostatics 20 Å). The 5-substituents are shown with space-filling. | ||
 | ||
| Fig. 7 Global minimum structures obtained by 5 ns MD simulations of selected DNA:RNA duplexes from Table 1: monomer X (entries 2 + 5), monomer Y (entries 6 + 9), and monomer Z (entries 10 + 11). The calculations were performed using the all-atom AMBER* force field in GB/SA solvation (van der Waals 8 Å and electrostatics 20 Å). The 5-substituents are shown with space-filling. | ||
As expected, the modeled DNA:DNA duplexes are B-type helices with the 5-substituents of X, Y and Z situated in the major groove. In line with the CD spectra, no significant perturbation of the helix structure occurs when introducing single incorporations of X, Y or Z. As expected from the conformational analysis, the Watson-Crick face of the additional uracil moiety in Y is placed toward the core of the duplex. However, while the aromatic rings of X and Y are aligned in the plane of the nucleobases (perpendicular to the axis of the duplex), the phenyl group of monomer Z points away from the duplex core. This conformation of Z corresponds precisely to the energetically favored syn periplanar torsion of the nucleobase–triazole bond. In the duplex with 2× Z, the 5-substituents are not aligned in parallel planes and do not appear to engage in any stacking interactions. In fact, one of the Z residues adopts the less-favored anti periplanar spatial orientation in order to accommodate both modifications. This behavior appears to induce a slight unwinding of the helix. In the duplexes with 4× X or 4× Y, the 5-substituents appear to strongly stack in the major groove, although they appear to shift the helix toward the more compact A/B-type. This is particularly apparent in the duplex with 4× Y, where the bending of the duplex appears to enable a few additional 1.8–2.0 Å hydrogen bonding interactions between the additional uracil and the nucleobases, while at the same time conserving all Watson-Crick base pairs.
Essentially the same picture was observed for X, Y and Z in DNA:RNA structures. Monomers X and Y are well-accommodated in the major grooves of the duplexes, and consecutive incorporations stack strongly and induce a slight compression of the duplex. But while the Z residue almost retains the same conformation as in the case of DNA:DNA, even a single incorporation appears to induce a slight unwinding of the helix. Notably, the two adjacent incorporations of Z stack efficiently on the outside of the duplex, however, in doing so, they induce unwinding of the duplex.
Discussion
Although some excellent artificial DNA building blocks,23 such as locked nucleic acids,24 have pushed the stability of nucleic acid duplexes to a new level, the strategy of using simple 5-substituted pyrimidine nucleotides benefits from their easy technical preparation and their less perturbing consequences on the sugar pucker and helix geometry, which is important for many biological applications. With our previously-published monomer X,6,7a we have devised a feasible technique to significantly stabilize duplexes by enhancing the inherent stacking interactions of the DNA:RNA duplexes, and recently we have shown that 5-substituted phenylpyrazole dC analogs stack particularly strongly with X,17 allowing efficient targeting of homo-purine sequences.
In the present study, we have evaluated the consequences of replacing the apolar phenyl substituent of X with the very polar uracil substituent (monomer Y), and the consequences of swapping the 1,4-disubstituted triazole in X with a 1,5-disubstituted derivative (monomer Z). The thermal and structural effects imposed by these two new analogs of X in the center of 9-mer DNA:DNA and DNA:RNA duplexes were examined by UV and CD spectroscopy. Monomers Y and Z were conveniently synthesized in a few synthetic steps starting from 5-ethynyl-2′-deoxyuridine by employing high-yielding Cu(I)- and Ru(II)-catalyzed alkyne–azide cycloadditions, respectively, and their incorporation into oligonucleotides as DMT-protected phosphoramidites 3 and 8 was very efficient.
Compared to X, the oligonucleotide featuring a single polar Y residue was found to have a slightly higher affinity toward complementary RNA and a significantly higher affinity toward complementary DNA, which appears to arise from the fact that the uracil substituent on Y is much better solvated in the duplex than the hydrophobic phenyl group on X. With multiple modifications, monomer Y appears to be equipotent to X in terms of increasing the affinity toward complementary RNA. That is, for each triazole moiety stacking on top of another triazole moiety, an increase in the Tm of the duplex of about +7 °C was observed. In general, fine base-pairing fidelity was observed with the oligonucleotide containing four Y residues, although a central A to C mismatch discrimination was smaller compared to the case of X. According to CD spectroscopy and molecular modelling, some differences in the duplex structure were indicated. Notably, 4× Y residues induce a slight bending of the duplex, possibly due to more efficient stacking interactions and hydrogen bonding interactions across the major groove, and the CD spectrum supported this deviation from the standard A/B-type duplex. Indeed, these secondary interactions via the additional uracil moiety makes monomer Y a so-called double-headed nucleotide.25,26 We have previously decorated DNA duplexes with additional nucleobases in the major groove26 for use as double-coding DNA systems that could turn out to be important tools in nucleic acid nanotechnology in the future. With monomer Y, with its triazole linker between the nucleobases, we now have a double-headed nucleotide with unprecedented thermostabilizing properties.
In the case of targeting DNA, oligonucleotides containing at least three consecutive Y were found to display increased affinities toward complementary DNA relative to the unmodified oligonucleotide; and, in doing so, they significantly outperform X. This stacking affinity inequivalence between X and Y in DNA:DNA but not in DNA:RNA indicates that the higher vertical rise of B-type helices houses the stacking of polar aromatic moieties better than apolar moieties. Nevertheless, it is interesting to note that Kool and co-workers found that halogenated nucleoside isosteres stack more efficiently than the canonical nucleobases when positioned as dangling residues,14c,27 and therefore they concluded that nonpolar analogues stack more strongly, in general, than the more polar natural bases.14c We now conclude that this correlation appears to be reversed, when the modification is situated in the major groove. Upon each incorporation of Y in DNA:DNA, a gradual shift toward an A/B-type helix geometry was observed, however, this geometry impact is similar to X and is presumably negligible in longer sequences.
Surprisingly, oligonucleotides bearing monomer Z were found to have extraordinarily low affinities toward both complementary DNA and RNA compared to the unmodified oligonucleotide. This result is most precisely explained in terms of structural issues, since the helical geometries of the resulting duplexes were severely affected by the presence of Z according to the molecular modeling. Either way, this is indicative of the extraordinary destabilizing effects of Z in duplexes. Remarkably, this complete depression in duplex formation arises from just a delicate positional isomerism of the triazole ring (1,4 vs. 1,5).
Conclusions
In this study, the efficiency of stacking interactions in the major groove of our previously published 1-phenyltriazol-4-yl scaffold (monomer X) have been altered by, respectively, replacing the phenyl substituent with a polar uracil-5-yl substituent (monomer Y), and replacing the 1,4-triazole with its 1,5-derivative (monomer Z). While consecutive incorporations of monomer X thermally stabilizes DNA:RNA duplexes significantly due to efficient stacking, the corresponding 1-phenyltriazol-5-yl modification (monomer Z) does not engage in stacking interactions and effectively undermines duplex formation. Thus, this study signifies that the precise spatial orientation of the phenyltriazole moieties is absolutely crucial for optimal duplex stability. Concerning the polarity of the 5-substituent, the single incorporation of a polar 1-uracil-5-yltriazol-4-yl modification (monomer Y) was found to be energetically much more favored than the apolar 5-substituent of monomer X. Comparing the thermal effects of multiple incorporations, the stacking of Y residues appears, in general, marginally better than the stacking of X residues in terms of increasing the melting temperatures of the DNA:RNA duplexes, and Y is clearly superior to X in terms of stabilizing homo-DNA duplexes. As a result, monomer Y is a promising candidate in RNA- and DNA-targeting oligonucleotides.
Experimental section
All reagents were used as supplied except CH2Cl2, which was distilled prior to use. Microwave irradiated reactions were conducted in sealed reaction vessels in a Biotage Initiator+ instrument using external surface sensor probes. All reactions were monitored by TLC using silica gel (60 F254) precoated plates. Flash chromatography was performed using silica gel 60 (particle size 0.040–0.063 mm). For the purification of DMT-protected nucleosides, the silica gel was pretreated with 1% pyridine in CH2Cl2 (v/v). HRMS-ESI was recorded on a quadrupole-time of flight instrument in positive ion mode with an accuracy of ±5 ppm. 1H, 13C and 31P NMR spectra were recorded at 400.12 MHz, 100.62 MHz and 161.97 MHz, respectively. Chemical shifts are reported in ppm relative to tetramethylsilane (δH,C 0 ppm) or the deuterated solvents (DMSO δc 39.5 ppm, CDCl3δc 77.16 ppm). For 31P NMR spectra, 85% H3PO4 was used as external standard. 2D spectra (HSQC, COSY and HMBC) have been used in assigning 1H and 13C NMR signals.5-Azidouracil16
To a magnetically stirred solution of 5-aminouracil (2.25 g; 17.7 mmol) in H2O (150 mL) was added TsOH·H2O (30.3 g; 160 mmol) and NaNO2 (11.0 g; 160 mmol). The reaction mixture was stirred at rt for 1 h, upon which NaN3 (1.84 g; 28.4 mmol) was slowly added. After the bubbles had settled (∼15 min), the reaction mixture was cooled to 5 °C, and the precipitate was filtered through a sintered glass funnel to obtain 5-azidouracil as a light beige solid (1.28 g; 8.4 mmol). Yield: 47%. Rf 0.4 (50% MeOH in CH2Cl2). M.p. 98–99 °C (decomp.) 1H NMR (400 MHz, DMSO-d6): δ 11.55 (br, 1H, NH), 10.97 (br, 1H, NH), 7.31 (s, 1H; H6). 13C NMR (101 MHz, DMSO-d6): δ 160.7 (C4), 150.0 (C2), 130.0 (C6), 112.0 (C5). HRMS-ESI+: calcd for [C4H3N5O2]2-Na+m/z 329.0466, found m/z 329.0433.:1), sodium ascorbate (107 mg; 0.54 mmol) and CuSO4·5H2O (40 mg; 162 μmol) were added, and the reaction mixture was stirred at rt for 3 h. The reaction mixture was diluted with EtOAc (100 mL) and washed with brine (50 mL). The mixture was filtered through a short Celite pad, and the organic phase was separated and concentrated under reduced pressure. The residue was purified by flash chromatography (0–10% MeOH in CH2Cl2) to obtain nucleoside 2 as a white solid (288 mg; 0.41 mmol). Yield: 75%. Rf 0.5 (10% MeOH in CH2Cl2). 1H NMR (400 MHz, DMSO-d6): δ 11.79 (br, 2H, NH), 11.55 (br, 1H, NH), 8.51 (s, 1H, triazole H5), 8.38 (s, 1H, H6), 8.06 (s, 1H, H6 in external uracil), 7.39 (d, J = 7.4 Hz, 2H, DMT), 7.32–7.23 (m, 6H, DMT), 7.18 (t, J = 7.3 Hz, 1H, DMT), 6.88–6.82 (m, 4H, DMT), 6.19 (t, J = 6.5 Hz, 1H, H1′), 5.36 (br, 1H, 3′-OH), 4.24–4.19 (m, 1H, H3′), 3.96 (q, J = 4.3 Hz, 1H, H4′), 3.71 (s, 3H, DMT), 3.70 (s, 3H, DMT), 3.34 (br, 1H, 3′-OH), 3.23 (d, J = 4.3 Hz, 2H, H5′, H5′′), 2.30–2.25 (m, 2H, H2′, H2′′). 13C NMR (101 MHz, DMSO-d6): δ 161.0 (C4), 159.3 (C4 in external uracil), 157.9 (DMT), 150.2 (C2), 149.4 (C2 in external uracil), 144.7 (DMT), 138.53 (triazole C4), 138.52 (C6 in external uracil), 135.7 (C6), 135.4, 129.6, 127.7, 127.6, 126.5 (DMT), 123.6 (triazole C5), 113.1 (DMT), 112.3 (C5), 104.8 (C5 in external uracil), 85.7 (C4′), 85.6 (DMT), 85.2 (C1′), 70.3 (C3′), 63.6 (C5′), 54.9 (DMT), 48.5 (C2′). HRMS-ESI+: calcd for C36H33N7O9-Na+m/z 730.2232, found m/z 730.2218.
Synthesis of oligonucleotides
Oligonucleotides were synthesized on a fully-automated DNA synthesizer in ∼0.2 μmol scale loaded on 500 Å controlled-pore glass (CPG) supports using the phosphoramidite approach and following the manufacturer's protocol. Double coupling (2 × 5 min) cycles were used for commercial phosphoramidites and prolonged coupling times (20 minutes) were used for the modified phosphoramidites 3 and 8. The phosphoramidites were activated using 1H-tetrazole, and incorporated into oligonucleotides via manual couplings: 10 μmol of the modified phosphoramidite was dissolved in anhydrous MeCN (2 mL) and treated with 1H-tetrazole (3 mL, 0.45 M solution in MeCN), and infused into the reaction compartment. The stepwise coupling efficiencies were monitored by measuring the absorbance of the trityl cation at 495 nm, which in all cases were 98–100% for the commercial phosphoramidites, and 95–100% for the modified phosphoramidites. The final 5′-terminal DMT group in the oligonucleotides was retained for purification purposes. The final crude oligonucleotides on solid support were treated with NH3 (28% in H2O, 1 mL) at 55 °C for 16 h. The mixture was filtered and the filtrate was evaporated to dryness at 45 °C by a steady N2 flow, and dissolved in an aqueous triethylammonium acetate buffer (500 μL, 0.05 M, pH 7.4). Analytically pure oligonucleotides were obtained by reversed-phase HPLC purification on a Waters 600 system using Xterra MS C18 10 μm (7.8 × 50 mm) columns and Xterra MS C18 10 μm (7.8 × 10 mm) precolumns. Elution was performed with 100% eluent A over 2 min, followed by a linear gradient down to 30% eluent A over 38 min, and then washed with 100% eluent B over 10 min, and 100% eluent A over 10 min. (Eluent A = triethylammonium acetate (0.05 M, pH 7.4). Eluent B = 75% MeCN/H2O (3:1, v/v)). The pure fractions were pooled and evaporated at 45 °C. The 5′-terminal DMT group was removed by treatment with acetic acid (80% in H2O, 100 μL) for 30 min, upon which an aqueous solution of NaOAc (15 μL, 3 M), an aqueous solution of NaClO4 (15 μL, 5 M), and pure acetone (1 mL) were added. The oligonucleotides precipitated overnight at −20 °C. The supernatant was removed from the sedimented solid (centrifugation, 12000 rpm, 10 min at 2 °C), and the remaining pellet was washed with cold acetone (3 × 1 mL) and dissolved in 500 μL pure water. Mass spectra of the oligonucleotides were recorded on a MALDI-TOF MS instrument in ES+ mode. The concentrations of the purified oligonucleotides were determined by the optical density at 260 nm, assuming that the molar absorptivities of the oligonucleotides equal the sum of each constituent nucleotide monomer. The extinction coefficients of the modified monomers in mL μmol−1 cm−1, ε260 (X) = 7.8, ε260 (Y) = 5.8 and ε260 (Z) = 5.2, were determined from the slopes of the absorbance of the fully deprotected nucleosides at 25, 50, 75 and 100 μM concentrations (R2 > 0.9995).
Thermal denaturation experiments
The duplex samples consisted of 1.5 μM concentrations of each oligonucleotide in phosphate buffered saline solution (2.5 mM Na2HPO4, 5.0 mM NaH2PO4, 100 mM NaCl, 0.1 mM EDTA, pH 7). The strands were annealed by heating the sample to 80 °C followed by a slow cooling to 10 °C. The increase in absorbance at 260 nm as a function of temperature from 10 °C to 75 °C (1 °C min−1) was recorded on a UV/Vis spectrometer using a Peltier Temperature Programmer. The listed Tm values were determined as the first-derivative maximum of the absorbance vs. temperature curves, and are averages of at least duplicate determinations that agreed with each other within 0.5 °C.Circular dichroism spectroscopy
CD spectra were recorded on a Jasco J-715 spectropolarimeter as a digital average of 5 scans from 200–350 nm using a split width of 2.0 nm and a scan speed of 50 nm min−1. The samples were prepared similarly to the samples used for the melting temperature studies with 1.5 μM concentrations of each strand. Quartz optical cells with an optical path length of 5.0 mm were used.Molecular modelling
The global minimum structures were found from 5 ns molecular dynamics simulations using the all-atom AMBER* force field applying the GB/SA continuum solvation model22 with extended cut-offs for non-bonded interactions (van der Waals 8 Å and electrostatics 20 Å). Calculations were performed in MacroModel V10.4 (within Maestro V9.8.017). The hybrid duplexes were built in B-type helical geometries, and relaxed with AMBER*. Initial Monte Carlo torsional samplings (MCMM) were performed to generate 1000 structures, which were individually minimized into local minima. The lowest energy structure of each simulation was used for the subsequent MD simulations. The MD simulations were performed at 300 K. The SHAKE all bonds to hydrogen setting was imposed in order to increase the time step to 2.2 fs, and an equilibrium time of 100 ps was used to stabilize the calculations. A multiple minimization of the 500 sample structures was performed to obtain a converged global minimum structure.The torsional energy profiles were calculated on the geometry optimized structures (DFT/B3LYP/6-31G**) by a series of fully relaxed coordinate scans of the dihedral angles φ and ψ. The bonds defining the φ and ψ dihedral angles are shown in Fig. 5. The computations were performed in MacroModel V10.4 using the OPLS-2005 force field21 applying the GB/SA solvation model. The torsional sampling was performed by 1° increments of each of the dihedral angles (from 0° to 360°) to obtain a total of 130321 structures per search. The collection of conformations was energy minimized by allowing all degrees of freedom except φ and ψ which were frozen, the relative energies of which were plotted in the (φ,ψ) space using isoenergy contours at 2 kJ mol−1 intervals.
Acknowledgements
The research was supported by The Danish Council for Independent Research | Natural Sciences (FNU).Notes and references
- M. Ahmadian and D. E. Bergstrom, in Modified Nucleosides, ed. P. Herdewijn, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 249–276 Search PubMed.
- L. C. Sowers, B. R. Shaw and W. D. Sedwick, Biochem. Biophys. Res. Commun., 1987, 148, 790–794 CrossRef CAS PubMed.
- P. P. Seth and E. E. Swayze, in Natural Products in Medicinal Chemistry, ed. S. Hanessian, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 403–440 Search PubMed.
- (a) B. C. Froehler, S. Wadwani, T. J. Terhorst and S. R. Gerrard, Tetrahedron Lett., 1992, 33, 5307–5310 CrossRef CAS; (b) J. Sági, A. Szemzö, K. Ébinger, A. Szabolcs, G. Sági, É. Ruff and L. Ötvös, Tetrahedron Lett., 1993, 34, 2191–2194 CrossRef; (c) T. W. Barnes and D. H. Turner, J. Am. Chem. Soc., 2001, 123, 4107–4118 CrossRef CAS PubMed.
- A. J. Gutierrez, T. J. Terhorst, M. D. Matteucci and B. C. Froehler, J. Am. Chem. Soc., 1994, 116, 5540–5544 CrossRef CAS.
- P. Kočalka, N. K. Andersen, F. Jensen and P. Nielsen, ChemBioChem, 2007, 8, 2106–2116 CrossRef PubMed.
- (a) N. K. Andersen, N. Chandak, L. Brulíková, P. Kumar, M. D. Jensen, F. Jensen, P. K. Sharma and P. Nielsen, Bioorg. Med. Chem., 2010, 18, 4702–4710 CrossRef CAS PubMed; (b) N. K. Andersen, H. Døssing, F. Jensen, B. Vester and P. Nielsen, J. Org. Chem., 2011, 76, 6177–6187 CrossRef CAS PubMed.
- M. E. Østergaard, P. Kumar, J. Nichols, A. Watt, P. K. Sharma, P. Nielsen and P. P. Seth, Nucleic Acid Ther., 2015, 25, 266–274 CrossRef PubMed.
- M. J. Robins and P. J. Barr, Tetrahedron Lett., 1981, 22, 421–424 CrossRef CAS.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- (a) P. Kumar, N. Chandak, P. Nielsen and P. K. Sharma, Bioorg. Med. Chem., 2012, 20, 3843–3849 CrossRef CAS PubMed; (b) P. Kumar, M. Hornum, L. J. Nielsen, G. Enderlin, N. K. Andersen, C. Len, G. Hervé, G. Sartori and P. Nielsen, J. Org. Chem., 2014, 79, 2854–2863 CrossRef CAS PubMed.
- (a) P. Yakovchuk, E. Protozanova and M. D. Frank-Kamenetskii, Nucleic Acids Res., 2006, 34, 564–574 CrossRef CAS PubMed; (b) M. Petersheim and D. H. Turner, Biochemistry, 1983, 22, 256–263 CrossRef CAS PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- (a) C. A. Hunter, J. Mol. Biol., 1993, 230, 1025–1054 CrossRef CAS PubMed; (b) R. Luo, H. S. R. Gilson, M. J. Potter and M. K. Gilson, Biophys. J., 2001, 80, 140–148 CrossRef CAS PubMed; (c) K. M. Guckian, B. A. Schweitzer, R. X.-F. Ren, C. J. Sheils, D. C. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 2000, 122, 2213–2222 CrossRef CAS PubMed; (d) R. A. Friedman and B. Honig, Biophys. J., 1995, 69, 1528–1535 CrossRef CAS PubMed.
- D. J. Hurley and Y. Tor, J. Am. Chem. Soc., 1998, 120, 2194–2195 CrossRef CAS.
- K. Kutonova, M. Trusova, P. Postnikov, V. Filimonov and J. Parello, Synthesis, 2013, 2706–2710 CAS.
- M. Hornum, P. Kumar, P. Podsiadly and P. Nielsen, J. Org. Chem., 2015, 9592–9602 CrossRef CAS PubMed.
- (a) L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998–15999 CrossRef CAS PubMed; (b) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923–8930 CrossRef CAS PubMed.
- (a) U. Pradere, V. Roy, T. R. McBrayer, R. F. Schinazi and L. A. Agrofoglio, Tetrahedron, 2008, 64, 9044–9051 CrossRef CAS; (b) A. Montagu, V. Roy, J. Balzarini, R. Snoeck, G. Andrei and L. A. Agrofoglio, Eur. J. Med. Chem., 2011, 46, 778–786 CrossRef CAS PubMed.
- G. R. Bishop and J. B. Chaires, in Curr. Protoc. Nucleic Acid Chem, ed. S. Beaucage, et al., 2003, ch. 7, unit 7.11 Search PubMed.
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed.
- D. Qiu, P. S. Shenkin, F. P. Hollinger and W. C. Still, J. Phys. Chem. A, 1997, 101, 3005–3014 CrossRef CAS.
- G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 19, 937–954 CrossRef CAS PubMed.
- (a) A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS; (b) S. Obika, D. Nanbu, Y. Hari, K. Morio, Y. In, T. Ishida and T. Imanishi, Tetrahedron Lett., 1997, 38, 8735–8738 CrossRef CAS.
- (a) T. Y. Shen, Angew. Chem., Int. Ed., 1970, 9, 678–688 CrossRef CAS PubMed; (b) P. Kielkowski, R. Pohl and M. Hocek, J. Org. Chem., 2011, 76, 3457–3462 CrossRef CAS PubMed; (c) P. Kielkowskia, H. Cahováa, R. Pohla and M. Hocek, Bioorg. Med. Chem., 2016, 24, 1268–1276 CrossRef PubMed.
- (a) P. Kumar, A. F. Sorinas, L. J. Nielsen, M. Slot, K. Skytte, A. Nielsen, M. Dalager, P. K. Sharma, B. Vester, M. Petersen and P. Nielsen, J. Org. Chem., 2014, 79, 8020–8030 CrossRef CAS PubMed; (b) M. Dalager, N. K. Andersen, P. Kumar, P. Nielsen and P. K. Sharma, Org. Biomol. Chem., 2015, 13, 7040–7049 RSC; (c) P. Kumar, P. K. Sharma, J. Hansen, L. Jedinak, C. Reslow-Jacobsen, M. Hornum and P. Nielsen, Bioorg. Med. Chem., 2016, 24, 742–749 CrossRef CAS PubMed.
- (a) K. M. Guckian, B. A. Schweitzer, R. X.-F. Ren, C. J. Sheils, P. L. Paris, D. C. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 1996, 118, 8182–8183 CrossRef CAS PubMed; (b) T. W. Kim and E. T. Kool, J. Org. Chem., 2005, 70, 2048–2053 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected NMR spectra and MALDI-TOF of oligonucleotides. See DOI: 10.1039/c6ob00609d |
| This journal is © The Royal Society of Chemistry 2016 |